

Abstract
Ordered protein complexes are often formed from partially ordered fragments that are difficult to structurally characterize by conventional NMR and crystallographic techniques. We show that concentration-dependent hydrogen exchange studies of a fragment complex can provide structural information about the solution structures of the isolated fragments. This general methodology can be applied to any bimolecular or multimeric system. The experimental system used here consists of Ribonuclease S, a complex of two fragments of Ribonuclease A. Ribonuclease S and Ribonuclease A have identical three-dimensional structures but exhibit significant differences in their dynamics and stability. We show that the apparent large dynamic differences between Ribonuclease A and Ribonuclease S are caused by small amounts of free fragments in equilibrium with the folded complex, and that amide exchange rates in Ribonuclease S can be used to determine corresponding rates in the isolated fragments. The studies suggest that folded RNase A and the RNase S complex exhibit very similar dynamic behavior. Thus cleavage of a protein chain at a single site need not be accompanied by a large increase in flexibility of the complex relative to that of the uncleaved protein.
Structural characterization of partially folded proteins and peptides is an important but difficult task. Such partially folded structures are often formed during protein folding. In several protein/protein and protein/peptide complexes, the complex is well structured, but one or both of the individual partners (or part of the polypeptide chain) is partially or completely unfolded before binding (1, 2). Structural characterization of partially and completely unfolded polypeptides can therefore provide considerable insight into protein folding and macromolecular recognition (3–5). Conventional methods of structure determination such as multidimensional NMR and crystallography cannot easily be applied to determine structures of partially folded molecules (6–8). Such molecules are difficult to crystallize and often aggregate at the high concentrations required for NMR.
Equilibrium and kinetic experiments, which couple hydrogen exchange with two-dimensional (2D) NMR, provide useful information on partially folded states (9–11). We show here that it is possible to use concentration-dependent hydrogen exchange studies of a bimolecular complex to provide structural information about the individual uncomplexed fragments that are present in small quantities in equilibrium with the folded complex. This is a method to structurally characterize such partially folded structures.
Fragment complementation systems are two or more fragments of a protein that can be reconstituted to give a complex with similar structure and activity to that of the native uncleaved protein. Fragment complementation systems are powerful tools in the study of protein folding and stability (1, 12). Ribonuclease S (RNase S) is a well studied fragment complementation system. It consists of a noncovalent complex between S peptide (S pep; residues 1–20) and S protein (S pro; residues 21–124), two proteolytic fragments of the 124-aa residue protein, Ribonuclease A (RNase A; ref. 13). An NMR structure is available (14, 15) for RNase A with preliminary data for RNase S. Refined 3D x-ray structures of RNase A and RNase S are available at high resolution (16, 17). The two proteins have very similar structures and enzymatic activity but exhibit significant differences in their dynamics and stability (18–21). RNase S has been used as a model system to study the thermodynamics of protein folding and stability (22–25). Although RNase A and RNase S have identical crystal structures (17), it is believed that RNase S shows much larger fluctuations about its time-averaged structure in solution. The evidence for the larger fluctuation is as follows. At room temperature, RNase S is readily cleaved by trypsin, whereas RNase A is indefinitely stable (26, 27). Both tritium exchange (28) and Fourier-transformed IR-deuterium exchange studies (21, 29, 30) have reported that rates of exchange of amide protons from RNase S are faster than those of RNase A (31). Data from thermodynamic studies have also been used to support the assertion that RNase S is more flexible than RNase A (20). However, a comparative analysis of RNase A and RNase S by using x-ray crystallography (17) and molecular dynamic simulations (27) did not show any differences in either structure or dynamics between RNase A and RNase S. It was recently suggested (27) that the apparent observed differences in the dynamics of the two proteins can be ascribed to the small amounts of free S pep and S pro that are in equilibrium with the RNase S complex (Fig. 1). The dynamic properties of RNase S should therefore be concentration dependent (27, 32). In the present work, we provide quantitative support for this hypothesis by carrying out concentration-dependent hydrogen exchange studies of RNase A and RNase S by using one-dimensional (1D) and 2D NMR. A simple model was used to estimate the exchange rates of individual protons in the isolated fragments from the measured rates in RNase S. The model was validated by predicting the concentration dependence of hydrogen exchange in RNase S, as measured by 2D NMR. The data indicated that a specific subdomain of the isolated S pro fragment was protected from exchange.
Figure 1.

Schematic model for the exchange of two hypothetical protons in RNase S by two parallel pathways. Pathway I involves exchange in the RNase S complex (k1ex), and pathway II involves exchange through the dissociated fragments (k2ex).
MATERIALS AND METHODS
Sample Preparation.
RNase A (Type XII A) and Subtilisin Carlsberg were purchased from Sigma. RNase S was prepared from RNase A by Subtilisin digestion (33). S pro and S pep were separated by gel filtration (G-50, Fine, Amersham Pharmacia) in 10% Formic acid. S pro, RNase S, and RNase A were purified further via cation exchange on a Resource S column as described (27). The samples were dialyzed extensively against MilliQ grade water to remove the salt or formic acid used during the purification. All proteins were found to be >99% pure based on silver-stained SDS/PAGE gels and mass spectroscopy. Protein and peptide concentrations were estimated as described (22).
Hydrogen Exchange by Using 1D and 2D NMR.
The protonated protein was dissolved in water and the pH adjusted to 6.0 with acetic acid. The required amount of protein (estimated optically) from the above was aliquoted and lyophilized. Exchange of the protonated protein was initiated by adding 100% 2H2O (D2O) to the lyophilized protein. The time of addition of D2O was taken as the zero time of the experiment. The pH of the solution (uncorrected for isotope effects) was adjusted to pH 6 by using small amounts of deuterated acetic acid. Samples were centrifuged and then transferred to the NMR tube. 1D NMR experiments for RNase A were carried out at 298 K in a Bruker (Billerica, MD) AMX-400 MHz spectrometer with sample concentrations of 2.5 mM and 0.5 mM. Each spectrum was the average of 50 scans for 2.5 mM and 819 scans for 0.5 mM (each scan takes 5 s). 1D NMR experiments for RNase S were carried out at 298 K in a Bruker DRX-500 MHz spectrometer at concentrations of 2.5 mM, 1 mM, and 0.1 mM. Each spectrum was the average of eight scans for 2.5 mM, 200 scans for 1 mM, and 700 scans for 0.1 mM (each scan takes 4 s). Around ten time points (30 for 0.1 mM) were collected over a course of 100 hr at all the concentrations. The number of amide protons protected at each time point were obtained by comparing the area under the amide region from 9.75 to 7.38 ppm with respect to a single well-resolved peak at 7.41 ppm. The error in estimation of total amide protons was determined from repeated integrations of the same spectrum as well as from repeat experiments and is about three protons.
Total correlation spectroscopy (TOCSY) experiments were carried out to determine the exchange rates of the individual amide protons of RNase S at 2.5 mM, 1 mM, and 0.5 mM. Presaturation of the water signal was used for water suppression. Data sets consisted of 256 t1 increments with 8, 16, and 24 scans at each t1 point. A spectral width of 6,000 Hz was used in both the dimensions. Each TOCSY spectrum was acquired in 45 min, 1.5 hrs and 3 hrs for 2.5 mM, 1 mM and 0.5 mM RNase S respectively. Eleven TOCSY spectra for 2.5 mM, ten TOCSY spectra for 1 mM, and twelve TOCSY spectra for 0.5 mM RNase S were acquired on a Bruker DRX-500 MHz spectrometer. Previously known assignments of RNase A (14, 34, 35) were used to assign the present TOCSY spectra for RNase S. 2D double quantum filtered correlation experiments were carried out on a Varian Unity + 600-MHz spectrometer to determine the exchange rates of individual protons of RNase A. Each data set consisted of 512 t1 increments with 32 scans and took 8 hr for completion. 2D nuclear Overhauser effect spectroscopy and TOCSY experiments on both RNase A and RNase S were used to resolve any ambiguities in the assignments. Volumes of positive crosspeaks in the 2D spectra were calculated by using the package xwinnmr to determine the exchange rates of the amide protons. The volumes were normalized with respect to the crosspeak volume of a nonexchangeable aromatic proton from residue Y97. Exchange rates (kexSi) for RNase S amide protons (Table 1) were calculated by fitting the normalized volume of the protons over time to the equation I = Io exp(−kex⋅t) + I(∞), where I is the normalized volume at time t and kex is the rate constant of exchange for the proton.
Table 1.
Experimental exchange rate constants determined by 2D NMR at 25°C for protons common to RNase A and RNase S.
| Residue | *krci (hr−1) | RNase A
|
RNase S
|
S pro
|
|||
|---|---|---|---|---|---|---|---|
| kexAi (hr−1) | PF × 10−6 | kexSi, hr−1 | PF × 10−6 | k2exi, hr−1 | PF × 100 | ||
| M30 | 6327 | 0.11 | 0.059 | 0.12 | 0.052 | 1.54 | 4104 |
| F46 | 4584 | 0.0009 | 5.1 | 0.2 | 0.023 | 20 | 228 |
| V54 | 663 | 0.00037 | 1.8 | 0.036 | 0.018 | 3.6 | 183 |
| Q55 | 4180 | 0.0032 | 1.3 | 0.5 | 0.0083 | 50 | 83 |
| V57 | 1003 | 0.00032 | 3.1 | 0.067 | 0.015 | 6.7 | 149 |
| C58 | 12918 | 0.00024 | 54 | 0.084 | 0.15 | 8.5 | 1520 |
| S59 | 33979 | 0.1 | 0.34 | 0.32 | 0.11 | 31 | 1080 |
| Q60 | 11514 | 0.012 | 0.99 | 0.23 | 0.049 | 22 | 512 |
| K61 | 7265 | 0.00001 | 726 | 0.27 | 0.027 | 27 | 265 |
| V63 | 2095 | 0.00079 | 2.7 | 0.08 | 0.026 | 8.1 | 258 |
| C65 | 17832 | 0.01 | 1.8 | 0.27 | 0.065 | 26 | 667 |
| C72 | 37257 | 0.018 | 2.0 | 0.12 | 0.3 | 10 | 3480 |
| Y73 | 7784 | 0.000096 | 81 | 0.2 | 0.039 | 20 | 385 |
| Q74 | 6475 | 0.000083 | 78 | 0.23 | 0.028 | 24 | 273 |
| M79 | 7784 | 0.00013 | 58 | 0.11 | 0.066 | 12 | 654 |
| I81 | 1867 | 0.00001 | 186 | 0.10 | 0.018 | 10 | 176 |
| T82 | 2519 | 0.002 | 1.3 | 0.18 | 0.014 | 17 | 143 |
| D83 | 3992 | 0.032 | 0.13 | 0.18 | 0.023 | 14 | 272 |
| C84 | 11782 | 0.0016 | 7.4 | 0.14 | 0.085 | 13 | 853 |
| R85 | 17427 | 0.026 | 0.67 | 0.11 | 0.16 | 8 | 2107 |
| K91 | 9146 | 0.01 | 0.92 | 0.37 | 0.025 | 36 | 251 |
| Y97 | 2699 | 0.01 | 0.27 | 0.25 | 0.011 | 24 | 110 |
| K98 | 5143 | 0.018 | 0.28 | 0.5 | 0.01 | 48 | 106 |
| I100 | 6780 | 0.00001 | 677 | 0.12 | 0.057 | 11 | 569 |
| K104 | 9577 | 0.00089 | 11 | 0.20 | 0.046 | 20 | 460 |
| I106 | 6327 | 0.000055 | 114 | 0.089 | 0.071 | 9 | 700 |
| I107 | 551 | 0.00001 | 55 | 0.074 | 0.0074 | 7.5 | 73 |
| V108 | 590 | 0.000036 | 16 | 0.087 | 0.0068 | 8.8 | 67 |
| A109 | 3641 | 0.000047 | 77 | 0.048 | 0.076 | 4.844 | 752 |
| C110 | 17832 | 0.0012 | 14 | 0.075 | 0.24 | 7.42 | 2403 |
| E111 | 4476 | 0.017 | 0.27 | 0.15 | 0.03 | 13.918 | 322 |
| V116 | 1125 | 0.00012 | 9.5 | 0.029 | 0.04 | 2.927 | 384 |
| V118 | 577 | 0.00012 | 4.8 | 0.33 | 0.0018 | 33.04 | 17 |
| H119 | 22973 | 0.00054 | 42 | 0.25 | 0.09 | 25.73 | 893 |
For RNase A DQFC spectra, the volumes of 25 protons did not decrease appreciably even after 80 hr of exchange. Exchange rates for all RNase A protons under the present conditions were therefore estimated by using the hydrogen exchange data of Wang et al. (35), who have measured the rates of 45 protons, both intermediate and slow exchanging, of RNase A by collecting data at a higher temperature (35°C) and at two different pH values (pH 6.5 and pH 7.4). These measured rates were extrapolated to pH 6.0, 25°C, by correcting for the difference in chemical exchange (36) as a function of pH and temperature. The chemical exchange rates were calculated at 25°C by using low salt conditions (37). In the case of a few protons (M30, S59, and A56) for which rates could be directly measured under the present conditions, the measured rates agreed well with those extrapolated from the data of Wang et al. (35). The extrapolated rates also correlate well with the experimental measurements of Neira et al. (37), who recently measured the exchange rates for RNase A and RNase S in 200 mM NaCl and 50 mM phosphate buffer, pH 6.0. All RNase A rates listed in Table 1 are therefore obtained by extrapolation of the data of Wang et al. (35) to pH 6. The chemical exchange rates (krci in units of hr−1) at 25°C, low salt, pH 6 (uncorrected for isotope effect or pH 6.4 if corrected for the isotope effect) are also listed. The chemical exchange rates at 35°C for pH 7.4 and pH 6.5 (krc in units of min−1) are listed in Table 1 of ref. 35.
RESULTS
Concentration-Dependent Hydrogen Exchange.
1D NMR was used to determine the total number of unexchanged protons in the amide region as a function of time in both RNase A and RNase S at pH 6, 298 K. pH 6 was chosen for the experiments because extensive thermodynamic data on S pep binding to S pro are available under these conditions (22, 23, 25). At higher temperatures, S pro undergoes thermal unfolding (23), and it was therefore necessary to carry out the experiments at 298 K. Experiments were carried out at three different protein concentrations. Fig. 2 shows the concentration dependence of overall exchange for RNase S and RNase A determined by using 1D NMR. For RNase A the curves at two different concentrations (2.5 mM and 0.5 mM) overlap. Thus the overall exchange rate of amide protons in RNase A is independent of concentration, as one would expect for a single polypeptide chain. In contrast, for RNase S, the exchange shows a distinct concentration dependence at all the concentrations investigated, unlike RNase A. It was not possible to collect 1D NMR exchange data for S pro because of sample aggregation (26, 38). However, exchange rates of individual protons in S pro could be predicted from the corresponding rates of RNase A and RNase S protons by using a simple model described in a later section.
Figure 2.

Concentration-dependent overall exchange by using 1D NMR in RNase S at three concentrations, 2.5 mM (●), 1 mM (Δ), and 0.1 mM (■). (Inset) Hydrogen exchange for RNase A at two concentrations, 2.5 mM (□, ■), and 0.5 (▴).
Comparative Exchange of RNase A and RNase S at 1 mM.
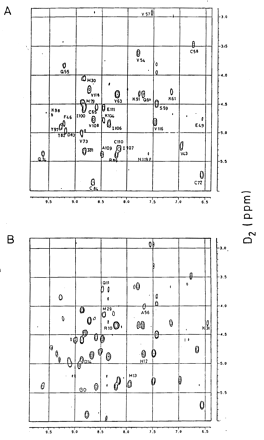
The 2D NMR spectra for RNase A and RNase S were used to calculate the individual exchange rates for the amide protons of RNase S and RNase A (see Materials and Methods). TOCSY spectra of RNase A and RNase S at 1 mM after 1 hr of exchange are shown (see the supplemental data on the PNAS web site, www.pnas.org). None of the amide protons in the S pep region are visible in the RNase S spectrum, whereas R10, Q11, H12, M13, and D14 are clearly visible in the RNase A spectrum. Amide protons 11–13 are slow-exchanging protons and remain protected even after 80 hr of exchange. Rates of exchange of protons common to both RNase A and RNase S are listed in Table 1. These exchange rates have been used to calculate the protection factors (PF) for the individual protons. Although the x-ray structures as well as NMR spectra for RNase A and RNase S are similar (14, 17), most protons in RNase A have protection factors 10 to 100-fold higher than in RNase S. Fig. 3 A and B show the PFs for amide protons of RNase A and RNase S. The lower protection factors in RNase S relative to RNase A have been generally ascribed to an increased dynamic flexibility of the RNase S complex relative to folded RNase A (20, 21). In the present work, we show that this view is incorrect and propose an alternative model to account for the hydrogen exchange data.
Figure 3.

Protection factors for RNase S (A), RNase A (B), and S pro (C). (D) Residues of S pro that are occluded (48) by S pep in the RNase S complex.
Model for Exchange in RNase S.
A fundamental difference between RNase A and RNase S is that RNase A is a unimolecular system, whereas RNase S is a bimolecular system. The ratio of folded to unfolded RNase A is independent of the total (folded + unfolded) RNase A concentration. In contrast, the ratio of undissociated to dissociated RNase S will be a function of the total (folded RNase S + S pro) RNase S concentration (39). The dissociation is described by the following equations:
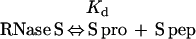 |
1 |
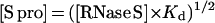 |
2 |
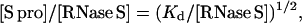 |
3 |
where Kd is the dissociation constant and the S pep and S pro concentrations are equal. Eq. 3 shows that the ratio [S pro]/[RNase S] decreases with increase in RNase S concentration. In the 1D NMR studies discussed above, the ratio [S pro]/[RNase S] ranges from 3% at 0.1 mM RNase S to 0.6% at 2.5 mM RNase S.
Because the dissociation constant of RNase S is about 10−7 M, it has previously been assumed that the small amount of S pro present in most experiments would not affect the observed exchange rates (26–28), and the fast exchange in RNase S is a reflection of the flexibility of the RNase S complex. Recent molecular dynamics simulations (27) of RNase A and RNase S showed that the RNase S complex and folded RNase A appear to have very similar dynamic behavior. To explain these contradictory results, we propose a model in which the hydrogen exchange in RNase S can take place via two parallel pathways (Fig. 1). The first pathway involves exchange from the undissociated RNase A complex, whereas in the second pathway exchange occurs through the small amounts of free S pep and S pro present in solution at equilibrium.
The S pro fragment is a partially folded structure, whereas S pep is a random coil at 25°C. Two hypothetical amide protons from the S pep and S pro regions have been indicated in Fig. 1. Both protons are inaccessible to solvent in the RNase S complex. However, one proton lies in a region of S pro that remains folded even after dissociation, whereas the other is in S pep. If exchange took place primarily through the first pathway, both protons should exchange slowly. However, if the second pathway were important, the proton on S pep should exchange much faster than the proton on S pro. The fact that there are several slow-exchanging protons in the S pep region in RNase A but none in RNase S strongly suggests that the second pathway is important.
Both pathways can contribute to exchange for every amide proton in RNase S. k1 and k−1 are the rate constants for dissociation and association of S pep from S pro (40). k1ex and k2ex are the rate constants of exchange in the RNase S complex and dissociated fragments, respectively. The value of k−1 is 1.2 × 106 M−1 s−1 as measured by stopped-flow studies (40) at pH 6.8 and 30°C. Under similar conditions, the measured dissociation constant (Kd) for RNase S is 10−7 M (23, 40). Hence k1 is estimated to be approximately 1.2 × 10−1 s−1. Stopped-flow experiments at pH 6.0, 25°C give similar kinetic parameters (R.V., unpublished results). From the model outlined in Fig. 1, the equations describing exchange kinetics at an amide proton ‘i ’ in the S pro region of RNase S are given by:
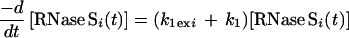 |
4 |
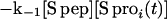 |
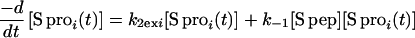 |
5 |
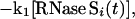 |
where the subscript i indicates the site of exchange, and the rate constants are as described in Fig. 1. The concentration of S pep is the total concentration of free peptide present at equilibrium. The exchange for a proton in the S pep region can be described by equations identical to Eqs. 4 and 5, except that [S pep] is replaced by [S pro] (although the two quantities are numerically equal) and [S proi(t)] by [S pepi(t)]. Exchange at site i in the RNase S complex is described by a single exponential with an apparent rate constant kexSi and hence:
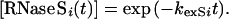 |
6 |
It can be shown that the solution to Eqs. 4 and 5 is given by
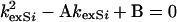 |
7 |
with A = k1exi + k1 + k−1 [S pep] + k2exi, B = k−1 k1exi [S pep] + k2exi k1exi + k2exi k1.
Solving Eq. 7, one obtains,
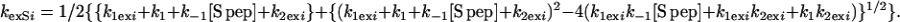 |
8 |
Thus kexSi, the apparent exchange rate of the ith proton in the RNase S complex, can be calculated based on the exchange rates of the same proton in the dissociated fragment (k2exi), and k1exi ⋅ k1exi is the rate of exchange in the RNase S complex in the absence of exchange through its fragments (see Fig. 1) and is assumed to be equal to kexAi, the experimentally determined rate of exchange of the proton in RNase A. The value of [S pep] at any concentration of RNase S can be calculated from Kd (Eq. 2). Values of k2exi for protons in the S pep region are assumed to be equal to the corresponding random coil values because S pep is unstructured at room temperature (22), and the exchange rates are consistent with the observed lack of protection for S pep protons in RNase S. The value of k2exi, the exchange rate of the ith proton in the S pro fragment of RNase S, can be determined from Eq. 9.
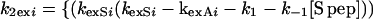 |
9 |
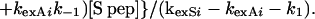 |
Eq. 9 is obtained by substituting the values of A, B, and kexSi in Eq. 7 and assuming that kexAi is equal to k1exi. Both kexAi and kexSi, the experimentally determined rates of individual protons in RNase A and RNase S at 1 mM, are tabulated in Table 1. From these data, the exchange rates of various amide protons of S pro, k2exi, can be predicted by using Eq. 9. It is not possible to obtain values of k2exi from conventional NMR experiments on S pro, as the sample aggregates at concentrations suitable for NMR studies. Under conditions where only the second pathway of exchange in Fig. 1 is important, then under the assumption of EX2 conditions for exchange, Eq. 9 can be simplified to:
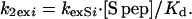 |
10 |
Eq. 10 provides a convenient method of predicting the concentration dependence of exchange but is subject to the validity of the two assumptions made above.
Prediction of Exchange Kinetics as a Function of RNase S Concentration.
The 1D exchange curves in Fig. 2 show that the overall exchange rate is concentration dependent in RNase S but, as expected, is independent of concentration in RNase A. The concentration dependence suggests that the second pathway in Fig. 1 strongly contributes to exchange in RNase S, which was confirmed by the results of 2D exchange measurements. The measured values of kexsi for 1 mM RNase S were substituted into Eq. 9 to derive values of k2exi for S pro. These values of k2exSi were substituted into Eq. 8 and used to predict values of kexSi for RNase S protons at protein concentrations of 2.5 and 0.5 mM. The predicted values of kexSi are listed in Table 2 (see the supplemental data on the PNAS web site, www.pnas.org) and are very similar to values predicted from Eq. 10. The predicted and experimental values of kexSi at these concentrations are compared in Fig. 4. There is good agreement between the experimental and calculated rates for these concentrations of RNase S that validates the model used. The agreement also validates one of the assumptions of the model, namely that exchange rates in the RNase S complex are similar to those of RNase A, and the reason for the faster exchange in RNase S is exchange through dissociated fragments of RNase S, in contrast to what has been widely believed for almost 40 years about this important fragment complementation system.
Figure 4.

Comparison of experimental and calculated individual proton exchange rates for 2.5 mM (□), 1 mM (●), and 0.5 mM (Δ) RNase S. The exchange rates at 2.5 mM and 0.5 mM are calculated from the experimentally measured 1 mM rates as described in the text. The solid line represents a line with slope equal to 1.
A Subdomain of S pro Is Protected from Exchange.
The hydrogen exchange rates derived for S pro (k2exi) provide valuable structural information about this protein fragment. The S pro is a folded (42) stable structure that undergoes a two-state reversible thermal transition at neutral pH characterized by the following thermodynamic parameters: Tm = 39°C, ΔH = 170 kJ/mol and ΔCp= 2.2 kJ/mol (43). The low value of ΔCp is much lower than the value of 5.5 kJ/mol calculated by using the coordinates of the S pro region in the RNase S complex. This suggests that dissociation of S pro from S pep is accompanied by an appreciable loss of S pro tertiary structure (42).
S pro has a faster overall hydrogen exchange rate than RNase S (28) and unlike RNase A (26) is susceptible to proteolytic attack by trypsin (27). Although S pro is 20 residues shorter than RNase A, it has a similar Stokes radius and is therefore believed to be a less compact structure. The aggregation of S pro above 10 μM (26, 27) has impeded efforts to crystallize the protein or solve its structure by NMR methods (G.S.R. and R.V., unpublished observations). The aggregation has been confirmed by concentration-dependent differential scanning calorimetry studies (43).
The deduced exchange rates of the S pro amide protons (Table 1) can be used to calculate the PFs of these protons (Fig. 3C). The slowest exchanging protons in S pro have protection factors in the 103 range, compared with 108 for RNase S and 106 for RNase A. The protection factors for S pro are higher than those of molten globules (101) (44) and A-states (45) and are comparable to those of native monomeric globular proteins such as α-lactalbumin (44), barstar (46), and chymotrypsin inhibitor 2 (47).
The surface occluded by the S pep on S pro in RNase S is shown in Fig. 3D by using the intchos routine of the occluded surface algorithm (48). With the exception of residues 30 and 110, none of these residues have high protection factors in S pro. The locations of highly protected regions of S pro are mapped onto the structure of the S pro region (Fig. 5) in the molscript (49) representation of the RNase S complex. Fig. 5 indicates that the N-terminal part of the S pro molecule is not well protected. Many of the highly protected protons (PFs >0.5 × 103) are in a single region of the tertiary structure. This protected region includes the third helix (residues 56–60 are in 310 conformation), two beta strands (residues 61–64; 72–76), which follow the 310 helix and also the beta strands (residues 105–111; 116–119) near the C terminus. These regions are well separated from each other in the primary sequence of the protein but are close together in the tertiary structure. Two disulfide bridges (58–110; 65–72) are part of this well protected region and may play a role in the stabilization of this domain. The ΔCp (50) calculated (ΔCp = 0.25 ± 0.02 × 4.18 × ΔAnp/1,000) by using the coordinates of the residues in the protected region of S pro (darker shading) is 1.4 kJ/mol and accounts for a significant amount (60%) of the experimentally determined ΔCp of S pro.
Figure 5.

Protected protons mapped onto the tertiary structure of a MOLSCRIPT (48) representation of the S pro fragment of RNase S. (●) Protons with PF > 500; (•) protons with PF < 500. Highly protected protons are clustered in a single region of tertiary structure (darker shade). Disulfide bonds are shown as thin black lines.
The methodology developed in the present work can be extended readily to any bimolecular complex. Concentration-dependent studies of hydrogen exchange in the complex by using either NMR, tritium exchange, or mass spectrometry can be used to determine whether exchange through the dissociated fragments is significant. In most cases, it should be possible to manipulate solution conditions and protein concentrations to ensure that this is indeed the case. If significant concentration dependence is observed, the observed exchange rates in the complex can be used to deduce exchange rates in the isolated fragments by making the assumption that exchange through the complex is negligible and that exchange takes place exclusively through the dissociated fragments. The assumption would correspond to having k1ex in Fig. 1 equal to zero and by using Eq. 10 if EX2 conditions apply. The protection factors for the isolated fragments calculated by using this simplifying assumption would thus represent a lower limit of the true protection factors. In the present case, if we assumed that k1ex was zero instead of the values observed in RNase A, the calculated protection factors differ by only a small amount (about 20% overall) from the true values. Such exchange studies are therefore a powerful tool to obtain structural and dynamic information about disordered protein subunits and protein fragments.
Supplementary Material
Acknowledgments
We are grateful to the Sophisticated Instruments Facility (SIF), Indian Institute of Science, for the use of the 400-MHz and 500-MHz Bruker NMR spectrometers. We thank Virod Nayak and Chandrashekar (SIF) for assistance in NMR data collection. We thank Dr. Jayant Udgaonkar, Dr. Abani Bhuyan, National Center for Biological Sciences, Tata Institute of Fundamental Research (TIFR) Centre, Prof. Anil Kumar, and Dr Grace Cristhy Rani (Physics Dept, IISc) for useful discussions. G.C. is a Council for Scientific and Industrial Research (CSIR), India, fellow. This work was supported by grants CSIR/37(913)96-EMR-II and DST/SP/SO/D-21/93 to R.V.
ABBREVIATIONS
- S pep
S peptide
- S pro
S protein
- 1D
one-dimensional
- 2D
two-dimensional
- TOCSY
total correlation spectroscopy
- PF
protection factor
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.de Prat-Gay G. Protein Eng. 1996;9:843–847. doi: 10.1093/protein/9.10.843. [DOI] [PubMed] [Google Scholar]
- 2.Dyson H J, Wright P E. Nat Struct Biol (NMR Suppl) 1988;5:499–503. doi: 10.1038/739. [DOI] [PubMed] [Google Scholar]
- 3.Dobson C M. Curr Biol. 1994;4:636–640. doi: 10.1016/s0960-9822(00)00141-x. [DOI] [PubMed] [Google Scholar]
- 4.Dotsch V, Wider G, Siegal G, Wuthrich K. FEBS Lett. 1995;366:6–10. doi: 10.1016/0014-5793(95)00459-m. [DOI] [PubMed] [Google Scholar]
- 5.Shortle D R. Curr Opin Struct Biol. 1996;46:171–177. doi: 10.1016/s0959-440x(96)80091-1. [DOI] [PubMed] [Google Scholar]
- 6.Evans P A, Kautz R A, Fox R O, Dobson C M. Biochemistry. 1989;28:362–370. doi: 10.1021/bi00427a050. [DOI] [PubMed] [Google Scholar]
- 7.Ni F, Scheraga H A. J Am Chem Soc. 1994;27:257–264. [Google Scholar]
- 8.Wuthrich K. Curr Opin Struct Biol. 1994;4:93–99. [Google Scholar]
- 9.Englander S W, Mayne L. Annu Rev Biophys Biomol Struct. 1992;21:243–265. doi: 10.1146/annurev.bb.21.060192.001331. [DOI] [PubMed] [Google Scholar]
- 10.Baldwin R L. Curr Opin Struct Biol. 1993;3:84–91. [Google Scholar]
- 11.Mayo S L, Baldwin R L. Science. 1993;262:873–876. doi: 10.1126/science.8235609. [DOI] [PubMed] [Google Scholar]
- 12.Taniuchi H T, Parr G R, Juillerat M A. Methods Enzymol. 1986;131:185–217. doi: 10.1016/0076-6879(86)31042-5. [DOI] [PubMed] [Google Scholar]
- 13.Richards F M, Vithyathil P J. J Biol Chem. 1959;234:1459–1465. [PubMed] [Google Scholar]
- 14.Rico M, Bruix M, Santoro J, Gonzalez C, Neira J L, Nieto J L, Herranz J. Eur J Biochem. 1989;183:623–638. doi: 10.1111/j.1432-1033.1989.tb21092.x. [DOI] [PubMed] [Google Scholar]
- 15.Santoro J, Gonzalez C, Bruix M, Neira J L, Nieto J L, Herranz J, Rico M. J Mol Biol. 1993;229:722–734. doi: 10.1006/jmbi.1993.1075. [DOI] [PubMed] [Google Scholar]
- 16.Wlodawer A, Svensson L A, Sjolin L, Gilliland G L. Biochemistry. 1988;27:2705–2717. doi: 10.1021/bi00408a010. [DOI] [PubMed] [Google Scholar]
- 17.Kim E E, Varadarajan R, Wyckoff H W, Richards F M. Biochemistry. 1992;31:12304–12314. doi: 10.1021/bi00164a004. [DOI] [PubMed] [Google Scholar]
- 18.Richards F M, Wyckoff H W. The Enzymes. 1971;4:647–806. [Google Scholar]
- 19.Blackburn P, Moore S. The Enzymes. 1982;15:317–433. [Google Scholar]
- 20.Catanzano F, Giancola C, Graziano G, Barone G. Biochemistry. 1996;35:13378–13385. doi: 10.1021/bi960855h. [DOI] [PubMed] [Google Scholar]
- 21.Dong A, Hyslop R M, Pringle D L. Arch Biochem Biophys. 1996;333:275–281. doi: 10.1006/abbi.1996.0391. [DOI] [PubMed] [Google Scholar]
- 22.Connelly P R, Varadarajan R, Sturtevant J M, Richards F M. Biochemistry. 1990;29:6108–6114. doi: 10.1021/bi00477a031. [DOI] [PubMed] [Google Scholar]
- 23.Varadarajan R, Connelly P R, Sturtevant J M, Richards F M. Biochemistry. 1992;31:1421–1426. doi: 10.1021/bi00120a019. [DOI] [PubMed] [Google Scholar]
- 24.Varadarajan R, Richards F M. Biochemistry. 1992;31:12315–12327. doi: 10.1021/bi00164a005. [DOI] [PubMed] [Google Scholar]
- 25.Thomson J, Ratnaparkhi G S, Varadarajan R, Sturtevant J M, Richards F M. Biochemistry. 1994;33:8587–8593. doi: 10.1021/bi00194a025. [DOI] [PubMed] [Google Scholar]
- 26.Allende J E, Richards F M. Biochemistry. 1962;1:295–304. doi: 10.1021/bi00908a017. [DOI] [PubMed] [Google Scholar]
- 27.Nadig G, Ratnaparkhi G S, Varadarajan R, Vishveshwara S. Protein Sci. 1996;5:2104–2114. doi: 10.1002/pro.5560051017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosa J J, Richards F M. J Mol Biol. 1981;145:835–850. doi: 10.1016/0022-2836(81)90318-1. [DOI] [PubMed] [Google Scholar]
- 29.Haris P I, Lee D C, Chapman D. Biochem Biophys Acta. 1986;874:255–265. doi: 10.1016/0167-4838(86)90024-5. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto T, Tasumi M. J Mol Biol. 1991;242:235–244. [Google Scholar]
- 31.Woodward C K, Rosenberg A. J Biol Chem. 1971;246:4105–4113. [PubMed] [Google Scholar]
- 32.Ide G J, Barksdale A D, Rosenberg A. J Am Chem Soc. 1976;98:1595–1596. doi: 10.1021/ja00422a060. [DOI] [PubMed] [Google Scholar]
- 33.Neumann U, Hofsteenge J. Protein Sci. 1994;3:248–256. doi: 10.1002/pro.5560030209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robertson A D, Purisima E O, Eastman M A, Scheraga H A. Biochemistry. 1989;28:5930–5938. doi: 10.1021/bi00440a033. [DOI] [PubMed] [Google Scholar]
- 35.Wang A, Robertson A D, Bolen D W. Biochemistry. 1995;34:15096–15104. doi: 10.1021/bi00046a016. [DOI] [PubMed] [Google Scholar]
- 36.Bai Y, Milne J S, Mayne L, Englander S W. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neira J L, Sevilla P, Menendez M, Bruix M, Rico M. J Mol Biol. 1999;285:627–643. doi: 10.1006/jmbi.1998.2365. [DOI] [PubMed] [Google Scholar]
- 38.Gawronski T H, Wold F. Biochemistry. 1972;11:442–448. doi: 10.1021/bi00753a023. [DOI] [PubMed] [Google Scholar]
- 39.Schreier A A, Baldwin R L. J Mol Biol. 1976;105:409–426. doi: 10.1016/0022-2836(76)90101-7. [DOI] [PubMed] [Google Scholar]
- 40.Labhardt A M, Ridge J A, Lindquist R N, Baldwin R L. Biochemistry. 1983;22:321–326. doi: 10.1021/bi00271a014. [DOI] [PubMed] [Google Scholar]
- 41.Hearn R P, Richards F M, Sturtevant J M, Watt G D. Biochemistry. 1971;10:806–810. doi: 10.1021/bi00781a013. [DOI] [PubMed] [Google Scholar]
- 42.Shindo H, Matsuura S, Cohen J S. Experientia. 1979;35:1284–1285. doi: 10.1007/BF01963958. [DOI] [PubMed] [Google Scholar]
- 43.Graziano G, Catanzano F, Giancola C, Barone G. Biochemistry. 1996;35:13386–13392. doi: 10.1021/bi960856+. [DOI] [PubMed] [Google Scholar]
- 44.Schulman B A, Redfield C, Peng Z, Dobson C M, Kim P S. J Mol Biol. 1995;253:651–657. doi: 10.1006/jmbi.1995.0579. [DOI] [PubMed] [Google Scholar]
- 45.Morozova-Roche L A, Haynie D T, Arico-Muendel C, Van Dael H, Dobson C M. Nat Struct Biol. 1995;2:871–875. doi: 10.1038/nsb1095-871. [DOI] [PubMed] [Google Scholar]
- 46.Wong K, Fersht A R, Freund S M V. J Mol Biol. 1997;268:494–511. doi: 10.1006/jmbi.1997.0989. [DOI] [PubMed] [Google Scholar]
- 47.Neira J L, Itzhakhi L S, Otzen D E, Davis B, Fersht A R. J Mol Biol. 1997;270:99–110. doi: 10.1006/jmbi.1997.1088. [DOI] [PubMed] [Google Scholar]
- 48.Pattabhiraman N, Ward K B, Fleming P J. J Mol Recognit. 1995;8:334–344. doi: 10.1002/jmr.300080603. [DOI] [PubMed] [Google Scholar]
- 49.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 50.Privalov P, Gill S J. Annu Rev Biophys Chem. 1988;39:191–234. doi: 10.1016/s0065-3233(08)60377-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}