

Summary
Hepatitis C virus (HCV) core protein, expressed with a Semliki forest virus (SFV) replicon, self-assembles into HCV-like particles (HCV-LPs) at the endoplasmic reticulum (ER) membrane, providing an opportunity to study HCV particle morphogenesis by electron microscopy. Various mutated HCV core proteins with engineered internal deletions were expressed with this system, to identify core domains required or dispensable for HCV-LP assembly. The HCV core protein sequence was compared with its counterpart in GB virus B (GBV-B), the virus most closely related to HCV, to identify conserved domains. GBV-B and HCV display similar tropism for liver hepatocytes and their core proteins are organized similarly into three main domains (I, II and III), although GBV-B core is smaller and lacks ≈ 35 amino acids (aa) in domain I. The deletion of short hydrophobic domains (aa 133-152 & 153-167 in HCV core) that appear highly conserved in domain II of both GBV-B and HCV core proteins resulted in loss of HCV core ER anchoring and self-assembly into HCV-LPs. The deletion of short domains found within domain I of HCV core protein but not in the corresponding domain of GBV-B core according to sequence alignment had contrasting effects. Amino acids 15-28 and 60-66 were shown to be dispensable for HCV-LP assembly and morphogenesis, whereas aa 88-106 were required for this process. The production of GBV-B core protein from a recombinant SFV vector was associated with specific ER ultrastructural changes, but did not lead to the morphogenesis of GBV-B-LPs, suggesting that different budding mechanisms occur in members of the Flaviviridae family.
Keywords: Amino Acid Sequence; Animals; Blotting, Western; Cell Line; Endoplasmic Reticulum; metabolism; ultrastructure; virology; GB virus B; genetics; growth & development; metabolism; Hepacivirus; genetics; growth & development; metabolism; Hydrophobicity; Microscopy, Confocal; Microscopy, Electron, Transmission; Molecular Sequence Data; Mutation; Sequence Deletion; Sequence Homology, Amino Acid; Viral Core Proteins; chemistry; genetics; physiology
Keywords: hepatitis C virus, HCV, morphogenesis, viral budding, electron microscopy, virus-like particle, GBV-B, core protein, Flaviviridae
Introduction
Hepatitis C virus (HCV) is a small hepatotropic virus classified within the Flaviviridae family. An estimated 170–180 million people worldwide are infected with HCV. One of the major features of HCV infection is that more than 60 % of acutely infected patients become chronically infected (Alter et al., 1998). Current drug therapies are often poorly tolerated by patients and effective in about 50–55% of cases. In the absence of truly effective curative treatments, these chronic infections often lead to liver cirrhosis and fatal hepatocellular carcinoma (Feld et al., 2005). The viral genome consists of a 9.6-kb single-stranded positive-sense RNA encoding a precursor polyprotein of about 3000 amino-acid residues. Polyprotein translation is initiated via binding of ribosomes to an internal ribosome entry site (IRES), located within the 5′ untranslated region (5′UTR). During translation, the mature viral products are generated from the polyprotein by a series of cleavage events (Lindenbach and Rice, 2005). The structural components [one core (C) and two envelope (E1 and E2) proteins] are produced by cellular protease-mediated cleavages. Processing of the non-structural proteins (NS2-NS5B) that are involved in polyprotein processing and viral RNA replication requires virus-encoded proteases (Lohmann et al., 1996). The nonstructural proteins are separated from the structural proteins by a short hydrophobic polypeptide (p7), the function of which is unknown.
The HCV core protein is produced at the N-terminal end of the polyprotein and is followed by the signal sequence of the E1 envelope glycoprotein. The signal sequence targets the nascent HCV polyprotein to the endoplasmic reticulum (ER), allowing the translocation of E1 to the ER lumen, an essential step in the membrane-dependent processing of the core protein (Liu et al., 1997). Cleavage by a signal peptidase in the ER lumen releases the N-terminal end of E1, leaving the 191-amino acids (aa) core protein anchored by the signal peptide (McLauchlan et al., 2002). This 191-aa polypeptide, also known as p23, is an immature form of the core protein, and is further processed by an intramembrane protease, the signal peptide peptidase (SPP), that cleaves within the C-terminal signal peptide and releases the N-terminal 173-179 aa of the core protein from the ER (McLauchlan et al., 2002). This cleaved core protein, also known as p21, is then available for association with lipid droplets (McLauchlan et al., 2002) or morphogenesis of the viral particle (Ait-Goughoulte et al., 2006). Thus, p21 is the mature form of the core protein found in the secreted viral particle, as shown by the predominance of p21 in serum of patients infected with HCV (Yasui et al., 1998). The role of core association with lipid droplets in the viral life cycle remains however largely unknown. Three domains have been identified in HCV core protein, based on predicted structural and functional characteristics (Hope and McLauchlan, 2000). Domain I, corresponding to the N-terminal approximately 120 aa, is a highly basic domain that is probably involved in the recruitment of viral RNA during particle morphogenesis. Domain II, located between aa ≈120 and aa ≈175, is a hydrophobic region predicted to form one or two α-helices that are probably involved in the association of core with the ER membrane and lipid droplets. Domain III, corresponding to the C-terminal ≈20 aa of the protein, is a highly hydrophobic region that serves as a signal sequence for the targeting of E1 to the ER (see Fig. 1).
Fig. 1. Sequence alignment and design of the HCV core deletion mutant proteins.

A. Amino acid sequence comparison of HCV (1a genotype) and GBV-B core proteins. Highlighted dark and light gray sequences correspond to identical and conservative residues, respectively. Numbering corresponds to amino acid (aa) position in the HCV core protein.
B. Design of the various constructs expressed with SFV vectors (del: deletion). The HCV core mutants contained unique or combined deletions of three short conserved domains present in the HCV core sequence but not in the GBV-B core sequence (del 1, 2 and 3), or deletion of either or both of two short relatively conserved sequences in domain II of the core proteins of both viruses (del 4 and 5). Numbers on the right correspond to the aa removed in the HCV core deletion mutants.
Recent advances in the development of cell culture systems have made it possible for the first time to produce infectious HCV that can be efficiently propagated in cultured, naïve hepatoma cells (Lindenbach et al., 2005; Wakita et al., 2005; Zhong et al., 2005). These new systems will clearly provide powerful tools for analyzing host cell-virus interactions, but it remains not possible to visualize HCV assembly and morphogenesis with these models (Rouillé et al., 2006). We have established a model based on Semliki forest virus (SFV) replicon vectors, which can be used to produce HCV core protein in mammalian cells, leading to the assembly of this protein into HCV-like particles (HCV-LPs) (Blanchard et al., 2002; Blanchard et al., 2003). This model displays abortive HCV-LP budding, but remains a useful tool for electron microscopy (EM) studies of the early events of HCV assembly in mammalian cells (Ait-Goughoulte et al., 2006; Roingeard et al., 2004). In this study, we used this HCV core assembly model for the production of several deletion mutants of the HCV core protein, in order to identify domains of the core protein that are required or dispensable for virus morphogenesis. We identified sequences of potential interest by comparing the core protein sequence of HCV (1a genotype) with that of GB virus B (GBV-B), the virus most closely related to HCV (Fig. 1). Both GBV-B and HCV display tropism for liver hepatocytes, and GBV-B was first isolated in tamarins after serial passages of serum from a surgeon with nonA-nonB hepatitis. GBV-B is infectious in tamarins and has been proposed as a surrogate model for HCV (Beames et al., 2000; Beames et al., 2001; Martin et al., 2003). GBV-B and HCV core protein sequences display significant similarities, in particular they both contain a domain (domain II) that is absent from the core proteins of other members of the Flaviviridae family (Boulant et al., 2005; Hope et al., 2002). The core mutants engineered allowed us to identify sequences in domain II, as well as a short sequence in domain I of the HCV core protein that are likely required for HCV budding. In addition, the comparison of GBV-B and HCV core protein expression from SFV vectors suggests that different budding mechanisms occur in members of the Flaviviridae family.
Results
Expression of HCV core proteins, HCV core deletion mutants, and GBV-B core protein
Several plasmids encoding HCV core mutants that contain unique or combined deletions of three short sequences present in the domain I of HCV core but not in the corresponding domain of GBV-B core (del 1, 2 and 3), or deletion of either or both of two short relatively conserved sequences in domain II of the core protein of both viruses (del 4 and 5) were generated (Fig. 1). Sixteen hours after transfection with the SFV recombinant constructs expressing each of these HCV core deletion mutants, HCV core protein, or GBV-B core protein (Fig. 1), BHK-21 cells were harvested and lysed for western blot analysis. Consistent with our previous results (Ait-Goughoulte et al., 2006; Blanchard et al., 2003), transfection of the wild-type (WT) C191 construct in BHK-21 resulted in the production of higher amounts of core protein than did the transfection of the C173 construct (Fig. 2A, lanes 1 and 3). Also consistent with our previous work (Ait-Goughoulte et al., 2006; Blanchard et al., 2003), the WT C191 core protein was fully cleaved by SPP, the cleaved WT C191 comigrating with the C173 core protein as a 21 kDa product (Fig. 2A, lanes 1 and 3). Differences in the amounts of the WT C191 and C173 core proteins probably resulted from a lack of targeting of C173 to the ER membrane, leading to a defect in membrane anchoring and potentially higher levels of degradation due to ubiquitination and targeting to the proteasome (Suzuki et al., 2001). The HCV core mutants produced from the del 1, del 2, del 3, del 5 and del 1/2 constructs had slightly lower apparent molecular weights than the WT C191 core protein, which is consistent with the deletions introduced (Fig. 2A). HCV core deletion mutants expressed from the del 2 and del 3 constructs were produced in similar quantities as the WT protein, whereas those expressed from the del 1, del 5 or del 1/2 constructs were produced in smaller amounts, as confirmed in repeated experiments. This might be due to higher instability of the latter series of deletion mutant proteins. Lower molecular weight products, correlated to the production of large amounts of predominant products were observed in some cases (Fig. 2A, lanes 1, 5, 6 and 8). These minor additional products might represent degradation products, as we previously reported in this model (Blanchard et al., 2003). The HCV core mutant encoded by the del 4 construct was barely detected (Fig. 2A, lane 7), and the HCV core mutant encoded by the del 4/5 construct (lane 10) was detected only after prolonged exposure of the film (data not shown). This was not linked to weak detection of del 4 and del 4/5 core deletion mutants by the C1856 anti-HCV core monoclonal antibody, as various monoclonal and polyclonal anti-HCV core antibodies gave similar results (data not shown). We observed marked cell death in cultures transfected with del 4 and del 4/5 constructs, suggesting that the expression of these core deletion mutants was toxic to cells. The GBV-B core protein was produced efficiently as a protein of approximately 16 kDa (Fig. 2B, lane 2), consistent with results of other studies (Hope et al., 2002).
Fig. 2.

Analysis of GBV-B core, HCV core and HCV core deletion mutants produced in BHK-21 cells. Proteins expressed at 16hrs post-transfection with SFV recombinant RNAs were separated, blotted onto PVDF membranes, and identified after incubation of the membrane with the C1856 anti-HCV core mouse monoclonal antibody (A) or with rabbit polyclonal anti-GBV-B core antibodies then with the C1856 antibody, for side-by-side comparison of the size of WT HCV and GBV-B core proteins (B). Molecular weight markers (M) are indicated on the left of the blots. Cells transfected with a recombinant SFV RNA encoding β-galactosidase (β-gal) were used as a negative control. In panel C, GBV-B core, WT C191 HCV core or HCV core del 3 and del 5 mutants were expressed in BHK-21 cells in the presence (+) or absence (−) of (Z-LL)2-ketone, an inhibitor of the signal peptide peptidase (SPP), and identified by immunobloting.
To check whether the core/E1 signal peptide is cleaved off by SPP, GBV-B core, HCV core and some of the HCV core deletion mutants were expressed in BHK-21 cells treated with (Z-LL)2-ketone, a commercially available inhibitor of SPP. As previously reported (Ait-Goughoulte et al., 2006), the immunoblot analysis of cells transfected with the WT C191 construct and treated with the SPP inhibitor showed that SPP cleavage was only partially inhibited, leading to the detection of both p23 and p21 (Fig. 1C). Similar results were obtained with GBV-B core protein and the HCV core del 3 mutant, whereas a complete inhibition of SPP cleavage was observed for the HCV core del 5 mutant (Fig. 1C). These data showed that HCV and GBV-B WT core or HCV core deletion mutants were efficiently cleaved by SPP in the system used here.
Colocalization of GBV-B core, HCV core and HCV core deletion mutants with lipid droplets
Experiments aiming at characterizing the colocalization of GBV-B core and HCV core deletion mutants with lipid droplets gave similar results in either BHK-21 or FLC4 cell line. We report here results in FLC4 hepatoma cells, since the lower efficiency of the SFV vectors in these cells resulted in the production of lower amounts of protein than in BHK-21 cells, allowing a more precise analysis of subcellular distribution of core proteins. Nile red labeling in control FLC4 cells transfected with β-Gal RNA showed that lipids were evenly distributed throughout the cytoplasm (Fig. 3A, bottom). Transfection with the WT C191 and C173 constructs induced clustering in the perinuclear area of large lipid droplets, staining strongly for HCV core protein (Fig. 3A), as previously reported (Ait-Goughoulte et al., 2006; Hope and McLauchlan, 2000; McLauchlan et al., 2002). HCV core deletion mutants encoded by the del 1, del 2, del 3 and del 1/2 constructs displayed a similar distribution as C191 and C173 and mostly colocalized with lipid droplets (Fig. 3A). Thus, all HCV core proteins with deletions within the first 120 amino acids retained their ability to associate with lipid droplets, which is in agreement with previous work using other deletion core mutants (Hope and McLauchlan, 2000). In contrast, the HCV core deletion mutant encoded by the del 5 construct clearly showed lack of colocalization with lipid droplets (Fig. 3A). Deletion 5 corresponds to a sequence within domain II of the core protein that was reported to fold as a putative α-helix (Boulant et al., 2005; Hope et al., 2002). Our data thus suggest that this putative α-helix is probably required for lipid droplet association. Very weak signals were obtained for mutant proteins encoded by the del 4 and del 4/5 constructs (data not shown), consistent with our western blotting results (Fig. 2), making it impossible to investigate colocalization of these mutant proteins with lipid droplets (this was also the case when these constructs were expressed in BHK-21 cells). As previously reported (Hope et al., 2002), the GBV-B core protein was cytoplasmic, displayed a reticulate pattern compatible with association with ER membranes, and colocalized with lipid droplets (Fig. 3B).
Fig. 3.
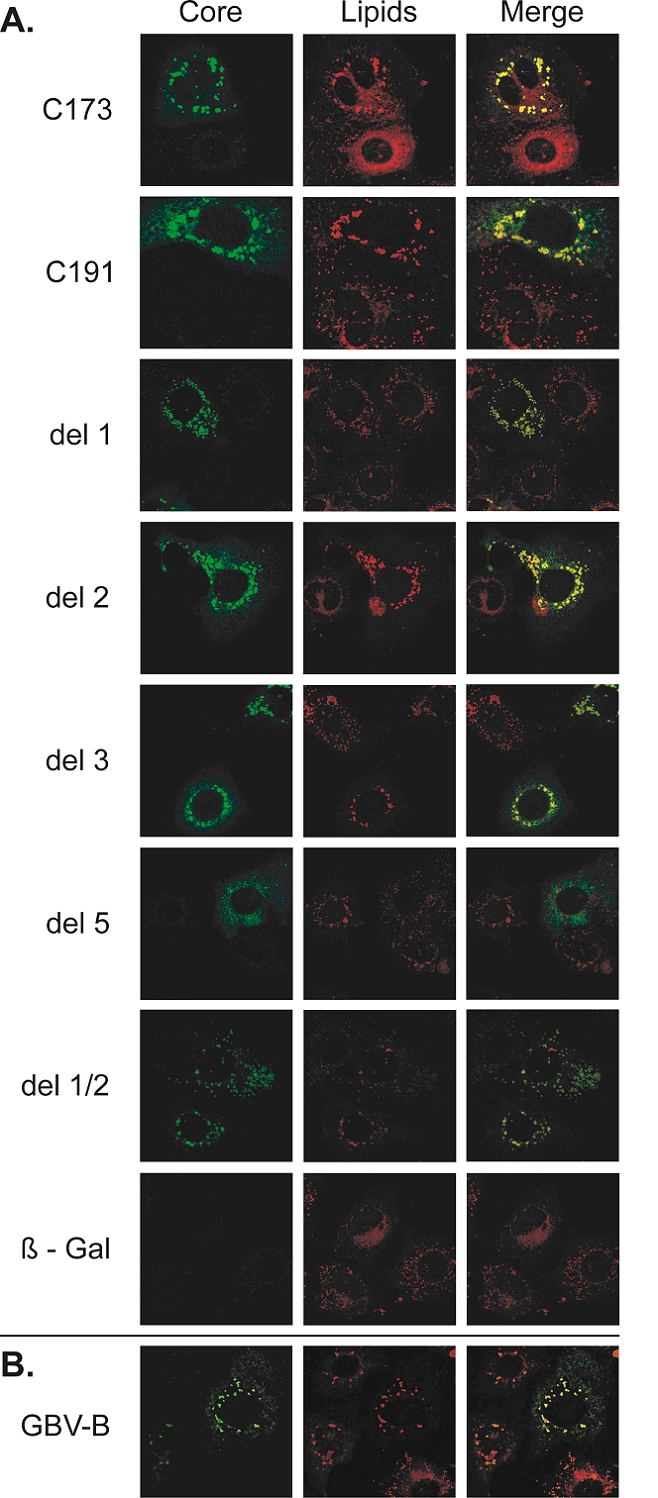
Combined detection of GBV-B core, HCV core, HCV core deletion mutants and lipid droplets in FLC4 cells. HCV WT and deleted core proteins (A) or GBV-B core protein (B) were detected at 16 hrs post-transfection in cells transfected with the corresponding SFV recombinant RNAs by immunofluorescence with the C1856 anti-HCV core mouse monoclonal antibody and rabbit polyclonal anti-GBV-B core antibodies, respectively (left panels). Lipid droplets were visualized after staining with Nile red (central panels). Colocalization of viral proteins with lipids was seeked in merged images (right panels). Significant clusters of lipid droplets colocalizing with HCV core proteins were observed in the perinuclear area of cells producing the WT C173 and C191, del 1, del 2, del 3, or del 1/2 HCV mutant core proteins, but not in cells producing the del 5 HCV core protein deletion mutants. Clusters of lipid droplets were also found to colocalize with the GBV-B core protein in cells producing this protein. Cells expressing β-galactosidase (β-gal) were used as a negative control.
Ultrastructural changes and VLP formation induced by the expression of GBV-B core, HCV core, or HCV core deletion mutants
Ultrastructural changes observed in BHK-21 cells transfected with recombinant SFV vectors, as studied by low-magnification EM, were consistent with results obtained by confocal microscopy: all HCV core mutants harboring deletions in domain I (del 1, del 2, del 3 and del 1/2) displayed a similar pattern as that of WT core proteins C191 and C173, showing clusters of lipid droplets in the perinuclear area (Figs. 4a, 4b, 5a, 5b and 6a ; data not shown for del 1/2). Similar results were obtained in BHK-21 cells producing GBV-B core protein (Fig. 8a). In cells producing WT C191 core, del 1, del 2, del 1/2, or del 3 HCV core mutants or the GBV-B core protein, these clusters of lipid droplets were always surrounded by electron-dense, convoluted ER membranes (Figs. 4a, 5a, 5,b, 6a and 8a; data not shown for del 1/2). The frequency of cells with such ER alterations was similar for HCV WT or core deletion mutants, whereas ER changes were observed less frequently in cells expressing GBV-B core protein (Table 1). The Ultrastructural modification of the ER in cells producing HCV core mutant proteins encoded by the del 1 (Fig. 5a), del 2 (Fig. 5b) and del 1/2 (not shown) constructs was similar to that observed in cells producing the WT C191 protein (Fig. 4a), whereas the ER of cells producing the HCV del 3 core mutant protein or the GBV-B core protein formed a tubular web (Figs. 6 and 8). In contrast, consistent with our previous findings (Ait-Goughoulte et al., 2006; Blanchard et al., 2003), the WT HCV C173 core protein did not cause any specific ultrastructural modification of the ER (Fig. 4b), as did the expression of irrelevant β-Gal (not shown). Similarly, no specific ultrastructural change of the ER was observed in cells producing the HCV core mutants encoded by the del 4 (Fig. 7a), del 5 (Fig. 7c) and del 4/5 (not shown at low magnification) constructs, but the cytoplasm of these cells contained non-structured electron-dense material. A particularly marked cell injury, as indicated by the presence of large vacuoles, was observed in cells producing the del 4 (Fig. 7a) or the del 4/5 (not shown at low magnification) HCV core deletion mutants. This cell injury most probably explained the poor detection of these mutant proteins by western blot (Fig. 2).
Fig. 4.
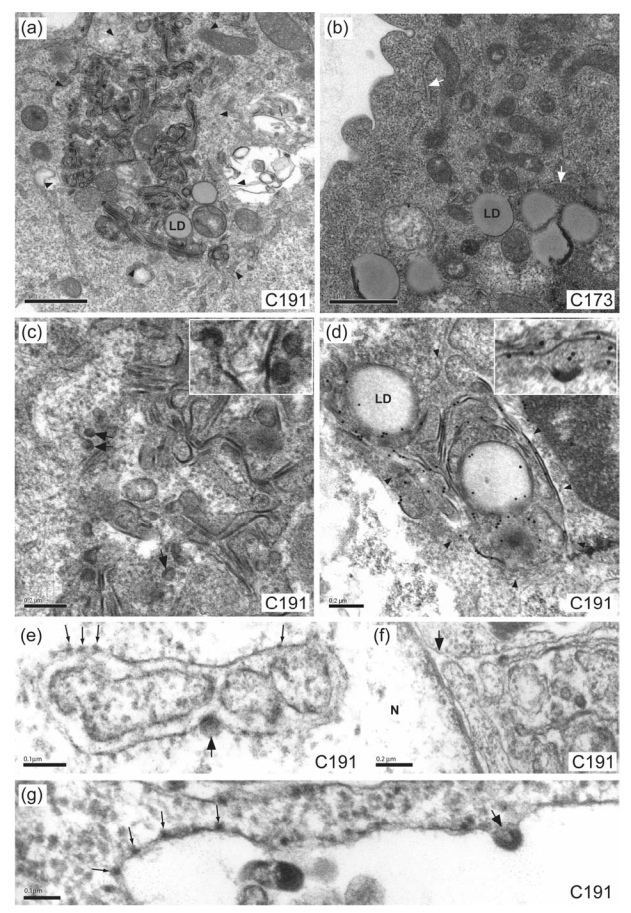
Electron micrographs of BHK-21 cells producing HCV C191 or C173 proteins. At low magnification, the expression of WT C191 HCV core protein (a) led to the presence in the perinuclear area of convoluted ER membranes (delimited by arrowheads) surrounding clusters of lipid droplets (LD). Some lipid droplet accumulation was also observed in cells producing the WT C173 HCV core protein (b), but these cells displayed no ultrastructural change of ER membranes (white arrows indicate normal ER structures). HCV-LP assembly and budding (arrows and inset) was frequently observed on high magnification electron micrographs of cells producing the HCV C191 protein (c). The HCV core protein was shown to be present in the convoluted ER membranes (delimited by arrowheads), at the surface of lipid droplets (LD), as well as in the budding HCV-LPs (inset), by immuno-EM using the monoclonal C1856 anti-HCV core antibody (d). High magnification electron micrographs of cells producing the HCV C191 protein (e and g) showed that HCV-LP budding (large arrows) occurs at rough ER membrane that carries ribosomes (thin arrows). These convoluted membranes showing HCV-LP budding were also found to be continuous with the outer membrane (arrow) of the nuclear envelope (f). Bars, 1 μm (a and b), 0.2 μm (c, d and f), 0.1 μm (e and g); N, nucleus.
Fig. 5.

Electron micrographs of BHK-21 cells producing the del 1, del 2 or del1/2 HCV core mutants showing similar ultrastructural changes as in cells expressing the WT C191 HCV core protein. At low magnification (panels a and b for del 1 and del 2 mutants, respectively) convoluted ER membranes (delimited by arrowheads) surrounded clusters of lipid droplets (LD). On high-magnification electron micrographs, HCV-LP budding at the ER membrane (arrows and insets) was frequently observed in cells producing the del 1 (c), del 2 (d) or del 1/2 (e) HCV mutant core proteins. The HCV core protein was shown to be present in these HCV-LPs by immuno-EM (see arrows for the del 2 HCV core protein in panel f and inset), using the monoclonal C1856 anti-HCV core antibody. Bars, 1 μm (a and b), 0.2 μm (c to f); N, nucleus.
Fig. 6.

Electron micrographs of BHK-21 cells producing the del 3 HCV core mutant. As for the WT C191 HCV core protein, the production of this mutant was also associated with convoluted ER membranes (delimited by arrowheads) surrounding lipid droplets (LD), but the ER presented a very different pattern, characterized by a tubular web (a). These tubular membranes were also found to be continuous with the outer membrane (arrows) of the nuclear envelope (b). No HCV-LP was detected in the lumen or at the membrane of this tubular ER (c) in spite of the presence of the protein in these membranous structures as demonstrated by immuno-EM, using the monoclonal C1856 anti-HCV core antibody (d). Bars, 1 μm (a), 0.5 μm (b), 0.2 μm (d), 100 nm (c); N, nucleus.
Fig. 8.

Electron micrographs of BHK-21 cells producing the GBV-B core protein. In these cells, dense convoluted ER membranes (delimited by arrowheads) surrounding large clusters of lipid droplets (LD) were frequently found in the perinuclear area (a). No VLP was observed in the lumen or at the ER membrane of these cells (b). Bars, 1 μm; N, nucleus.
Table 1.
Summary and frequency of the observations related to the expression of HCV C191 or C173 WT core proteins, HCV C191 deletion mutants or GBV-B WT core protein.
| Core construct | aa positions framing the deleted domain(s) | Lipid droplets association | Cytoplasmic electron-dense aggregatesa | Production of convoluted ER membranesa | Assembly into VLPs at the ER membraneb |
|---|---|---|---|---|---|
| HCV C173 WT | − | + | − | − | − |
| HCV C191 WT | − | + | − | + (63%) | + (11) |
| HCV C191 del 1 | [15–28] | + | − | + (48%) | + (7) |
| HCV C191 del 2 | [60–66] | + | − | + (51%) | + (10) |
| HCV C191 del 1/2 | [15–28]+[60–66] | + | − | + (35%) | + (8) |
| HCV C191 del 3 | [88–106] | + | − | + (56%) | − |
| HCV C191 del 4 | [133–152] | ND | + (11%) | − | − |
| HCV C191 del 5 | [153–167] | − | + (29%) | − | − |
| HCV C191 del 4/5 | [133–167] | ND | + (10%) | − | − |
| GBV-B WT | − | + | − | + (18%) | − |
Percentage of cell sections showing these specific ultrastructural changes.
Mean number of viral particles/section observed in the positive cell sections. (a and b were determined on 100 consecutive EM cell sections).
ND: Not determined.
Fig. 7.

Electron micrographs of BHK-21 cells producing the del 4, del 5 or del 4/5 HCV core mutants. At low magnification (panels a and c for del 4 and del 5 mutant core proteins, respectively), the production of these mutants did not lead to specific ER ultrastructural change, but was associated with the presence of large amounts of cytoplasmic electron-dense material (arrows). Cytoplasmic disorganization and the presence of large vacuoles (V) were observed, especially in cells producing the del 4 HCV core mutant (a). On high-magnification electron micrographs, no HCV-LP assembly was observed in cells producing the del 4 (b), del 5 (d) or del 4/5 (e) HCV core mutants, but high-magnification EM confirmed the presence of large amounts of electron-dense, non-structured cytoplasmic material (arrows in b, d and e and insets) in these cells. Immuno EM performed with the monoclonal C1856 anti-HCV core antibody demonstrated the presence of the HCV deletion mutant proteins in these cytoplasmic structures (arrows), as shown for the del 5 HCV core mutant in (f). Bars, 1 μm (a and c), 0.2 μm (other micrographs).
At higher magnification, cells producing the HCV core deletion mutants encoded by the del 1, del 2 and del 1/2 constructs demonstrated high levels of HCV-LP assembly at convoluted ER membranes (arrows and insets on Figs. 5c, 5d and 5e), like in cells producing WT C191 (Fig. 4c and Table 1). The association of core protein with these membranes was confirmed by immuno-electron microscopy (as shown as examples for the HCV WT and del 2 core proteins in Figs. 4d and 5f, respectively). The presence of ribosomes at their surface (Figs 4e and 4g) and their continuum with the outer membrane of the nuclear envelope (Fig 4f) showed unambiguously that these membranes were ER-related. This is in agreement with previous studies, showing that the HCV core protein colocalizes in part with lipid droplets and in part with ER markers such as calregulin (Suzuki et al., 2005). Also, electron microscopic studies of flavivirus-infected cells have consistently observed morphogically mature virions within the lumen of a compartment that appear to be rough ER or in vesicles linked to this compartment (Rice, 1996).
In contrast, cells producing the HCV core deletion mutant encoded by the del 3 construct showed no HCV-LP budding at their tubular, electron-dense ER membranes (Figs. 6b and 6c), although large amounts of HCV del 3 core were present in these tubular ER membranes, as confirmed by immuno-electron microscopy (Fig. 6d). These tubular membranes were also continuous with the outer membrane of the nuclear envelope (Fig 6b), showing that they were ER-related. Cells producing the HCV core deletion mutants encoded by the del 4, del 5 and del 4/5 constructs showed no HCV-LP assembly (Figs. 7b, 7d and 7e). However, the presence of the HCV core deletion mutants in a cytoplasmic, non-structured electron-dense material in these cells (arrows and insets in Figs. 7b, 7d and 7e) was unambiguously demonstrated by immunoelectron microscopy (as shown for example for the HCV del 5 core protein in Fig. 7f). No virus-like particles were observed in these cytoplasmic structures containing HCV core deletion mutants. Finally, no virus-like particle was detected in the electron-dense ER membranes nor in the lumen of cells expressing the GBV-B core protein (Fig. 8b), despite extensive investigation. The presence of GBV-B core protein in these dense ER membranes could not be confirmed by immunoelectron microscopy due to poor activity of the anti-GBV-B core antibody in resin-embedded cells.
Confocal and electron microscopy observations related to the expression of HCV C191 or C173 WT core proteins, HCV C191 deletion mutants or GBV-B WT core protein are summarized in Table 1. The Table 1 reports also the frequency of the ultrastructural changes encountered in cells producing the different proteins.
Discussion
In addition to its putative role in viral morphogenesis, the HCV core protein has been shown to interact with various cellular proteins involved in multiple cellular and/or pathogenesis processes, such as gene transcription, apoptosis, cell signaling and lipid retention (for a review, see Giannini et al., 2003). Steatosis often occurs in patients infected with HCV, and the core protein seems to play a critical central role in this phenotype, as demonstrated in transgenic mice expressing the HCV core protein (Moriya et al., 1998; Moriya et al., 1997; Perlemuter et al., 2002). Studies in cell cultures have clearly shown that HCV core protein interacts specifically with phospholipid bilayers and is probably involved in the biogenesis of lipid droplets (Barba et al., 1997), through a well described specific interaction (Hope and McLauchlan, 2000). These findings drew increased interest in this protein. Previously identified correlations between core protein structure and functions led to the mapping of three distinct domains (Hope et al., 2002) : an N-terminal basic domain (domain I), followed by an hydrophobic domain (domain II), and a C-terminal signal peptide (domain III). A recent study compiling several hundred sequences of the HCV core protein from viruses of various genotypes has confirmed such an organization in three domains for this protein (Boulant et al., 2005). The GBV-B core protein, although smaller in size, exhibits the same organization, in contrast to core proteins of other members of the Flaviviridae family.
Using our SFV/HCV-LP model (Blanchard et al., 2002), we previously showed that the HCV core protein, expressed alone or as a fusion with E1-E2, drives the budding of the HCV-LP and that the targeting of core to the ER by means of its domain III is essential for this process (Blanchard et al., 2003). This SFV/HCV-LP model was recently used to demonstrate that HCV core processing by SPP is required both for trafficking of the protein to the lipid droplets, and for assembly and viral budding at the ER membrane (Ait-Goughoulte et al., 2006). In contrast, it was not possible to document viral assembly and morphogenesis by similar EM studies in cell culture systems that efficiently propagate the JFH-1 virus (Rouillé et al., 2006). Thus, although HCV-LPs seem to undertake abortive budding in the SFV system, this model remains a useful tool for EM studies of the early events in HCV assembly and morphogenesis in mammalian cells. In the present study, HCV core deletion mutants were designed on the basis of sequence alignment of HCV and GBV-B core proteins. These mutants contained unique or combined deletions of three short domains present in the domain I of the HCV core sequence but not in the GBV-B core sequence, and thus presumably dispensable for the HCV budding process (del 1, 2 and 3), or deletion of either or both of two short, relatively conserved sequences in domain II of core proteins of both viruses, which were hypothesized to be involved in viral morphogenesis (del 4 and 5).
All HCV core mutant proteins bearing deletions within domain I were able to associate with lipids droplets, confirming that deletions within the N-terminal 106 aa of the HCV core protein have no effect on lipid droplet interaction (Table 1), as reported earlier (Hope and McLauchlan, 2000). In contrast, the del 5 core mutant bearing a deletion within domain II (deletion of aa 154-166) was clearly unable to associate with lipid droplets (Table 1). Deletion del 5 includes a conserved motif “YATG” that has been postulated to be essential for lipid association of HCV core protein as well as oleosin (Hope and McLauchlan, 2000). It was not possible to draw definitive conclusions concerning the ability of the del 4 and del 4/5 core deletion mutants to associate with lipid droplets, as these proteins were produced in very small amounts in cells. Low levels of expression may be due to high toxicity of these particular deletion mutants, or possibly to higher instability of these proteins. The constructs encoding these proteins shared a deletion domain (del 4), corresponding to a hydrophobic sequence located between aa 133 and 152 in domain II. It has been reported that an HCV core mutant bearing a deletion of aa 125-166 is rapidly degraded by the proteasome (Hope and McLauchlan, 2000). EM analysis reported in our study showed accumulation of large amounts of electron-dense material containing HCV core deletion mutants in the cytoplasm of cells producing del 4 or del 4/5 proteins (Fig. 7, Table 1). The cell injury observed with the del 4 and del 4/5 mutants may therefore be due to the saturation of the degradation pathway. Similar HCV core-positive, cytoplasmic electron-dense material was observed by EM in cells producing the del 5 core mutant, but no such toxic effect was documented. Like the del 4 and del 4/5 deletions, deletion 5 in domain II probably prevents the mutated HCV core protein from interacting with the ER membrane, leading to the accumulation of the protein in the cytoplasm. Our immuno EM analysis of these deletion mutants confirms a previous hypothesis (Hope and McLauchlan, 2000) suggesting that a mutated HCV core protein deleted of its domain II would not remain anchored to the ER membrane after cleavage by SPP. Our EM study also showed that the del 4, del 5 and del 4/5 mutants cannot form HCV-LPs (Table 1). This lack of HCV-LP budding could not be attributed to a defect in SPP cleavage of these core mutants. Although it was not possible to draw definite conclusions as to whether the del 4 and del 4/5 core deletion mutants were cleaved by SPP, due to their low amounts in cells, the del 5 core mutant was found to be efficiently cleaved by SPP (Fig. 2C). Thus, after SPP cleavage, the residual association of the HCV core protein to the ER membrane via domain II appears essential for viral particle formation. This confirms our earlier hypothesis that membrane-binding properties of the HCV core protein are essential for HCV-LP assembly (Blanchard et al., 2002). However, the C 173 core protein, which contains a complete domain II and mimics the mature HCV core protein, forms no VLP in this system. Immunoelectron microscopy in cells producing the C 173 core protein confirmed results obtained by confocal microscopy, showing that C 173 is mostly associated with the membrane of lipid droplets in these cells, and not with the ER membrane (data not shown). The absence of cytoplasmic or ER-associated HCV-LP in cells producing C 173 contrasts with results obtained in cell-free assays, in which the C 173 protein was found to self-assemble into nucleocapsid-like particles (Klein et al., 2005; Klein et al., 2004). This may reflect differences between cell-free and cellular assays. In cellular assays, our data suggest that the initial targeting of the HCV core to the ER by domain III and the subsequent association of the protein with the ER via domain II appear to be required to trigger the formation of HCV-LPs. However, the SPP cleavage site in HCV core protein has not been precisely determined, and may lie between residues 173 and 179 (McLauchlan et al., 2002; Ogino et al., 2004). We therefore cannot exclude the possibility that C 173 represents a truncated form of the mature HCV core protein, and that aa 173-179 may help anchor the protein in the ER membrane, thereby playing a role in HCV-LP assembly and morphogenesis.
The mutated HCV core proteins harboring deletions in domain I gave two sharply contrasting ultrastructural patterns by EM. Cells producing mutated HCV core proteins with deletion 1, 2, or 1/2 and cells producing HCV WT C191 core protein displayed intense HCV-LP assembly at convoluted ER membranes (Fig. 5). This suggests that the two short domains corresponding to aa 15-28 and 60-66 of the HCV core protein are not required for HCV-LP assembly and morphogenesis. This finding again contrasts with results obtained in cell-free assays, in which truncations of various lengths within the N-terminal 68 aa of the HCV core protein partly or completely abolished nucleocapsid-like assembly (Klein et al., 2005; Klein et al., 2004). However, in such cell-free assays, VLP assembly depends on interactions between the core protein and nucleic acid, and the deletion of basic residue clusters in the N-terminal domain of the HCV core protein is likely to hamper these interactions. It would be interesting to assay the del 1, del 2 and del 1/2 core deletion mutants in the background of the infectious JFH-1 virus, to investigate whether mutated core proteins assemble into virus particles, and whether the viral genome is packaged in these particles. Unlike these three deletion mutants, the HCV del 3 mutated core protein did not lead to HCV-LP formation (Fig. 6), although it was cleaved by SPP (Fig. 2C). Immuno EM clearly showed that this deletion mutant was targeted to the ER membrane, where it induced specific ultrastructural changes, characterized by an electron-dense tubular ER with no HCV-LP budding. Thus a short central segment of 18 aa in domain I (aa 88-106) in the HCV core protein seems essential for HCV-LP assembly and budding. The defect in HCV core del 3 mutant assembly may result from conformational changes altering the structure of the protein, and thereby compromising its assembly. Alternatively, this short domain may be directly involved in the multimerization of the core protein, which may be a prerequisite for the budding of HCV-LPs from the ER membrane. Support for this hypothesis is provided by the results of yeast two-hybrid experiments, showing the presence of a central homodimerization domain between aa 82 and 102 of the HCV core protein (Nolandt et al., 1997).
As previously described (Hope et al., 2002), the GBV-B core protein was mostly associated with lipid droplets, confirming that HCV and GBV-B core proteins share structural and functional features. However, although GBV-B core protein was efficiently cleaved by SPP and its production induced specific ER ultrastructural changes, these changes were not associated with the formation of GBV-B-like particles (Fig. 8, Table 1). The GBV-B sequence expressed in this study is likely functional since it is derived from an infectious GBV-B molecular clone (Martin et al., 2003) and the cleavage site between core and E1 has been mapped by sequencing (Ghibaudo et al., 2004). It is interesting to note that, according to GBV-B and HCV core protein alignment (Fig. 1A), GBV-B core lacks a sequence corresponding to the segment deleted in the HCV core mutant del 3, which seems required for HCV-LP assembly. In addition, it is striking that the ER ultrastructural changes observed in cells expressing GBV-B core or HCV del 3 mutant are very similar (compare Figs 6 and 8), suggesting that the absence of this strech of aa may be directly linked to the lack of core assembly in this system. Alternatively, we cannot rule out that, unlike with HCV, the coexpression of E1 and E2 with core might be a prerequisite for GBV-B-LP assembly and budding. The lack of GBV-B-LP formation may thus reflect differences in the mechanisms for assembly and morphogenesis of these two viruses. It is generally believed that, in members of the flavivirus genus, viral particle formation is coordinated by the membrane-associated core protein and the envelope proteins (recently reviewed in Mukhopadhyay et al., 2005). The envelope proteins certainly play a key role in these budding mechanisms, as subviral envelope particles lacking core are routinely observed in flavivirus infections (Hunt et al., 2001; Lorenz et al., 2003; Scholle et al., 2004). In the case of HCV, our data suggest that the core protein may play a central role in viral particle formation, driving the budding process. Interestingly, subviral HCV envelope particles have not been described in serum of HCV-infected patients. Furthermore, we were unable to generate HCV subviral envelope particles by overproducing HCV E1 and E2 proteins in various cell lines (unpublished results). These data suggest that budding mechanisms may differ in different members of the Flaviviridae family. Altogether, our data suggest that the short domain deleted in the del 3 HCV core mutant, which is present in the HCV core but not in the GBV-B core protein, may be involved in the budding properties of HCV core protein. An understanding of the roles played by HCV proteins in virus assembly and budding might help develop novel antiviral strategies in the future.
Experimental procedures
HCV core deletion mutant and GBV-B core constructs
All HCV core constructs were obtained from our previously described (Blanchard et al., 2002) genotype 1a cDNA clone (Dj6.4; Genbank accession number AF529293), containing the C-E1-E2 coding sequence. The HCV core sequence was aligned with the GBV-B core sequence (Genbank accession number AY243572) (Martin et al., 2003), using the clustal algorithm based AlignX software of the Vector Nti 9.0 package (Invitrogen). The sequence encoding HCV core protein was amplified from the Dj6.4 sequence by PCR, using a specific set of oligonucleotide primers, consisting of one forward primer (5′ GTGGATCCTGCACCATGAGCACGAATCCT 3′) and two different reverse primers: (5′ GGATCCTACTAGGCTGACGCGGGC 3′) for the 191 aa form of the core protein, and (5′ GGATCCCTACTAAGAGCAACCAGGAAGG 3′) for the 173 aa form of the core protein. The start codon and inserted stop codons are indicated in bold, italic typeface. BamH1 sites were also included in the primer sequences (underlined), for subcloning in the pSFV1 vector (Invitrogen). PCR was carried out with the Pfu Turbo DNA polymerase (Stratagene, La Jolla CA), and clones were checked by sequencing. The deletion constructs (del) were obtained by PCR, using additional central pairs of primers corresponding to sequences framing the deletion (Fig. 1): 5′-GAAAAACCAAACGTAACCAGATCGTTGGTGG-3′ and 5′-CCACCAACGATCTGGTTACGTTTGGTTTTTC-3′ for del 1 (Δaa 15-28), 5′-GCGGTCGCAACCTCGAAAGGCGCGTCGGCCC-3′ and 5′-GGGCCGACGCGCCTTTCGAGGTTGCGACCGC-3′ for del 2 (Δ aa 60-66), 5′-CCTTGGCCTCTCTATGGCTGGGGCCCCACAGAC-3′ and 5′-GTCTGTGGGGCCCCAGCCATAGAGAGGCCAAGG-3′ for del 3 (Δ aa 88-106), 5′-GCGGCTTCGCCGACCATGGCGTCCGGG-3′ and 5′-CCCGGACGCCATGGTCGGCGAAGCCGC-3′ for del 4 (Δ aa 133-152), 5′-CGCTGCCAGGGCCCTGGCGAACCTTCCTGGTTGCTC-3′ and 5′-GCAACCAGGAAGGTTCGCCAGGGCCCTGGCAGCGC-3′ for del 5 (Δ aa 153-167). Two additional constructs were also designed: del 1/2, which combines del 1 and del 2 in the same construct, and del 4/5, which combines del 4 and del 5 in the same construct. For del 4/5, two additional primers were used: 5′-CGTGCGGCTTCGCCGACAACCTTCCTGGTTGCTC-3′ and 5′-GCAACCAGGAAGGTTGTCGGCGAAGCCGCACG-3′. The GBV-B core protein nucleotide sequence was amplified by PCR from pVL1392-GB containing sequences encoding structural proteins C-E1-E2 (Martin et al., unpublished), using the following nucleotide primers: 5′-AGGGGATCCGCGAGGGGATCTGGGAG-3′ and 5′-GATGGATCCCTATTAGTCTGGGTCAGTGACCCGC-3′ (Fig. 1). After subcloning in pSFV1, the resulting plasmid drives the expression of aa 1-163 of GBV-B polyprotein including the N-terminal 156 aa of core protein.
Cell culture and RNA transfection
Baby hamster kidney cells (BHK-21) were cultured at 37°C in Glasgow Minimal Essential Medium (GMEM) supplemented with 5% fetal calf serum and 2 % tryptose phosphate. The human hepatocellular carcinoma cell line FLC4 (a gift from Dr Yoshiharu Matsuura) was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10 % fetal calf serum. BHK-21 or FLC4 cells were electroporated with recombinant SFV RNA encoding the various HCV or GBV-B core proteins, as previously described (Blanchard et al., 2002). Briefly, for recombinant RNA synthesis, the various pSFV1 constructs were linearized by digestion at the unique SpeI restriction site located downstream from the 3′ non coding region of the SFV replicon. These constructs were then transcribed in vitro using SP6 RNA polymerase, making use of the SP6 promoter located upstream from the 5′ extremity of the SFV replicon, as recommended by the manufacturer (Invitrogen). As a control, we synthesized recombinant RNA encoding β-galactosidase, using the pSFV3 (Invitrogen) expression vector. For transfection, 8 × 106 cells were mixed with 5 μg of recombinant SFV RNA and electroporated by a single pulse at 350 V, 750 μF (Easyject One Electroporator, Eurogentec, Belgium). Following electroporation, cells were diluted in growth medium, plated in 75 cm2 culture dishes, and cultured for 16 h at 37°C before treatment with trypsin for subsequent analysis. In some experiments, HCV or GBV-B core proteins were expressed in BHK-21 cells in the presence of 100 μM of (Z-LL)2-ketone (Calbiochem), an inhibitor of the signal peptide peptidase (SPP), as previously described (Ait-Goughoulte et al, 2006). For confocal microscopy, transfected cells were directly cultured on glass coverslips in 24-well plates, at a density of 2 × 104 cells per coverslip.
Western blotting
Cells were rinsed twice with PBS and treated with a lysis buffer (1 M Tris pH8, 1 mM EDTA, 1% NP40) supplemented with protein inhibitor cocktail (1 mM phenylmethylsulfonyl fluoride, 2 μg/ml aprotinin, 2 μg/ml leupeptin). Samples were boiled for 10 min in loading buffer before separation by SDS-PAGE in 15 % polyacrylamide gels and transfer to a polyvinylidene difluoride membrane (Amersham Bioscience). Membranes were blocked by incubation for one hour at room temperature in PBS containing 0.05 % (vol/vol) Tween 20 (PBS-T) and 5 % (wt/vol) non-fat milk powder. Membranes were then incubated overnight at 4°C with the anti-HCV core C1856 antibody (mouse monoclonal antibody, Virostat, Portland, ME) diluted 1:5000 in PBS-T or the anti-GBV-B core antibody (polyclonal rabbit antibody generously provided by Dr Eric Gowans) diluted 1:1000 in PBS-T. Membranes were then washed five times for 10 minutes in PBS-T and incubated at room temperature for 2 h with the corresponding horseradish peroxidase-conjugated secondary antibody diluted 1:10000 in PBS-T. Membranes were washed five times in PBS-T, rinsed once in distilled water, and antibody-binding was detected by enhanced chemiluminescence (ECL plus; Amersham Bioscience) on Kodak Biomax Light films.
Core protein and lipid droplet staining for confocal microscopy
Cells grown on glass coverslips were washed with PBS and fixed by incubation for 30 min at room temperature in 4% paraformaldehyde in PBS. The reaction was blocked by incubation for 10 minutes at room temperature with 100 mM glycine in PBS, and cells were permeabilized by incubation for 30 min in 0.05% saponin, 0.2 % bovine serum albumin (BSA) in PBS. Cells were then incubated for 30 minutes with anti-HCV or anti-GBV-B core antibodies (1:50 and 1:100, respectively) in permeabilization buffer, in a dark, humid chamber. Cells were then washed in PBS for 15 min and incubated with the corresponding secondary antibody coupled to Alexa Fluor 488 (Molecular Probes, Eugene, OR), diluted 1:1000 in permeabilization buffer. For lipid staining, cells were treated with Nile red (Sigma Aldrich), diluted 1:1000 (from a 1 mg/ml stock solution in acetone) in permeabilization buffer, during incubation with the secondary antibody. Cells were washed and mounted in 25 mM Tris pH 8.8, 5% glycerol, 2.5% 1,4-diazabicyclo [2,2,2] octane (DABCO), 10 % poly(vinyl alcohol) (PVA) MW range 31000–50000 (Sigma Aldrich). Indirect immunofluorescence and lipid droplet staining were analyzed under an Olympus Fluoview 500 confocal laser scanning microscope (Olympus, Japan).
Electron microscopy and immunogold labeling
Transfected cells were fixed by incubation for 48 h in 4% paraformaldehyde and 1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) and were then postfixed by incubation for 1 h with 1% osmium tetroxide. They were dehydrated in a graded acetone series, and cell pellets were embedded in Epon resin, which was allowed to polymerize for 24 h at 60°C. Ultrathin sections were cut, stained with 1 % uranyl acetate 1 % lead citrate, placed on EM grids coated with collodion membrane, and observed with a Jeol 1010 electron microscope (Tokyo, Japan). To quantify the frequency of specific ultrastructural modifications induced by the expression of a given protein, we recorded these changes in 100 consecutive EM sections of cells transfected with each construct. When present, the HCV-LPs were counted in each positive section, to determine their mean number per cell section.
For immunogold labeling, ultrathin sections were treated for 10 min with 10% hydrogen peroxide to dissolve the resin polymer. After several washes in PBS, grids were incubated with anti-HCV C1856 monoclonal antibody or anti-GBV-B polyclonal antibody diluted 1:50 in PBS supplemented with 1% bovine serum albumin (BSA) for 90 min at room temperature. Grids were washed several times in PBS and were then incubated for 90 minutes at room temperature with the corresponding secondary anti-mouse or anti-rabbit antibody conjugated to gold particles (15 nm in diameter, British Biocell International, Cardiff, UK), diluted 1:40 in PBS. Sections were washed in PBS, fixed in 4% glutaraldehyde in PBS and stained as described above.
Acknowledgments
We thank Eric Gowans (Macfarlane Burnet Institute for Medical Research and Public Health, Melbourne, Australia) and Yoshiharu Matsuura (Research Institute for Microbial Diseases, Osaka, Japan) for providing us with the polyclonal anti-GBV-B core reagent and the FLC4 cell line, respectively. We thank Alain Moreau and Sylvie Trassard for technical assistance with DNA sequencing and immunoEM, respectively. This work was supported by grants from ANRS (Agence Nationale de Recherche sur le SIDA et les hepatites virales), and the Ligue Contre le Cancer (Comite d’Indre & Loire). Our research is supported by the Region Centre (Equipe ESPRI). M.A.-G. and R.P. were supported by fellowships from the French Ministry of Research. Our data were generated with the help of the RIO Electron Microscopy Facility of François Rabelais University.
References
- Ait-Goughoulte M, Hourioux C, Patient R, Trassard S, Brand D, Roingeard P. Core protein cleavage by signal peptide peptidase is required for hepatitis C virus-like particle assembly. J Gen Virol. 2006;87:855–860. doi: 10.1099/vir.0.81664-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter MJ, Mast EE, Moyer LA, Margolis HS. Hepatitis C. Infect Dis Clin North Am. 1998;12:13–26. doi: 10.1016/s0891-5520(05)70405-0. [DOI] [PubMed] [Google Scholar]
- Barba G, Harper F, Harada T, Kohara M, Goulinet S, Matsuura Y, et al. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc Natl Acad Sci U S A. 1997;94:1200–1205. doi: 10.1073/pnas.94.4.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beames B, Chavez D, Guerra B, Notvall L, Brasky KM, Lanford RE. Development of a primary tamarin hepatocyte culture system for GB virus-B: a surrogate model for hepatitis C virus. J Virol. 2000;74:11764–11772. doi: 10.1128/jvi.74.24.11764-11772.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beames B, Chavez D, Lanford RE. GB virus B as a model for hepatitis C virus. Ilar J. 2001;42:152–160. doi: 10.1093/ilar.42.2.152. [DOI] [PubMed] [Google Scholar]
- Blanchard E, Brand D, Trassard S, Goudeau A, Roingeard P. Hepatitis C virus-like particle morphogenesis. J Virol. 2002;76:4073–4079. doi: 10.1128/JVI.76.8.4073-4079.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard E, Hourioux C, Brand D, Ait-Goughoulte M, Moreau A, Trassard S, et al. Hepatitis C virus-like particle budding: role of the core protein and importance of its Asp111. J Virol. 2003;77:10131–10138. doi: 10.1128/JVI.77.18.10131-10138.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulant S, Vanbelle C, Ebel C, Penin F, Lavergne JP. Hepatitis C virus core protein is a dimeric alpha-helical protein exhibiting membrane protein features. J Virol. 2005;79:11353–11365. doi: 10.1128/JVI.79.17.11353-11365.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feld JJ, Hoofnagle JH. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- Ghibaudo D, Cohen L, Penin F, Martin A. Characterization of GB virus B polyprotein processing reveals the existence of a novel 13-kDa protein with partial homology to hepatitis C virus p7 protein. J Biol Chem. 2004;279:24965–24975. doi: 10.1074/jbc.M401148200. [DOI] [PubMed] [Google Scholar]
- Giannini C, Brechot C. Hepatitis C virus biology. Cell Death Differ. 2003;10(Suppl 1):S27–38. doi: 10.1038/sj.cdd.4401121. [DOI] [PubMed] [Google Scholar]
- Hope RG, McLauchlan J. Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis C virus core protein. J Gen Virol. 2000;81:1913–1925. doi: 10.1099/0022-1317-81-8-1913. [DOI] [PubMed] [Google Scholar]
- Hope RG, Murphy DJ, McLauchlan J. The domains required to direct core proteins of hepatitis C virus and GB virus-B to lipid droplets share common features with plant oleosin proteins. J Biol Chem. 2002;277:4261–4270. doi: 10.1074/jbc.M108798200. [DOI] [PubMed] [Google Scholar]
- Hunt AR, Cropp CB, Chang GJ. A recombinant particulate antigen of Japanese encephalitis virus produced in stably-transformed cells is an effective noninfectious antigen and subunit immunogen. J Virol Methods. 2001;97:133–149. doi: 10.1016/s0166-0934(01)00346-9. [DOI] [PubMed] [Google Scholar]
- Klein KC, Dellos SR, Lingappa JR. Identification of residues in the hepatitis C virus core protein that are critical for capsid assembly in a cell-free system. J Virol. 2005;79:6814–6826. doi: 10.1128/JVI.79.11.6814-6826.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein KC, Polyak SJ, Lingappa JR. Unique features of hepatitis C virus capsid formation revealed by de novo cell-free assembly. J Virol. 2004;78:9257–9269. doi: 10.1128/JVI.78.17.9257-9269.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature. 2005;436:933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- Liu Q, Tackney C, Bhat RA, Prince AM, Zhang P. Regulated processing of hepatitis C virus core protein is linked to subcellular localization. J Virol. 1997;71:657–662. doi: 10.1128/jvi.71.1.657-662.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann V, Koch JO, Bartenschlager R. Processing pathways of the hepatitis C virus proteins. J Hepatol. 1996;24:11–19. [PubMed] [Google Scholar]
- Lorenz IC, Kartenbeck J, Mezzacasa A, Allison SL, Heinz FX, Helenius A. Intracellular assembly and secretion of recombinant subviral particles from tick-borne encephalitis virus. J Virol. 2003;77:4370–4382. doi: 10.1128/JVI.77.7.4370-4382.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Bodola F, Sangar DV, Goettge K, Popov V, Rijnbrand R, et al. Chronic hepatitis associated with GB virus B persistence in a tamarin after intrahepatic inoculation of synthetic viral RNA. Proc Natl Acad Sci U S A. 2003;100:9962–9967. doi: 10.1073/pnas.1731505100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLauchlan J, Lemberg MK, Hope G, Martoglio B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 2002;21:3980–3988. doi: 10.1093/emboj/cdf414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065–1067. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Ishibashi K, Matsuura Y, et al. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J Gen Virol. 1997;78(Pt 7):1527–1531. doi: 10.1099/0022-1317-78-7-1527. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Kuhn RJ, Rossmann MG. A structural perspective of the flavivirus life cycle. Nat Rev Microbiol. 2005;3:13–22. doi: 10.1038/nrmicro1067. [DOI] [PubMed] [Google Scholar]
- Nolandt O, Kern V, Muller H, Pfaff E, Theilmann L, Welker R, Krausslich HG. Analysis of hepatitis C virus core protein interaction domains. J Gen Virol. 1997;78(Pt 6):1331–1340. doi: 10.1099/0022-1317-78-6-1331. [DOI] [PubMed] [Google Scholar]
- Ogino T, Fukuda H, Imajoh-Ohmi S, Kohara M, Nomoto A. Membrane binding properties and terminal residues of the mature hepatitis C virus capsid protein in insect cells. J Virol. 2004;78:11766–11777. doi: 10.1128/JVI.78.21.11766-11777.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chretien Y, et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. Faseb J. 2002;16:185–194. doi: 10.1096/fj.01-0396com. [DOI] [PubMed] [Google Scholar]
- Rice CM. Flaviviridae: The viruses and their replication. In: Fields BN, Knipe DM, Howley PM, et al., editors. Fields Virology. 3. Lippincott-Raven Publ; Philadelphia: 1996. pp. 931–959. [Google Scholar]
- Roingeard P, Hourioux C, Blanchard E, Brand D, Ait-Goughoulte M. Hepatitis C virus ultrastructure and morphogenesis. Biol Cell. 2004;96:103–108. doi: 10.1016/j.biolcel.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Rouille Y, Helle F, Delgrange D, Roingeard P, Voisset C, Blanchard E, et al. Subcellular localization of hepatitis C virus structural proteins in a cell culture system that efficiently replicates the virus. J Virol. 2006;80:2832–2841. doi: 10.1128/JVI.80.6.2832-2841.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholle F, Girard YA, Zhao Q, Higgs S, Mason PW. trans-Packaged West Nile virus-like particles: infectious properties in vitro and in infected mosquito vectors. J Virol. 2004;78:11605–11614. doi: 10.1128/JVI.78.21.11605-11614.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Tamura K, Li J, Ishii K, Matsuura Y, Miyamura T, Suzuki T. Ubiquitin-mediated degradation of hepatitis C virus core protein is regulated by processing at its carboxyl terminus. Virology. 2001;280:301–309. doi: 10.1006/viro.2000.0785. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Sakamoto S, Tsutsumi T, Rikimaru A, Tanaka K, Shimoike T, Moriishi K, Iwasaki T, Mizumoto K, Matsuura Y, Miyamura T, Suzuki T. Molecular determinants for subcellular localization of hepatitis C virus core protein. J Virol. 2005;79:1271–1281. doi: 10.1128/JVI.79.2.1271-1281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui K, Wakita T, Tsukiyama-Kohara K, Funahashi SI, Ichikawa M, Kajita T, et al. The native form and maturation process of hepatitis C virus core protein. J Virol. 1998;72:6048–6055. doi: 10.1128/jvi.72.7.6048-6055.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]