

Abstract
The final step of ethylene biosynthesis in plants is catalyzed by the enzyme 1-aminocyclopropane-1-carboxylic acid (ACC) oxidase (ACCO). In addition to ACC, Fe(II), O2, CO2, and ascorbate are required for in vitro enzyme activity. Direct evidence for the role of the Fe(II) center in the recombinant avocado ACCO has now been obtained through formation of enzyme⋅(substrate or cofactor)⋅NO complexes. These NO adducts convert the normally EPR-silent ACCO complexes into EPR-active species with structural properties similar to those of the corresponding O2 complexes. It is shown here that the ternary Fe(II)ACCO⋅ACC⋅NO complex is readily formed, but no Fe(II)ACCO⋅ascorbate⋅NO complex could be observed, suggesting that ascorbate and NO are mutually exclusive in the active site. The binding modes of ACC and the structural analog alanine specifically labeled with 15N or 17O were examined by using Q-band electron nuclear double resonance (ENDOR). The data indicate that these molecules bind directly to the iron through both the α-amino and α-carboxylate groups. These observations are inconsistent with the currently favored mechanism for ACCO, in which it is proposed that both ascorbate and O2 bind to the iron as a step in O2 activation. We propose a different mechanism in which the iron serves instead to simultaneously bind ACC and O2, thereby fixing their relative orientations and promoting electron transfer between them to initiate catalysis.
The common industrial chemical ethylene plays an important role in nature as a plant hormone that regulates fruit ripening and other aspects of the growth process (1, 2). Plants synthesize ethylene to time development, and, as a consequence, exogenous control of its production would be a powerful tool with far-reaching environmental and economic ramifications. The ultimate step of ethylene biosynthesis is the oxidation of 1-aminocyclopropane-1-carboxylic acid (ACC) catalyzed by ACC oxidase (ACCO) (3, 4), which in vitro requires the addition of Fe(II), O2, CO2, and ascorbate (5–7). Previous studies of ACCO invoked a catalytic role for Fe(II) (8–10), but the nature of this role has remained obscure. The dominant proposal posits the initiating step in catalysis to be ascorbate-driven oxygen activation. In this scenario, ascorbate association with the metal ion activates the bound O2 to yield a high-valent iron-oxo species that could readily oxidize ACC to release ethylene (11, 12). Here, we report the use of the O2 analog nitric oxide (NO) to convert the electron paramagnetic resonance (EPR)-silent Fe(II) active site of recombinant avocado ACCO into a form that can be probed by EPR and electron nuclear double resonance (ENDOR) spectroscopies. This investigation provides direct evidence for the role of the Fe(II) center in ACCO catalysis and suggests a different mechanism for ethylene formation in plants.
MATERIALS AND METHODS
All chemicals used in this study were analytical grade or better (Sigma, Fisher, and Pharmacia) and were used without further purification. NO (Matheson) was bubbled through concentrated NaOH prior to introduction into samples. Argon gas, used for anaerobic procedures, was passed over copper catalyst (BASF) at 170°C to remove trace O2. All protein concentrations were determined by using the Sigma bicinchoninic acid protein assay kit, which is based on a modified Lowry procedure (13).
Growth, Overexpression, and Purification of ACCO in Escherichia coli BL21(DE3)pLysS.
E. coli BL21(DE3)pLysS/pETe3–4C2 possessing the AVOe3 plasmid was grown either in 1-liter shaker flasks as described in ref. 14 or in a New Brunswick Scientific MF114 fermentor (10-liter working volume). The pH was maintained at 7.0 ± 0.1 by the controlled addition of NH4OH.
In the fermentor, E. coli cells were grown in 2× Luria–Bertani (LB) medium supplemented with 2.5% glucose, 0.4 mg/ml ampicillin, and 50 μg/ml chloramphenicol at 37°C. The dissolved oxygen level was maintained by agitation (400 rpm) and by sparging with air at a vessel pressure of 7 psi (48 kPa). Foaming was suppressed by the addition of antifoam (Sigma). One half of the ampicillin was added at the beginning of the growth and the second half was added when the OD550 reached approximately 1. ACCO production was induced by the addition of isopropyl β-d-thiogalactoside (IPTG) to a final concentration of 2 mM. IPTG was added while the cultures were still in logarithmic phase and had depleted the glucose (OD550 ≈ 12). Induction was carried out at 27°C to increase the amount of enzyme expressed in the soluble fraction. The cells were batch fed glucose to 0.25% every 30 min for 4 h, at which time cells were harvested by centrifugation. The cell pellet was frozen and stored at −80°C.
Cells were resuspended in 20 mM Bis-Tris, pH 6.0/1.0 mM EDTA/1.0 mM DTT/10% (vol/vol) glycerol, disrupted by sonication, and spun at 40,000 × g for 30 min. Supernatant was loaded onto Q-Sepharose (Pharmacia) equilibrated with 20 mM Bis-Tris, pH 6.0/1.0 mM EDTA/1.0 mM DTT/50 mM NaCl/10% glycerol. ACCO was eluted with a gradient of 0–0.5 M NaCl. The sample was mixed with an equal volume of 50 mM Tris pH 7.5 buffer containing 1.0 mM EDTA, 1.0 mM DTT, 10% glycerol, and 1 M ammonium sulfate and loaded onto a phenyl-Sepharose (Pharmacia) column preequilibrated with similar buffer but with 0.5 M ammonium sulfate. Protein was eluted with a gradient of 0.5–0 M ammonium sulfate. Pooled fractions were loaded onto a Superdex-75 (Pharmacia) gel filtration column equilibrated with 200 mM Mops, pH 7.2/10% glycerol. Active fractions were pooled, concentrated if necessary, and stored at −80°C (refer to Table 1).
Table 1.
Purification of recombinant ACCO
| Purification step | Total protein, mg | Total activity, nmol C2H4/min | Specific activity, nmol C2H4/min per mg protein | Activity recovery, % | Purification, fold |
|---|---|---|---|---|---|
| Lysate | 9,262 | 96,320 | 10.4 | 100 | 1 |
| Q-Sepharose | 1,558 | 71,028 | 45.6 | 73.7 | 4.4 |
| Phenyl-Sepharose | 452 | 33,208 | 73.4 | 34.5 | 7.1 |
| Superdex-75 | 370 | 28,176 | 75.8 | 29.3 | 7.3 |
Cell pellet (100 g) was lysed and recombinant ACCO was purified as described in Materials and Methods.
ACCO activity was measured in the presence of 100 mM Mops at pH 7.2, 10 mM NaHCO3, 12.5 mM ascorbate, 20 μM Fe(NH4)2(SO4)2, 2 mg/ml BSA, and 1.0 mM ACC. Evolved C2H4 was measured by injecting head-space gas samples onto a Hewlett Packard 5890 gas chromatograph outfitted with a 30 m × 0.25 mm DB-1 hydrophobic column (Alltech). Separations were carried out at 50°C and gas peaks were detected with a flame ionization detector. Peak integration was performed with a Hewlett Packard 3396 Series II integrator, and the peak area of a given sample was compared with a five-point standard C2H4 curve.
The enzyme obtained from this purification protocol (see Table 1) had a specific activity in the range of 2.3–3.1 mol of ethylene per min per mol of ACCO, which is comparable to other reports of recombinant ACCO purification protocols (15).
EPR and ENDOR Spectroscopy of ACCO.
All biochemical procedures were performed at 4°C. All samples were made from deoxygenated stock solutions by using gas-tight syringes. Stocks were deoxygenated through repeated cycles of evacuation and flushing with argon. All samples were prepared in 200 mM Mops, pH 7.2/50 mM NaHCO3/10% glycerol. Fe(II)ACCO was prepared by reconstitution of apo-ACCO with 0.8 molar equivalent of ferrous ammonium sulfate. For the enzyme complexes with NO and substrates or substrate analogs, the following concentrations were used: 10 mM ACC, 10 mM α-aminoisobutyric acid (AIB), 100 mM alanine, 100 mM cyclopropane carboxylic acid (CCA), 100 mM cyclopropylamine (CPA), or 100 mM glycine.
EPR spectra were obtained with a Bruker E500 spectrometer equipped with an Oxford ESR10 cryostat and using the following conditions for the nitrosyl complexes: temperature, 8 K; microwave frequency, 9.23 GHz; microwave power, 0.160 mW; modulation frequency, 100 kHz; and modulation amplitude, 10 G. EPR quantifications were performed by double integration under nonsaturating conditions versus Cu(II)EDTA and Fe(II)EDTA–NO standards.
EPR spectra of the S = 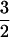 complexes were analyzed according to the spin Hamiltonian:
complexes were analyzed according to the spin Hamiltonian:
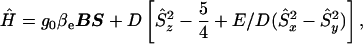 |
where D and E/D are zero field-splitting parameters and other parameters have their usual definitions. The term E/D is a measure of the departure of the electronic environment of the iron from axial symmetry, and changes in the E/D are diagnostic of changes in the environment of the iron. E/D values were determined by using rhombo version 1.0 from W. R. Hagen of the Agricultural University in the Netherlands. This program calculates all effective g values for high-spin Kramers systems. Note that a g0 of 2.017 was required to obtain the exact g values for a given E/D.
Q-band (35-GHz) ENDOR spectra were recorded in either continuous wave (CW) (16) or pulsed (17) mode with instruments of local design. CW and pulsed experiments gave virtually identical data. For a single molecular orientation, the first-order ENDOR response from a nucleus with I = ½ (e.g., 1H, 15N) is a doublet with frequencies ν± = |νN ± AN/2|, where A is its orientation-dependent hyperfine coupling and νN the nuclear Larmor frequency. For nuclei with I > ½, (e.g. 14N, I = 1; 17O, I = 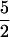 ) each peak of the doublet is further split into 2I lines, each separated by 3P, with P being the quadrupole coupling parameter. The ν- feature is often absent in Q-band ENDOR spectra (15). An ENDOR experiment performed at the low- and high-field edges (“single-crystal like” positions) of an EPR signal interrogates only a single orientation, or subpopulation; at intermediate fields a well-defined subset of orientations is examined; analysis of such “orientation-selective” spectra can permit complete analysis of hyperfine and quadrupole interaction tensors (18).
) each peak of the doublet is further split into 2I lines, each separated by 3P, with P being the quadrupole coupling parameter. The ν- feature is often absent in Q-band ENDOR spectra (15). An ENDOR experiment performed at the low- and high-field edges (“single-crystal like” positions) of an EPR signal interrogates only a single orientation, or subpopulation; at intermediate fields a well-defined subset of orientations is examined; analysis of such “orientation-selective” spectra can permit complete analysis of hyperfine and quadrupole interaction tensors (18).
Isotopic Labeling of d-Alanine.
Carboxyl oxygens of dl-alanine were labeled with 17O (I = 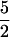 ) by using procedures described in ref. 19. dl-Alanine was dissolved in H217O (isotopic enrichment approximately 39%), acidified to 1 M HCl, and incubated in vacuo at 121°C for 24 h. Isotopic enrichment of dl-alanine was confirmed with mass spectrometry.
) by using procedures described in ref. 19. dl-Alanine was dissolved in H217O (isotopic enrichment approximately 39%), acidified to 1 M HCl, and incubated in vacuo at 121°C for 24 h. Isotopic enrichment of dl-alanine was confirmed with mass spectrometry.
RESULTS
EPR Spectroscopy.
Reconstitution of as-isolated enzyme with Fe(II) followed by anaerobic exposure to NO afforded a nearly axial (E/D = 0.008) EPR spectrum (Fig. 1, upper trace) characteristic of an S = 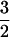 [Fe–NO]7 complex, similar to those reported for a number of Fe(II)-dependent enzymes (19–22). Addition of ascorbate to the preformed Fe(II)ACCO⋅NO complex caused no change in the spectrum. However, preincubation of Fe(II)ACCO with ascorbate followed by exposure to NO led to a markedly lower intensity of the S =
[Fe–NO]7 complex, similar to those reported for a number of Fe(II)-dependent enzymes (19–22). Addition of ascorbate to the preformed Fe(II)ACCO⋅NO complex caused no change in the spectrum. However, preincubation of Fe(II)ACCO with ascorbate followed by exposure to NO led to a markedly lower intensity of the S = 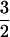 spectrum. The EPR intensity decreased in a hyperbolic fashion with increasing ascorbate concentration, with an apparent dissociation constant (Kd) of ≈1.5 mM; this value compares well with a Km of 2.1 mM for ascorbate during turnover (unpublished data). These results suggest that ascorbate excludes NO from the active site iron. If NO is a good model for O2, then it is unlikely that ascorbate binding to the iron initiates the O2 activation process as proposed by others in the earlier studies (11, 12).
spectrum. The EPR intensity decreased in a hyperbolic fashion with increasing ascorbate concentration, with an apparent dissociation constant (Kd) of ≈1.5 mM; this value compares well with a Km of 2.1 mM for ascorbate during turnover (unpublished data). These results suggest that ascorbate excludes NO from the active site iron. If NO is a good model for O2, then it is unlikely that ascorbate binding to the iron initiates the O2 activation process as proposed by others in the earlier studies (11, 12).
Figure 1.

Nitrosyl complexes of Fe(II)ACCO. Upper trace, X-band EPR spectrum (low-field region) of Fe(II)ACCO⋅NO; lower trace, Fe(II)ACCO⋅ACC⋅NO (10 mM ACC). (Inset) Fraction of substrate-bound enzyme plotted versus [ACC]free. Conditions: T = 8 K; frequency, 9.23 GHz; power, 0.160 mW; modulation frequency, 100 kHz.
In contrast to the case just described, ACC and NO readily bind simultaneously to the active-site Fe(II) of ACCO. The pronounced effect of ACC on the electronic structure of the ACCO nitrosyl complex is easily observed as a substantial increase in the rhombicity of the S = 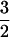 EPR spectrum (E/D = 0.035; Fig. 1, lower trace). The order of the addition of ACC and NO has no effect on the EPR spectrum of the resulting ACCO complex. The microwave power required to cause half-saturation (P1/2) of the EPR signal increases by an order of magnitude over that of the Fe(II)ACCO⋅NO complex, further indicating that substrate binding perturbs the metal site. The shift in spectral line shape with ACC binding showed saturation behavior (Fig. 1 Inset) with Kd = 0.14 mM. This value compares favorably with the Km found for ACC (0.07 mM) during steady-state turnover (unpublished data). Ascorbate added before or after NO and ACC to ACCO had no effect on the EPR spectrum of the resulting Fe(II)ACCO⋅ACC⋅NO complex, demonstrating that the Fe(II)ACCO active site has a much higher affinity for ACC than for ascorbate.
EPR spectrum (E/D = 0.035; Fig. 1, lower trace). The order of the addition of ACC and NO has no effect on the EPR spectrum of the resulting ACCO complex. The microwave power required to cause half-saturation (P1/2) of the EPR signal increases by an order of magnitude over that of the Fe(II)ACCO⋅NO complex, further indicating that substrate binding perturbs the metal site. The shift in spectral line shape with ACC binding showed saturation behavior (Fig. 1 Inset) with Kd = 0.14 mM. This value compares favorably with the Km found for ACC (0.07 mM) during steady-state turnover (unpublished data). Ascorbate added before or after NO and ACC to ACCO had no effect on the EPR spectrum of the resulting Fe(II)ACCO⋅ACC⋅NO complex, demonstrating that the Fe(II)ACCO active site has a much higher affinity for ACC than for ascorbate.
Several substrate analogs were added to Fe(II)ACCO⋅NO to evaluate the structural requirements for proper interaction of the substrate with the Fe(II) active site (Table 2). The E/D = 0.035 species was obtained only when the analog had both α-amino and α-carboxylate groups. Inhibitors lacking either functionality afforded EPR spectra similar in appearance to the spectrum of substrate-free Fe(II)ACCO⋅NO. However, their P1/2 values were closer to the value of Fe(II)ACCO⋅ACC⋅NO, suggesting that these analogs do bind to and perturb the metal center.
Table 2.
EPR-derived data for ACCO⋅NO complexes
| Compound* | E/D | %† | P1/2,‡ mW |
|---|---|---|---|
| No added compound | 0.008 | 0 | 0.95 |
| ACC | 0.035 | 100 | 8.5 |
| ACC analogs | |||
| Cyclopropane carboxylic acid | 0.008 | 9.1 | |
| Cyclopropylamine | 0.011 | 11.2 | |
| α-Aminoisobutyric acid | 0.035/0.008 | 84.3 | 10.4 |
| d-Alanine | 0.035/0.008 | 80.7 | ND |
| l-Alanine | 0.035/0.008 | 40.2 | ND |
| Glycine | 0.035/0.008 | 5.3 | ND |
All samples contain 0.21 mM Fe(II)ACCO, 50 mM HaHCO3, 200 mM Mops at pH 7.2, and 10% glycerol.
ACC and α-aminoisobutyric acid concentrations were 10 mM and the concentration of all other compounds was 100 mM.
This value refers to the percent of S = 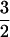 spins possessing an E/D = 0.035.
spins possessing an E/D = 0.035.
Power at half-saturation. ND, not determined.
These spectroscopic studies are consistent with in vivo turnover studies (23–25), which indicated that there are binding sites in the ACCO active site for both the α-amino and α-carboxyl groups of substrate analogs. However, no serious consideration has been given to the idea that substrate orientation within the active site is achieved by binding both the amino and carboxylate groups of ACC to the Fe(II) center itself. Such a configuration would be analogous to the bidentate coordination of catechols to the Fe(II) center of extradiol dioxygenases (19) or the α-keto acid cofactor to the Fe(II) center of α-ketoglutarate-dependent oxygenases (26–28), both of which have recently been crystallographically demonstrated (29–31).
ENDOR Spectroscopy.
The iron ligation of the Fe(II)ACCO⋅substrate⋅NO complex was directly examined by ENDOR spectroscopy. The Fe(II)ACCO⋅ACC⋅NO complex and the corresponding complex with the ACC analog alanine (with natural abundance 14,15N) exhibit essentially identical 35-GHz Davies ENDOR spectra, as illustrated by those recorded at the low-field (high-g) edge of the EPR envelope (g = 4.23; Fig. 2, traces A and B, respectively). A number of strong signals can be seen in the range of 2–16 MHz, arising from several distinct nitrogenous ligands. Readily detectable changes occur when 15N-alanine (I = ½) is used (Fig. 2, trace C). The [15N − 14N] difference spectrum (Fig. 2, trace D) reveals the gain of a doublet from a single 15N, centered at A(15N)/2 = 9.2 MHz, with the concomitant loss of a four-line pattern from the corresponding 14N, centered at A(14N)/2 = 7.1 MHz. The data are consistent with the expected reduction in AN on going from 15N to 14N [gN(14N)/gN(15N) = 0.7]. Such large hyperfine interactions require that the amino group of alanine be directly coordinated to the Fe–NO center of the Fe(II)ACCO⋅alanine⋅NO adduct. The equivalence of the 14N spectra for complexes with natural-abundance alanine and ACC suggests that these two species bind similarly to the iron, including binding of the amine nitrogen. This is unambiguously confirmed by the difference spectrum for the complexes with 15N-alanine and 14N-ACC (Fig. 2, trace E). This too reveals the gain of a 15N-alanine doublet with the loss of a four-line pattern from the amino group of ACC, whose parameters, including A(14N)/2 = 7.0 MHz, differ only slightly from those of the bound amino group of alanine.
Figure 2.

35-GHz Davies pulsed-ENDOR of Fe(II)ACCO⋅NO. Trace A, with natural-abundance ACC (g1 = 4.23). Trace B, with natural-abundance dl-alanine (g1 = 4.23). Trace C, with 15N-substituted dl-alanine (g1 = 4.23). Trace D, C − B difference spectrum. Trace E, C − A difference spectrum. Trace F, with natural-abundance dl-alanine (g2 = 3.82). Trace G, with 17O-substituted dl-alanine (g2 = 3.82). Conditions: T = 2 K, νMW = 34.72 GHz, MW pulse lengths = 80, 40, and 80 ns, RF pulse length = 60 μs, repetition rate = 50 Hz. Each spectrum consists of 256 points, with each point an average of 1,000 transients.
Labeling the carboxyl oxygens of alanine with 17O (I = 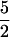 ) leads to the appearance of a broad ENDOR line from the 17O label (g ≈ 3.82; Fig. 2, trace G), while leaving unaffected the 14N signals of the natural-abundance sample (Fig. 2, trace F). The center frequency of the broad 17O line is assigned as ν+(17O) = A(17O)/2 + ν(17O) ≈ 16 MHz, which gives A(17O) ≈ 24 MHz; the breadth of the feature corresponds to ≈12 times the quadrupole splitting, and gives P ≈ 0.5 MHz. The alternative assignment of this feature as ν-(17O) would give A(17O) ≈ 40 MHz, which is notably larger than we have seen for 17O species bound to iron ions (32). In either case, the value of A(17O) is far too large for a noncoordinated 17O and requires that the carboxylate group of alanine be coordinated to the iron center. The similarity of the EPR and 14,15N ENDOR spectra for the alanine and ACC adducts of Fe(II)ACCO⋅NO shows that the results obtained from the labeled alanine complexes can be extended to the ACC complexes. Therefore, the ENDOR data unequivocally indicate the simultaneous coordination to the Fe(II) of both the amino and carboxyl groups of ACC, as well as the inhibitor alanine.
) leads to the appearance of a broad ENDOR line from the 17O label (g ≈ 3.82; Fig. 2, trace G), while leaving unaffected the 14N signals of the natural-abundance sample (Fig. 2, trace F). The center frequency of the broad 17O line is assigned as ν+(17O) = A(17O)/2 + ν(17O) ≈ 16 MHz, which gives A(17O) ≈ 24 MHz; the breadth of the feature corresponds to ≈12 times the quadrupole splitting, and gives P ≈ 0.5 MHz. The alternative assignment of this feature as ν-(17O) would give A(17O) ≈ 40 MHz, which is notably larger than we have seen for 17O species bound to iron ions (32). In either case, the value of A(17O) is far too large for a noncoordinated 17O and requires that the carboxylate group of alanine be coordinated to the iron center. The similarity of the EPR and 14,15N ENDOR spectra for the alanine and ACC adducts of Fe(II)ACCO⋅NO shows that the results obtained from the labeled alanine complexes can be extended to the ACC complexes. Therefore, the ENDOR data unequivocally indicate the simultaneous coordination to the Fe(II) of both the amino and carboxyl groups of ACC, as well as the inhibitor alanine.
The remaining ENDOR signals in the 2- to 16-MHz region Fe(II)ACCO⋅ACC⋅NO and Fe(II)ACCO⋅alanine⋅NO complexes (Fig. 2) must arise from other nitrogenous ligands in the enzyme active site. These features presumably derive from the iron-bound NO and endogenous amino acid ligands. The latter are very likely His residues found in the His-Xaa-Asp-(Xaa)53–57-His sequence conserved among ACCOs and also for related iron enzymes such as isopenicillin N synthase (IPNS) and deacetoxycephalosporin C synthase (DAOCS) (33–35). IPNS and DAOCS have been characterized by x-ray crystallography, and their Fe(II) centers are found to be coordinated to the three conserved residues in this common sequence (29, 36). The three residues occupy the face of an octahedron in what is termed a “2-His-1-carboxylate facial triad” that is emerging as a common structural motif among mononuclear nonheme Fe(II) enzymes (26). This facial triad is proposed to hold the Fe(II) center in the active site, leaving the three remaining sites available for exogenous ligand binding.
DISCUSSION
Fig. 3 summarizes our current view of the substrate-bound Fe(II)ACCO active site and also depicts our proposed mechanism for ACCO. The studies presented here show that ACC and NO, or by extension O2, can simultaneously occupy the three available coordination sites opposite the three endogenous ligands. A similar coordination pattern can be associated with other enzymes having a 2-His-1-carboxylate facial triad (26). For example, there is strong evidence from either crystallography or spectroscopy that substrate or cofactor binds at the same time as NO for IPNS, DAOCS, and the extradiol-cleaving catechol dioxygenases (21, 22, 29–31, 36–38). With such an arrangement, we propose that the iron center can play two roles. First, binding to the iron in this manner juxtaposes the two reactants in a suitable geometry for interaction with each other, and second, it allows facile electron transfer between them.
Figure 3.

Proposed structure of the ternary Fe(II)ACCO⋅ACC⋅O2 complex and catalytic mechanism.
Past considerations of the mechanism of ACCO (11, 12) have led to the proposal that ascorbate binds to the iron before O2 as part of the oxygen activation process.‖ Clearly, there is ample precedent indicating that the enediolate unit of ascorbate might occupy two of the exogenous ligand sites on the Fe(II) that are revealed in the current study (21, 22, 30, 31, 37, 38). However, the current data exclude the simultaneous binding of ascorbate and NO, suggesting that the oxygen activation process does not proceed along this path. Instead our unequivocal demonstration that ACC and NO simultaneously bind to the iron suggests a different mechanism. We propose that the initial bidentate binding of the anionic ACC lowers the redox potential of the Fe(II) center and primes it for O2 binding. Charge is then delocalized from the metal center to the bound O2 in the resulting oxy adduct, affording an Fe(III)–superoxide complex. A similar charge delocalization is used to describe the S = 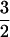 spin state of the corresponding [Fe–NO]7 adduct (40).
spin state of the corresponding [Fe–NO]7 adduct (40).
Earlier mechanistic studies of ACCO clearly show that the oxidation of ACC involves a substrate radical (12, 23). From the putative Fe(III)ACCO⋅ACC⋅superoxo intermediate, a number of pathways could be envisioned to yield an ACC radical. For example, direct abstraction of the amino hydrogen atom of ACC by the nascent superoxide could occur as depicted in Fig. 3 to form Fe(III)ACCO⋅ACC radical·hydroperoxide. The same intermediate could also form by electron transfer from ACC to the superoxide via the iron center followed by proton transfer. The role of the ascorbate in these mechanisms would be to reduce the peroxide by-product to prevent the formation of diffusible reactive oxygen species that could inactivate the enzyme. Alternatively, the ascorbate may donate two electrons to the Fe(III)ACCO⋅ACC⋅superoxo intermediate by an outer-sphere mechanism to cleave the O—O bond and form an Fe(IV)⩵O species; the latter would then be responsible for oxidizing ACC to its radical form. Once formed, the ACC amine radical spontaneously disintegrates into the observed products of ACC oxidation: ethylene, CO2, and HCN. These various mechanistic alternatives require further investigation.
In summary, the unexpected finding by ENDOR spectroscopy of bidentate coordination by the amine and carboxylate of substrate ACC or inhibitor alanine to the iron of ACCO requires a revision of the previously proposed enzymic mechanism. Our proposals for the active-site structure and mechanism share key features with those of other nonheme, Fe(II)-containing enzymes. This class exploits the ability of iron to dynamically coordinate multiple exogenous ligands and promote communication between them. The result is a catalytic center of nearly unparalleled versatility, which can promote reactions ranging from the biodegradation of arenes and the formation of antibiotics, to the biosynthesis of ethylene by ACCO.
Acknowledgments
This work was supported by National Institutes of Health Grants GM33162 to L.Q., GM24689 to J.D.L., and HL13531 to B.M.H., and U.S. Department of Agriculture Grant NRICGP 93–37304-9161 to R.E.C. and N.O.R.
ABBREVIATIONS
- ACC
1-aminocyclopropane-1-carboxylic acid
- ACCO
ACC oxidase
- ENDOR
electron nuclear double resonance
Footnotes
This mechanistic proposal was based, in part, on the observation that in vitro, one mole of ascorbate is utilized for each mole of ethylene produced (39). However, there is no direct evidence that the reaction is initiated by ascorbate binding to the iron or that ascorbate is the reductant utilized in vivo.
References
- 1.Abeles F B, Morgan P W, Saltveit M E., Jr . Ethylene in Plant Biology. San Diego: Academic; 1992. [Google Scholar]
- 2.Kende H. Annu Rev Plant Physiol Plant Mol Biol. 1993;44:283–307. [Google Scholar]
- 3.Adams D O, Yang S F. Proc Natl Acad Sci USA. 1979;76:170–174. doi: 10.1073/pnas.76.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamilton A J, Lycett G W, Grierson D. Nature (London) 1990;346:284–287. [Google Scholar]
- 5.Ververidis P, John P. Phytochemistry. 1991;30:725–727. [Google Scholar]
- 6.Fernandez-Maculet J C, Dong J G, Yang S F. Biochem Biophys Res Commun. 1993;193:1168–1173. doi: 10.1006/bbrc.1993.1748. [DOI] [PubMed] [Google Scholar]
- 7.Smith J J, John P. In: Cellular and Molecular Aspects of the Plant Hormone Ethylene. Pech J C, Latche A, Balague C, editors. Dordrecht, the Netherlands: Kluwer; 1993. pp. 33–38. [Google Scholar]
- 8.Baldwin J E, Adlington R M, Lajoie G A, Lowe C, Baird P D, Prout K. J. Chem. Soc. Chem. Commun. 1988. 775–777. [Google Scholar]
- 9.Shaw J F, Chou Y S, Chang R C, Yang S F. Biochem Biophys Res Commun. 1996;225:697–700. doi: 10.1006/bbrc.1996.1237. [DOI] [PubMed] [Google Scholar]
- 10.Lay V J, Prescott A G, Thomas P G, John P. Eur J Biochem. 1996;242:228–234. doi: 10.1111/j.1432-1033.1996.0228r.x. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Z, Barlow J N, Baldwin J E, Schofield C J. Biochemistry. 1997;36:15999–16007. doi: 10.1021/bi971823c. [DOI] [PubMed] [Google Scholar]
- 12.Pirrung M C, Cao J, Chen J. Chem Biol. 1998;5:49–57. doi: 10.1016/s1074-5521(98)90086-2. [DOI] [PubMed] [Google Scholar]
- 13.Lowry O H, Rosebrough N J, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 14.McGarvey D J, Yu H, Christoffersen R E. Plant Mol Biol. 1990;15:165–167. doi: 10.1007/BF00017736. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Schofield C J, Baldwin J E, Thomas P, John P. Biochem J. 1995;307:77–85. doi: 10.1042/bj3070077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Werst M W, Davoust C E, Hoffman B M. J Am Chem Soc. 1991;113:1533–1538. [Google Scholar]
- 17.Davoust C E, Doan P E, Hoffman B M. J Magn Reson A. 1996;119:38–44. [Google Scholar]
- 18.Hoffman B M, DeRose V J, Doan P E, Gurbiel R J, Houseman A L P, Telser J. In: Biological Magnetic Resonance. Berliner L, Reuben J, editors. Vol. 13. New York: Plenum; 1993. pp. 151–218. [Google Scholar]
- 19.Arciero D M, Lipscomb J D. J Biol Chem. 1986;261:2170–2178. [PubMed] [Google Scholar]
- 20.Nelson M J. J Biol Chem. 1987;262:12137–12142. [PubMed] [Google Scholar]
- 21.Chen V, Orville A M, Harpel M R, Frolik C A, Surerus K K, Münck E, Lipscomb J D. J Biol Chem. 1989;264:21677–21681. [PubMed] [Google Scholar]
- 22.Harpel M R, Lipscomb J D. J Biol Chem. 1990;265:22187–22196. [PubMed] [Google Scholar]
- 23.Baldwin J E, Adlington R M, Lajoie G A, Rawlings B J. J. Chem. Soc. Chem. Commun. 1985. 1496–1498. [Google Scholar]
- 24.Hoffman N E, Yang S F, Ichihara A, Sakamura S. Plant Physiol. 1982;70:195–199. doi: 10.1104/pp.70.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang S F, Hoffman N E. Annu Rev Plant Physiol. 1984;35:155–189. [Google Scholar]
- 26.Hegg E L, Que L., Jr Eur J Biochem. 1997;250:625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- 27.Que L, Jr, Ho R Y N. Chem Rev. 1996;96:2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 28.Pavel E G, Zhou J, Busby R W, Gunsior M, Townsend C A, Solomon E I. J Am Chem Soc. 1998;120:743–747. [Google Scholar]
- 29.Valegard K, Vanscheltinga A C T, Lloyd M D, Hara T, Ramaswamy S, Perrakis A, Thompson A, Lee H J, Baldwin J E, Schofield C J, Hajdu J, Andersson I. Nature (London) 1998;394:805–809. doi: 10.1038/29575. [DOI] [PubMed] [Google Scholar]
- 30.Han S, Eltis L D, Timmis K N, Muchmore S W, Bolin J T. Science. 1995;270:976–980. doi: 10.1126/science.270.5238.976. [DOI] [PubMed] [Google Scholar]
- 31.Senda T, Sugiyama K, Narita H, Yamamoto T, Kimbara K, Fukuda M, Sato M, Yano K, Mitsui Y. J Mol Biol. 1996;255:735–752. doi: 10.1006/jmbi.1996.0060. [DOI] [PubMed] [Google Scholar]
- 32.Burdi D, Willems J-P, Riggs-Gelasco P, Antholine W E, Stubbe J, Hoffman B M. J Am Chem Soc. 1998;120:12910–12919. [Google Scholar]
- 33.Prescott A G. J Exp Botany. 1993;44:849–861. [Google Scholar]
- 34.Pirrung M C, Kaiser L M, Chen J. Biochemistry. 1993;32:7445–7450. doi: 10.1021/bi00080a015. [DOI] [PubMed] [Google Scholar]
- 35.Borovok H, Landman O, Kreisberg-Zakarin R, Aharonowitz Y, Cohen G. Biochemistry. 1996;35:1981–1987. doi: 10.1021/bi951534t. [DOI] [PubMed] [Google Scholar]
- 36.Roach P L, Clifton I J, Hensgen C M, Shibata N, Schofield C J, Hajdu J, Baldwin J E. Nature (London) 1997;387:827–830. doi: 10.1038/42990. [DOI] [PubMed] [Google Scholar]
- 37.Arciero D M, Orville A M, Lipscomb J D. J Biol Chem. 1985;260:14035–14043. [PubMed] [Google Scholar]
- 38.Lipscomb J D, Orville A M. Met Ions Biol Syst. 1992;28:243–298. [Google Scholar]
- 39.Dong J G, Fernandez-Maculet J C, Yang S F. Proc Natl Acad Sci USA. 1992;89:9789–9793. doi: 10.1073/pnas.89.20.9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown C A, Pavlosky M A, Westre T E, Zhang Y, Hedman B, Hodgson K O, Solomon E I. J Am Chem Soc. 1995;117:715–732. [Google Scholar]