

Abstract
The monovalent cation selective channel formed by a dimer of the polypeptide gramicidin A has a single-stranded, right-handed helical motif with 6.5 residues per turn forming a 4-Å diameter pore. The structure has been refined to high resolution against 120 orientational constraints obtained from samples in a liquid-crystalline phase lipid bilayer. These structural constraints from solid-state NMR reflect the orientation of spin interaction tensors with respect to a unique molecular axis. Because these tensors are fixed in the molecular frame and because the samples are uniformly aligned with respect to the magnetic field of the NMR spectrometer, each constraint restricts the orientation of internuclear vectors with respect to the laboratory frame of reference. The structural motif of this channel has been validated, and the high-resolution structure has led to precise models for cation binding, cation selectivity, and cation conductance efficiency. The structure is consistent with the electrophysiological data and numerous biophysical studies. Contrary to a recent claim [Burkhart, B. M., Li, N., Langs, D. A., Pangborn, W. A. & Duax, W. L. (1998) Proc. Natl. Acad. Sci. USA 95, 12950–12955], the solid-state NMR constraints for gramicidin A in a lipid bilayer are not consistent with an x-ray crystallographic structure for gramicidin having a double-stranded, right-handed helix with 7.2 residues per turn.
Orientational constraints derived from solid-state NMR can be used to determine high-resolution three-dimensional structures. Such an approach has been used to define the structure of the ion channel, gramicidin A, in lamellar phase lipids (ref. 1; PDB accession no. 1MAG). Although a reasonable model of this structure has been extant for nearly 30 years (2) and a structure was determined by solution NMR spectroscopy in SDS micelles (3, 4), crystallographic and solution NMR methods have not been successful in a lipid environment. Recently, the validity of the solid-state NMR structure has been challenged (5). In this report, the structural fold of the channel is validated by comparing predicted and observed values for structural constraints not used quantitatively in solving for the structural fold. Furthermore, the NMR observables are compared with predicted values from several structures in the Protein Data Bank. The results establish the high resolution of the solid-state structure and the clear validity of this motif in a lipid environment.
Gramicidin A is a polymorphic structure and the dominant sequence in gramicidin D, the biosynthetic product from Bacillus brevis: HCO-Val-Gly-Ala-dLeu-Ala-dVal-Val-Val-Trp-dLeu-Trp-dLeu-Trp-dLeu-Trp-NHCH2CH2OH. In isotropic organic solvents, this peptide typically forms a double-stranded dimer that may be parallel or antiparallel, left-handed or right-handed and has a range of residues per turn from 5.6 to 7.2 (5–10). In the heterogeneous anisotropic lipid environment, the structure is almost exclusively single-stranded. Because of the short helix length, a single-stranded monomer buries one of its termini in the bilayer. Exposure of the carboxyl terminus to the bilayer surface and lack of exposure for the amino terminus was documented by shift reagent NMR experiments (11). It has been observed that the native monovalent cation selective channel function is maintained when formal charges are introduced at the carboxyl terminus but not when they are introduced at the amino terminus (12). Circular dichroism can distinguish between single-stranded and double-stranded conformers (13, 14), and bilayer preparations of gramicidin have been shown, based on this technique, to be single-stranded. Low-angle x-ray scattering of bilayer preparations has characterized the helical pitch as single-stranded, not double-stranded (15). Moreover, the structure in the membrane-mimetic SDS micelles is definitively single-stranded, and the solid-state NMR structure described here in lamellar phase lipids is also definitively single-stranded. The only structurally characterized double-stranded conformer of gramicidin A in a lipid bilayer was shown to be in a kinetically trapped state that, on heating at 68°C for 3 days, was converted to the single-stranded channel state (16).
The helical sense of the channel state was originally thought to be left-handed (17), but both the SDS micellar structure (3) and the solid-state NMR data (18) in hydrated lipid bilayers clearly showed that the gramicidin A conformation in membrane mimetic environments was right-handed.
Structural Determination from Orientational Constraints.
Orientational constraints are derived from the anisotropic nuclear spin interactions observed by solid-state NMR of uniformly aligned samples. Isotopic labeling has been achieved by using solid-phase peptide synthesis with Fmoc blocking chemistry and HPLC purification when necessary, but typical purity before HPLC purification is >95% (19, 20). As shown by NMR, alignment of the samples with a mosaic spread as small as 0.3° (21) has been obtained for gramicidin A in dimyristoyl phosphatidylcholine bilayers (1:8 molar ratio and ≈50% by weight water). The small mosaic spread has been achieved by preparing samples on thin glass slides, and, in the NMR magnet, the peptides, through their diamagnetic susceptibility, have a tendency to align parallel to the magnetic field axis.
Observed dipolar splittings, such as 15N–1H, 15N–13C, and 15N–2H have a cos2θ dependence with respect to the magnetic field axis. The observed dipolar splitting can be interpreted in light of the magnitude of the dipolar coupling, ν∥. This magnitude depends on physical constants, the distance separating the two nuclei, and a characterization of motional averaging. The motional averaging can be assessed independently and has been for gramicidin throughout the molecule (22–24). This interpretation of the observed dipolar splitting, therefore, leads directly to the orientation of the internuclear vector with an error that is dominated by the error in the dipolar observations.
The dipolar and 2H quadrupolar interactions are essentially axially symmetric interactions, and hence, their magnitude is characterized by a single number. The anisotropic chemical shift is an axially asymmetric interaction characterized by three tensor elements whose magnitudes can be assumed along with a substantial error bar or experimentally characterized from the observation of an unoriented powder pattern. Experimental characterization has been done for each of the 15N chemical shift tensors in the gramicidin backbone (25). In addition, the orientations of approximately half of the chemical shift tensors have been characterized with respect to the molecular frame (25, 26). Based on these characterizations, reasonable assumptions were made about the tensor orientation for the other sites. Therefore, the error for chemical shifts reflects not only the error in observation of the anisotropic chemical shift but also a small contribution from tensor characterization.
The interpretation of dipolar and quadrupolar data, Δνobs, as orientational constraints leads to several ambiguities in the orientation of the internuclear vectors, represented by unit vectors, u.
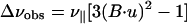 |
1 |
B is a unit vector in the direction of the magnetic field and B⋅u has values between −1 and 1. Therefore, Δνobs is positive if Δνobs > ν∥, but if Δνobs ≤ ν∥, it can be either positive or negative, a sign that is not readily obtained by experiment. Even when Δνobs is positive, B⋅u can be either positive or negative. Hence, there are either two or four possible orientations for each internuclear vector consistent with the dipolar constraint. The interpretation of spin interactions with axially asymmetric tensors, such as the chemical shift, does not give rise to discrete solutions for the internuclear vectors,
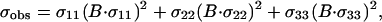 |
2 |
where σii are unit vectors that define the orientation of the chemical shift tensor elements and σobs is the chemical shift in an aligned sample. Because
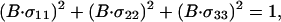 |
3 |
there is not a unique solution or even a discrete set of solutions to these two equations with three unknowns. For 15N chemical shift tensors, two tensor elements, σ11 and σ33, are typically in the peptide plane. Therefore, σ11 and σ33 can be rewritten as
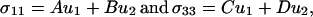 |
4 |
where A, B, C, and D are known from the covalent geometry and the orientation of the tensor relative to the molecular frame. If u1 and u2 are chosen, such that B⋅u1 and B⋅u2 are known, albeit with ambiguity, from dipolar or quadrupolar interactions, then the use of
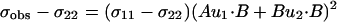 |
5 |
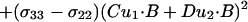 |
will greatly reduce the number of possible orientations for the peptide plane containing u1 and u2 (Fig. 1).
Figure 1.

(A) The primary structural constraints for the polypeptide backbone are derived from the 15N–1H and 15N–13C dipolar interactions, the Cα–2H quadrupolar interaction, and the anisotropic 15N chemical shift. The initial structure is developed by determining each peptide-plane orientation with respect to the magnetic field axis with two dipolar interactions. The relative orientations of the peptide planes is then determined (i.e., the φ and ψ angles) for a diplane structure. (B) The initial structure is assembled sequentially with overlapping diplanes. The initial structure is not a unique structure because of chirality ambiguities; however, the molecular fold, hydrogen-bonding pattern, helical sense, and residues per turn are uniquely defined.
In gramicidin for an isolated peptide plane, two solutions remain, identical orientations with respect to the +B and −B directions. This ambiguity is not a problem for two reasons. First, this ambiguity reflects whether the molecule as a whole is oriented with respect to the +B or −B field directions. Because both molecular orientations exist in our aligned samples and because the NMR observables are independent of the sign, this ambiguity is not a problem. However, when considering the adjacent peptide plane, this ambiguity affects the relative orientation of the peptide planes and hence the conformation. Fortunately, the relative orientation is typically defined through a combination of the Cα–2H orientational constraint and the covalent geometry surrounding the Cα site.
Although a unique set of bond orientations is hereby defined with respect to B, a final ambiguity remains, the orientation of the normal to the peptide plane, B⋅u1xu2. The sign of this triple product is not defined. Because the angle of B to the peptide plane is small and because the Cα–Cα axis is nearly perpendicular to B, the sign of this triple product has a very small effect on the position of the Cα carbons. Consequently, this ambiguity has little effect on the helical parameters and on the determination of the molecular fold. As will be shown later, this ambiguity, known as a chirality ambiguity, can be resolved in the refinement procedure for the molecular structure. The unique nature of the molecular fold has been illustrated by assembling four initial structures (Fig. 1B) with differing chirality patterns: (i) all chiralities positive, (ii) all chiralities negative, (iii) chiralities alternating +/−, and (iv) chiralities alternating −/+ (24). The four structures all have the same helical sense, number of residues per turn, and hydrogen-bond pattern. This gramicidin A channel conformation is a β-strand in which, because of the alternating pattern of d and l amino acid stereochemistry, all of the side chains radiate toward the lipid environment, leaving the polypeptide backbone to line the channel. The 6.5 residues per turn result in a nominal channel diameter of 4 Å, which permits just a single-file column of water molecules. There are 10 parallel β-sheet-type hydrogen bonds stabilizing this monomeric structure. In addition, six intermolecular hydrogen bonds stabilize the dimer at the bilayer center through antiparallel β-sheet-type hydrogen bonds. Although the monomeric unit of the channel structure has been characterized previously, only recently has there been direct solid-state NMR distance measurements confirming this model of the monomer–monomer interface (R. Fu, M. Cotten, and T.A.C., unpublished results).
Indeed, in building these structures several important features of the orientational constraints are illustrated. The initial structures have hydrogen-bond distances within an rms deviation of 0.5 Å of the ideal β-sheet hydrogen bonds (27). The orientational constraints must be both precise and accurate, because 14 separate dipolar orientational constraints are used quantitatively to define a turn of the helix. Furthermore, given the initial assumption of Engh and Huber (28) covalent geometry and 180° ω-torsion angles seem very reasonable. Finally, errors associated with each orientational constraint, even errors of only a couple of degrees, would result in far worse hydrogen-bond distances if the errors accumulated; however, because the constraints fix each site independently with respect to the laboratory z axis, the errors do not sum. In other words, the orientational constraints are absolute as opposed to relative constraints.
Structural Refinement.
The orientational constraints have not been used optimally in the development of the initial structures. Although the dipolar constraints have been used quantitatively, the chemical shifts and the Cα–2H quadrupolar constraints have been used only as filters to eliminate certain possible peptide-plane orientations. In a refinement protocol, the structure will be refined against a generalized global penalty function including all of the orientational constraints, as well as ideal hydrogen-bond geometry and the Chemistry at Harvard Macromolecular Mechanics (CHARMM) force field. The refined structure is obtained through a geometrical search in which the NMR observables and conformational parameters are calculated for each structural modification and compared with the observed data, ideal hydrogen-bond geometry, and the CHARMM energy of the previous structure. The conformational search and evaluation is particularly difficult with the accurate orientational constraints. The possible conformations are separated by very high-penalty barriers, and therefore, an adequate search of the conformational space required a different approach. Three types of structural modifications were implemented: (i) random atom moves with a diffusion parameter of 5 × 10−4 Å; (ii) compensating torsional moves for ψi and φi+1 of equal magnitude (≤3°) and opposite sign; and (iii) tunneling moves, a specialized form of compensating torsional moves, designed to approximate a change in chirality. Simulated annealing was used to perform the minimization of the penalty function (29) and to generate a structure with minimized energy and optimized fit to the experimental data. Moreover, initial assumptions, such as uniform covalent geometry and ω = 180°, were relaxed.
For this refinement, the experimental data were weighed heavily compared with the CHARMM force field energy, because the experimental constraints were obtained from samples within a lipid bilayer environment, whereas the CHARMM energy was calculated in the absence of both water and lipid. The balance of the contributions to the penalty function represented a difficult choice between accurate experimental constraints and an important force field used to maintain appropriate covalent geometry and van der Waals contacts. Actually, a few significant distortions in bond angles have been identified by procheck (1), indicating that further development of the refinement protocol is needed.
In refining the four initial structures with differing chiralities, a unique chirality solution was achieved for nearly all of the peptide planes. The rms deviation between all 40 refinements was just 0.48 Å. To achieve the structure deposited in the Protein Data Bank (Fig. 2A), these 40 structures were averaged, and a final refinement was performed by using only atom moves and not torsional moves in the simulated annealing.
Figure 2.

Gramicidin A structures in different environments. In addition to the atomic ball-and-stick structures, a ribbon is added to accentuate the handedness and strandedness of the helix. The two monomers have different colored ribbons: one yellow and one orange. The backbone carbonyl oxygens that line the pore of the channel are highlighted in red, and the indole 15N sites, important in dictating the strandedness of the structure in a membrane environment, is shown in blue. For each structure the Ala3–Leu4 peptide-plane orientation is shown with respect to B. (A) The solid-state NMR-derived structure from a bilayer environment: single-stranded, right-handed, and 6.5 residues per turn (ref. 1; PDB accession no. 1MAG). (B) An x-ray crystallographic structure of crystals prepared from Cs+/MeOH solution: double-stranded, right-handed, and 7.2 residues per turn (ref. 5; PDB accession no. 1AV2). (C) A solution NMR structure from an SDS micellar environment: single-stranded, right-handed, and 6.3 residues per turn (ref. 4; PDB accession no. 1GRM). (D) An x-ray crystallographic structure of crystals prepared from benzene/methanol solution: double-stranded, left-handed, and 5.6 residues per turn (ref. 7; PDB accession no. 1ALZ).
Validation of the Structural Fold.
Although there are opportunities to modify and potentially improve the refinement protocol, the solid-state NMR structure in lamellar phase lipids is a high-resolution structure precisely constrained by the orientational constraints. Cross-validation of solution NMR and x-ray crystallographic structures can be achieved by leaving some of the data out of the structure determination followed by a comparison of predicted values from the resultant structure and the observed values for the data not used in the structure determination. Because of the more limited number of constraints in solid-state NMR, the opportunities for cross-validation are less. However, the initial backbone structure determination was achieved with just the quantitative use of the dipolar constraints. A calculation of the chemical shifts from the initial structure and comparison to the experimental data generate a penalty contribution of less than one error bar per constraint. Moreover, the Cα–2H quadrupolar splittings lend additional validity to this fold as will be discussed later with Fig. 3. The solid-state NMR structure is both a high-resolution structure and an accurate structure. Furthermore, the experimental constraints have been obtained from samples of gramicidin A solubilized in liquid crystalline phase lipid bilayers. Because this environment is dynamic, it is important to recognize that this structural solution is a time-averaged structure, for which the characterized molecular motions have been taken into consideration through averaging of the nuclear spin interaction tensors.
Figure 3.

Predicted NMR observables from the four structures shown in Fig. 2 are compared with the experimental values. The vertical scale is the difference in predicted and observed values of the NMR observables for the backbone divided by the error bar for each class of observables: 5 ppm for the 15N chemical shift, 100 Hz for the 15N–13C dipolar interaction; 2 kHz for the 15N–1H dipolar interaction; and 5 kHz for the Cα–2H quadrupolar interaction. In addition to the deviations for the four structures shown in Fig. 2 (letters A–D here correspond to structures A–D in Fig. 2), the deviations are also shown for the solid-state NMR derived initial structure (A1). This structure is calculated based on the dipolar constraints and not on the 15N chemical shift or Cα–2H quadrupolar interactions; therefore, the deviations displayed for these data represent a validation of the initial structure that defines the structural motif.
Gramicidin Structural Polymorphism.
Recently, Duax and coworkers (5) published a crystal structure of gramicidin, a right-handed, antiparallel, double-stranded structure with 7.2 residues per turn. The authors mistakenly claim that their structure agrees with 15N-NMR data on the functional gramicidin D channel in lipid bilayers (5) and that the solid-state NMR characterized structure “does not have an open channel for ion passage” (5).
The referenced solid-state NMR data were resonances from a chemical shift spectrum of uniformly 15N-labeled gramicidin D in oriented dimyristoyl phosphatidylcholine bilayers (30). Resonance assignments and single-site resolution were not available in 1987, although they have been for the past 5 years. To claim agreement with the NMR data, the authors fabricate a new linear scale in θN–H, the angle between the N–H internuclear axis and B, drawn parallel to the chemical shift scale. Only with significant assumptions, as described in Nicholson et al. (30), is the anisotropic chemical shift proportional to cos2θ, and never is the chemical shift linearly proportional to θ. Furthermore, this scale for θ from 0 to 90° is displayed over a 230-ppm chemical shift range, rather than the maximum amide 15N chemical shift anisotropy in gramicidin A of 170 ppm. On their scale, the authors have presented values of θN–H from their structure and from the single-stranded structure defined by solid-state NMR for comparison to the chemical shift.
Here, we have redone this invalid analysis by accurately predicting the NMR observables for four different gramicidin structures (Fig. 2). These represent structures determined by solution NMR (Fig. 2C), x-ray crystallography (Fig. 2 B and D), and solid-state NMR (Fig. 2A). They also represent right-handed (Fig. 2 A–C) and left-handed (Fig. 2D) helices, as well as single-stranded (Fig. 2 A and C) and double-stranded (Fig. 2 B and D) helices. Although these are major differences and the residues per turn vary from 5.6 to 7.2, the secondary structure is a β-strand for all of them. The predicted values of orientational constraints are directly compared by using a normalized difference between predicted and observed values. Normalization was achieved by dividing the differences by the observed error-bar magnitude. These results are presented in Fig. 3 as a histogram for each structure and organized by constraint type.
Clearly, the structure developed from and refined against the solid-state NMR data (Fig. 2A) is most consistent with the observed data (Fig. 3A). Among the other three structures, the next best fit to the solid-state NMR data is the Arseniev structure from SDS micelles (Figs. 2C and 3C) that has the same fold. Although there are very significant deviations from the observed data, there is no consistent pattern of deviation as there is for both of the double-stranded structures, suggesting that the fold is correct but that details of the peptide-plane orientations tilting into and away from the channel axis are in error. Indeed, such characterizations are beyond the resolution of this solution NMR structure. Note that the scale for the analysis of the 1ALZ structure (Fig. 3D) has been compressed by a factor of four to save space; this structure is the most inconsistent with the solid-state NMR data. The alternating sign pattern in the chemical shift deviations is the result of the opposite helical sense for this structure. Because these structures are all β-strand-type structures, the repeat unit is a dipeptide, and the orientations of the two planes are quite different. A change in handedness results in an inversion of the peptide planes as shown in Fig. 2. The systematic error in the 15N–13C dipolar data for both of the double-stranded structures is indicative of an error in the helical pitch. The axis of the N–C1 bond in the peptide backbone is very sensitive to the helical pitch. Consequently, the correct fold of the solution NMR structure has an average deviation from the observed 15N–13C dipolar data that is less than 1 (in units of the error bar; Fig. 3C). The Cα–2H deviations are some of the largest, but they are also the most precise constraints; the error bar used to normalize these deviations is small compared with the magnitudes of the observables. Furthermore, these data are very sensitive to the angle formed between peptide planes, resulting from a combination of the helical pitch and residues per turn. The average deviations from the solution NMR structure (Fig. 3C) nearly cancel, but the deviations for the double-stranded structures (Fig. 3 B and D) have a very significant average value, indicative of systematic errors.
Fig. 3A1 shows the deviations for one of the initial solid-state NMR structures. Because the initial structures differ only by chirality ambiguities, the deviations between predicted and observed data are the same for all of the initial structures. The deviations for the 15N chemical shift and the Cα–2H quadrupolar splittings are even smaller than those for the solution NMR structure. These data were not used in the calculation of the initial structure, and consequently, they represent a validation of the initial structure and hence the fold of the polypeptide in lipid bilayers.
It is also possible to refine the other structures against the NMR observables. In refining the 1AV2 structure (Fig. 2B) with full atom and torsional moves, 22 of the 30 N–C1–Cα and C1–N–Cα bond angles are three or more standard deviations removed from the Engh and Huber (28) geometry as assessed by procheck, as the refinement protocol attempts to change the N–C1 bond orientation to be consistent with the experimental data. Furthermore, if atom moves are inactivated so that the covalent geometry remains fixed and if the hydrogen-bond distances are inactivated in one of the two groves of the double-stranded structure, then the number of residues per turn changes from 7.2 to ≈10 to accommodate the N–C orientations. Finally, if all hydrogen-bond distances are constrained, the experimental data are not well fit, and the structure is still substantially distorted (≫1-Å rms deviation) with respect to the crystallographic coordinates. Moreover, the initial solid-state NMR structure and the solution NMR structure can be refined readily against the experimental data to a good fit even when atom moves are turned off.
The primary argument presented by Duax and coworkers (5) for claiming that their structure is the membrane active form is that their structure was consistent with the solid-state NMR data obtained in a bilayer environment. Because this statement is inaccurate, there is no reason to think that this structure crystallized from methanol solution is the channel conformation that occurs in lipid bilayers. Moreover, it is well understood why the membrane active form is single-stranded. Indoles are much more stable in the hydrophilic/hydrophobic bilayer interface than in the hydrophobic core of the bilayer (21, 31–33). When the tryptophans are completely replaced by phenylalanines, the predominant conformation in the bilayer is the left-handed, double-stranded structure, the same fold as shown in Fig. 2D (34, 35). The indoles are distributed along the molecular axis in both double-stranded structures (Fig. 2 B and D) as opposed to the interface location in both single-stranded structures. Therefore, it has been argued that the tryptophan’s propensity for the bilayer interface is the primary reason for the structural conversion from double-stranded to single-stranded (35). Clearly, the polypeptide environment is an important factor in dictating the molecular fold of this structure. Consequently, modeling this very heterogeneous bilayer environment with a homogeneous model, such as an isotropic organic solvent, may not be adequate.
Although the structure described by Duax and coworkers (5) is not the membrane active form, it does illustrate the polymorphic structural nature of this molecule, reflecting the numerous environments in which it has been studied. Indeed, this polymorphism provides a clear example that the amino acid sequence is not the sole determinant of conformation. For instance, a correlation between the antiparallel double-stranded conformation and nonpolar organic solvents has been established (36). In more polar organic solvents, the parallel double-stranded conformation with a net axial dipole is much more stable. It should be noted that this “new fold” was previously described by solution NMR (37).
Gramicidin A has proven to be an excellent model channel for understanding cation selectivity and conductance efficiency. The cation binding site in the channel conformation has been shown to include three or more water ligands (38). Such flexibility results in only modest selectivity among monovalent cations, as the binding site can accommodate cations of various size. Delocalized cation binding leads to a shallow potential-energy well, a minimized entropic penalty for cation binding, and a stepwise dehydration mechanism leading to high cation-association rates. In the very extensive literature from electrophysiologists, molecular dynamicists, and other biophysicists, gramicidin has developed into a great tool for understanding cation conductance. There is little question that the cation conducting conformation for gramicidin is the single-stranded, right-handed structure with 6.5 residues per turn and a 4-Å pore that supports a single-file column of water molecules and monovalent cation transport across membranes.
Moreover, it is shown here that solid-state NMR-derived orientational constraints can lead to both a precise and accurate high-resolution structure in an environment that requires neither isotropic solution nor crystallization. Indeed, this approach has many advantages for characterizing structures in liquid crystalline lipid bilayer environments.
Acknowledgments
This work was supported by National Institutes of Health Grant AI-23007 and National Science Foundation Grant MCB 9603935. The work was largely performed at the National High Magnetic Field Laboratory supported by the National Science Foundation Cooperative Agreement DMR-9527035 and the State of Florida.
ABBREVIATION
- CHARMM
Chemistry at Harvard Macromolecular Mechanics
References
- 1.Ketchem R R, Roux B, Cross T A. Structure (London) 1997;5:1655–1669. doi: 10.1016/s0969-2126(97)00312-2. [DOI] [PubMed] [Google Scholar]
- 2.Urry D W. Proc Natl Acad Sci USA. 1971;68:672–676. doi: 10.1073/pnas.68.3.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bystrov V F, Arseniev A S, Barsukov I L, Lomize A L. Bull Magn Reson. 1987;8:84–94. [Google Scholar]
- 4.Lomize A L, Orekhov V, Arseniev A S. Bioorg Khim. 1992;18:182–200. [PubMed] [Google Scholar]
- 5.Burkhart B M, Li N, Langs D A, Pangborn W A, Duax W L. Proc Natl Acad Sci USA. 1998;95:12950–12955. doi: 10.1073/pnas.95.22.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veatch W R, Fossel E T, Blout E R. Biochemistry. 1974;13:5249–5256. doi: 10.1021/bi00723a001. [DOI] [PubMed] [Google Scholar]
- 7.Langs D A. Science. 1988;241:188–191. doi: 10.1126/science.2455345. [DOI] [PubMed] [Google Scholar]
- 8.Arseniev A S, Bystrov V F, Ivnov V T, Ovchinnikov Y A. FEBS Lett. 1984;165:51–56. [Google Scholar]
- 9.Pascal S M, Cross T A. J Mol Biol. 1992;226:1101–1109. doi: 10.1016/0022-2836(92)91055-t. [DOI] [PubMed] [Google Scholar]
- 10.Doyle D A, Wallace B A. J Mol Biol. 1997;266:963–977. doi: 10.1006/jmbi.1996.0837. [DOI] [PubMed] [Google Scholar]
- 11.Weinstein S, Wallace B A, Blout E R, Morrow J S, Veatch W R. Proc Natl Acad Sci USA. 1979;76:4230–4234. doi: 10.1073/pnas.76.9.4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bamberg E, Apell H J, Alpes H. Proc Natl Acad Sci USA. 1977;74:2402–2406. doi: 10.1073/pnas.74.6.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace B A, Veatch W R, Bout E R. Biochemistry. 1981;20:5754–5760. doi: 10.1021/bi00523a018. [DOI] [PubMed] [Google Scholar]
- 14.Bano M C, Braco L, Abad C. FEBS Lett. 1989;250:67–71. doi: 10.1016/0014-5793(89)80686-6. [DOI] [PubMed] [Google Scholar]
- 15.Katsaras J, Prosser R S, Stinson R H, Davis J H. Biophys J. 1992;61:827–830. doi: 10.1016/S0006-3495(92)81888-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arumugam S, Pascal S, North C L, Hu W, Lee K C, Cotten M, Ketchem R R, Xu F, Brenneman M, Kovacs F. Proc Natl Acad Sci USA. 1996;93:5872–5876. doi: 10.1073/pnas.93.12.5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Urry D W, Walker J T, Trapane T L. J Membr Biol. 1982;69:225–231. doi: 10.1007/BF01870401. [DOI] [PubMed] [Google Scholar]
- 18.Nicholson L K, Cross T A. Biochemistry. 1989;28:9379–9385. doi: 10.1021/bi00450a019. [DOI] [PubMed] [Google Scholar]
- 19.Fields C G, Fields G B, Noble R L, Cross T A. Int J Pept Protein Res. 1989;33:298–303. doi: 10.1111/j.1399-3011.1989.tb01285.x. [DOI] [PubMed] [Google Scholar]
- 20.Fields G B, Fields C G, Petefish J, Van Wart H E, Cross T A. Proc Natl Acad Sci USA. 1988;85:1384–1388. doi: 10.1073/pnas.85.5.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu W, Lazo N D, Cross T A. Biochemistry. 1995;34:14138–14146. doi: 10.1021/bi00043a019. [DOI] [PubMed] [Google Scholar]
- 22.North C L, Cross T A. Biochemistry. 1995;34:5883–5895. doi: 10.1021/bi00017a018. [DOI] [PubMed] [Google Scholar]
- 23.Lee K-C, Huo S, Cross T A. Biochemistry. 1995;34:857–867. doi: 10.1021/bi00003a020. [DOI] [PubMed] [Google Scholar]
- 24.Ketchem R R, Lee K-C, Huo S, Cross T A. J Biomol NMR. 1996;8:1–14. doi: 10.1007/BF00198135. [DOI] [PubMed] [Google Scholar]
- 25.Mai W, Hu W, Wang C, Cross T A. Protein Sci. 1993;2:532–542. doi: 10.1002/pro.5560020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teng Q, Iqbal M, Cross T A. J Am Chem Soc. 1992;114:5312–5321. [Google Scholar]
- 27.Jeffrey G A, Saenger W. Hydrogen Bonding in Biological Systems. Berlin: Springer; 1994. [Google Scholar]
- 28.Engh R A, Huber R. Acta Crystallogr A. 1991;47:392–400. [Google Scholar]
- 29.Kirkpatrick S, Gelatt C D, Jr, Vecchi M P. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- 30.Nicholson L K, Moll F, Mixon T E, LoGrasso P V, Lay J C, Cross T A. Biochemistry. 1987;26:6621–6626. doi: 10.1021/bi00395a009. [DOI] [PubMed] [Google Scholar]
- 31.O’Connell A M, Koeppe R E, II, Andersen O S. Science. 1990;250:1256–1259. doi: 10.1126/science.1700867. [DOI] [PubMed] [Google Scholar]
- 32.Jacobs R E, White S H. Biochemistry. 1989;28:3421–3437. doi: 10.1021/bi00434a042. [DOI] [PubMed] [Google Scholar]
- 33.Michel M, Deisenhofer J. Curr Top Membr Transp. 1990;36:53–69. [Google Scholar]
- 34.Cotten M, Xu F, Cross T A. Biophys J. 1997;73:614–623. doi: 10.1016/S0006-3495(97)78097-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cotten M, Fu R, Cross T A. Biophys J. 1999;76:1179–1189. doi: 10.1016/S0006-3495(99)77282-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu F, Cross T A. Magn Reson Chem. 1998;36:651–655. [Google Scholar]
- 37.Arseniev A S, Barsukov I L, Bystrov V F. FEBS Lett. 1985;180:33–39. doi: 10.1016/0014-5793(85)80702-x. [DOI] [PubMed] [Google Scholar]
- 38.Tian F, Cross T A. J Mol Biol. 1999;5:1993–2003. doi: 10.1006/jmbi.1998.2434. [DOI] [PubMed] [Google Scholar]