

Abstract
Single intradermal or intramuscular inoculations of GM-CSF DNA with the DNA prime for a simian-human immunodeficiency virus (SHIV)-89.6 vaccine, which consists of DNA priming followed by modified vaccinia Ankara (MVA) boosting, increased protection of both the blood and intestines against the acute phase of an intrarectal SHIV-89.6P challenge. GM-CSF appeared to contribute to protection by enhancing two antibody responses: the avidity maturation of anti-Env IgG in blood (p=<0.01) and the presence of long lasting anti-viral IgA in rectal secretions (p<0.01). The avidity of anti-Env IgG showed strong correlations with protection both pre and post challenge. Animals with the highest avidity anti-Env Ab had 1000-fold reductions in peak viremia over those with the lowest avidity anti-Env Ab. The enhanced IgA response was associated with the best protection, but did not achieve significance.
Keywords: Immunodeficiency virus, Vaccine, GM-CSF Adjuvant, Ab Avidity, Rectal IgA
Introduction
HIV/AIDS, currently the leading cause of death in Africa and the 4th leading cause of death worldwide, is projected to join heart disease and stroke as the three top causes of death worldwide by 2030 (Mathers and Loncar, 2006). Antiretroviral drugs, although effective for most patients, are available to only a fraction of infected humans, require treatment for life and have side effects. The solution to the pandemic is a vaccine capable of both preventing disease and limiting transmission. The current pandemic is spread primarily by intravenous drug use and sexual intercourse, with heterosexual intercourse being the dominant method for transmission worldwide (UNAIDS and WHO, 2006). Therefore, an effective vaccine will need to protect the mucosal surfaces of the vagina and rectum as well as blood against viral transmission.
Here we report the ability of GM-CSF DNA, delivered with the DNA prime of a vaccine that consists of DNA priming and modified vaccinia Ankara (MVA) boosting (DNA/MVA vaccine) to enhance protection of the blood as well as the intestines against the acute phase of a SHIV-89.6P infection. GM-CSF was tested for adjuvant activity because of its demonstrated ability to enhance the protective efficacy of cancer vaccines (Borrello and Pardoll, 2002; Dranoff et al., 1993) and its early success as an adjuvant for DNA vaccines in mouse models (Barouch et al., 2002; Lee, Cho, and Sung, 1998; Xiang and Ertl, 1995). GM-CSF is a critical factor in the development and differentiation of dendritic cells and monocytes (Inaba et al., 1992). Both of these cell types are “professional antigen presenting cells” capable of initiating immune responses by presenting antigens to the immune system (Banchereau and Steinman, 1998).
The study was done using SHIV-89.6 sequences for immunization (Reimann et al., 1996b) and its highly pathogenic neutralization-escape variant, SHIV-89.6P, for challenge (Karlsson et al., 1997; Reimann et al., 1996a). Both the DNA and MVA immunogens expressed SHIV-89.6 virus-like-particles (VLPs). These particles contained SIV structural (Gag) and enzymatic proteins (protease and reverse transcriptase) surrounded by a viral membrane displaying a native form of the 89.6 HIV-1 envelope glycoprotein (Env). Tests for protection focused on peak viremia, because peak titers of virus show greater differences than set point levels of virus (many of which are below the level of detection) for SHIV-89.6P challenges in our DNA/MVA vaccinated macaques (Amara et al., 2001). In SIV/macaque models early viral replication is a critical determinant of the virulence of an infection (Lifson et al., 1997) and peak viremia shows a better correlation than set point viremia with progression to disease within one year (p=0.03) (Smith et al., 1999). Thus, for SHIV-89.6P infections that kill the vast majority of animals within one year, peak viremia is a relevant correlate for protection.
In a prior study in the SHIV-89.6/SHIV-89.6P model, the use of GM-CSF as an adjuvant reduced peak viremia by 4-fold and increased the stringency of control during the chronic phase of infection as evidenced by reduced frequencies for brief appearances of re-emergent virus (p<0.01) (Robinson et al., 2006). In this study, GM-CSF appeared to have enhanced the elicitation of neutralizing Ab for SHIV-89.6P by enhancing the avidity maturation of anti-Env Ab. The current study was undertaken to further study the effects of GM-CSF on the avidity maturation of anti-Env Ab and the contribution of the avidity of anti-Env IgG to protection. At the time of challenge, assays were extended to include tests for mucosal immune responses and protection of mucosal CD4 T cells.
Our results reveal a single inoculation of GM-CSF DNA consistently enhancing the avidity maturation of the anti-Env Ab response. They also reveal a strong direct correlation between the avidity of anti-Env IgG and protection against peak viremia (p<0.01). Interestingly, the mucosal studies revealed GM-CSF also supporting the elicitation of long lasting anti-viral IgA in rectal secretions (p<0.01). Anti-viral IgA was present in the secretions of animals with the lowest peak viremias, but did not show a significant correlation with protection.
Results
Immunogens and trial design
The ability of GM-CSF DNA to serve as an adjuvant for the DNA prime of a DNA/MVA vaccine was tested both by co-inoculating a dedicated GM-CSF-expressing plasmid with a VLP expressing SHIV-89.6 vaccine DNA (GM-CSF DNA in trans) and by expressing GM-CSF DNA in the vaccine DNA in the place of nef (GM-CSF DNA in cis) (Fig. 1A). In transient transfections, the dedicated GM-CSF expression vector expressed 15-times higher levels of GM-CSF than the GM-CSF expressed in cis (Fig. 1A). However, the cis expression achieved levels found to be effective in GM-CSF-adjuvanted cancer vaccines (∼1 μg per 106 expressing cells) (Borrello and Pardoll, 2002) and was directly targeted to cells expressing the vaccine antigen.
Fig. 1.

Immunogens, immunization schedules, and the elicitation of similar protection in groups receiving GM-CSF DNA in cis and trans. (A) Expression cassettes for the DNA prime. The schematics indicate the proteins expressed by the SHIV-89.6 vaccine. CMVIE and IA, the CMV immediate early promoter and intron A; gag and RT, SIV239 genes for group specific antigen and reverse transcriptase; X, inactivating point mutation in PR; tat, vpu, rev, and env; HIV-1 89.6 auxiliary and envelope genes; BGHpA, the bovine growth hormone polyadenylation sequence. Levels of expressed GM-CSF are for transiently transfected 293T cells. (B) Immunization and challenge schedule. Immunizations in trans used DNA/89.6.VLP plus DNA/GM-CSF. Immunizations in cis used DNA/89.6.VLP.GM-CSF. The challenge was an intra-rectal challenge with SHIV-89.6P (see Materials and Methods for details). (C) Similar protection in plus and minus GM-CSF groups. Boxplots provide a graphic summary for peak viral loads in the trial groups. The white line within the box is the median, the upper and lower limits of the boxes indicate the 75th and 25th percentiles respectively. Minimum and maximum values are indicated by the upper and lower brackets. Vaccine groups and the number of animals in each group (n) are indicated below the box plots.
A single immunization with the DNA vaccine in the presence or absence of GM-CSF DNA was followed by three MVA boosts (Fig. 1B). For animals receiving GM-CSF DNA in trans, a mixture of equal amounts of DNA/89.6.VLP and DNA/GM-CSF (1.5 mg of each) were delivered intradermally (i.d.) to achieve at least some co-localization of cells expressing the vaccine and adjuvant. Inoculations of GM-CSF DNA in cis were conducted both i.d. and intramuscularly (i.m.) by inoculating 3 mg of the GM-CSF-expressing vaccine DNA (DNA/89.6.VLP.GM-CSF). Three boosts with 108 pfu of MVA/89.6 (Amara et al., 2001) were delivered by the same route used for the prime at weeks 8, 24 and 40 (Fig. 1B). Challenges were conducted intra-rectally with SHIV-89.6P at 32 weeks post the last MVA inoculation in animals receiving no GM-CSF DNA or inoculations of the GM-CSF DNA in trans, and at 60 weeks post the last MVA immunization in animals given the GM-CSF DNA in cis (Fig. 1B). Unvaccinated control animals were included with both challenges.
Groups receiving GM-CSF DNA in both cis and trans showed 10-fold lower levels of peak viremia than the unadjuvanted vaccine group following the SHIV-89.6P challenge (Fig. 1C). No differences were found in the extent of viral control in groups that had received GM-CSF in cis or in trans (Fig. 1C). Therefore, to gain statistical power, immune response data for all of the GM-CSF-adjuvanted animals are considered together.
Protection
The GM-CSF-adjuvanted group had better control of peak viremia and a more rapid reduction of viremia to the background for detection than the non-adjuvanted group (Fig. 2A). At peak viremia, the adjuvanted group had reduced viremia by 10-fold over that in the nonadjuvanted group (p=0.01), a reduction that was 30-fold over that in the unvaccinated controls (p<0.001) (Fig 2E). By 5 weeks post challenge, 6 out of the 16 GM-CSF-adjuvanted animals had viral loads below 100 copies per ml of plasma, whereas none of the non-adjuvanted animals had achieved this level of control. By 12 weeks post challenge, the geometric mean viremia for the adjuvanted animals was at our level of detection (100 copies per ml) and the mean for nonadjuvanted animals was approaching our level of detection (260 copies per ml), whereas the mean for the unvaccinated controls remained high (5.2×104). Shedding of virus in the stool was monitored for the challenge at 100 weeks, a challenge which included only adjuvanted animals and unvaccinated controls. These analyses revealed that the adjuvanted vaccine also had reduced viral shedding (P=0.008) (Fig. 2B).
Fig. 2.

Post challenge control of SHIV-89.6P viral RNA and protection of CD4 T cells. (A) and (B) Temporal titers of viral RNA in the blood and stool are presented as geometric means ± standard deviations. Stool samples are for 8 macaques vaccinated in the presence of GM-CSF and 4 control macaques. Data are not available for the no GM-CSF group because stool samples were first harvested at the 100 week challenge, a time when only control and plus GM-CSF animals underwent challenge. (C) and (D) Temporal frequencies of CD4 T cells as a % of total CD3 cells in blood and colorectal biopsies are presented as averages ± standard deviations. Data for colorectal CD4 T cells are available for only part of the animals in each group at the different time points due to restrictions on the frequency of biopsies and the changing of harvest times as data were obtained for temporal responses. Symbols for the different groups are given in (A). Pre, samples taken 2-3 weeks prior to challenge but plotted at the time of challenge. (E) and (F). Statistical analyses for protection against peak viremia and preservation of colorectal CD4 T cells. The statistical analysis for colorectal CD4 T cells was done for 8 weeks post challenge, a time for which more data were available than for week 12. Explanation of the boxplots is given in Figure 1.
The GM-CSF adjuvant also showed increased protection against CD4 T cell loss, with this achieving significance in the intestines (p=0.05) but not the blood (Fig. 2C D and F). At their lowest point, colorectal CD4 T cells were reduced to 18% of CD3 T cells in the adjuvanted group, 13% of CD3 T cells in the non-adjuvanted group and 3% of CD3 T cells in the unvaccinated group. These levels represented 42%, 31% and 5% of the normal levels of colorectal CD4 T cells for the three groups respectively. By 12 weeks post challenge, CD4 T cells had recovered to 36%, 24% and 8% of CD3 T cells (86%, 57% and 19% of normal colorectal levels) in the three trial groups, respectively. In the blood, CD4 T cells were reduced to 36% of total CD3 cells, or 68% of their normal levels in the two vaccinated groups whereas reduction was to 8% of CD3 T cells, or 15% of normal levels in the unvaccinated animals. By 12 weeks post challenge, blood had recovered 84% of the normal level of CD4 T cells in the GM-CSF-adjuvanted group, 66% of the normal level in the non-adjuvanted vaccine group, and only 24% of the normal level in the unvaccinated controls. The rapid loss of CD4 T cells in both blood and intestines in the unvaccinated group likely reflects the ability of SHIV-89.6P to use both CXCR-4 and CCR-5 co-receptors. Naïve CD4 T cells bearing CXCR-4 (Zhang et al., 2000) predominate in the blood, whereas memoryCD4 T cells displaying CCR-5 as well as CXCR-4 predominate in the intestines (Pitcher et al., 2002).
Serum IgG Antibody responses
The GM-CSF adjuvant did not affect the titers of elicited IgG for the 89.6 or 89.6P Envs in serum (Fig. 3A). Binding Ab was first detected for the 89.6 Env following the 1st MVA boost; and for the 89.6P Env, after the 2nd MVA boost. The titers of binding Ab rose and fell with boosts, with each boost increasing peak titers. Post challenge, both adjuvanted and non-adjuvanted vaccine groups exhibited strong anamnestic expansions of anti-Env IgG. This expansion was highest in the non-GM-CSF group, a finding consistent with the higher titers of post challenge virus in this group. Binding Ab also appeared in the serum of the unvaccinated controls, but at peak viremia had 1000-times lower titers than in the vaccinated animals (Fig. 3A).
Fig. 3.
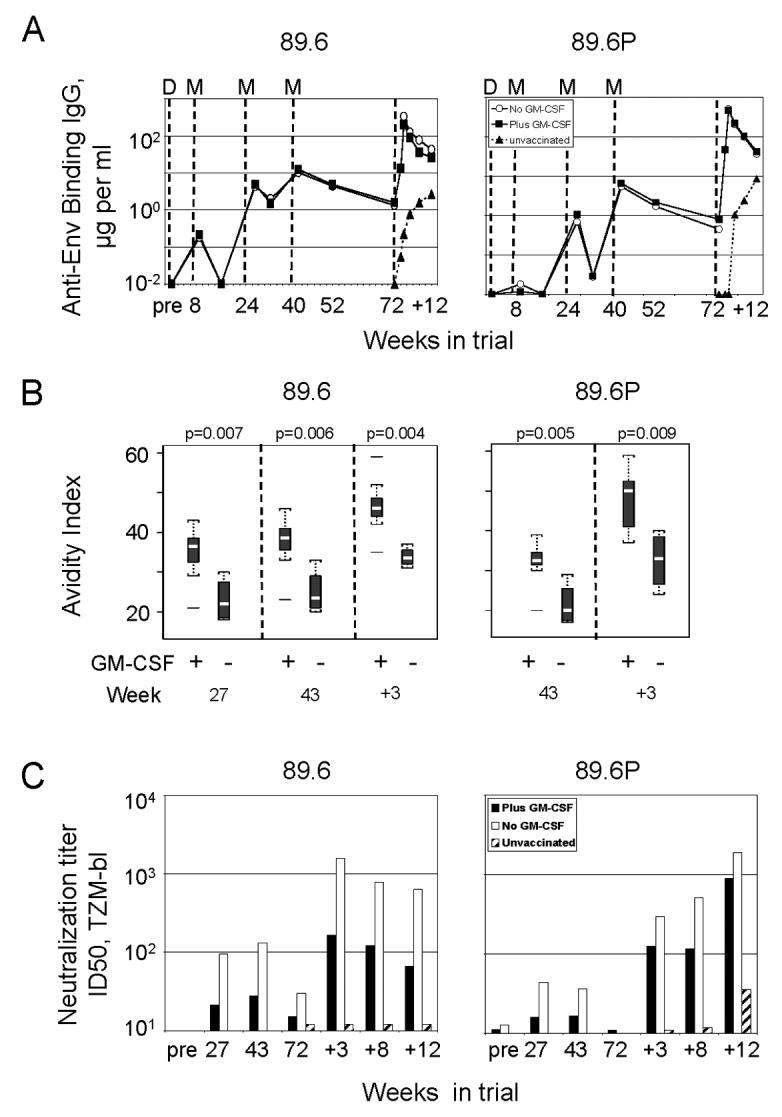
Post challenge Ab responses in the blood. The left column shows data for the 89.6 Env and the right column, for the 89.6P Env. (A) Geometric mean titers for anti-Env binding IgG determined using ELISAs. Vertical dashed lines indicate the times of D (DNA) and M (MVA) immunizations and 89.6P (SHIV-89.6P) challenge. Pre, samples taken prior to immunization but plotted at the time of the 1st immunization. Weeks post challenge are indicated by “+” preceding the week number. (B) Boxplots showing avidity indices for anti-Env binding Ab harvested at different times in the trial. See legend to Fig. 1 for explanation of graphic. Horizontal lines above or below plots indicate outliers. (C) Geometric mean titers for neutralizing activity for SHIV-89.6 and SHIV-89.6P. Titers are serum dilutions at which relative luminescence units were reduced 50% compared to virus control wells.
Although GM-CSF did not serve as an adjuvant for higher titers of anti-Env binding Ab, it did serve as an adjuvant for the avidity maturation of anti-Env Ab. The avidity of the elicited anti-Env IgG was consistently higher as it rose over time for the plus GM-CSF than the minus GM-CSF group (p=0.004 to p=0.009) (Fig. 3B). The higher avidity was seen for both the 89.6 and 89.6P Envs. Post challenge, the range of values observed for the 89.6P Env broadened, a finding that would be consistent with the challenge boosting the primed Ab responses as well as eliciting new Ab that had not yet undergone avidity maturation.
Serum neutralizing Ab for both SHIV-89.6 and SHIV-89.6P was higher in the nonadjuvanted than the adjuvanted group (Fig. 3C). Neutralizing Ab for SHIV-89.6 and SHIV-89.6P was 1st detected after the 2nd MVA boost and underwent a further boost following the challenge. The unvaccinated controls showed much lower titers of post challenge neutralizing Ab than the vaccinated groups, with neutralizing activity not appearing in the controls until 12 weeks post challenge.
IgA antibody responses
Unexpectedly, the GM-CSF-adjuvanted, but not the nonadjuvanted group, had a long-lived anti-SIV IgA response in rectal secretions (Fig. 4). At the time of challenge, 8 of the 16 GM-CSF-adjuvanted animals, but none of the non-adjuvanted animals, had anti-SIV IgA in rectal secretions (Fig. 4B). The anti-SIV IgA was not present in the blood of GM-CSF adjuvanted animals at the time of challenge, indicating that the specific IgA in the secretions had originated from the rectal mucosa rather than from serum transudate or blood contaminant (Fig. 4A). The anti-SIV IgA was likely against SIV Gag/Pol because the SIV lysate contained very limited levels of Env. The anti-SIV IgA response was present in secretions collected at 32 (GM-CSF delivered in trans) and 60 weeks (GM-CSF delivered in cis) after the last MVA boost indicating that the GM-CSF adjuvanted vaccine had supported a long lasting IgA response.
Fig. 4.
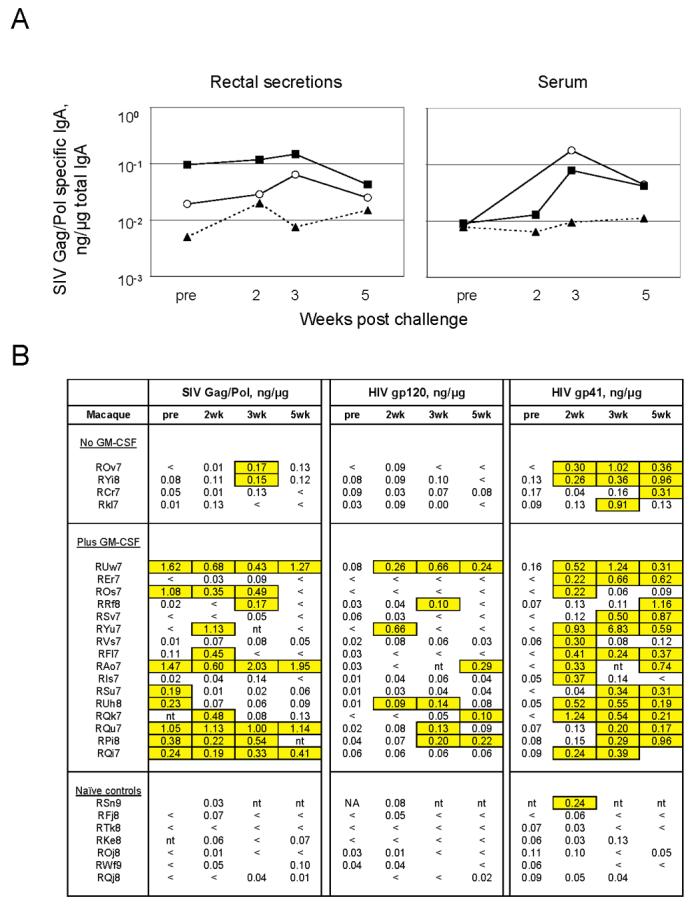
Temporal anti-viral IgA in rectal secretions and serum. Data are the specific activity of an anti-viral response (ng of anti-viral IgA per μg of total IgA). (A) Temporal geometric means for anti-SIV Gag/Pol IgA for the different groups. The left hand panel shows data for rectal secretions and the right hand panel, for serum. Closed square, plus GM-CSF; Open circle, minus GM-CSF; Closed triangle, unvaccinated (B) Temporal data for individual animals for anti-Gag/Pol (SIV-239) IgA, anti-gp120 (MN) IgA and anti-gp41 (MN) IgA in rectal secretions. Significant values (mean plus 3 SD of 15 naïve macaques) are highlighted and boxed. One outlier in the unvaccinated controls with constitutive high levels of IgA in rectal secretions is not included in the data set. <, values below the background for detection (0.005 ng of IgA/μg of total IgA); pre, samples taken 2-3 weeks prior to challenge but plotted at the time of challenge; wk, week ; nt, no test; blanks, no sample.
Post challenge, the GM-CSF-adjuvanted group continued to have a good frequency of animals with anti-SIV IgA in rectal secretions. By two weeks post challenge anti Mn gp120 IgA had appeared in the rectal secretions of the GM-CSF-adjuvanted animals. This response was detected in 8 of the 16 animals in the adjuvanted group, but none of the 4 in the non-adjuvanted group. Post challenge, anamestic responses for Anti-SIV IgA and anti-Mn gp120 IgA also occurred in the bloods of the vaccinated animals (Fig. 4A and data not shown). Following the challenge, an anti-gp41 IgA response appeared in all groups, including one unvaccinated control, suggesting that gp41 is a particularly good target for anti-viral IgA. When both pre and post challenge IgA responses for SIV and gp120 were considered, the difference in the elicitation of IgA between the adjuvanted and unadjuvanted vaccine groups was highly significant (p=0.003).
The IgA antibodies were measured using the specific activity of the elicited IgA (ng of SIV-specific IgA/total IgA) to normalize for differences in the recovery of IgA in rectal secretions and minimize artifacts that can occur in tests for IgA. Data on the presence of anti-viral IgA in rectal secretions are not available for the immunization phase of the trial because these assays were not undertaken until the time of challenge.
T cell responses
Intracellular cytokine (ICS) and tetramer analyses revealed overall similar patterns of expansion and contraction for responding CD4 and CD8 T cells in the blood from the plus and minus-GM-CSF groups (Fig 5A and 6A). In contrast to the sequential increases in titers of anti-Env IgG with the three MVA boosts, both CD8 and CD4 T cell responses achieved their highest values following the 1st MVA boost. Following the 2nd and 3rd MVA boosts, the CD8 responses underwent some expansion, whereas the CD4 responses underwent undetectable to minimal increases. Post challenge, the frequencies of anti-viral CD8 and CD4 T cells rapidly expanded, achieving frequencies approximating their original peak. By 5 weeks post challenge these responses were contracting, likely in response to reduced viral loads. Anti-viral CD8 and CD4 T cells were present at much lower levels in the unvaccinated controls than in the vaccinated groups following challenge.
Fig. 5.
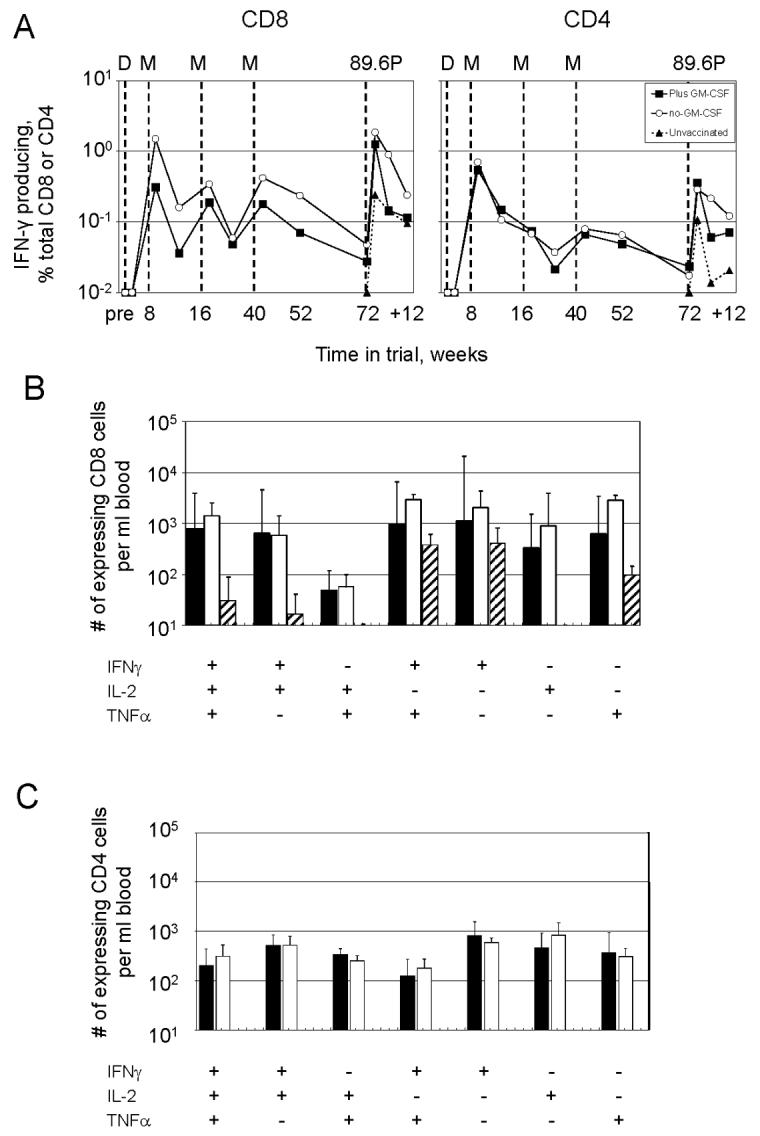
Analyses for responding T cells using ICS assays. (A) Temporal CD8 and CD4 T cell measured as IFN-γ producing cells as a % of total CD8 or CD4 T cells. The times of immunization and challenge are indicated by vertical dotted lines (see legend to Fig. 3 for detail). Pre, samples taken prior to immunization but plotted at the time of the 1st immunization. Weeks indicated with a “+” are weeks post challenge. (B) and (C) Characterization of cytokine co-production profiles at 3 weeks post challenge for CD8 T cells and CD4 T cells for the different trial groups. Data are expressed as number of expressing cells per ml of blood. Filled bar, plus GM-CSF group; open bar, minus GM-CSF group; and hatched bar, unvaccinated controls. Sufficient CD4 T cells were not present in the unvaccinated controls for analyses.
Fig. 6.
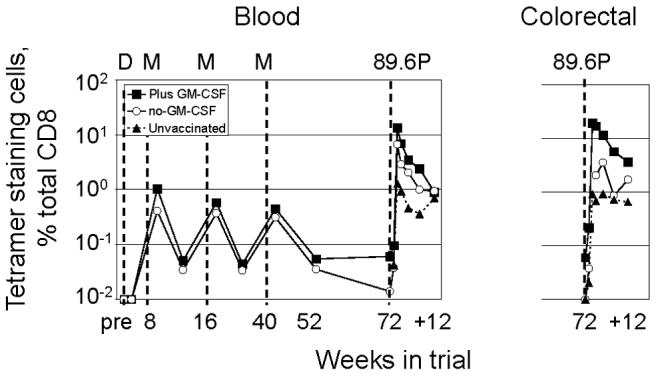
Temporal CD8 T cells in blood and rectal biopsies for the immunodominant Gag-CM9 epitope. (A) Temporal frequency of tetramer staining cells throughout the trial. Weeks indicated with a “+” are weeks post challenge. Pre, samples taken prior to immunization but plotted at the time of the 1st immunization. Analyses for colorectal cells were initiated at the time of challenge and are not available for the vaccination period of the trial.
Multicolor flow cytometry conducted on responding T cells at 3 weeks post challenge revealed no differences in cytokine co-expression patterns between the adjuvanted and nonadjuvanted vaccine groups (Fig. 5 B and C). Both vaccine groups differed from the unvaccinated group in having much higher proportions of CD8 T cells co-expressing IFN-γ, IL-2, and TNF-α, or co-expressing IFN-γ and IL-2. In the unvaccinated group, the predominant responding cells expressed IFN-γ and TNF-α, only IFN-γ or only TNF-α (Fig. 5B). Expression patterns for anti-viral CD4 T cells were also similar in the adjuvanted and non-adjuvanted vaccine groups (Fig. 5D). Sufficient anti-viral CD4 T cells were not available for analyses for post challenge co-expression profiles in the unvaccinated controls.
Post challenge, the GM-CSF-adjuvanted group had as high frequencies of tetramer-staining cells in colorectal biopsies as in the blood (Fig. 6). The responding cells in the intestines showed the same temporal expansion as those in blood. During the 1st week post challenge, limited to no expansion of CD8 T cells was observed in the blood, or the intestines. However, by 2 weeks post challenge, rapid expansions had taken place at all 3 of these sites (Fig. 6). In contrast, in the no-GM-CSF group, the responding cells in the intestines showed a slower temporal appearance than in blood, and were present at lower frequencies (Fig. 6). Statistics were not done on the relative appearance of responding cells in the intestines in the GM-CSF-adjuvanted and nonadjuvanted groups because of small sample sizes, which were smaller than group sizes due to restrictions on the frequency of biopsy collection and the adjustment of sampling times as information was gained on the tempo of mucosal responses. Data on the relative presence of tetramer-staining cells in colorectal samples from adjuvanted and non-adjuvanted groups are not available for the immunization phase of the trial because these assays were not instituted until the time of challenge.
Correlations
Correlations were undertaken to determine the role of GM-CSF adjuvanted (Fig. 7 A, B) as well as non-GM-CSF adjuvanted immune responses in protection (Fig. 7, C). A response was considered to be GM-CSF adjuvanted if it had shown a significant difference between the GM-CSF and non-GM-CSF groups during the immunization phase of the trial. Two antibody responses met these criteria, the avidity of anti-Env IgG and the presence of anti-viral IgA in rectal secretions (Fig. 3 and 4). Correlations for protection utilized data from the vaccinated, but not unvaccinated groups.
Fig. 7.
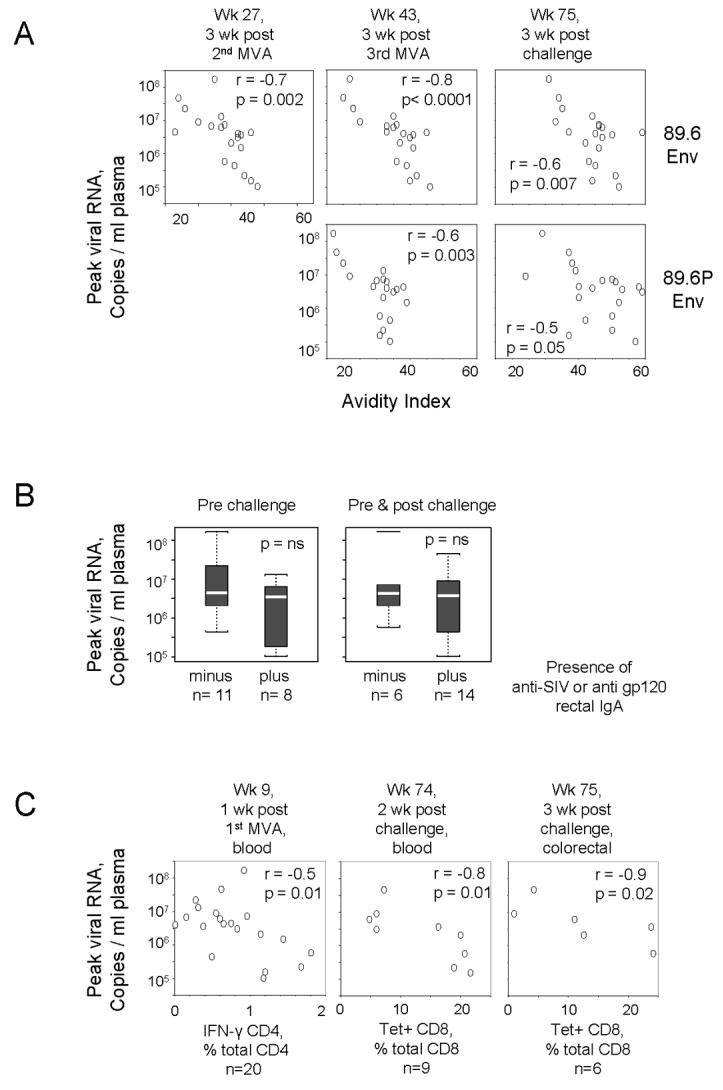
Correlations between adjuvanted and non-adjuvanted vaccine-elicited responses and protection. (A) and (B) Correlations between GM-CSF-adjuvanted Ab responses and protection. (C) Correlations between non-adjuvanted T cell responses and protection. All correlations use reductions in peak viremia as a correlate for protection. The time in the trial for each correlation is given above its graphic. (A) Correlation between avidity of anti-Env IgG with protection. The Env being tested as a target for anti-Env IgG binding is shown on the right. Note the increasing avidity of the anti-Env IgG response with time in the trial. (B) Test for a correlation between the presence of anti-viral IgA in rectal secretions and protection. The plus and minus groups were defined based on the presence of anti-SIV IgA or anti-gp120 IgA. See legend to Fig. 1 for explanation of the graphic. C. Correlations between T cells responses and protection. The type of T cell being used for the correlation is shown below its graphic. 20 samples were used in all tests in (A). For (B) and (C) the number of samples (n) in each test are indicated below the graphics. Wk, week; Tet+, tetramer staining;
The pre-challenge as well as post challenge avidity of anti-Env IgG in serum showed consistent correlations with protection (Fig. 7A). These correlations revealed animals with the highest avidity anti-Env Ab having 1000-times lower titers of virus at peak virema than those with the lowest avidity anti-Env Ab. Pre-challenge, r values ranged from −0.6 to −0.7 and p values from <0.0001 to 0.003. Post challenge, the r and p values for the challenge Env (89.6P), but not the immunogen Env (89.6) decreased, consistent with the lowering of the avidity due to the challenge Env eliciting new Ab as well as expanding the primed Ab.
Correlations for the presence of anti-viral IgA in rectal secretions and protection were done using data for anti-SIV IgA and anti-Mn gp120, because these specificities distinguished the vaccine and control groups. These correlations were done for the presence of anti-SIV IgA pre challenge and for the presence of anti-SIV or anti-gp120 IgA either pre or post challenge. The anti-viral IgA response, which was scored as present or absent, did not show a significant correlation with protection. However, animals with the lowest post challenge peak viral loads were uniformly IgA positive.
Three other immune responses, which were not affected by GM-CSF, also showed correlations with protection (Fig. 7C). The 1st of these, the frequency of IFN-γ producing anti-viral CD4 T cells, showed a weak correlation with protection (r=−0.5, p=0.01) at one week following the 1st MVA boost. The other two correlations were observed post challenge. At 2 weeks post challenge, tetramer staining cells in the blood showed a strong correlation with protection (r=−0.8, p=0.01) and at 3 weeks post challenge, colorectal tetramer staining cells showed a strong correlation with protection (r=−0.9, p=0.02).
Discussion
Our studies reveal co-delivery of GM-CSF DNA with the DNA prime of a DNA/MVA vaccine enhancing protection against the acute phase of a SHIV-89.6P challenge. Analyses of immune responses during the vaccine and challenge phases of the trial revealed Ab, CD4 T cells and CD8 T cells all contributing to protection against the acute infection. The adjuvant effect of GM-CSF enhanced the protective efficacy of the elicited Ab, but not of the elicited T cells.
The most important protective effect of GM-CSF on the elicited Ab was the enhancement of the avidity of anti-Env binding Ab. Animals with high avidity anti-Env Ab had up to 1000-times lower titers of virus at peak viremia than animals with low avidity Ab responses. The avidity indices for anti-Env Ab response underwent a gradual increase throughout the trial from indices of less than 20 to more than 60. At each time point, the anti-Env IgG in the GM-CSF adjuvanted group had indices ∼10 points higher than the indices in the non-adjuvanted animals. Throughout the trial, the avidity indices of the vaccine-elicited anti-Env IgG showed strong inverse correlations with peak viral loads (r values as high as −0.8 and p values as low as <0.0001).
The avidity-mediated protection could reflect a number of non-neutralizing Ab activities. Ab bound to virus could have mediated complement (C') dependent virolysis and opsonization (Montefiori, 1997). Ab bound to infected cells could have led to cell death by Fc receptor-mediated functions such as complement mediated lysis and Ab dependent cellular cytotoxicity (ADCC) (Gomez-Roman et al., 2005). Both virolysis by C' lysis and killing of infected cells by ADCC contribute to the control of HIV in acutely infected humans (Connick et al., 1996; Forthal et al., 2007; Huber et al., 2006). The importance of avidity has long been recognized for neutralizing monoclonals (Parren and Burton, 2001; Parren et al., 1998). This study extends these findings to non-neutralizing mechanisms of Ab-mediated control of immunodeficiency virus infections
Surprisingly, the titers of neutralizing Ab did not correlate with protection. Neutralizing Ab was not present at sufficient titers pre-challenge to prevent the challenge infection. However, at the time of challenge, it was present at different titers in different animals. Why did differences in avidity but not differences in neutralizing Ab correlate with differences in peak viral loads? Immunodeficiency viruses are known to rapidly escape from neutralizing Ab that is present at insufficient titers to completely block an infection (Parren et al., 1999). Could avidity have had a greater effect on viral control than neutralizing Ab by virtue of its being harder to escape than neutralizing Ab? Such could reflect binding Ab having more targets on the surface of a virus than neutralizing Ab, which specifically targets entry functions.
Our detection of avidity playing an important role in Ab-mediated protection likely reflects a number of our experimental conditions. Our immunization schedule fostered avidity maturation by providing pulses of antigen at 4 month intervals. We used native forms of Env for our immunogens and our binding assays to elicit and test for Ab recognizing the forms of Env on virions and infected cells. We consider the use of native forms of Env to have been critical because non-neutralizing mechanisms of Ab-mediated protection depend on Ab recognizing the forms of Env that are present on virus and infected cells.
Unexpectedly, the GM-CSF adjuvant also enhanced the levels of anti-viral IgA in rectal secretions. This enhanced IgA response showed a trend towards better protection, a trend that did not achieve significance in our data set. The presence of anti SIV IgA in rectal secretions at the time of challenge (32 to 60 weeks after the last immunization) suggests that the GM-CSFadjvuanted vaccine had established long-lived IgA secreting cells in the rectal mucosa. In general, parenteral immunizations with inactivated or replication-defective vaccines have been poor at establishing IgA-secreting plasma cells in the mucosa (Neutra and Kozlowski, 2006). Thus, the identification of GM-CSF as an adjuvant for fostering rectal IgA responses may have considerable use for the development of vaccines that protect mucosal surfaces.
A single inoculation of GM-CSF DNA with the single DNA prime enhanced both the avidity maturation of the anti-Env IgG response and the presence of mucosal IgA. These findings imply that the GM-CSF DNA, delivered at the time of the DNA prime, initiated an immune response that enhanced both the avidity maturation of Ab and mucosal immune responses. GM-CSF is well known for its ability to support the expansion and differentiation of dendritic cells and monocytes (Inaba et al., 1992). GM-CSF is also known to stimulate the production of IL-6(Evans et al., 1998). In mouse studies, the use of GM-CSF as an adjuvant has consistently enhanced Ab and CD4 T cell responses ( Xiang and Ertl, 1995; Geissler et al., 1997; Kamath et al., 1999; Lee, Cho, and Sung, 1998; Pasquini et al., 1997; Sin et al., 1998; Weiss et al., 1998; Barouch et al., 2002). In vaccine studies in primates, it has affected the height of Ab responses(Kumar et al., 2002; Rogers et al., 2002). In a study in humans, in which Ab responses were not measured, it appeared to reduce CD8 T cell responses(Wang et al., 2005). We are the first to have recognized its potential to foster the avidity maturation of Ab responses (see also Robinson et al., 2006), and this is the 1st study suggesting that GM-CSF can foster long-lived colorectal IgA responses.
GM-CSF likely enhanced the avidity maturation of Ab responses by enhancing germinal center reaction. Interestingly, IL-6 influences germinal center development(Kopf et al., 1998) and paracrine IL-6 can promote B cell growth and differentiation in germinal centers(Burdin et al., 1996). Thus the known ability of GM-CSF to stimulate Il-6 production could affect its ability to enhance avidity maturation of anti-Env Ab. As for enhancing colorectal IgA responses, recent studies in mice show blood monocytes preferentially giving rise to mucosal dendritic cells (Varol et al., 2007). In the macaque model, GM-CSF expands and matures blood monocytes. By analogy to the mouse model, these monocytes could preferentially become mucosal dendritic cells (work in progress). Studies in mice also show IL-6 stimulating the presence of IgA-secreting B cells (Mora et al., 2006). Thus GM-CSF could enhance the presence of mucosal IgA secreting cells by enhancing the presence of mucosal dendritic cells as well as by providing IL-6 for the differentiation of IgA-secreting B cells.
The temporal appearance of binding and neutralizing Ab was more similar in the plus and minus GM-CSF groups in this study than in our prior GM-CSF study(Robinson et al., 2006). In the prior study, both binding and neutralizing Ab appeared in the GM-CSF-adjuvanted group prior to its appearance in the non-adjuvanted group. The current study was designed to enhance Ab responses and used a DNA vaccine that expressed virus like particles (VLP) and a vaccine regimen that included 3 MVA boosts. The earlier study used a DNA that expressed a plasma membrane form of Env and a regimen with only a single MVA boost. The VLP-expressed Env plus the increased number of MVA boosts enhanced the elicitation of Ab by about 100-fold, and likely minimized effects of GM-CSF on the temporal appearance of anti-Env Ab. However, in both studies, GM-CSF enhanced the avidity of the anti-Env Ab response. The fact that enhanced avidity did not enhance neutralizing Ab in this study likely reflected the elicited avidity in both groups being sufficient for neutralizing 89.6 and 89.6P. Our hope had been that the avidities we would achieve in this study would extend the neutralizing activity to incident isolates. This hope was not realized.
Two non-adjuvanted T cell responses also showed correlations with protection against the acute phase of protection. These included a weak correlation with the magnitude of the CD4 T cell response measured as IFN-γ producing cells at one week post the 1st MVA boost and strong correlations between the heights of tetramer staining CD8 T cells in the blood and intestines early post challenge. Thus, all arms of the adaptive immune response contributed to protection: the adjuvanted Ab response and the non-adjuvanted CD4 and CD8 T cell responses. Of these, only the adjuvanted Ab response showed a strong pre-challenge correlation with protection.
In conclusion, our studies reveal a remarkable ability of GM-CSF DNA, when used as an adjuvant for the DNA prime of a DNA/MVA vaccine, to enhance the avidity maturation of anti-Env IgG and increase the presence of antiviral IgA in rectal secretions. Although the presence of rectal IgA was associated with the best viral control, this trend did not achieve significance and will require additional studies to clearly define the role of anti-viral IgA in protection. The strong correlation of the avidity of anti-env IgG with protection against peak viremia did achieve significance. Peak viremia was reduced by 1000-fold in animals with the highest avidity anti-Env Ab.
Materials and Methods
Vaccines
DNA/SHIV.89.6.VLP was constructed from SHIV-89.6 sequences (kindly provided by J. Sodroski) to express SIV239 gag/pol sequences and HIV-1 89.6 env, tat, rev, and vpu sequences (Fig. 1A). The integrase region of pol was deleted and an inactivating point mutation was added to the active site in protease (D25A) to achieve the production of VLPs rather than the formation of intracellular aggregates by premature cleavage of over-expressed Gag (Smith et al., 2004). Two DNA/89.6VLP constructs were made, one with a full length Env and one with a cytoplasmic tail truncated Env that enhances the exposure of conserved targets for neutralization in the ectodomain (Edwards et al., 2002). The truncation was encoded by a frameshift mutation at amino acid 706 in the cytoplasmic domain (Edwards et al., 2002). The pGA1 expression cassette was used for constructs (Fig. 1A) (Genbank AF424297). Rhesus GMCSF was expressed either in a dedicated DNA, DNA/GM-CSF (Robinson et al., 2006); or in the VLP vaccine DNA in the position of Nef, DNA/89.6.VLP.GM-CSF (Fig. 1A). Expression of the DNA constructs was tested in transiently transfected 293T cells using western blots for the expression of Gag and Env and electron microscopy for the production of VLP (data not shown). Supernatants were tested for GM-CSF protein using an antigen capture ELISA for human GMCSF (BioSource Inc.) (Fig. 1A). MVA/89.6 was the same as previously described and expressed SIV239 Gag/Pol in deletion II and a 115 aa C-terminal truncated SHIV-89.6 Env in deletion III (Amara et al., 2001).
Macaque trials
Vaccinations were conducted in 20 young adult rhesus macaques, 9 of which contain the A*01 histocompatibility type that presents an immunodominant epitope in Gag (Gag-CM9) (Allen et al., 1998). Macaques were randomized into 5 trial groups of four animals each based on weight and A*01 status. Trial groups were then randomized into four inoculation and sampling groups. A “no GM-CSF” group received a mixture of 1.5 mg of DNA/89.6.VLP and 1.5 mg of pGA1 vector DNA intradermally (id), two “GM-CSF in trans” groups received a mixture of either 1.5 mg of DNA/89.6.VLP, or 1.5 mg of DNA/89.6.VLP with the truncated Env, mixed with 1.5 mg of GM-CSF DNA id. Two additional “GM-CSF in cis” groups received 3 mg of DNA/89.6/VLP.GM-CSF administered id or intramuscularly (im). Intradermal DNA inoculations were delivered as twelve 0.1 ml shots, 3 to each limb. Intramuscular DNA inoculations consisted of a single 1 ml inoculation to the thigh. MVA boosts, 1×108 pfu of MVA/SHIV-89.6, used the same route as the prime. Intradermal MVA boosts consisted of one 0.1 ml shot of 2.5×107 pfu to each limb; and intramuscular boosts, of a single 1 ml shot in the thigh. The immunization schedule consisted of a DNA inoculation at week 0 followed by three MVA boosts delivered at weeks 8, 24 and 40, (1B). Intrarectal challenge with an estimated 20 intrarectal infectious units of SHIV-89.6P (1.1×1010 copies of viral RNA) was conducted as previously described (Amara et al., 2001). Three groups, the no-GM-CSF group and the two GM-CSF in trans groups, were challenged at 72 weeks, and the two GM-CSF in cis groups at 100 weeks (Fig 1b). At each challenge, 4 naive animals served as unvaccinated controls, giving a total of 8 unvaccinated controls. Six of these were A*01-positive. The challenge was staggered to allow for more intensive sampling and T cell analyses for individual animals. Animals were cared for under guidelines established by the Animal Welfare Act and the NIH “Guide for the Care and Use of Laboratory Animals” using protocols approved by the Emory University Institutional Animal Care and Use Committee.
Anti-Env IgG
Antigen for detecting anti-Env IgG in serum was produced in transient transfections of 293T cells with DNA/89.6.VLP. Lipofectamine-assisted transfections (Invitrogen) were conducted using the supplier's directions and DMEM supplemented with 10% fetal bovine serum (D-10). At 4 hours, cultures were changed to DMEM without serum. Culture supernatants were harvested at about 36 hrs, clarified by low speed centrifugation, dissociated by adjusting to 1% Triton-X-100 and tested for the ability to bind anti-Env Ab following capture with concanavilin A (Con A) (Vector Laboratories) (Cole et al., 1997). ELISA plates (Costar, Corning Life Sciences) were coated with ConA by incubating 100 μl of 25 μg per ml Con A in 10 mM Hepes buffer (pH 7.5) with 0.15 M NaCl, 1 mM CaCl2 and 1 mM MnCl2 overnight at 4°. Plates were washed six times with PBS containing 0.05% Tween -20 (PBS-tween) following which 100 μl of undiluted VLP supernatant was added, incubated for one hr at room temperature, washed six times with PBS-tween, blocked for 1 hr at room temperature with 100 μl blocking buffer (PBS-tween with 4% whey and 5% dry milk). Each lot of VLP supernatant was verified to achieve the same binding as a standard VLP supernatant using a standard anti-Env serum. The Triton-treated VLP supernatant was then divided into aliquots and frozen at −80°. For ELISA assays, test sera were diluted in PBS-tween with 4% whey and 100 μl of serial 3-fold dilutions added to duplicate wells and incubated for 1 hr at room temperature. The plates were then washed 6 times with PBS-tween and bound Ab detected using peroxidase conjugated anti-monkey IgG (Accurate Chemical and Scientific Corp) and TMB substrate (KPL). Reactions were stopped with 100 μl of 2 N H2SO4. Each plate included a standard curve generated using goat anti-monkey IgG and rhesus IgG (both from Accurate Chemicals) as previously described (Robinson et al., 2006). Standard curves were fitted and sample concentrations interpolated as μg of Ab per ml of serum using SOFTmax 2.3 software (Molecular Devices). The concentrations of IgG are relative to our standard curve, not absolute values.
A NaSCN displacement ELISA assay modeled after that described by Vermont was used for determining avidity (Vermont et al., 2002). This assay was conducted using parallel titrations of test sera in our standard ELISA assay. Following the binding of the test sera, the parallel titrations were treated for 10 minutes at room temperature with PBS or 1.5M NaSCN (prepared fresh in PBS). Then, the relative levels of bound Ab were determined using the standard ELISA procedure (see above). The avidity index was calculated by dividing the dilution of the serum that gave an OD of 0.5 with NaSCN treatment by the dilution of the serum that gave an OD of 0.5 without NaSCN treatment and multiplying by 100. Each assay included one plate with a standard serum with known avidity. Between assay variation for a reference standard included in each assay was ± 3 for an index of 27.
Neutralization assay
Neutralization was measured as a function of reductions in luciferase reporter gene expression after a single round of infection in TZM-bl cells as described (Li et al., 2005; Montefiori, 2004). TZM-bl cells were obtained from the NIH AIDS Research and Reference Reagent Program, as contributed by John Kappes and Xiaoyun Wu. Briefly, 200 TCID50 of virus was incubated with serial 3-fold dilutions of serum sample in triplicate in a total volume of 150 μl for 1 hr at 37°C in 96-well flat-bottom culture plates. Freshly trypsinized cells (10,000 cells in 100 μl of growth medium containing 75 μg/ml DEAE dextran) were added to each well. One set of control wells received cells + virus (virus control) and another set received cells only (background control). After a 48 hour incubation, 100 μl of cells was transferred to 96-well black solid plates (Costar) for measurements of luminescence using the Britelite Luminescence Reporter Gene Assay System (PerkinElmer Life Sciences). Assay stocks of SHIV-89.6 and SHIV-89.6P were prepared in human PBMC (2) and titrated in TZM-bl cells. Assay stocks of Env-pseudotyped viruses (incident clade B isolates) were prepared by transfection in 293T cells and were titrated in TZM-bl cells as described (2).
IgA
Rectal secretions were collected using premoistened Weck-Cel Sponges (Medtronic ENT) as described (Kozlowski et al., 2000). Sponges with absorbed secretions were placed in 15 ml Falcon tissue culture tubes and maintained on ice until storage at −80°C. Secretions were extracted from matched sponges by sequential centrifugation in the presence of 100 μl PBS containing protease inhibitors and 0.5% Igepal detergent (Kozlowski et al., 2000).
Anti-SIV gag/pol, HIV gp120, and HIV gp41 IgA antibodies were measured by ELISA as described (Bertley et al., 2004) using Fisherbrand high protein binding microtiter plates coated with 250ng SIVmac251 viral lysate (Advanced Biotechnologies Inc), 100ng HIV rgp120MN, and 100 ng HIV rgp41MN (both ImmunoDiagnostics) in PBS. Positive controls were pooled sera obtained from SIV-infected or vaccinated macaques (Bertley et al., 2004). Serum samples collected after challenge were depleted of IgG before addition to plates as previously described (Wright et al., 2002). The secondary antibody was affinity-purified biotinylated goat anti-monkey IgA (Rockland Immunochemicals). Plates were developed with 0.5μg/ml avidin-labeled peroxidase and ABTS as described (Bertley et al., 2004). To account for the variable immunoglobulin concentration in secretions, the concentration of antigen-specific IgA in each secretion was divided by the total IgA concentration to obtain its specific activity. Total IgA was measured by ELISA (Bertley et al., 2004) using plates coated with 0.5 μg/ml affinity-purified goat anti-monkey IgA antibody (Rockland). The standard for total IgA was pooled rhesus macaque serum containing a known amount of IgA (Bertley et al., 2004). Plates were developed as above. IgA specific activity was considered significant if it was equal to or greater than the mean + 3 SD determined from analysis of the specific antibody in specimens collected from nonvaccinated normal macaques. Mn Env was used for the study because it is a purified Env that has good binding activity for anti-Env sera and purified 89.6 Env was not available.
T cells
Cells for T cell assays were collected from blood, axillary lymph nodes and colorectal biopsies. Blood was collected into CPT sodium citrate tubes (BD Pharmaceuticals) for PBMC fractionation according to the directions of the provider. Lymph nodes were minced, ground using a Medimachine (BD Pharmaceuticals) and then washed in RPMI-1640 plus 10% fetal bovine serum (R-10). 15 to 20 mucosal biopsies were obtained from the rectum or distal colon using biopsy forceps and placed in ice cold RPMI-1640 for transport to the lab. In the lab, intestinal biopsies were washed twice with ice cold Hank's buffered saline solution (HBSS) and digested with 350 units per ml of collagenase IV (Worthington) in HBSS for 2 hours at room temperature on a shaker adjusted to slow speed. The digested tissue was passed 5-times through a syringe with a 16 gauge needle, followed by 5-times through an 18 gauge needle, followed by five-times through a 23 gauge needle and then filtered through a 70 μm nylon filter (BD BioScience). Cells were washed with room temperature R-10. In some experiments, harvested cells were enriched for lymphocytes by resuspending in 35% percoll (Sigma), layering over 60% percoll, and centrifuging at 400 g for 30 minutes at room temperature to enrich for lymphocytes. Cells from all sources were stained with trypan-blue for counting prior to T cell assays. The intestinal biopsies typically yielded several million cells, 5 to 10% of which were lymphocytes as evidenced by later staining with CD3.
All sources of T cells were subjected to tetramer staining and typing for the presence of CD4 and CD8 T cells. This was done using a panel of CD3 (Alexa Fluor 700, clone SP34-2, BD Biosciences PharMingen), CD4 (Percp, clone L200), CD8 (PE-Texas Red, clone 3B5, Invitrogen,), CD28 (FITC, clone CD28.2, Beckman Coulter), CD95 (Pacific blue, clone DX2,), CCR5 (PE, clone 3A9, BD Pharmingen) and CXCR4 (PE-Cy7, clone 12G5, eBiosciences). The levels of CD4 T cells in both the blood and intestinal biopsies are presented as a % of total CD3 T cells. Losses of CD4 T cells were calculated using the average % for each group at a given time divided by the normal %. The normal levels of CD4 T cells as a % of CD3 T cells were 53±9% for blood and 42±11% for the intestinal biopsies.
During the immunization phase, PBMC were tested for intracellular cytokine staining for IFN-γ and IL-2 as previously described (Amara et al., 2002) but using a single pool of 125 SIV239 Gag peptides (NIH AIDS Research and Reference Reagent Program) and a single pool of 221 SHIV-89.6 Env peptides (Emory Microchemical Facility) for stimulations. All peptides were 15-mers overlapping by 11. Following challenge, PBMC were tested for intracellular cytokine staining for IFN-γ, IL-2 and TNF-α using an LSR-II multicolor flow cytometer (BD Immunocytometry Systems). These stimulations were conducted for Gag and Env using two million PBMC in a total volume of 200 μl in the presence of anti-CD28 antibody and anti-CD49d antibody (1μg/ml; BD Pharmingen). Golgi-stop (BD Pharmingen) was added after 2 hrs of incubation. Tubes were mixed briefly and then incubated for an additional 4 hrs. Cells were washed once with cold phosphate-buffered saline (PBS) containing 2% FBS, surface stained with anti-human CD4 PerCP (clone L200; BD Pharmingen), anti-human CD8 Alexa Fluor 405 (clone 3B5; Caltag), fixed with cytofix/cytoperm (BD Pharmingen) and permeabilized with 1x permwash (BD Pharmingen). Cells were then incubated for 30 min at 4°C with a cocktail of monoclonal antibodies conjugated to different fluorochromes for the detection of CD3 (FITC, clone FN-18, BioSource) and cytokines IFN-γ (APC Alexa Fluor 750, clone B27, Invitrogen), IL-2 (APC, clone MQ1-17H12, BD Pharmingen) and TNF-α (PE-CY7, clone Mab11, eBiosciences). Cells were washed twice with 1 X Permwash, once with 2% FBS in PBS and resuspended in 1% formalin in PBS. Approximately 500,000 lymphocytes were acquired and analyzed using FlowJo software (Treestar, Inc.). Lymphocytes were identified based on their scatter pattern, CD3+, CD8−, CD4+ cells were considered as CD4 T cells and CD3+, CD8+, CD4− cells were considered as CD8 T cells. These CD4 or CD8 T cells were then gated for respective cytokine positive cells. Boolean gating was performed to calculate the frequencies of the seven different combinations of cytokines using the FlowJo software. After subtracting the background, the absolute numbers of the seven cytokine subsets per ml of blood was determined and plotted for each antigen per individual.
Viral RNA
Assays for viral RNA were conducted using a quantitative real time PCR as previously described (Amara et al., 2001; Hofmann-Lehmann et al., 2000). All specimens were extracted and assayed in duplicate, with the mean results reported. Nucleic Acid extractions from feces used the RNAqueous-Midi kit (Ambion) according to the directions of the manufacturer.
Statistical analyses
The Wilcoxon rank-sum test was used for pairwise comparisons between groups for levels of peak viral RNA, survival of CD4 T cells, avidity indices of anti-Env IgG and levels of peak viral RNA in animals that did, or did not, score for anti-viral IgA in rectal secretions. To assess the relationship between viral load and avidity of anti-Env IgG, the Pearson correlation test was used. The Spearman rank-correlation method was used to determine correlations between T cell responses and protection. The Fischer's exact test was used to test for differences in the presence of anti-viral IgA in plus and minus GM-CSF groups. For all comparisons between groups, a two-sided p<0.05 was considered statistically significant. Statistical analyses were performed using software programs S-PLUS 7.0 and SAS 9.1.
Acknowledgements
This research was supported by Integrated Preclinical/Clinical AIDS Vaccine Development program project, P01 AI 49364 to H. Robinson; Emory Center for AIDS Research, P30 DA 12121; Yerkes National Primate Research Center base grant, P51 RR00165; R01 AI 58896 to P. Kozlowski; and AI 30034 to D. Montefiori.
We are grateful to The Yerkes Division of Research Resources for the consistent excellence of veterinary care and pathology support, Helen Drake-Perrow for outstanding technical assistance, Dr. J. Sodroski for provision of SHIV-89.6 plasmid DNA and the NIH AIDS Research and Reference Reagent Program for the provision of peptides.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen TM, Sidney J, del Guercio MF, Glickman RL, Lensmeyer GL, Wiebe DA, DeMars R, Pauza CD, Johnson RP, Sette A, Watkins DI. Characterization of the peptide binding motif of a rhesus MHC class I molecule (Mamu-A*01) that binds an immunodominant CTL epitope from simian immunodeficiency virus. Journal of Immunology. 1998;160(12):6062–71. [PubMed] [Google Scholar]
- Amara RR, Smith JM, Staprans SI, Montefiori DC, Villinger F, Altman JD, O'Neil SP, Kozyr NL, Xu Y, Wyatt LS, Earl PL, Herndon JG, McNicholl JM, McClure HM, Moss B, Robinson HL. Critical role for Env as well as Gag-Pol in control of a simian-human immunodeficiency virus 89.6P challenge by a DNA prime/recombinant modified vaccinia virus Ankara vaccine. J Virol. 2002;76(12):6138–46. doi: 10.1128/JVI.76.12.6138-6146.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara RR, Villinger F, Altman JD, Lydy SL, O'Neil SP, Staprans S, Montefiori DC, Xu Y, Herndon JG, Wyatt LS, Candido MA, Kozyr NL, Earl PL, Smith JM, Ma H-L, Grimm BD, Hulsey ML, Miller J, McClure HM, McNicholl JM, Moss B, Robinson HL. Control of a Mucosal Challenge and Prevention of AIDS by a Multiprotein DNA/MVA Vaccine. Science. 2001;292:69–74. doi: 10.1126/science.1058915. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Barouch DH, Santra S, Tenner-Racz K, Racz P, Kuroda MJ, Schmitz JE, Jackson SS, Lifton MA, Freed DC, Perry HC, Davies ME, Shiver JW, Letvin NL. Potent CD4+ T cell responses elicited by a bicistronic HIV-1 DNA vaccine expressing gp120 and GM-CSF. J Immunol. 2002;168(2):562–8. doi: 10.4049/jimmunol.168.2.562. [DOI] [PubMed] [Google Scholar]
- Bertley FM, Kozlowski PA, Wang SW, Chappelle J, Patel J, Sonuyi O, Mazzara G, Montefiori D, Carville A, Mansfield KG, Aldovini A. Control of simian/human immunodeficiency virus viremia and disease progression after IL-2-augmented DNA-modified vaccinia virus Ankara nasal vaccination in nonhuman primates. J Immunol. 2004;172(6):3745–57. doi: 10.4049/jimmunol.172.6.3745. [DOI] [PubMed] [Google Scholar]
- Borrello I, Pardoll D. GM-CSF-based cellular vaccines: a review of the clinical experience. Cytokine Growth Factor Rev. 2002;13(2):185–93. doi: 10.1016/s1359-6101(01)00034-x. [DOI] [PubMed] [Google Scholar]
- Burdin N, Galibert L, Garrone P, Durand I, Banchereau J, Rousset F. Inability to produce IL-6 is a functional feature of human germinal center B lymphocytes. J Immunol. 1996;156(11):4107–13. [PubMed] [Google Scholar]
- Cole KS, Rowles JL, Jagerski BA, Murphey-Corb M, Unangst T, Clements JE, Robinson J, Wyand MS, Desrosiers RC, Montelaro RC. Evolution of envelope-specific antibody responses in monkeys experimentally infected or immunized with simian immunodeficiency virus and its association with the development of protective immunity. Journal of Virology. 1997;71(7):5069–79. doi: 10.1128/jvi.71.7.5069-5079.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connick E, Marr DG, Zhang XQ, Clark SJ, Saag MS, Schooley RT, Curiel TJ. HIV-specific cellular and humoral immune responses in primary HIV infection. AIDS Res Hum Retroviruses. 1996;12(12):1129–40. doi: 10.1089/aid.1996.12.1129. [DOI] [PubMed] [Google Scholar]
- Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan RC. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90(8):3539–43. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards TG, Wyss S, Reeves JD, Zolla-Pazner S, Hoxie JA, Doms RW, Baribaud F. Truncation of the cytoplasmic domain induces exposure of conserved regions in the ectodomain of human immunodeficiency virus type 1 envelope protein. J Virol. 2002;76(6):2683–91. doi: 10.1128/JVI.76.6.2683-2691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R, Shultz LD, Dranoff G, Fuller JA, Kamdar SJ. CSF-1 regulation of Il6 gene expression by murine macrophages: a pivotal role for GM-CSF. J Leukoc Biol. 1998;64(6):810–6. doi: 10.1002/jlb.64.6.810. [DOI] [PubMed] [Google Scholar]
- Forthal DN, Gilbert PB, Landucci G, Phan T. Recombinant gp120 Vaccine-Induced Antibodies Inhibit Clinical Strains of HIV-1 in the Presence of Fc Receptor-Bearing Effector Cells and Correlate Inversely with HIV Infection Rate. J Immunol. 2007;178(10):6596–603. doi: 10.4049/jimmunol.178.10.6596. [DOI] [PubMed] [Google Scholar]
- Geissler M, Gesien A, Tokushige K, Wands JR. Enhancement of cellular and humoral immune responses to hepatitis C virus core protein using DNA-based vaccines augmented with cytokine-expressing plasmids. Journal of Immunology. 1997;158(3):1231–7. [PubMed] [Google Scholar]
- Gomez-Roman VR, Patterson LJ, Venzon D, Liewehr D, Aldrich K, Florese R, Robert-Guroff M. Vaccine-elicited antibodies mediate antibody-dependent cellular cytotoxicity correlated with significantly reduced acute viremia in rhesus macaques challenged with SIVmac251. J Immunol. 2005;174(4):2185–9. doi: 10.4049/jimmunol.174.4.2185. [DOI] [PubMed] [Google Scholar]
- Hofmann-Lehmann R, Swenerton RK, Liska V, Leutenegger CM, Lutz H, McClure HM, Ruprecht RM. Sensitive and robust one-tube real-time reverse transcriptase- polymerase chain reaction to quantify SIV RNA load: comparison of one-versus two-enzyme systems. AIDS Research & Human Retroviruses. 2000;16(13):1247–57. doi: 10.1089/08892220050117014. [DOI] [PubMed] [Google Scholar]
- Huber M, Fischer M, Misselwitz B, Manrique A, Kuster H, Niederost B, Weber R, von Wyl V, Gunthard HF, Trkola A. Complement lysis activity in autologous plasma is associated with lower viral loads during the acute phase of HIV-1 infection. PLoS Med. 2006;3(11):e441. doi: 10.1371/journal.pmed.0030441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693–702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath AT, Hanke T, Briscoe H, Britton WJ. Co-immunization with DNA vaccines expressing granulocyte-macrophage colony-stimulating factor and mycobacterial secreted proteins enhances T-cell immunity, but not protective efficacy against Mycobacterium tuberculosis. Immunology. 1999;96(4):511–6. doi: 10.1046/j.1365-2567.1999.00703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson GB, Halloran M, Li J, Park IW, Gomila R, Reimann KA, Axthelm MK, Iliff SA, Letvin NL, Sodroski J. Characterization of molecularly cloned simian-human immunodeficiency viruses causing rapid CD4+ lymphocyte depletion in rhesus monkeys. Journal of Virology. 1997;71(6):4218–25. doi: 10.1128/jvi.71.6.4218-4225.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf M, Herren S, Wiles MV, Pepys MB, Kosco-Vilbois MH. Interleukin 6 influences germinal center development and antibody production via a contribution of C3 complement component. J Exp Med. 1998;188(10):1895–906. doi: 10.1084/jem.188.10.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski PA, Lynch RM, Patterson RR, Cu-Uvin S, Flanigan TP, Neutra MR. Modified wick method using Weck-Cel sponges for collection of human rectal secretions and analysis of mucosal HIV antibody. J Acquir Immune Defic Syndr. 2000;24(4):297–309. doi: 10.1097/00126334-200008010-00001. [DOI] [PubMed] [Google Scholar]
- Kumar S, Villinger F, Oakley M, Aguiar JC, Jones TR, Hedstrom RC, Gowda K, Chute J, Stowers A, Kaslow DC, Thomas EK, Tine J, Klinman D, Hoffman SL, Weiss WW. A DNA vaccine encoding the 42 kDa C-terminus of merozoite surface protein 1 of Plasmodium falciparum induces antibody, interferon-gamma and cytotoxic T cell responses in rhesus monkeys: immuno-stimulatory effects of granulocyte macrophage-colony stimulating factor. Immunol Lett. 2002;81(1):13–24. doi: 10.1016/s0165-2478(01)00316-9. [DOI] [PubMed] [Google Scholar]
- Lee SW, Cho JH, Sung YC. Optimal induction of hepatitis C virus envelope-specific immunity by bicistronic plasmid DNA inoculation with the granulocyte-macrophage colony-stimulating factor gene. J Virol. 1998;72(10):8430–6. doi: 10.1128/jvi.72.10.8430-8436.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol. 2005;79(16):10108–25. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifson JD, Nowak MA, Goldstein S, Rossio JL, Kinter A, Vasquez G, Wiltrout TA, Brown C, Schneider D, Wahl L, Lloyd AL, Williams J, Elkins WR, Fauci AS, Hirsch VM. The extent of early viral replication is a critical determinant of the natural history of simian immunodeficiency virus infection. Journal of Virology. 1997;71(12):9508–14. doi: 10.1128/jvi.71.12.9508-9514.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montefiori DC. Role of complement and Fc receptors in the pathogenesis of HIV-1 infection. Springer Seminars in Immunopathology. 1997;18(3):371–90. doi: 10.1007/BF00813504. [DOI] [PubMed] [Google Scholar]
- Montefiori DC. Evaluating neutralizing antibodies against HIV, SIV and SHIV in a luciferase reporter gene assay. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, Coico R, editors. Current Protocols in Immunology. John Wiley and Sons; New York: 2004. pp. 12.11.1–12.11.15. [DOI] [PubMed] [Google Scholar]
- Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, Otipoby KL, Yokota A, Takeuchi H, Ricciardi-Castagnoli P, Rajewsky K, Adams DH, von Andrian UH. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science. 2006;314(5802):1157–60. doi: 10.1126/science.1132742. [DOI] [PubMed] [Google Scholar]
- Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6(2):148–58. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- Parren PW, Burton DR. The antiviral activity of antibodies in vitro and in vivo. Adv Immunol. 2001;77:195–262. doi: 10.1016/S0065-2776(01)77018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parren PW, Mondor I, Naniche D, Ditzel HJ, Klasse PJ, Burton DR, Sattentau QJ. Neutralization of human immunodeficiency virus type 1 by antibody to gp120 is determined primarily by occupancy of sites on the virion irrespective of epitope specificity. J Virol. 1998;72(5):3512–9. doi: 10.1128/jvi.72.5.3512-3519.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parren PW, Moore JP, Burton DR, Sattentau QJ. The neutralizing antibody response to HIV-1: viral evasion and escape from humoral immunity. AIDS. 1999;13(Suppl A):S137–62. [PubMed] [Google Scholar]
- Pasquini S, Xiang Z, Wang Y, He Z, Deng H, Blaszczyk-Thurin M, Ertl HC. Cytokines and costimulatory molecules as genetic adjuvants. Immunol Cell Biol. 1997;75(4):397–401. doi: 10.1038/icb.1997.62. [DOI] [PubMed] [Google Scholar]
- Pitcher CJ, Hagen SI, Walker JM, Lum R, Mitchell BL, Maino VC, Axthelm MK, Picker LJ. Development and homeostasis of T cell memory in rhesus macaque. J Immunol. 2002;168(1):29–43. doi: 10.4049/jimmunol.168.1.29. [DOI] [PubMed] [Google Scholar]
- Reimann KA, Li JT, Veazey R, Halloran M, Park IW, Karlsson GB, Sodroski J, Letvin NL. A chimeric simian/human immunodeficiency virus expressing a primary patient human immunodeficiency virus type 1 isolate env causes an AIDS-like disease after in vivo passage in rhesus monkeys. Journal of Virology. 1996a;70(10):6922–8. doi: 10.1128/jvi.70.10.6922-6928.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann KA, Li JT, Voss G, Lekutis C, Tenner-Racz K, Racz P, Lin W, Montefiori DC, Lee-Parritz DE, Lu Y, Collman RG, Sodroski J, Letvin NL. An env gene derived from a primary human immunodeficiency virus type 1 isolate confers high in vivo replicative capacity to a chimeric simian/human immunodeficiency virus in rhesus monkeys. Journal of Virology. 1996b;70(5):3198–206. doi: 10.1128/jvi.70.5.3198-3206.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson HL, Montefiori DC, Villinger F, Robinson JE, Sharma S, Wyatt LS, Earl PL, McClure HM, Moss B, Amara RR. Studies on GM-CSF DNA as an adjuvant for neutralizing Ab elicited by a DNA/MVA immunodeficiency virus vaccine. Virology. 2006;352(2):285–94. doi: 10.1016/j.virol.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Rogers WO, Weiss WR, Kumar A, Aguiar JC, Tine JA, Gwadz R, Harre JG, Gowda K, Rathore D, Kumar S, Hoffman SL. Protection of rhesus macaques against lethal Plasmodium knowlesi malaria by a heterologous DNA priming and poxvirus boosting immunization regimen. Infect Immun. 2002;70(8):4329–35. doi: 10.1128/IAI.70.8.4329-4335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin JI, Kim JJ, Ugen KE, Ciccarelli RB, Higgins TJ, Weiner DB. Enhancement of protective humoral (Th2) and cell-mediated (Th1) immune responses against herpes simplex virus-2 through co-delivery of granulocyte-macrophage colony-stimulating factor expression cassettes. European Journal of Immunology. 1998;28(11):3530–40. doi: 10.1002/(SICI)1521-4141(199811)28:11<3530::AID-IMMU3530>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Smith JM, Amara RR, Campbell D, Xu Y, Patel M, Sharma S, Butera ST, Ellenberger DL, Yi H, Chennareddi L, Herndon JG, Wyatt LS, Montefiori D, Moss B, McClure HM, Robinson HL. DNA/MVA vaccine for HIV type 1: effects of codon-optimization and the expression of aggregates or virus-like particles on the immunogenicity of the DNA prime. AIDS Res Hum Retroviruses. 2004;20(12):1335–47. doi: 10.1089/aid.2004.20.1335. [DOI] [PubMed] [Google Scholar]
- Smith SM, Holland B, Russo C, Dailey PJ, Marx PA, Connor RI. Retrospective analysis of viral load and SIV antibody responses in rhesus macaques infected with pathogenic SIV: predictive value for disease progression. AIDS Res Hum Retroviruses. 1999;15(18):1691–701. doi: 10.1089/088922299309739. [DOI] [PubMed] [Google Scholar]
- UNAIDS . AIDS Epidemic Update. UNAIDS, and WHO; 2006. [Google Scholar]
- Varol C, Landsman L, Fogg DK, Greenshtein L, Gildor B, Margalit R, Kalchenko V, Geissmann F, Jung S. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J Exp Med. 2007;204:171–178. doi: 10.1084/jem.20061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermont CL, van Dijken HH, van Limpt CJ, de Groot R, van Alphen L, van Den Dobbelsteen GP. Antibody avidity and immunoglobulin G isotype distribution following immunization with a monovalent meningococcal B outer membrane vesicle vaccine. Infect Immun. 2002;70(2):584–90. doi: 10.1128/IAI.70.2.584-590.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Richie TL, Baraceros MF, Rahardjo N, Gay T, Banania JG, Charoenvit Y, Epstein JE, Luke T, Freilich DA, Norman J, Hoffman SL. Boosting of DNA vaccine-elicited gamma interferon responses in humans by exposure to malaria parasites. Infect Immun. 2005;73(5):2863–72. doi: 10.1128/IAI.73.5.2863-2872.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss WR, Ishii KJ, Hedstrom RC, Sedegah M, Ichino M, Barnhart K, Klinman DM, Hoffman SL. A plasmid encoding murine granulocyte-macrophage colony-stimulating factor increases protection conferred by a malaria DNA vaccine. Journal of Immunology. 1998;161(5):2325–32. [PubMed] [Google Scholar]
- Wright PF, Kozlowski PA, Rybczyk GK, Goepfert P, Staats HF, VanCott TC, Trabattoni D, Sannella E, Mestecky J. Detection of mucosal antibodies in HIV type 1-infected individuals. AIDS Res Hum Retroviruses. 2002;18(17):1291–300. doi: 10.1089/088922202320886334. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Ertl HC. Manipulation of the immune response to a plasmid-encoded viral antigen by coinoculation with plasmids expressing cytokines. Immunity. 1995;2(2):129–35. doi: 10.1016/s1074-7613(95)80001-8. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lou B, Lal RB, Gettie A, Marx PA, Moore JP. Use of inhibitors to evaluate coreceptor usage by simian and simian/human immunodeficiency viruses and human immunodeficiency virus type 2 in primary cells. J Virol. 2000;74(15):6893–910. doi: 10.1128/jvi.74.15.6893-6910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]