

Abstract
The noncovalent complex formed by the association of two fragments of chymotrypsin inhibitor-2 is reversibly denatured by pressure in the absence of chemical denaturants. On pressure release, the complex returned to its original conformation through a biphasic reaction, with first-order rate constants of 0.012 and 0.002 s−1, respectively. The slowest phase arises from an interconversion of the pressure-denatured state, as revealed by double pressure-jump experiments. Below 5 μM, the process was concentration dependent with a second-order rate constant of 1,700 s−1 M−1. Fragment association at atmospheric pressure showed a similar break in the order of the reaction above 5 μM, but both first- and second-order folding/association rates are 2.5 times faster than those for the refolding of the pressure-denatured state. Although the folding rates of the intact protein and the association of the fragments displayed nonlinear Eyring behavior for the temperature dependence, refolding of the pressure-denatured complex showed a linear response. The negligible heat capacity of activation reflects a balance of minimal change in the burial of residues from the pressure-denatured state to the transition state. If we add the higher energy barrier in the refolding of the pressure-denatured state, the rate differences must lie in the structure of this state, which has to undergo a structural rearrangement. This clearly differs from the conformational flexibility of the isolated fragments or the largely unfolded denatured state of the intact protein in acid and provides insight into denatured states of proteins under folding conditions.
Kinetic studies aiming at the elucidation of protein-folding mechanisms are most frequently based on the reaction that takes place on the transfer of an unfolded polypeptide from strong denaturing conditions to those that favor a folded conformation. Pressure is an ideal perturbant for folding equilibria in that it gives valuable thermodynamic information without the need of altering the chemical composition of the solution. High-pressure techniques were applied in folding studies at the equilibrium (1–3) and more recently in the study of the transition state for protein folding (4). The possibility of monitoring folding kinetics by simply releasing the pressure is very attractive, and the method is particularly suitable for slow reactions. Pressure-jump relaxation kinetics was used to investigate the folding mechanism of pressure-denatured staphylococcal nuclease (5, 6). The effect of pressure on the rate of a reaction is described by (7)
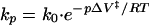 |
1 |
where kp corresponds to the rate of the reaction at pressure p, k0, the rate at atmospheric pressure, and ΔV‡, the activation molar volume for the transition state. According to Le Chatelier’s principle, the increase of pressure in a system will favor the state with the smaller volume. Depending on the nature of the transition state involved (positive or negative ΔV‡), pressure may accelerate or decrease the rate of the reaction.
CI-2 is a member of a class of small fast-folding proteins that follow a simple two-state model for folding, which constitutes a paradigm for the study of experimental and theoretical aspects of protein folding (8–13). CI-2 can be cleaved into two fragments by cyanogen bromide, and these reassociate/refold to give a native-like structure (14, 15). The noncovalent complex [CI-2(1–40)⋅(41–64)] forms readily through a folding pathway that is equivalent to that of the uncleaved inhibitor, except that it is a second-order process (16–19). We took advantage of the lower stability of the complex compared with uncleaved CI-2 and described the reversible pressure denaturation of [CI-2(1–40)⋅(41–64)] to an undissociated denatured state with residual structure (20).
In the present work, we describe the kinetic folding mechanism of the pressure-denatured noncovalent complex [CI-2(1–40)⋅(41–64)]. We propose a model that accounts for the overall equilibrium and kinetic mechanism and discuss its implications for protein folding in the absence of denaturants.
MATERIALS AND METHODS
All materials, including chemicals, protein, and fragment purification, were described (20).
Pressure Denaturation–Renaturation Experiments.
Pressure experiments were carried out in a high-pressure cell (21) fitted with sapphire windows and adapted to an ISS K2 fluorimeter (Champaign, IL). The [CI-2(1–40)⋅(41–64)] complex was produced by mixing equimolar concentrations of the fragments and incubated for 14 hr at 25°C in 50 mM [bis(2-hydroxyethyl)amino]tris(hydroxymethyl)methane (Bistris) HCl buffer, pH 6.0, before use, as described previously (20). Kinetic experiments of fragment association at atmospheric pressure were carried out with a Hitachi F-4500 fluorimeter (Tokyo). The fluorescence intensity at 356 nm was monitored, with excitation fixed at 280 nm.
For the renaturation, pressure was applied to the sample until the center of mass stabilized to values corresponding to a solvent accessible tryptophan. The pressure was released (taking 60–90 s) and the data within the dead time were collected but not considered for the analysis. In the pressure-dependence experiments, pressure was brought to indicated values and fluorescence was monitored. The same considerations apply to the kinetic pressure denaturation experiments in terms of experimental dead time. For “double-jump” experiments, the sample was pressurized and, after the indicated times, the pressure was returned to atmospheric values. Unless stated, the temperature was kept at 25 ± 0.2°C.
Analysis of the Data.
The activation volume for the transition state of refolding of the pressure-denatured complex (ΔV‡) is calculated from the logarithmic form of Eq. 2:
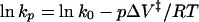 |
2 |
Temperature dependence of CI-2 refolding, fragment association, and renaturation of pressure-denatured complex was studied over the range 10–35°C. When the Eyring plots (ln k/T vs. 1/T) were linear, we used the following equation, derived from the transition-state theory (22):
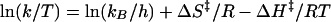 |
3 |
where k is the measured rate of the reaction, ΔS‡, the activation entropy, ΔH‡, the activation enthalpy, h is the Planck’s constant, kB is the Boltzmann’s constant, R, the gas constant, and T, the absolute temperature. When nonlinear Eyring plots were observed, we used the modified equation derived by Chen et al. (23). The data were fitted by using kaleidagraph (Abelbeck Software) to calculate the activation parameters.
RESULTS
Kinetics of Pressure Denaturation and Renaturation of the [CI-2(1–40)⋅(41–64)] Complex.
The equilibrium pressure denaturation of the [CI-2(1–40)⋅(41–64)] complex showed a shift in the unique tryptophan residue to values corresponding to large solvent accessibility and a 2.5-fold increase in the fluorescence spectral area (20). We applied the maximal pressure attainable in our equipment [3.5 kbar (1 bar = 100 kPa)], and we took fluorescence spectra at different time intervals, monitoring the change in the center of spectral mass (CM). After 20 min of application of pressure, the CM changed over 95% to values compatible with complete solvent accessibility of the tryptophan (Fig. 1a Inset). When the change in fluorescence intensity was monitored, unfolding was best described by a single exponential trace with an approximately 2-fold change (Fig. 1a). The unfolding rate at the highest pressure was measured at different protein concentrations, and the rate remained invariant, with a first-order rate of 0.009 s−1.
Figure 1.

Denaturation–renaturation kinetics of [CI2(1–40)⋅(41–64)] by pressure. (a) The complex was denatured at high pressures, and the fluorescence intensity was recorded immediately after the maximum pressure was attained (small black dots). In a similar experiment, fluorescence spectra were recorded at longer time intervals and the center of mass calculated (Inset). (b) After 45 min at 3.5 kbar, the pressure was released and the fluorescence monitored. The concentration of the complex was 10 μM.
After pressure release, the fluorescence slowly decreases to a basal value, consistent with the large quenching observed in the folded conformation (14) (Fig. 1b). The total change observed is a 2.3-fold decrease, in excellent agreement with the change observed at equilibrium (20). The data adjusts only to two exponential decays, corresponding to two phases with observed rates of 0.01 and 0.002 s−1, respectively, at 10 μM concentration of the complex (Fig. 1b Inset).
The effect of pressure on refolding rates was tested by releasing the pressure to fixed low pressures, i.e., at the beginning of the unfolding transition, where the contribution of unfolding is minimal. Data were fitted to double exponentials and the observed rates plotted against final pressure (Fig. 2a). The major phase is not significantly affected by pressure, in the range and conditions of the experiment. Small positive or negative changes ΔV‡ for folding cannot be measured accurately because of the limitations of our equipment and the inherent error in measuring such a small change (Fig. 2a). The first-order rate of the minor phase, accounting for 15% of the amplitude at atmospheric pressure, is decreased as the final pressure increases (Fig. 2b), indicating that the molar volume of the transition state for this reaction is small but positive. From the logarithmic form of the equation for the analysis of the effect of pressure on reaction rates (Eq. 2), we calculate a ΔV‡ of 22 ± 4 ml mol−1 (Fig. 2b Inset).
Figure 2.

Effect of pressure on the refolding rates of the pressure-denatured [CI2(1–40)⋅(41–64)] complex. The complex was released from the high pressures as described in Fig. 1 to different final pressures lower than the p1/2, in which the refolding rate is predominant. a corresponds to the observed rates of the major phase, b the minor phase (Inset shows a logarithmic plot) and c, the percent relative amplitudes.
The amplitudes for the two phases were analyzed on a percent basis at the different final pressures used and are shown in Fig. 2c. Pressure exerts a significant effect on the amplitudes; above 400 bar, the contribution of each phase goes from 85:15% fast:slow phase ratio to a 50:50% ratio.
Parallel Break in the Reaction Molecularity of Fragment Association/Folding and Renaturation of the High-Pressure State.
Association of the fragments in equimolar concentrations at atmospheric pressure is accompanied by a 6-fold decrease in fluorescence intensity (14), and the time course change can be approached by two exponential decays, as a simplification to the underlying second-order process. This procedure was repeated at different concentrations of the fragments, and the observed rates of the major phase were represented in Fig. 3, showing a concentration dependence that deviates from linearity above 5 μM. From the slope of the plot, we estimated the second-order rate constant to be 4,400 s−1 M−1, in excellent agreement with previous values from pseudo-first-order experiments (16). The distribution of the amplitudes of both phases indicates that at high concentrations, the amplitudes stabilize at approximately 80:20% fast:slow phase ratio (not shown), corresponding to the folding or first-order-driven reaction as opposed to association or second-order-driven reaction. This ratio is similar to that observed in pseudo-first-order conditions (16). The rate of the major folding/association phase product of mixing both fragments in equimolar concentrations at atmospheric pressure stabilizes with a first-order rate of 0.039 s−1.
Figure 3.

Concentration dependence of the renaturation of pressure-denatured complex. The experiment of Fig. 1b was repeated at different complex concentrations, and the observed rates from the double exponential analysis are plotted (open circles). For the concentration dependence of the association rates of the free fragments in equimolar concentrations at atmospheric pressure, the fragments were mixed at the concentrations indicated in 50 mM [bis(2-hydroxyethyl)amino]tris(hydroxymethyl)methane (Bistris) buffer, pH 6.0. The fluorescence intensity of the tryptophan probe was monitored, the data fitted by using a double-exponential equation, and the observed rates plotted (closed circles). The second-order rate constants were estimated from the initial slopes.
On the other hand, after pressure release, the rate of the slowest of the two observable phases is independent of the protein concentration along the range tested with a resulting first-order rate constant of 0.002 s−1, corresponding to 15% of the amplitude, while the major phase accounts for 85% of the amplitude (not shown). Both distribution of amplitudes as well as the concentration dependence are similar to what was observed in the association/folding of the [CI-2(1–40)⋅(41–64)] complex (16). The observed rate of the major phase shows concentration dependence below 5 μM, reaching a constant value at 0.011 s−1 (Fig. 3, open circles). From the initial slope of the concentration dependence of pressure refolding, we can estimate a second-order rate constant of 1,700 s−1 M−1, 2.5-fold slower than the association and folding of the fragments at atmospheric pressure. The first-order value reached at high protein concentration in the pressure release experiment is also slower than that reached in the fragment association/folding experiment (0.012 against 0.039 s−1). These results are in excellent agreement with equilibrium denaturation experiments, which showed that the fragments remained associated at the highest pressures, above 5 μM concentration (20). The amplitudes stabilized at 5 μM, coincident with the break in the concentration dependence (not shown).
Double Pressure-Jump Kinetics: Nature of the Two Phases.
Defining a folding mechanism involves the understanding of the nature of the phases and the sequence of the events and species involved. We wanted to investigate whether the two phases are present at short times after attaining the maximum pressure, and we approached the problem by using a double-jump pressure experiment. When the complex was pressurized for 30 min, the refolding process that followed the release of the pressure followed a biphasic behavior, in a similar manner to our standard kinetic experiments after pressure release (cf. Fig. 1b, Fig. 4a). After only 5 min of pressurization, the reaction proceeded to 80% (Fig. 1a), the pressure was released, and the fluorescence change monitored (not shown). In this case, the renaturation process takes place through a single phase with the expected rate for the major phase (Fig. 4b).
Figure 4.

Double-pressure denaturation–reneturation jump. The complex (10 μM) was pressure denatured for (a) 30 min or (b) 5 min, based on the time trace in Fig. 1a. The rate constants are similar to those obtained in Fig. 1a, but a fits well only to two exponentials, while b fits well to a single exponential decay.
Effect of Temperature on Folding Kinetics of Intact CI-2, Fragment Association, and the Pressure-Denatured State.
The transition state for the folding pathway of intact CI-2 and that for the fragment association were characterized by protein engineering methods (24, 17). Transition states for protein-folding reactions can also be analyzed from temperature dependence of the reaction rates, where thermodynamic activation parameters can be obtained from Eyring plots (23).
We set out to analyze comparatively the effect of temperature on: (i) folding rates of intact CI-2, (ii) folding/association of fragments, and (iii) renaturation of the pressure-denatured [CI-2(1–40)⋅(41–64)] complex. In the case of intact CI-2, we started from acid-denatured protein and mixed it with a buffer at neutral pH (8), monitoring the corresponding fluorescence decrease at different temperatures. As described previously, the process is characterized by a major phase that shows a rate increase with temperature. The data are represented as ln k/T vs. 1/T in Fig. 5a and are clearly nonlinear (Fig. 5a Inset), indicative of a significant ΔCp‡. The activation parameters obtained are shown in Table 1. The prefactor in the Eyring equations used for the analysis of these reactions, although unknown, was assumed to be kBT/h (23). If we assume that the prefactor, although unknown, is similar in the three cases we discuss here, the parameters obtained can be used in a comparative manner. Therefore, the activation parameters in Table 1 are not correct in absolute terms.
Figure 5.

Temperature dependence of the refolding of intact CI-2, the association of CI2(1–40) and CI2(41–64) fragments, and renaturation of pressure denatured state. (a) The major rate of folding of acid-unfolded CI-2 (10 μM) was measured at different temperatures, and the data represented in the figure were fitted to a nonlinear Eyring equation (cf. ref. 23). (b) CI2(1–40) (1 μM) was mixed with 10 μM of CI2(41–64), the fluorescence change was monitored at different temperatures, and the rate of the major phase represented and fitted as in a. (c) The complex (10 μM) was denatured by pressure at the indicated temperatures until the center of spectral mass was equilibrated at the corresponding “denatured” value. The pressure was released and the fluorescence was monitored. Data for each temperature were analyzed as two exponentials as in Fig. 1 and the observed rates of the major phase plotted. The residuals of the single-exponential fitting are shown at the bottom of each plot.
Table 1.
Activation parameters for folding of CI-2, fragment association at atmospheric pressure, and pressure-denatured [C12(1-40)⋅(41-64)]
| ΔCp‡, | ΔH‡, | ΔS‡, | ΔG‡, | |
|---|---|---|---|---|
| kcal⋅mol−1⋅K−1 | kcal⋅mol−1 | cal⋅mol⋅K−1 | kcal⋅mol−1⋅K−1 | |
| CI-2 | −0.75 ± 0.06 | +12.7 ± 1.2 | −7.4 ± 1.2 | +14.9 |
| Fragment association | −0.41 ± 0.05 | +16.4 ± 0.3 | −8.0 ± 0.9 | +18.8 |
| Pressure denatured [C12(1-40)⋅(41-64)] | – | +19.1 ± 0.5 | −3.1 ± 1.7 | +20.2 |
Thermodynamic parameters at 25°C.
A similar approach was undertaken for the temperature dependence of fragment association, except that this reaction consists of mixing fragment CI-2(1–40) with a 10-fold excess of CI-2(41–64), in pseudo-first-order conditions (16). The rate of the major phase was plotted at the different temperatures (Fig. 5b) and the corresponding thermodynamic parameters obtained (Table 1). The Eyring plot for fragment association is also nonlinear (Fig. 5b Inset).
The [CI-2(1–40)⋅(41–64)] complex was pressurized as described in previous sections and equilibrated at the indicated temperatures before release of the pressure and monitoring of the accompanying fluorescence decrease. The rates of the major phase thus obtained were plotted as ln k/T vs. 1/T (Fig. 5c). In this case, the data fit to the linear form of the Eyring equation, without the ΔCp‡ component (Fig. 5c Inset).
DISCUSSION
Equilibrium pressure denaturation experiments showed a lack of protein concentration dependence in the 5 to 100 μM range, indicating that the high-pressure state of the complex was denatured but remained associated (20). However, the nature of the interactions that hold together this “artificial dimer” is different from that of nature-designed dimers (3). The biological function of CI-2 as a proteinase slow-release inhibitor requires that the fragments remain bound after cleavage by the enzyme. We have previously discussed our system in comparison with other known natural dimers (20).
Folding kinetics after pressure release was biphasic, coincident with the behavior of the intact protein and the association of the fragments (8, 17). We were able to follow the folding rates below the minimal concentration that the sensitivity of the system allowed us in previous equilibrium experiments and found concentration dependence below 5 μM. The presence of a unimolecular process above 5 μM with a rate constant of 0.012 s−1 further supports an undissociated pressure-denatured state. How can the fragments remain associated if the complex is largely unfolded? The key argument is that full solvent accessibility to the unique tryptophan residue may not necessarily mean extensive unfolding; it may instead indicate a fluctuating molten globule-type structure (25, 26). CI-2 fragments containing 94% of the amino acid sequence showed full solvent accessibility to the tryptophan, binding of 1-anilino-8-naphtalene sulfonate, and lack of cooperativity, but unequivocal native-like folded chemical shift of the residue by NMR (27). Further evidence comes from the dissociation at low, but not at high, concentrations: if the pressure-denatured state was indeed dissociated, there could be no possibility of tertiary long-range interactions, with the consequence that the quenching would be completely suppressed in the free CI-2(1–40) fragment (14).
Kinetic-pressure unfolding is monophasic in the time frame of our experiments, but we cannot rule out the possibility of a much slower reaction involving dissociation with a consequent release of the tryptophan quenching. The unfolding rate is 20-fold faster than the unfolding of the complex (16), but clearly the final state is not the same. The chemical denaturant causes dissociation that can be attained only by complete unfolding (20).
A double pressure-jump experiment indicated that the second phase appears only after longer times under high pressure, strongly suggesting the presence of a slow interconversion of the pressure-denatured state, presumably an isomerization around a peptide bond. Nevertheless, above 10 μM at atmospheric pressure, 85% of the amplitude corresponds to the fastest phase. In these conditions, the association appears to be fast, and a first-order folding step becomes rate limiting. As the pressure increases, the phases are no longer “major” or “minor,” but approach similar percent amplitudes values. Assuming that changes in the center of mass and spectral area are separable, it means that the quenching of the tryptophan and its exposure to the solvent do not necessarily take place in parallel. Furthermore, there may not be a linear relationship between the fluorescence intensity change and the contribution of that species to the total amplitude. Percent amplitudes taken directly from fluorescence intensities must, therefore, be interpreted with caution.
Fragment association in equimolar concentrations at atmospheric pressure displays concentration dependence in the same range as pressure renaturation of the complex and a deviation from linearity above that range. However, both the second-order rate at low concentration and the first-order folding rate at high concentration are approximately 3-fold faster in the absence of high pressure. A higher energy barrier in the refolding after pressure release may come from a stable structure in the pressure-denatured state that needs to undergo rearrangements. This structured state would differ from the flexibility of the two peptide fragments that encounter one another in solution to yield a folding reaction when no pressure was applied. The striking similarity in the switch of the reaction order indicates that this phenomenon is inherent to the system and independent of the perturbation applied to it (Scheme I).
 |
Curvature in Eyring plots can be observed when there is a significant change in heat capacity between the ground and the transition states (23). This is the case of transition states for folding of intact CI-2 and the association/folding of the fragments. Of these two, the fragment association/folding reaction has a larger overall activation energy, influenced by a larger ΔH‡ because the activation entropies (ΔS‡) are of similar magnitude. This activation enthalpy difference could be caused by interactions broken in the isolated fragments in solution. Alternatively, the fragments should be able to associate rapidly to form an unfolded complex product of the initial collision that could be the starting point of the reaction, and this has to undergo rearrangements different from the intact protein, which causes the extra difference in ΔH‡. Similarly, we interpret the smaller change in ΔCp‡ of the fragment association as a residual structure in the free fragments (15, 17) or associated denatured complex that has to undergo rearrangements to reach the transition state (20).
The folding kinetics from the pressure-denatured state takes place at atmospheric pressure, after the pressure is released. Thus, conditions of pressure, temperature, and solvent are essentially similar to those for the time-dependent fragment folding/association (16), suggesting energetically and structurally related ensembles of transition states. Therefore, differences observed in the activation parameters may not lie in the structure of the transition states, but rather in the denatured states. The Arrhenius behavior for the pressure-jump refolding experiment is linear, which indicates a very small change in heat capacity and, therefore, in solvent-accessible surface area (ΔASA) (28, 29). The intact CI-2 buries most of its ASA in the transition state (8); however, we show here that the transition state for the fragment association buries less ASA and has to overcome a higher energy barrier. Because mutagenesis suggests similar transition states (17, 18), and they have an identical number of residues, the difference must lie either in the initial, i.e., denatured state and free fragments or final, i.e., intact folded CI-2 and folded noncovalent complex. In the intact CI-2, we start from the acid-denatured protein and in the fragments, we simply mix the two peptides in aqueous buffer at neutral pH.
In the case of the transition state after pressure release, the key difference is the smaller activation entropy compared with the fragment association and the folding of intact CI-2, which we interpret as a small overall change in the conformational freedom on going from denatured to transition states, because of a more ordered or structured denatured state at high pressure. The highest overall free energy barrier in the transition state for refolding after pressure release (slower reaction) adds to the body of evidence concerning the type of structure present in the pressure-denatured state. Whereas heat or chemical denaturation corresponds to the transfer of nonpolar groups into solution, pressure denaturation is the result of incorporation of water molecules into the interior of the protein (30). Pressure-denatured states of proteins retain considerably more structure than any other means of denaturation (31).
From the energy landscape perspective (32, 33), pressure-denatured states are located closer to the bottom of the funnel, i.e., after the collapse of the polypeptide chain, but retain solvent in the interior of the protein as the result of the effect of pressure. The conformational freedom is narrowed to a limited number of ensembles, much lower than the chemically denatured polypeptides, or, in the present case, the conformational freedom of the free fragments. A slower refolding rate of the pressure-denatured state compared with the reaction at atmospheric pressure supports the idea of a rough landscape near the energy minima of the native state (4, 30). Theoretical studies pointed at pressure as an ideal tool to explore this region of the folding funnel (34).
We can summarize the equilibrium and kinetic evidence for a compact and undissociated molten globule-like pressure-denatured state from: (i) the undissociated nature of the high-pressure state that calls for some type of structure holding the fragments together but not extensive unfolding, (ii) the persistent quenching in the fluorescence of the unique tryptophan residue, (iii) the solvent accessibility of the hydrophobic core in the pressure-denatured state as probed by its structure nucleating tryptophan residue, (iv) small ΔV and ΔV‡, (v) previous evidence that described large molten globule-like CI-2 fragments with a fully solvent accessible tryptophan residue but folded-like chemical shifts of the tryptophan and other key residues, (vi) a clear-cut switch in the order of the reaction from bi- to unimolecular, and (vii) activation parameters that evidence a higher energy barrier, very small amount of solvent accessible area buried, and a smaller activation entropy.
An ultimate goal in the study of the protein-folding problem is its biological relevance, besides the challenge it represents for chemistry or polymer physics. Protein folding in vivo can be assisted by accessory proteins, but the folded conformation of proteins are in equilibrium with their denatured states in solution. The complete absence of chemical denaturants whilst using a subtle but otherwise fully reversible and readily interpretable perturbant such as pressure takes us one step forward toward understanding the structural, kinetic, and thermodynamic grounds for the folding reactions occurring in the mild solvent conditions compatible with the natural environment of proteins.
Acknowledgments
This work was partly supported by grants from Programa de Apoio ao Desenvolvimento Científico e Tecnologico (PADCT) and Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq). R.M.B. holds a fellowship from CNPq. J.L.S. is an International Research Scholar of the Howard Hughes Medical Institute. G.P.G. is a John Simon Guggenheim Fellow and wishes to acknowledge the support of Fundación Bunge y Born and Fundación Antorchas.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Heremans K. Annu Rev Biophys Bioeng. 1982;11:1–21. doi: 10.1146/annurev.bb.11.060182.000245. [DOI] [PubMed] [Google Scholar]
- 2.Weber G, Drickamer H G. Q Rev Biophys. 1983;16:89–112. doi: 10.1017/s0033583500004935. [DOI] [PubMed] [Google Scholar]
- 3.Silva J L, Weber G. Annu Rev Phys Chem. 1993;44:89–113. doi: 10.1146/annurev.pc.44.100193.000513. [DOI] [PubMed] [Google Scholar]
- 4.Vidugiris G J A, Markley J, Royer C A. Biochemistry. 1995;34:4909–4912. doi: 10.1021/bi00015a001. [DOI] [PubMed] [Google Scholar]
- 5.Vidugiris G J A, Truckses, Markley J, Royer C A. Biochemistry. 1996;35:3857–3864. doi: 10.1021/bi952012g. [DOI] [PubMed] [Google Scholar]
- 6.Panick G, Malessa R, Winter R, Rapp G, Frye K J, Royer C A. J Mol Biol. 1998;275:389–402. doi: 10.1006/jmbi.1997.1454. [DOI] [PubMed] [Google Scholar]
- 7.Glasstone S, Laidler K J, Eyring H. The Theory of Rate Processes. New York: McGraw–Hill; 1941. [Google Scholar]
- 8.Jackson S E, Fersht A R. Biochemistry. 1991;30:10436–10443. doi: 10.1021/bi00107a011. [DOI] [PubMed] [Google Scholar]
- 9.Otzen D, Itzhaki L, elMasry N, Jackson S E, Fersht A R. Proc Natl Acad Sci USA. 1994;91:10422–10425. doi: 10.1073/pnas.91.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fersht A R. Curr Opin Struct Biol. 1994;5:79–84. doi: 10.1016/0959-440x(95)80012-p. [DOI] [PubMed] [Google Scholar]
- 11.Li A J, Dagget V. J Mol Biol. 1996;257:412–429. doi: 10.1006/jmbi.1996.0172. [DOI] [PubMed] [Google Scholar]
- 12.Shakhnovich E I, Abkevich V, Ptitsyn O. Nature (London) 1996;379:96–98. doi: 10.1038/379096a0. [DOI] [PubMed] [Google Scholar]
- 13.Shoemaker B A, Wang J, Wolynes P. Proc Natl Acad Sci USA. 1997;94:777–782. doi: 10.1073/pnas.94.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prat Gay G de, Fersht A R. Biochemistry. 1994;33:7957–7963. doi: 10.1021/bi00191a024. [DOI] [PubMed] [Google Scholar]
- 15.Neira J L, Davis B, Prat Gay G de, Ladurner A G, Buckle A, Fersht A R. Folding Des. 1996;1:189–208. doi: 10.1016/s1359-0278(96)00031-4. [DOI] [PubMed] [Google Scholar]
- 16.Prat Gay G de, Ruiz-Sanz J, Fersht A R. Biochemistry. 1994;33:7964–7970. doi: 10.1021/bi00191a025. [DOI] [PubMed] [Google Scholar]
- 17.Prat Gay G de, Ruiz-Sanz J, Davis B, Fersht A R. Proc Natl Acad Sci USA. 1994;91:10943–10946. doi: 10.1073/pnas.91.23.10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruiz-Sanz J, Prat Gay G de, Otzen D E, Fersht A R. Biochemistry. 1995;34:1695–1701. doi: 10.1021/bi00005a026. [DOI] [PubMed] [Google Scholar]
- 19.Prat Gay G de. Protein Eng. 1996;9:843–847. doi: 10.1093/protein/9.10.843. [DOI] [PubMed] [Google Scholar]
- 20.Mohana-Borges R, Silva J L, Prat Gay G de. J Biol Chem. 1998;274:7732–7740. doi: 10.1074/jbc.274.12.7732. [DOI] [PubMed] [Google Scholar]
- 21.Paladini A A, Weber G. Rev Sci Instrum. 1981;52:419–427. [Google Scholar]
- 22.Laidler K J. Chemical Kinetics. New York: McGraw–Hill; 1950. [Google Scholar]
- 23.Chen B, Baase W A, Schellman J A. Biochemistry. 1989;28:691–699. doi: 10.1021/bi00428a042. [DOI] [PubMed] [Google Scholar]
- 24.Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 25.Silva J L, Foguel D, Da Poian A T, Prevelige P E. Curr Opin Struct Biol. 1996;6:166–175. doi: 10.1016/s0959-440x(96)80071-6. [DOI] [PubMed] [Google Scholar]
- 26.Foguel D, Silva J L, Prat Gay G de. J Biol Chem. 1998;273:9050–9057. doi: 10.1074/jbc.273.15.9050. [DOI] [PubMed] [Google Scholar]
- 27.Prat Gay G de, Ruiz-Sanz J, Neira J L, Corrales F, Otzen D O, Ladurner A G, Fersht A R. J Mol Biol. 1995;254:968–979. doi: 10.1006/jmbi.1995.0669. [DOI] [PubMed] [Google Scholar]
- 28.Livingstone J R, Spolar R S, Record M T. Biochemistry. 1991;30:4237–4244. doi: 10.1021/bi00231a019. [DOI] [PubMed] [Google Scholar]
- 29.Myers J K, Pace C N, Scholtz J M. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hummer G, Garde S, García A E, Paulaitis M E, Pratt L R. Proc Natl Acad Sci USA. 1998;95:1552–1555. doi: 10.1073/pnas.95.4.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Peng X, Jonas A, Jonas J. Biochemistry. 1995;34:8631–8641. doi: 10.1021/bi00027a012. [DOI] [PubMed] [Google Scholar]
- 32.Dill K A, Chan H S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 33.Nymeyer H, Garcia A E, Onuchic J N. Proc Natl Acad Sci USA. 1998;95:5921–5928. doi: 10.1073/pnas.95.11.5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bryngleson J D, Onuchic J N, Socci N D, Wolynes P G. Proteins Struct Funct Genet. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]