

Abstract
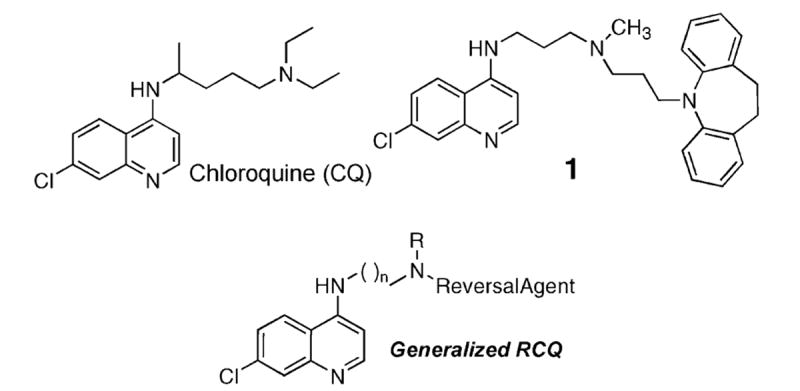
A class of hybrid molecules which we term ‘reversed chloroquines’ (RCQs) was designed, and a prototype molecule, N′-(7-chloroquinolin-4-yl)-N-[3-(10,11-dihydrodibenzo[b,f]azepin-5-yl)propyl]-N-methylpropane-1,3-diamine (1), was synthesized and tested against both chloroquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum. An in vitro assay against the two strains indicated that 1 was effective at low-nM concentrations against both strains. A preliminary study in mice demonstrated oral efficacy against P. chabaudi and the absence of obvious toxicity. The RCQ approach therefore appears to be feasible.
Introduction
In terms of human suffering, malaria is clearly the most important parasitic disease. Furthermore, the worldwide burden of malaria is increasing, in part due to the unfortunate spread of resistance to most, if not all, of the drugs that were once effective and safe.1,2 Among these drugs, chloroquine (CQ)1 had been the prime therapy for nearly half a century. CQ was safe, effective, widely available, and remarkably inexpensive and could be administered to pregnant women and infants, but P. falciparum, the cause of the most deadly variety of malaria, is now CQ-resistant (CQR) in nearly all malarious regions of the globe. The continuing spread of CQR and resistance to alternative drugs has helped fuel a strong increase in incidence and consequence of malaria worldwide.2 It has become generally accepted that in order to delay the emergence of resistance to new antimalarial agents, combination therapy is the way forward.2-4 In the search for new candidates, it seemed to us that the advantages of CQ are simply too strong to abandon; we thus set out to use its haloquinoline core, but linked to an entity that would overcome CQR.
CQ resistance in P. falciparum malaria has been found to be strongly associated with mutations in a parasite digestive vacuole (DV) membrane protein, termed PfCRT.5-8 Excessive export of CQ from its site of action in the DV is thought to result from these mutations. Many structurally diverse molecules, termed reversal agents (RA), have been identified that are known to inhibit P. falciparum chloroquine resistance transporter (PfCRT)-associated CQ export from the DV in CQR parasites.9-12 Others have deduced a pharmacophore for this RA activity.13 This pharmacophore may be described as a pair of aromatic rings, often with an aliphatic nitrogen atom a few angstroms removed from the aromatic rings. Such molecules include the Ca2+-channel blocker verapamil and the antidepressant imipramine, among many others. We hypothesized that linking a CQ-like moiety to a RA might yield a highly effective drug against malaria. The approach of linking two separately active drugs is rare, but does have positive precedent,14 yet to our knowledge this is the first attempt to block CQ export from the DV. Such a construct would deliver the RA in a one-to-one ratio with the quinoline moiety, thereby lowering the dose of required when the two molecules are given separately. This would be predicted to be the case since both low DV pH and binding of CQ to heme and/or hemozoin in the DV would favor obligatory accumulation of both components in the DV.15 In addition to promoting drug accumulation in the DV, we speculate that the RA component would interfere with export from the DV of the CQ moiety by the mutated CQR PfCRT. Together, these effects could allow a curative dose against CQR strains to be much lower than that of its components given separately, with resultant diminished cost and toxicity. Our objective thus became one of linking a CQ-like moiety to that of a RA (Scheme 1). We term such a combination molecule a ‘Reversed Chloroquine’ (RCQ; Schemes 1 and 2). This is our first report on RCQs, in which we demonstrate that the approach is valid.
Scheme 1.
Scheme 2.
Results and Discussion
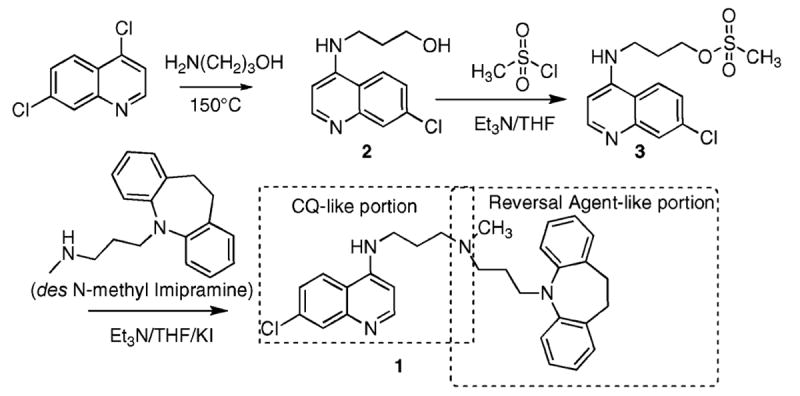
We synthesized 1 according to Scheme 2. The parent imipramine is among the better-studied PfCRT reversal agents known.13,16-18 The mode of action for CQ is not certain, but has widely been suggested as involving interaction with heme, a byproduct of hemoglobin digestion by the Plasmodium parasite. We therefore evaluated the binding of heme by both CQ and 1 using UV–vis spectroscopy (Figure 1). A binding stoichiometry of one drug molecule to one heme2 (μ-oxo dimer) was assumed throughout, and this assumption gave good fits to the data (e.g., the inset to Figure 1). At pH 5.7, near the DV pH,6 the measured binding constant (3 × 105 M−1) was found to be the same as measured for CQ (3 × 105 M−1). The measured binding constant at pH 7 (also 3 × 105 M−1) was also found to be comparable to that which we measured for CQ (1 × 106 M−1) under identical conditions. Our methods were validated in that the CQ-heme2 binding constants are similar to literature values.19,20 The conclusion is that the RA portion of 1 does not appear to affect the CQ-like binding to heme.
Figure 1.
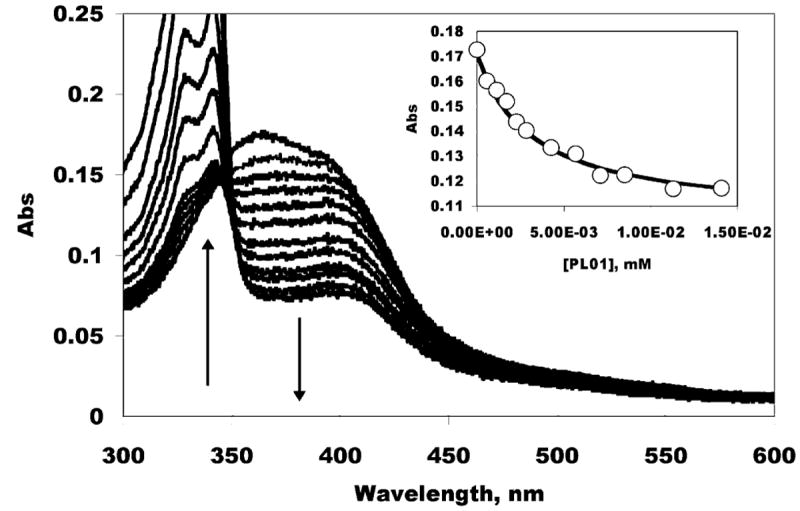
Optical titration of 1 into heme, pH 5.7. The increasing bands between 300 and 350 nm are from 1, the decreasing region from 350 to 450 nm is from heme. Inset: absorbance (circles) and fit described in the text.
The in vitro drug susceptibility assays of 1 against P. falciparum parasites are shown in Table 1, with the corresponding results for CQ as a reference. Compound 1 is at least as effective against either strain as is CQ against the CQS strain. The very low IC50 value for 1 against the CQS D6 strain means that attaching the RA moiety is not detrimental to its CQ-like activity, and its resistance-subverting activity against CQR Dd2 is a remarkable result for a first attempt at a lead compound. The observation that 1 has lower IC50 values than CQ for either strain may indicate that PfCRT in even the D6 strain is able to export CQ, but not as efficiently as does the Dd2 strain.
Table 1.
IC50 Values against P. falciparuma
| Drug | Cell line | CQR/CQS | IC50 (nM) |
|---|---|---|---|
| CQ | D6 | CQS | 6.5 |
| CQ | Dd2 | CQR | 102 |
| 1 | D6 | CQS | 2.9 |
| 1 | Dd2 | CQR | 5.3 |
The uncertainties are estimated to be ±15%, based on weighing uncertainties for 1 (which is an oil), as well as on variability between determinations that were performed on different weeks.
The prototype RCQ molecule 1 is probably too hydrophobic a molecule to be a strong drug candidate against malaria in humans; however, our pilot in vivo trial still demonstrated at least some oral bioavailability in mice. It was found that 64 mg/kg/day suppressed more than 99% of P. chabaudi growth after 4 days of dosing; lower dosing produced no observed suppression of the Plasmodium. Although CQ is effective at a lower dose in this animal model,21 the results serve as proof-of-concept and justification to pursue pharmacologic optimization. The reasons for the higher required dose of 1 are not known but may involve its lipophilic character, its metabolism in the liver, or other factors.
In conclusion, the RCQ approach seems to be viable. The prototype molecule 1 has demonstrated growth inhibition of P. falciparum CQR or CQS parasites in vitro and after oral dosing in vivo. We are synthesizing other candidate molecules that will address the practical concerns of oral availability and metabolism.
Experimental Section
General and Spectroscopy
All reagents and solvents were purchased from Aldrich and used as supplied. NMR spectral characterizations were done on a Tecmag Libra-modified NM-500 NMR spectrometer, operating at 499.8 MHz using simple one-pulse observation and C2HCl3 solvent, or with a Bruker AMX-400 NMR spectrometer operating at 400.14 MHz for 1H observation, or 100.62 MHz for 13C observation. UV–vis spectra were recorded with an Ocean Optics USB-2000 diode array spectrophotometer equipped with a cuvette accessory. The procedures for performing the titrations and processing the data were the same as published,20 after the data were exported directly into an Excel (Microsoft) spreadsheet for analysis.
3-(7-Chloroquinolin-4-ylamino)propan-1-ol (2)
A mixture of 4,7-dichloroquinoline (25.35 g, 0.128 mol) and 3-aminopropanol (120 mL, 1.57 mol) were heated with stirring at 130–140 °C for 24 h.22 After cooling, the reaction was poured into water (500 mL) and filtered, and the solid residue was dried then boiled in ethyl acetate (250 mL) to give 2 (27.3 g, 90%) as an off-white solid. 1H NMR (500 MHz, CDCl3) δ1.80 (bs, 1H, OH), 2.05 (m, J = 5.9, 5.6 Hz, 2H, CH2), 3.46 (td, J = 5.9, ~6 Hz, 2H, CH2), 3.98 (t, J = 5.4 Hz, 2H, CH2), 5.95 (bs, 1H, NH), 6.36 (d, J = 5.4 Hz, 1H, ClQ-C3-H), 7.31 (dd, J = 2.1, 8.9 Hz, 1H, ClQ-C6-H), 7.60 (d, J = 8.9 Hz, 1H, ClQ-C5-H), 7.92 (d, J = 2.0 Hz, 1H, ClQ-C8-H), 8.49 (d, J = 5.3 Hz, 1H, ClQ-C2-H).
3-(7-Chloroquinolin-4-ylamino)propyl Methanesulfonate (3)
To a suspension of 2 (0.5 g, 2.1 mmol) in anhydrous THF (10 mL) under a nitrogen atmosphere was added triethylamine (0.66 mL, 4.2 mmol).23 The mixture was cooled to below 0 °C. Methanesulfonyl chloride (0.17 mL, 2.2 mmol) was added slowly, keeping the temperature below 5 °C, and the reaction was stirred in an ice bath for 45 min. After dilution with saturated NaHCO3 solution (20 mL), the reaction was extracted with ether (20 mL then 2 × 10 mL). The organic extracts were dried over MgSO4, filtered, and evaporated to leave 3 (0.42 g, 63%) as a white solid. 1H NMR (500 MHz, CDCl3) δ2.18 (m, J = ~6.1 Hz, 2H, CH2), 3.04 (s, 3H, CH3), 3.59 (td, J = ~6.1 Hz, 2H, CH2), 4.43 (t, J = 5.7 Hz, 2H, CH2), 5.44 (bs, 1H, NH), 6.43 (d, J = 5.3 Hz, 1H, ClQ-C3-H), 7.40 (dd, J = 2.2, 8.9 Hz, 1H, ClQ-C6-H), 7.70 (d, J = 8.9 Hz, 1H, ClQ-C5-H), 7.97 (d, J = 2.2 Hz, 1H, ClQ-C8-H), 8.55 (d, J = 5.4 Hz, 1H, ClQ-C2-H).
N′-(7-Chloroquinolin-4-yl)-N-[3-(10,11-dihydrodibenzo-[b,f]azepin-5-yl)propyl]-N-methylpropane-1,3-diamine (1)
Desimipramine hydrochloride (0.65 g, 2.15 mmol) was dissolved in water (8 mL), and solid NaHCO3 (0.4 g, 4.9 mmol) was added with stirring.23 After addition of dichloromethane (10 mL), two clear layers resulted. The aqueous layer was removed and extracted with dichloromethane (2 × 10 mL). The combined organic layers were evaporated to leave desimipramine free base (0.63 g, 100%) as a yellow oil. To this oil were added anhydrous THF (15 mL) and 3 (0.42 g, 1.33 mmol) followed by triethylamine (0.4 mL, 2.9 mmol). After being stirred at 50–60 °C for 72 h, the reaction was allowed to cool to room temperature and the solvent evaporated under reduced pressure. The residue was partitioned between ethyl acetate (20 mL) and saturated NaHCO3 solution (20 mL). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic layers were dried over MgSO4, filtered, and evaporated. The residue was chromatographed on alumina (MCB type F20, 80–200 mesh), eluting with ethyl acetate:hexanes (30:70) to give 1 (0.53 g, 84%) as a yellow oil. Based on 1H NMR, 13C NMR, HR-MAS, and HPLC (Supporting Information), 1 was at least 98% pure. 1H NMR (500 MHz, CDCl3) δ 1.80 (m, J = ~2 Hz, 2H, CH2), 1.84 (m, J = ~2 Hz, 2H, CH2), 2.28 (s, 3H, CH3), 2.49 (t, J = 7.4 Hz, 2H, CH2), 2.54 (t, J = 5.3 Hz, 2H, CH2), 3.10 (s, 4H, 2×CH2), 3.28 (td, J = ~3.9, 2H, CH2), 3.72 (t, J = 6.7 Hz, 2H, CH2), 6.24 (d, J = 5.3 Hz, 1H, ClQ-C3-H), 6.83–7.10 (m, 8H, Ar), 7.13 (dd, J = 2.0, 8.9 Hz, 1H, ClQ-C6-H), 7.43 (d, J = 8.9 Hz, 1H, ClQ-C5-H), 7.67 (bs, 1H, NH), 7.91 (d, J = 2.0 Hz, 1H, ClQ-C8-H), 8.48 (d, J = 5.3 Hz, 1H, ClQ-C2-H). 13C NMR (100 MHz, CDCl3) δ 24.4, 26.0, 32.2, 42.3, 44.2, 48.7, 56.2, 57.9, 98.4, 117.6, 119.8, 121.6, 122.6, 124.9, 126.4, 128.6, 129.8, 134.1, 134.6, 148.0, 149.2, 150.5, 152.1. MS (ESI): m/z 485.2461 M + H (Calculated 485.2472).
In Vitro Drug Susceptibility Assays
Both CQS (D6) and CQR (Dd2) P. falciparum maintained continuously in culture were used. Asynchronous cultures were diluted with uninfected erythrocytes and complete medium (RPMI-1640 with 0.5% Albumax II) to achieve 0.2% parasitemia and 2% hematocrit. In 96-well microplates, chloroquine (positive control) or 1 diluted in complete medium from 10 mM stock in DMSO were added to the cell mixture to yield triplicate wells with drug concentrations ranging from 0 to 10−4 M in a final well volume of 100 μL. After 72 h of incubation under standard culture conditions, plates were harvested and read by the SYBR Green I fluorescence-based method24 using a 96-well fluorescence plate reader (Gemini-EM, Molecular Devices), with excitation and emission wavelengths at 497 and 520 nm, respectively. The fluorescence readings were plotted against log[drug], and the IC50 values were obtained from curve fitting performed by nonlinear regression using either Prism (GraphPad) or Excel (Microsoft) software.
In Vivo 4-Day Suppressive Test25
Female CF-1 mice, at 4–5 weeks of age, were injected intravenously with 106 erythrocytes infected with CQR P. chabaudi.26,27 The following day, and then daily for four total doses, four mice each were administered 64, 32, 16, or 0 mg/kg of compound 1 by gavage and evaluated by direct microscopic analysis of Giemsa-stained blood smears28 1 day after the final dose.
Acknowledgments
The authors thank the Medical Research Foundation of Oregon, the National Institutes of Health, and the U.S. Department of Defense for funding that enabled parts of this work.
a Abbreviations
- CQ
chloroquine
- CQR
chloroquine-resistant
- CQS
chloroquine-sensitive
- DV
digestive vacuole
- PfCRT
P. falciparum chloroquine resistance transporter
- RA
reversal agent
- RCQ
reversed chloroquine
Footnotes
Supporting Information Available: Structural characterization of compound 1, as well as an evaluation of its purity. This information consists of 1H and 13C NMR spectra and 13C NMR peak assignments, as well as HR-MS plots and HPLC traces. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Krogstad DJ. Malaria as a reemerging disease. Epidemiol Rev. 1996;18(1):77–89. doi: 10.1093/oxfordjournals.epirev.a017918. [DOI] [PubMed] [Google Scholar]
- 2.Greenwood BM, Bojang K, Whitty CJ, Targett GA. Malaria. Lancet. 2005;365(9469):1487–98. doi: 10.1016/S0140-6736(05)66420-3. [DOI] [PubMed] [Google Scholar]
- 3.Ashley EA, White NJ. Artemisinin-based combinations. Curr Opin Infect Dis. 2005;18(6):531–6. doi: 10.1097/01.qco.0000186848.46417.6c. [DOI] [PubMed] [Google Scholar]
- 4.Schellenberg D, Abdulla S, Roper C. Current Issues for Anti-Malarial Drugs to Control P. falciparum Malaria. Curr Mol Med. 2006;6(2):253–60. doi: 10.2174/156652406776055168. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Paguio M, Roepe PD. The antimalarial drug resistance protein Plasmodium falciparum chloroquine resistance transporter binds chloroquine. Biochemistry (Mosc) 2004;43(26):8290–6. doi: 10.1021/bi049137i. [DOI] [PubMed] [Google Scholar]
- 6.Bennett TN, Kosar AD, Ursos LM, Dzekunov S, Singh Sidhu AB, Fidock DA, Roepe PD. Drug resistance-associated pfCRT mutations confer decreased Plasmodium falciparum digestive vacuolar pH. Mol Biochem Parasitol. 2004;133(1):99–114. doi: 10.1016/j.molbiopara.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Martin RE, Kirk K. The malaria parasite’s chloroquine resistance transporter is a member of the drug/metabolite transporter superfamily. Mol Biol Evol. 2004;21(10):1938–49. doi: 10.1093/molbev/msh205. [DOI] [PubMed] [Google Scholar]
- 8.Ginsburg H. Should chloroquine be laid to rest? Acta Trop. 2005;96(1):16–23. doi: 10.1016/j.actatropica.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 9.Martin SK, Oduola AM, Milhous WK. Reversal of chloroquine resistance in Plasmodium falciparum by verapamil. Science. 1987;235(4791):899–901. doi: 10.1126/science.3544220. [DOI] [PubMed] [Google Scholar]
- 10.Krogstad DJ, Gluzman IY, Kyle DE, Oduola AM, Martin SK, Milhous WK, Schlesinger PH. Efflux of chloroquine from Plasmodium falciparum: mechanism of chloroquine resistance. Science. 1987;238(4831):1283–5. doi: 10.1126/science.3317830. [DOI] [PubMed] [Google Scholar]
- 11.van Schalkwyk DA, Walden JC, Smith PJ. Reversal of chloroquine resistance in Plasmodium falciparum using combinations of chemosensitizers. Antimicrob Agents Chemother. 2001;45(11):3171–4. doi: 10.1128/AAC.45.11.3171-3174.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Millet J, Torrentino-Madamet M, Alibert S, Rogier C, Santelli-Rouvier C, Mosnier J, Baret E, Barbe J, Parzy D, Pradines B. Dihydroethanoanthracene derivatives as in vitro malarial chloroquine resistance reversal agents. Antimicrob Agents Chemother. 2004;48(7):2753–6. doi: 10.1128/AAC.48.7.2753-2756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhattacharjee AK, Kyle DE, Vennerstrom JL, Milhous WK. A 3D QSAR pharmacophore model and quantum chemical structure—sactivity analysis of chloroquine(CQ)-resistance reversal. J Chem Inf Comput Sci. 2002;42(5):1212–20. doi: 10.1021/ci0200265. [DOI] [PubMed] [Google Scholar]
- 14.Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48(21):6523–43. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 15.Egan TJ, Mavuso WW, Ross DC, Marques HM. Thermodynamic factors controlling the interaction of quinoline antimalarial drugs with ferriprotoporphyrin IX. J Inorg Biochem. 1997;68(2):137–45. doi: 10.1016/s0162-0134(97)00086-x. [DOI] [PubMed] [Google Scholar]
- 16.Bhattacharjee AK, Kyle DE, Vennerstrom JL. Structural analysis of chloroquine resistance reversal by imipramine analogs. Antimicrob Agents Chemother. 2001;45(9):2655–7. doi: 10.1128/AAC.45.9.2655-2657.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menezes CM, Kirchgatter K, Di Santi SM, Savalli C, Monteiro FG, Paula GA, Ferreira EI. Antimalarial effect in vitro and lack of modulating effect of desipramine and imipramine. Trans R Soc Trop Med Hyg. 1997;91(6):697–700. doi: 10.1016/s0035-9203(97)90529-0. [DOI] [PubMed] [Google Scholar]
- 18.Miki A, Tanabe K, Nakayama T, Kiryon C, Ohsawa K. Plasmodium chabaudi: association of reversal of chloroquine resistance with increased accumulation of chloroquine in resistant parasites. Exp Parasitol. 1992;74(2):134–42. doi: 10.1016/0014-4894(92)90040-h. [DOI] [PubMed] [Google Scholar]
- 19.Dorn A, Vippagunta SR, Matile H, Jaquet C, Vennerstrom JL, Ridley RG. An assessment of drug-haematin binding as a mechanism for inhibition of haematin polymerisation by quinoline antimalarials. Biochem Pharmacol. 1998;55(6):727–36. doi: 10.1016/s0006-2952(97)00510-8. [DOI] [PubMed] [Google Scholar]
- 20.Xu Kelly J, Winter R, Riscoe M, Peyton DH. A spectroscopic investigation of the binding interactions between 4, 5- dihydroxy-xanthone and heme. J Inorg Biochem. 2001;86(2–3):617–25. doi: 10.1016/s0162-0134(01)00217-3. [DOI] [PubMed] [Google Scholar]
- 21.Hunt P, Cravo PV, Donleavy P, Carlton JM, Walliker D. Chloroquine resistance in Plasmodium chabaudi: are chloroquine-resistance transporter (crt) and multi-drug resistance (mdr1) orthologues involved? Mol Biochem Parasitol. 2004;133(1):27–35. doi: 10.1016/j.molbiopara.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 22.Bolte J, Demuynck C, Lhomme J. Synthetic models of DNA complexes with antimalarial compounds. 2. The problem of guanine specificity in chloroquine binding. J Med Chem. 1977;20(1):106–13. doi: 10.1021/jm00211a022. [DOI] [PubMed] [Google Scholar]
- 23.Fujita M, Seki T. Syntheses and bioactivities of novel carbamates combining platelet activating factor (PAF) receptor antagonist with thromboxane synthase inhibitor (TxSI) Bioorg Med Chem Lett. 2002;12(10):1383–6. doi: 10.1016/s0960-894x(02)00186-5. [DOI] [PubMed] [Google Scholar]
- 24.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother. 2004;48(5):1803–6. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peters W. The chemotherapy of rodent malaria, XXII. The value of drug-resistant strains of P. berghei in screening for blood schizontocidal activity. Ann Trop Med Parasitol. 1975;69(2):155–71. [PubMed] [Google Scholar]
- 26.Mackinnon MJ, Read AF. Virulence in malaria: an evolutionary viewpoint. Philos Trans R Soc London B Biol Sci. 2004;359(1446):965–86. doi: 10.1098/rstb.2003.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mackinnon MJ, Walker PR, Rowe JA. Plasmodium chabaudi: rosetting in a rodent malaria model. Exp Parasitol. 2002;101(2–3):121–8. doi: 10.1016/s0014-4894(02)00103-0. [DOI] [PubMed] [Google Scholar]
- 28.Hommel M. Diagnostic methods in malaria. 4. Arnold; London, New York: 2002. pp. 35–58. [Google Scholar]