

Abstract
The aim of this study was to clarify the association between the epigenetic instability phenotype and the chromosomal instability phenotype in primary hepatocellular carcinoma (HCC). Sixty primary HCC tumors were examined. Methylation status for nine CpG islands (the p16, COX2, GSTP1, RASSF1A, E-cadherin, and APC gene promoters, and the MINT 1, 25, and 31 clones) was evaluated by methylation-specific polymerase chain reaction. Chromosomal structural alterations of these 60 HCC tumors were characterized in our previous study by using whole genomic array-based comparative genomic hybridization. We found that the epigenetic instability phenotype and the chromosomal instability phenotype are not mutually exclusive in hepatocarcinogenesis and that they do not show a simple cause-and-effect relationship. Hepatitis virus infection in the background liver was significantly associated with these instability phenotypes. Furthermore, we identified an epigenetic instability-dependent HCC that shows frequent epigenetic aberrations without chromosomal instability. It was noteworthy that epigenetic instability-positive and -negative HCCs displayed distinctive combinations of chromosomal structural alterations. In summary, by combined analyses of genetic and epigenetic aberration profiles in HCC, we obtained a comprehensive view of genomic alterations in hepatocarcinogenesis. Our results have clinical relevance because epigenetic instability-dependent HCCs may respond well to methylation inhibitory therapies.
Hepatocellular carcinoma (HCC) is one of the most common human malignancies worldwide.1 Most HCC patients in Asian and African populations exhibit chronic hepatitis or cirrhosis caused by persistent infection with hepatitis B virus (HBV) or hepatitis C virus (HCV).1 In Western countries, the incidence of HCC has been increasing throughout the last decade, and it has been estimated that this trend will continue for 2 more decades, mainly because of the increase in hepatitis virus infection.2,3 Although such environmental risk factors have been clearly defined, the distinct molecular events that occur during HCC development are still not fully understood. Therefore, a clear definition of the genetic and epigenetic aberrations that characterize hepatocarcinogenesis would be of value.
Although both genetic alterations (eg, chromosomal deletions, amplifications, and point mutations) and epigenetic alterations (eg, regional CpG island hypermethylation and overall hypomethylation) play significant roles in hepatocarcinogenesis,4,5 the associations between these two major carcinogenesis pathways are far from clear. In other cancers the association between the CpG island methylator phenotype and microsatellite instability has been clearly established.6–8 Our previous study of HCC detected no aberrant hypermethylation of the hMLH1 gene and a very low frequency of microsatellite instability (8.0% or fewer of the cases analyzed).9 Therefore in HCC, epigenetic instability may play roles that are different from those in other types of tumors, and the functions of epigenetic abnormalities in relation to genetic aberrations remain to be clarified.
There have been reports claiming that aberrant DNA methylation status may trigger the alterations in chromosomal structures. Overall DNA hypomethylation that is frequently observed in cancers has been observed to cause chromosomal instability (CIN) in animal models.10–12 On the other hand, regional DNA hypermethylation has also been suggested to precede or even cause chromosomal structural alterations at the relevant loci.13–15 To the contrary, it has been proposed that CpG island methylator phenotype-negative HCCs may well show a higher incidence of loss of heterozygosity,16 and another report has claimed that CpG island methylator phenotype-positive pancreatic cancers show less frequent genetic alterations.17 Because both epigenetic instability and CIN have not been studied concurrently in an integrated and comprehensive manner, the associations between these genomic instability phenotypes remain unclear. The aim of this study was to investigate the associations between epigenetic instabilities and genome-wide chromosomal structural alterations in HCC using a substantial single cohort of primary HCCs.
Materials and Methods
Patient Materials
This study was approved by the institutional review board of the National Cancer Center. Methanol-fixed and paraffin-embedded HCC specimens from 60 patients who had undergone surgery between 1998 and 2001 at the National Cancer Center Hospital were examined. The clinicopathological data of the patients are listed in Table 1. We classified serum HBsAg- and HCVAb-positive patients as HBV- and HCV-positive, respectively. Cases with histopathological evidence of cancer invasion into the portal/hepatic veins were classified as vascular-invasion-positive. Cases were classified according to the TNM classification using the criteria of the American Joint Committee on Cancer.
Table 1.
Clinicopathological Parameters of 60 Cases Analyzed in This Study
| Gender | |
|---|---|
| Male/female | 46 (76.7%)/14 (23.3%) |
| Median age (Range) | 59.3 (23–78) |
| Viral infection | |
| HBV-positive | 17 (28.3%) |
| HCV-positive | 30 (50.0%) |
| negative | 13 (21.7%) |
| Grade of tumor differentiation | |
| well differentiated HCC | 6 (10.0%) |
| moderately differentiated HCC | 26 (43.3%) |
| poorly differentiated HCC | 28 (46.7%) |
| Maximum tumor diameter | |
| <5.0 cm/>5.0 cm | 37 (61.7%)/23 (38.3%) |
| TNM stage | |
| I | 12 (20.0%) |
| II | 30 (50.0%) |
| IIIA | 15 (25.0%) |
| IIIB | 0 (0.0%) |
| IIIC | 3 (5.0%) |
| IV | 0 (0.0%) |
| Vascular invasion | |
| positive/negative | 28 (46.7%)/32 (53.3%) |
| Intrahepatic metastasis | |
| positive/negative | 34 (56.7%)/26 (43.3%) |
Methylation-Specific Polymerase Chain Reaction (PCR) and Combined Bisulfite Restriction Enzyme Analysis for Multiple CpG Islands
For methylation analysis, methanol-fixed tumor specimens were scraped off slide preparations under a microscope and DNA was extracted by standard procedures using proteinase K digestion and phenol/chloroform extraction. Our preliminary experiments had revealed that frozen tissues and methanol-fixed tissues from the same tumors had the same methylation status of CpG islands examined in this study (data not shown). We qualitatively analyzed the methylation statuses of nine CpG islands that had been repeatedly investigated in previous studies of HCC.9,16,18–21 Bisulfite conversion was performed with 1 μg of tumor genomic DNA using a CpGenome DNA modification kit (Intergen, Purchase, NY) according to the manufacturer’s protocol. Methylation-specific PCR was performed to evaluate the DNA methylation status of CpG islands in the promoter regions of the p16, APC, E-cadherin, RASSF1A, COX2, and GSTP1 genes. DNA methylation status of the MINT 1, 25, and 31 clones was determined by combined bisulfite restriction enzyme analysis. The PCR primers and restriction enzymes for each CpG island are listed in Table 2 and in previous reports.9,16,18,20,21 The PCR products or enzyme-digested products were separated by 3% agarose gel electrophoresis (Figure 1A).
Table 2.
Primer Sequences and Restriction Enzymes for the MSP and the COBRA Analysis
| MINT clones | Forward primer | Reverse primer | Restriction enzyme |
|---|---|---|---|
| MINT 1 | F 5′ GGGTTGGAGAGTAGGGGAGTT 3′ | R 5′ CCATCTAAAATTACCTCRATAACTTA 3′ | TaqI |
| MINT 25 | F 5′ TYGGTGTTTGTAAAGGGTTGGAAT 3′ | R 5′ CCCRAACTAAAAACTAACTCRTAA 3′ | RsaI |
| MINT 31 | F 5′ GAYGGYGTAGTAGTTATTTTGTT 3′ | R 5′ CATCACCACCCCTCACTTTAC 3′ | MaeII |
| Gene promoters | |||
| p16-M | F 5′ TTATTAGAGGGTGGGGCGGATCGC 3′ | R 5′ GACCCCGAACCGCGACCGTAA 3′ | |
| p16-U | F 5′ TTATTAGAGGGTGGGGTGGATTGT 3′ | R 5′ CAACCCCAAACCACAACCATAA 3′ | |
| Cox2-M | F 5′ TTAGATACGGCGGCGGCGGC 3′ | R 5′ TCTTTACCCGAACGCTTCCG 3′ | |
| Cox2-U | F 5′ ATAGATTAGATATGGTGGTGGTGGT 3′ | R 5′ CACAATCTTTACCCAAACACTTCCA 3′ | |
| GSTP1-M | F 5′ TTCGGGGTGTAGCGGTCGTC 3′ | R 5′ GCCCCAATACTAAATCACGACG 3′ | |
| GSTP1-U | F 5′ GATGTTTGGGGTGTAGTGGTTGTT 3′ | R 5′ CCACCCCAATACTAAATCACAACA 3′ | |
| RASSF1A-M | F 5′ GTGTTAACGCGTTGCGTTGCGTATC 3′ | R 5′ AACCCCGCGAACTAAAAACGA 3′ | |
| RASSF1A-U | F 5′ TTTGGTTGGAGTGTGTTAATGTG 3′ | R 5′ CAAACCCCACAAACTAAAAACAA 3′ | |
| E-cadherin-M | F 5′ TTAGGTTAGAGGGTTATCGCGT 3′ | R 5′ TAACTAAAAATTCACCTACCGAC 3′ | |
| E-cadherin-U | F 5′ TAATTTTAGGTTAGAGGGTTATTGT 3′ | R 5′ CACAACCAATCAACAACACA 3′ | |
| APC-M | F 5′ TATTGCGGAGTGCGGGTC 3′ | R 5′ TCGACGAACTCCCGACGA 3′ | |
| APC-U | F 5′ GTGTTTTATTGTGGAGTGTGGGTT 3′ | R 5′ CCAATCAACAAACTCCCAACAA 3′ |
Figure 1.

Aberrant CpG island methylation profile of HCC. A: Representative results of methylation-specific PCR and combined bisulfite restriction enzyme analysis (COBRA). Bisulfite-modified DNA was amplified with primers specific to the methylated (M) or unmethylated (U) CpG islands of the p16, Cox2, GSTP1, RASSF1A, E-cadherin, and APC gene promoters. For the p16 gene promoter, we also performed PCR with primers (W) located outside the CpG islands to evaluate the efficiencies of the bisulfite modification; this PCR reaction never occurs if the modification is performed adequately. For the MINT 1, 25, and 31 clones, COBRA was performed in which only methylated samples were digested with restriction enzymes, and their locations are indicated by arrowheads. B: Before enzymatic digestion. A: After enzymatic digestion. B: Overview of the methylation analysis of nine CpG islands in 60 HCCs. Filled boxes indicate the presence of methylation and open boxes indicate the absence of methylation. The rows and columns indicate the nine CpG islands and the 60 HCC tumors, respectively.
Array-Based Comparative Genomic Hybridization (aCGH) Analysis of Primary HCC
We have previously conducted an aCGH analysis of 87 primary HCCs to clarify the whole genomic chromosomal structural alteration profile of HCC.22 In that study, all of the 60 HCC tumors analyzed in this study were also investigated and the chromosomal alteration profiles of these 60 HCCs have been clearly defined. Details of procedures of aCGH analysis have been previously described.22–24 We determined thresholds for chromosomal gain (ratio >1.25) and loss (ratio <0.75) by repeatedly performed normal versus normal control experiments.22,23,25
Statistical Analysis
Pearson’s correlation coefficients were calculated to investigate the correlations of methylation status for various combinations of CpG islands. Student’s unpaired t-test, χ2 test, and Fischer’s exact test were used for statistical analysis when comparing the frequencies of aberrations between groups. For analysis of survival probabilities, the log-rank test and Cox uni- and multiregression models were used. Simple linear regression analysis was performed to investigate the correlations between the numbers of aberrant CpG island methylations and those of chromosomal structural alterations.
Results
Methylation Status of Each CpG Island and Its Correlation with Clinicopathological Parameters
To redefine the overall picture of aberrant CpG island hypermethylation in HCC and its clinical significance, we investigated the methylation status of nine CpG islands that have been repeatedly investigated in previous studies of HCC (Tables 2 and 3).9,16,18–21 All of the nine CpG islands were found to be methylated in HCC at various frequencies ranging from 5.0 to 88.3% (Table 3), which were comparable to figures reported previously, especially for HCC in Asian populations.9,18,21 We then examined correlations between the methylation status of each CpG island and various clinicopathological features of the tumors (Table 4). Interestingly, the methylation statuses of the MINT31 clone and of the CpG islands of the p16, GSTP1, and RASSF1A gene promoters were all significantly correlated with viral infections in the background liver parenchyma (Table 4, P < 0.05). No significant association was observed between methylation status of any CpG islands and patient outcome (data not shown).
Table 3.
Incidence of Aberrant DNA Methylation in 60 HCCs
| CpG island | No. of methylated cases (%) |
|---|---|
| MINT1 | 13 (21.67) |
| MINT25 | 3 (5.00) |
| MINT31 | 40 (66.67) |
| p16 | 39 (65.00) |
| Cox2 | 23 (38.33) |
| GSTP1 | 32 (53.33) |
| RASSF1A | 45 (75.00) |
| E-cadherin | 26 (43.33) |
| APC | 53 (88.33) |
Table 4.
Promoter Methylation Status and its Clinicopathological Correlations in 60 HCCs
| CpG island | Methylation status | No. of samples | Gender
|
Age (average) | P value** | Viral infection
|
Histological differentiation
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Male | Fe- male | P value* | HBV- posi- tive | HCV- posi- tive | Nega- tive | P value*** | Well | Mode- rate | Poor | P value*** | |||||||
| MINT1 | Methylated | 13 | 9 | 4 | 0.47 | 61.23 | 0.39 | 3 | 9 | 1 | 0.24 | 1 | 6 | 6 | 0.94 | ||
| Unmethylated | 47 | 37 | 10 | 58.81 | 14 | 21 | 12 | 5 | 20 | 22 | |||||||
| MINT25 | Methylated | 3 | 3 | 0 | 0.33 | 57.33 | 0.40 | 0 | 3 | 0 | 0.21 | 1 | 0 | 2 | 0.19 | ||
| Unmethylated | 57 | 43 | 14 | 59.44 | 17 | 27 | 13 | 5 | 26 | 26 | |||||||
| MINT31 | Methylated | 40 | 29 | 11 | 0.28 | 58.68 | 0.56 | 13 | 24 | 3 | 0.00080 | 6 | 15 | 19 | 0.14 | ||
| Unmethylated | 20 | 17 | 3 | 60.65 | 4 | 6 | 10 | 0 | 11 | 9 | |||||||
| p16 | Methylated | 39 | 29 | 10 | 0.56 | 62.05 | 0.027 | 9 | 26 | 4 | 0.00092 | 5 | 18 | 16 | 0.40 | ||
| Unmethylated | 21 | 17 | 4 | 54.29 | 8 | 4 | 9 | 1 | 8 | 12 | |||||||
| Cox2 | Methylated | 23 | 18 | 5 | 0.82 | 58.13 | 0.52 | 7 | 11 | 5 | 0.95 | 2 | 9 | 12 | 0.80 | ||
| Unmethylated | 37 | 28 | 9 | 60.08 | 10 | 19 | 8 | 4 | 17 | 16 | |||||||
| GSTP1 | Methylated | 32 | 21 | 11 | 0.031 | 58.34 | 0.47 | 11 | 18 | 3 | 0.045 | 3 | 16 | 13 | 0.53 | ||
| Unmethylated | 28 | 25 | 3 | 60.46 | 6 | 12 | 10 | 3 | 10 | 15 | |||||||
| RASSF1A | Methylated | 45 | 36 | 9 | 0.29 | 60.67 | 0.20 | 13 | 26 | 6 | 0.019 | 5 | 18 | 22 | 0.65 | ||
| Unmethylated | 15 | 10 | 5 | 55.33 | 4 | 4 | 7 | 1 | 8 | 6 | |||||||
| E-cadherin | Methylated | 26 | 20 | 6 | 0.97 | 59.23 | 0.95 | 5 | 15 | 6 | 0.38 | 3 | 13 | 10 | 0.54 | ||
| Unmethylated | 34 | 26 | 8 | 59.41 | 12 | 15 | 7 | 3 | 13 | 18 | |||||||
| APC | Methylated | 46 | 42 | 4 | 0.19 | 60.79 | 0.13 | 15 | 28 | 10 | 0.31 | 6 | 24 | 23 | 0.33 | ||
| Unmethylated | 14 | 11 | 3 | 48.29 | 2 | 2 | 3 | 0 | 2 | 5 | |||||||
| Intrahepatic metastasis
|
Vascular invasion
|
TNM classification
|
Maximum diameter
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Posi- tive | Nega- tive | P value* | Posi- tive | Nega- tive | P value* | I to II | III to IV | P value* | <5.0 cm | 5.0 cm< | P value* | |||
| MINT1 | Methylated | 13 | 7 | 6 | 0.56 | 9 | 4 | 0.30 | 8 | 5 | 0.45 | 9 | 4 | 0.53 |
| Unmethylated | 47 | 21 | 26 | 25 | 22 | 34 | 13 | 28 | 19 | |||||
| MINT25 | Methylated | 3 | 1 | 2 | 0.63 | 2 | 1 | 0.44 | 2 | 1 | 0.90 | 2 | 1 | 0.85 |
| Unmethylated | 57 | 27 | 30 | 25 | 32 | 40 | 17 | 35 | 22 | |||||
| MINT31 | Methylated | 40 | 17 | 23 | 0.36 | 23 | 17 | 0.85 | 29 | 11 | 0.55 | 28 | 12 | 0.060 |
| Unmethylated | 20 | 11 | 9 | 11 | 9 | 13 | 7 | 9 | 11 | |||||
| p16 | Methylated | 39 | 16 | 23 | 0.23 | 18 | 21 | 0.025 | 32 | 7 | 0.0055 | 29 | 10 | 0.91 |
| Unmethylated | 21 | 12 | 9 | 16 | 5 | 10 | 11 | 8 | 3 | |||||
| Cox2 | Methylated | 23 | 13 | 10 | 0.23 | 14 | 9 | 0.60 | 13 | 10 | 0.072 | 15 | 8 | 0.66 |
| Unmethylated | 37 | 15 | 22 | 20 | 17 | 29 | 8 | 22 | 15 | |||||
| GSTP1 | Methylated | 32 | 13 | 19 | 0.32 | 17 | 15 | 0.55 | 25 | 7 | 0.14 | 23 | 9 | 0.082 |
| Unmethylated | 28 | 15 | 13 | 17 | 11 | 17 | 11 | 14 | 14 | |||||
| RASSF1A | Methylated | 45 | 21 | 24 | 1.00 | 25 | 20 | 0.76 | 34 | 11 | 0.10 | 30 | 15 | 0.17 |
| Unmethylated | 15 | 7 | 8 | 9 | 6 | 8 | 7 | 7 | 8 | |||||
| E-cadherin | Methylated | 26 | 13 | 13 | 0.65 | 15 | 11 | 0.89 | 20 | 6 | 0.31 | 18 | 8 | 0.29 |
| Unmethylated | 34 | 15 | 19 | 19 | 15 | 22 | 12 | 19 | 15 | |||||
| APC | Methylated | 46 | 24 | 29 | 0.55 | 28 | 25 | 0.099 | 39 | 14 | 0.095 | 35 | 18 | 0.055 |
| Unmethylated | 14 | 4 | 3 | 6 | 1 | 3 | 4 | 2 | 5 | |||||
P values were calculated by χ2 test for 2 × 2 squares.
P values were calculated by Student unpaired t-test.
Pvalues were calculated by χ2 test for 3 × 2 squares.
Subgrouping of HCC on the Basis of CpG Island Methylation Profile
We found significantly positive correlations of the methylation statuses among the investigated CpG islands. The CpG island methylation status of the MINT1 clone was significantly correlated with that of the MINT31 clone [Pearson correlation coefficient (r) = 0.286, P = 0.013], the COX2 gene promoter (r = 0.334, P = 0.005), and the GSTP1 gene promoter (r = 0.330, P = 0.005). Similarly, the methylation status of the p16 gene promoter was significantly correlated with that of the RASSF1A gene promoter (r = 0.222, P = 0.044) and the APC gene promoter (r = 0.386, P = 0.001), the methylation status of the RASSF1A gene promoter was significantly correlated with that of the E-cadherin gene promoter (r = 0.272, P = 0.018) and the APC gene promoter (r = 0.270, P = 0.019), the methylation status of the MINT31 clone was significantly correlated with that of the GSTP1 gene promoter (r = 0.402, P = 0.001), and the methylation status of the GSTP1 gene promoter was significantly correlated with that of the E-cadherin gene promoter (r = 0.211, P = 0.05). These concordant tendencies of hypermethylation in various CpG islands suggested the presence of a phenotype with simultaneous methylation of multiple CpG islands in HCC.
In a previous study, on the basis of examining eight CpG islands, Shen and colleagues16 have proposed that HCC can be divided into three degrees of methylation density: CpG island methylator phenotype-positive (32 of 85, 37.6% of the analyzed cases), -intermediate (36 of 85, 42.4%), and -negative (17 of 85, 20.0%) groups. Similarly, by examining nine CpG islands, Yang and colleagues20 have also proposed that HCC can be classified into three groups: dense methylation (14 of 47, 29.8%), scattered methylation (24 of 47, 51.1%), and nonmethylation (9 of 47, 19.1%) groups. On the basis of these criteria we categorized HCCs into three groups of CpG island methylation frequency: methylation intensity (METi)-high (18 of 60, 30.0%) and -low (16 of 60, 26.7%) groups with more than six and less than three methylated CpG islands out of nine, respectively, and we classified the rest as METi-intermediate HCCs (26 of 60, 43.3%) (Figure 1B).
Significant Correlation between METi and Hepatitis Viral Infection
We examined the correlations between METi status and various clinicopathological parameters (Table 5), and found that the presence of hepatitis viral infections was significantly more frequent in METi-high/intermediate tumors than in METi-low tumors (P < 0.005). Moreover, HBV- or HCV-positive HCCs showed more frequent hypermethylation of CpG islands than virus-negative ones (HCV, P < 0.0001; HBV, P < 0.01; Figure 2A). METi status showed no significant relationship with patient outcome in HCC (data not shown).
Table 5.
Methylation-Intensity (METi) Status vs Clinicopathological Features of 60 HCCs
| Gender
|
Age | Viral infection
|
Histological differentiation
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| METi-status | No. of cases | Male | Female | HBV | HCV | Negative | Well | Moderate | Poor | ||||
| METi-high | 18 | 11 | 7 | 60.0 | 5 | 13 | 0 | 3 | 6 | 9 | |||
| METi-intermediate | 26 | 22 | 4 | 59.27 | 7 | 14 | 5 | 3 | 14 | 9 | |||
| METi-low | 16 | 13 | 3 | 58.69 | 5 | 3 | 8 | 0 | 6 | 10 | |||
| Pvalue |
|
N.S.** |
|
|
|||||||||
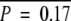
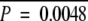
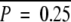
P values were calculated by χ2 test for 3 × 2 square.
Pvalues were calculated by Student′s unpaired t-test.
Pvalues were calculated by χ2 test for 3 × 3 square.
N.S., not significant.
Figure 2.

Aberrant CpG island hypermethylation and chromosomal structural alteration in HCC. A: Correlations between the types of hepatitis virus and the frequencies of CpG island hypermethylation. The y axis indicates the numbers of methylated CpG islands out of nine investigated loci. **P < 0.01, ****P < 0.00001. B: Correlations between the types of hepatitis virus and the extent of chromosomal loss (top) and gain (bottom). The y axis indicates the number of chromosomal losses or gains out of 800 investigated loci. **P < 0.01, ***P < 0.0001. C: Histogram of the frequency of chromosomal structural alterations in HCC. The x axis and the y axis indicate the numbers of chromosomal alterations out of 800 investigated loci and the numbers of cases out of 60 HCCs, respectively. The mean number of chromosomal alterations was 160.2 and the SD was 71.8. The black bars, gray bars, and white bars indicate CIN-positive, -intermediate, and -negative HCCs, respectively. D: Correlations between METi status and the extent of chromosomal loss (top) and gain (bottom). The y axis indicates the number of chromosomal losses or gains out of 800 investigated loci. E: Scatter plot for the numbers of methylated CpG islands out of nine investigated loci (x axis) and the numbers of chromosomal alterations out of 800 investigated loci (y axis). The regression line was determined by simple linear regression analysis. R2, coefficient of determination. P value was calculated by analysis of variance.
Chromosomal Structural Alterations in HCC and Their Correlation with Clinicopathological Parameters
We have previously analyzed the chromosomal structural alteration profile of these 60 HCCs using genome-wide array-based comparative genomic hybridization (aCGH) and found that HCC tumors with a high degree of chromosomal structural alteration more frequently showed malignant clinicopathological features such as invasiveness, metastatic ability, and a poorer grade of histological differentiation.22 Interestingly, as opposed to the METi classification that was significantly correlated with both HBV and HCV infection (Table 5, Figure 2A), the extent of chromosomal alteration was significantly correlated with HBV infection but not with HCV infection (HBV infection: chromosomal loss: P < 0.005, chromosomal gain: P < 0.001; HCV infection: chromosomal loss: P = 0.34, chromosomal gain: P = 0.10, compared with the frequencies in the virus-negative tumors; Figure 2B).
Epigenetic Instability and CIN in HCC
Our aCGH analysis of HCC revealed that the extent of chromosomal alteration varied to a significant degree among individual tumors (Figure 2C). Some tumors harbored considerable numbers of chromosomal losses/gains, whereas others exhibited just a few numbers of chromosomal alterations, suggesting that HCC, like other cancers, can also be divided into groups according to the degree of chromosomal structural alterations.26 According to the histogram for the frequency of chromosomal alterations (Figure 2C; mean, 160.2; SD, 71.8), we defined CIN phenotype (CIN)-positive (22 of 60, 36.7%) and -negative (18 of 60, 30.0%) as having more than 196 (mean + 0.5 SD) and less than 124 (mean −0.5 SD) chromosomal alterations out of 800 chromosomal loci, respectively, and classified the rest as CIN-intermediate (20 of 60, 33.3%).
To explore the relationships between the epigenetic and the chromosomal aberrations in HCCs, we then examined the correlation between the degree of chromosomal structural alteration and that of aberrant CpG island hypermethylation in 60 HCCs (Figure 2, D and E). There was no linear correlation between the frequencies of these alterations (coefficient of determination: r2 = 0.0025, P = 0.71, calculated by simple linear regression analysis), suggesting that the epigenetic instability phenotype and the CIN phenotype are not mutually exclusive and that they show a degree of interrelationship.
Interestingly, we found that there were meaningful associations between the epigenetic instability phenotype and the CIN phenotype in connection with viral infection in the background liver parenchyma (Table 6). Notably, 1) all of the METi-high HCCs were positive for either HBV or HCV (χ2 test, P < 0.005, compared among METi classifications). 2) All of the HBV-positive HCCs were classified as CIN-positive or intermediate (P < 0.01, compared among CIN groups). 3) All of the METi-high/CIN-negative tumors were HCV-positive (five of five, 100.0%). 4) Most of the METi-low/CIN-negative tumors were virus-negative (four of five, 80.0%).
Table 6.
Methylation-Intensity (METi) Status and the Chromosomal Instability (CIN) Status in 60 HCCs
| No. of cases | CIN-positive | CIN-intermediate | CIN-negative | Total | |||||
|---|---|---|---|---|---|---|---|---|---|
| METi-high | 9 | HCV: 5 | 4 | HCV: 3 | 5 | HCV: 5 | 18 | HCV: 13 | |
| HBV: 4 | HBV: 1 | HBV: 0 | HBV: 5 | ||||||
| Virus negative: 0 | Virus negative: 0 | Virus negative: 0 | Virus negative: 0 | ||||||
| METi-intermediate | 7 | HCV: 2 | 11 | HCV: 7 | 8 | HCV: 5 | 26 | HCV: 14 | |
| HBV: 5 | HBV: 2 | HBV: 0 | HBV: 7 | ||||||
| Virus negative: 0 | Virus negative: 2 | Virus negative: 3 | Virus negative: 5 | ||||||
| METi-low | 6 | HCV: 2 | 5 | HCV: 0 | 5 | HCV: 1 | 16 | HCV: 3 | |
| HBV: 2 | HBV: 3 | HBV: 0 | HBV: 5 | ||||||
| Virus negative: 2 | Virus negative: 2 | Virus negative: 4 | Virus negative: 8 | ||||||
| Total | 22 | HCV: 9 | 20 | HCV: 10 | 18 | HCV: 11 | 60 | HCV: 30 | |
| HBV: 11 | HBV: 6 | HBV: 0 | HBV: 17 | ||||||
| Virus negative: 2 | Virus negative: 4 | Virus negative: 7 | Virus negative: 13 | ||||||
To further clarify the associations between the epigenetic instability and specific chromosomal alterations, we next searched for chromosomal aberrations that were preferentially detected in METi-high or -low tumors (Figure 3, A and B). Chromosomal losses on 16p13.3-11 (between 4506 kb and 31,239 kb from the short-arm telomere of chromosome 16) and 16q22.1 (66,966 kb to 71,884 kb, containing the E-cadherin gene) and chromosomal gains on 1q44f (240,953 kb to telomere), 20p12.3 (5096 kb to 21,682 kb), and 20q13.2-13.3 (52,885 kb to 55,652 kb) were observed with significantly higher frequency in METi-high tumors than in METi-intermediate or -low tumors (P < 0.05). In contrast, chromosomal losses on 13q32-34d (90,495 kb to telomere) and chromosomal gains on 1p13.3-13 (112,548 kb to centromere) were detected significantly more frequently in METi-low tumors than in METi-intermediate or -high tumors (P < 0.05). Furthermore chromosomal losses on 6q21-22 (107,064 kb to 117,792 kb), 6q26-27 (160,238 kb to telomere) and 9p21 (21,853 kb to 27,220 kb, containing the p16 gene) were observed with significantly higher frequency in METi-low/intermediate tumors than in METi-high tumors (P < 0.05).
Figure 3.

METi status and the whole genomic chromosomal alteration profile of HCC. A: Overall chromosomal alteration profile (left: chromosomal losses; right: chromosomal gains) for METi-high, -intermediate, and -low HCCs. The frequencies of chromosomal alterations (%, y axis) are indicated from the 1p-telomere (left) to the Xq-telomere (right). Alterations of chromosomal loci that exhibited significantly (P < 0.05) higher and lower frequencies in METi-high tumors than in METi-low tumors are indicated by arrows and arrowheads, respectively. B: Chromosomal alterations that were more prevalent (P < 0.05) in METi-high or -low HCCs are indicated in detail. Each column indicates the bacterial artificial chromosome (BAC) clone on our CGH array and each row indicates an individual HCC tumor. The red boxes and green boxes indicate chromosomal losses and gains, respectively. Physical base positions of the BAC clones were determined with the aid of the National Center for Biotechnology Information (NCBI) Homo sapiens Map-view (http://www.ncbi.nlm.nih.gov/mapview/map_search.cgi?taxid = 9606).
Discussion
The aim of this study was to investigate the associations between epigenetic instability and CIN in HCC. To our knowledge, this is the first reported study to have investigated epigenetic alterations of multiple loci and whole genomic chromosomal structural alterations in a comprehensive manner in a substantial number of primary HCCs. Our study revealed that these two major carcinogenesis pathways are not mutually exclusive and do not show a simple cause-and-effect relationship. Interestingly, we clarified that HCCs with hepatitis virus infection has a statistically significant tendency to harbor more frequent epigenetic aberrations than virus-negative HCCs; at the same time HBV-positive HCCs tends to show the CIN phenotype. We clarified that there exists epigenetic instability-dependent HCC in which genomic alterations during multistep carcinogenesis are primarily due to epigenetic abnormalities. Furthermore, we found that the epigenetic instability-positive and -negative HCCs exhibit specific profiles of chromosomal structural alterations.
To identify the aberrant CpG island hypermethylation profile of HCCs we qualitatively examined the methylation status of nine CpG islands that had been repeatedly investigated in previous studies of HCC,9,16,18–21 and confirmed that they show frequent hypermethylation. We found that, in a subset of HCCs, there is a predisposition for simultaneous multiple hypermethylation of various CpG islands, as reported previously,16,20 and we classified HCCs into three groups—METi-high, -intermediate, and -low—in terms of their frequency of aberrant hypermethylation. The frequency of hypermethylation of each CpG island is, for the most part, consistent with previous studies of HCC in Asian populations, but partially inconsistent with studies conducted in Western populations.9,16,18–21 This discrepancy may be attributable to the differences in the environmental backgrounds of the patients: most HCCs in Asia occur in hepatitis virus-infected livers, whereas in Western countries the frequency of hepatitis virus infection is lower than in Asian countries.1 Because it has been estimated that the incidence of hepatitis virus infection is rising in Western countries,2,3 data from studies of HCC in Asia would be valuable for future application to Western populations.
In this study, we found that both the METi status and methylation status of each CpG island are rarely associated with the malignant features of HCC, as reported previously.9,16,18–21 This suggests that epigenetic instability is not a factor that determines the biological characteristics of the tumor cells, but plays a role mainly in the initial phase of hepatocarcinogenesis. This hypothesis is supported by the fact that aberrant CpG island methylations are detected at significantly high frequency even in precancerous lesions in the background liver, and that their frequency increases in a stepwise manner with progression of the precancerous pathological state.18,20,21
We found that the epigenetic instability phenotype and the CIN phenotype are not mutually exclusive in HCC and that they do not show a simple cause-and-effect relationship, implying that HCC precursor cells can acquire either or both of these genomic instability phenotypes during tumor progression. Interestingly, we discovered a correlation between METi status and CIN status in connection with viral infection. As opposed to the CIN phenotype that is significantly correlated only with HBV infection (Figure 2B; chromosomal loss, P < 0.005, and gain, P = 0.001, compared with virus-negative tumors),22 the frequency of CpG island methylation is significantly correlated with both HCV and HBV infection (Figure 2A; HCV, P < 0.0001, and HBV, P < 0.01, compared with virus-negative tumors). Previous reports have also suggested correlations between aberrant CpG island methylation and hepatitis viral infection,16,20 and our results confirmed that hepatitis virus infections have a tendency to induce epigenetic instability. Two possible hypotheses can explain this result. First, chronic hepatitis and cirrhosis have nonspecific effects on DNA methylation integrity as a consequence of persistent inflammatory stimulation, as it has been reported that inflammatory proliferative diseases such as ulcerative colitis, Barrett’s esophagitis, and Epstein-Barr virus-associated gastritis are strongly related to aberrant hypermethylation of various CpG islands.27–29 Second, it is also possible that hepatitis viruses may exert direct effects on the molecular machinery responsible for maintaining DNA methylation integrity, and that HCV and/or HBV infection might induce de novo CpG island hypermethylation through some unknown mechanism. A previous report has indicated that HBV X protein can repress E-cadherin gene expression by activating DNMT1 expression in vitro.30 Further studies to elucidate the association between the hepatitis virus infection and epigenetic instability will be required.
By comparing METi status with CIN status in HCC, we discovered that there existed METi-high/CIN-negative HCCs and that all of these tumors are HCV-positive (five of five, 100.0%; Table 6). We propose that this group of tumors represents an epigenetic instability-dependent type of HCC in which genomic alterations during multistep carcinogenesis are largely due to epigenetic abnormalities. This finding will be of clinical significance because various methylation-inhibitory agents have been investigated in clinical trials as potential therapies for a range of human malignancies, and have been producing favorable results.31,32 The epigenetic instability-dependent type of HCC would be a good target for these compounds. In multistep hepatocarcinogenesis, as aberrant DNA hypermethylation can be observed in precancerous lesions such as hepatitis and cirrhosis, such methylation-inhibitory therapy might also help to prevent the emergence of HCC in high-risk patients.
Our study also identified METi-low/CIN-negative tumors, most of which (four of five, 80.0%; Table 6) were virus-negative. These tumors may possess as yet unknown types of genomic instability. Alternatively, it is possible that a combination of a few critical genomic alterations is sufficient to generate HCC without acquiring a predisposition to genetic and epigenetic instability, although aCGH analysis failed to demonstrate any common chromosomal alterations in this group (data not shown). This may have been partly due to the limited number of chromosomal loci investigated, and therefore higher resolution analysis will be required to detect any such essential alterations, if present.
To investigate whether the epigenetic instability phenotype has any associations with chromosomal structural alterations of specific chromosomal loci, we searched for chromosomal losses/gains that were significantly more prevalent among METi-high or -low tumors. As shown in Figure 3, specific chromosomal alterations were correlated with METi status. Chromosomal losses on 16p13.3-11 and 16q22.1 and gains on 1q-telomere, 20p12.3, and 20q13.2-13.3 were observed more frequently in METi-high tumors than in METi-low/intermediate tumors. We have previously reported that CpG island methylation of the E-cadherin gene promoter (16q22.1) precedes loss of heterozygosity at the same locus,13 implying that aberrant CpG island hypermethylation may promote chromosomal structural aberrations of the relevant loci. Although the molecular mechanism underlying this phenomenon is not fully clarified, previous reports have suggested that the methylated DNA replicates later than the unmethylated DNA and that such uncoordinated replication time lags may render the chromosomal regions more susceptible to structural alteration.33,34 Interestingly, it has been reported that chromosome 16 is one of the most CpG island-rich chromosomes in the human genome.35 Therefore it can be considered that CpG island hypermethylation occurs with relatively higher density on chromosome 16 in METi-high HCCs, and that consequently frequent chromosomal losses will occur preferentially on this chromosome in the METi-high tumors. Another fascinating hypothesis is that genetic alterations of uncharacterized genes on these loci, 16p13.3-11, 16q22.1, 1q-telomere, 20p12.3, or 20q13.2-13.3, may cause epigenetic instability.
The frequency of chromosomal alterations on 6q21-22, 26-27, 9p21 (the p16 gene) and 13q3-34d and gains on 1p13.2-13 was negatively correlated with METi status. Among these alterations, loss of function of the p16 gene is caused by both genetic and epigenetic alterations in HCC,36 and it seems that under strong methylation pressure in METi-high conditions, the p16 gene tends to be inactivated epigenetically, whereas under METi-low conditions it may be inactivated genetically. It is interesting that chromosome 13 has a notable paucity of CpG islands.35 Therefore, CpG island hypermethylation may occur with relatively low density on this chromosome, even in METi-high tumors, and this chromosome may be more resistant to chromosomal alterations in the METi-high phenotype.
Even though we analyzed various kinds of CpG islands in HCC, the number investigated was small and our criteria for epigenetic instability (the METi classification) were not firm. Larger scale methylation profiling of HCC, such as genome-wide methylation profiling,37,38 would improve the current classification and help to reveal a detailed picture of the methylation aberrations at each chromosomal locus.
In summary, we have found that the epigenetic instability phenotype and the CIN phenotype are not mutually exclusive in HCC. Although the distinct molecular backgrounds that lead to genetic or epigenetic instability remain to be elucidated, our results suggest that persistent viral infection is one of the major causes of these genomic instabilities. It is clinically important to note that some HCCs exhibit an epigenetic instability-dependent phenotype that harbors frequent epigenetic abnormalities without CIN, implying that these tumors might be good therapeutic targets for methylation-inhibitory agents. Furthermore we found that epigenetic instability-positive and -negative HCCs exhibit distinctive profiles of chromosomal alteration. Susceptibility to chromosomal structural alterations resulting from DNA hypermethylation may differ among individual chromosomal loci.
Table 5A.
Continued
| Intrahepatic metasasis
|
Vascular invasion
|
TNM classification
|
Maximum diameter
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Positive | Negative | Positive | Negative | I to II | III to IV | <5.0 cm | <5.0 cm | ||||
| 10 | 8 | 6 | 12 | 15 | 3 | 15 | 3 | ||||
| 14 | 12 | 14 | 12 | 17 | 9 | 17 | 9 | ||||
| 10 | 6 | 8 | 8 | 10 | 6 | 7 | 9 | ||||
|
|
|
|
||||||||
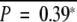
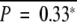
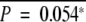
Footnotes
Address reprint requests to Setsuo Hirohashi, M.D., Pathology Division, National Cancer Center Research Institute, 5-1-1, Tsukiji, Chuo-ku, Tokyo, 104-0045, Japan. E-mail: shirohas@ncc.go.jp.
Supported in part by a grant-in-aid for the Comprehensive 10-Year-Strategy for Cancer Control from the Ministry of Health, Labor, and Welfare, Japan.
H.K. is a recipient of a research resident fellowship from the Foundation for the Promotion of Cancer Research in Japan.
References
- Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- El-Serag HB, Mason AC. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999;340:745–750. doi: 10.1056/NEJM199903113401001. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Hanada K, Mizokami M, Yeo AE, Shih JW, Gojobori T, Alter HJ. Inaugural article: a comparison of the molecular clock of hepatitis C virus in the United States and Japan predicts that hepatocellular carcinoma incidence in the United States will increase over the next two decades. Proc Natl Acad Sci USA. 2002;99:15584–15589. doi: 10.1073/pnas.242608099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman WB. Mechanisms of human hepatocarcinogenesis. Curr Mol Med. 2003;3:573–588. doi: 10.2174/1566524033479546. [DOI] [PubMed] [Google Scholar]
- Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–346. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota M, Itoh F, Imai K. DNA methylation and gastrointestinal malignancies: functional consequences and clinical implications. J Gastroenterol. 2000;35:727–734. doi: 10.1007/s005350070030. [DOI] [PubMed] [Google Scholar]
- Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Kanai Y, Sakamoto M, Mizokami M, Ueda R, Hirohashi S. Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis—a comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology. 2000;32:970–979. doi: 10.1053/jhep.2000.19797. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Ushijima S, Tsuda H, Sakamoto M, Hirohashi S. Aberrant DNA methylation precedes loss of heterozygosity on chromosome 16 in chronic hepatitis and liver cirrhosis. Cancer Lett. 2000;148:73–80. doi: 10.1016/s0304-3835(99)00316-x. [DOI] [PubMed] [Google Scholar]
- Makos M, Nelkin BD, Reiter RE, Gnarra JR, Brooks J, Isaacs W, Linehan M, Baylin SB. Regional DNA hypermethylation at D17S5 precedes 17p structural changes in the progression of renal tumors. Cancer Res. 1993;53:2719–2722. [PubMed] [Google Scholar]
- Makos M, Nelkin BD, Chazin VR, Cavenee WK, Brodeur GM, Baylin SB. DNA hypermethylation is associated with 17p allelic loss in neural tumors. Cancer Res. 1993;53:2715–2718. [PubMed] [Google Scholar]
- Shen L, Ahuja N, Shen Y, Habib NA, Toyota M, Rashid A, Issa JP. DNA methylation and environmental exposures in human hepatocellular carcinoma. J Natl Cancer Inst. 2002;94:755–761. doi: 10.1093/jnci/94.10.755. [DOI] [PubMed] [Google Scholar]
- Ueki T, Toyota M, Sohn T, Yeo CJ, Issa JP, Hruban RH, Goggins M. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000;60:1835–1839. [PubMed] [Google Scholar]
- Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S. Expression of mRNA for DNA methyltransferases and methyl-CpG-binding proteins and DNA methylation status on CpG islands and pericentromeric satellite regions during human hepatocarcinogenesis. Hepatology. 2001;33:561–568. doi: 10.1053/jhep.2001.22507. [DOI] [PubMed] [Google Scholar]
- Roncalli M, Bianchi P, Bruni B, Laghi L, Destro A, Di Gioia S, Gennari L, Tommasini M, Malesci A, Coggi G. Methylation framework of cell cycle gene inhibitors in cirrhosis and associated hepatocellular carcinoma. Hepatology. 2002;36:427–432. doi: 10.1053/jhep.2002.34852. [DOI] [PubMed] [Google Scholar]
- Yang B, Guo M, Herman JG, Clark DP. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol. 2003;163:1101–1107. doi: 10.1016/S0002-9440(10)63469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee HJ, Kim JH, Lee HS, Jang JJ, Kang GH. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am J Pathol. 2003;163:1371–1378. doi: 10.1016/S0002-9440(10)63495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh H, Shibata T, Kokubu A, Ojima H, Loukopoulos P, Kanai Y, Kosuge T, Fukayama M, Kondo T, Hosoda F, Sakiyama T, Ohki M, Imoto I, Inazawa J, Hirohashi S. Genetic profile of hepatocellular carcinoma revealed by array-based comparative genomic hybridization: identification of genetic indicators to predict patient outcome. J Hepatol. 2005;43:863–874. doi: 10.1016/j.jhep.2005.05.033. [DOI] [PubMed] [Google Scholar]
- Sonoda I, Imoto I, Inoue J, Shibata T, Shimada Y, Chin K, Imamura M, Amagasa T, Gray JW, Hirohashi S, Inazawa J. Frequent silencing of low density lipoprotein receptor-related protein 1B (LRP1B) expression by genetic and epigenetic mechanisms in esophageal squamous cell carcinoma. Cancer Res. 2004;64:3741–3747. doi: 10.1158/0008-5472.CAN-04-0172. [DOI] [PubMed] [Google Scholar]
- Shibata T, Uryu S, Kokubu A, Hosoda F, Ohki M, Sakiyama T, Matsuno Y, Tsuchiya R, Kanai Y, Kondo T, Imoto I, Inazawa J, Hirohashi S. Genetic classification of lung adenocarcinoma based on array-based comparative genomic hybridization analysis: its association with clinicopathologic features. Clin Cancer Res. 2005;11:6177–6185. doi: 10.1158/1078-0432.CCR-05-0293. [DOI] [PubMed] [Google Scholar]
- Inazawa J, Inoue J, Imoto I. Comparative genomic hybridization (CGH)-arrays pave the way for identification of novel cancer-related genes. Cancer Sci. 2004;95:559–563. doi: 10.1111/j.1349-7006.2004.tb02486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61:3573–3577. [PubMed] [Google Scholar]
- Eads CA, Lord RV, Kurumboor SK, Wickramasinghe K, Skinner ML, Long TI, Peters JH, DeMeester TR, Danenberg KD, Danenberg PV, Laird PW, Skinner KA. Fields of aberrant CpG island hypermethylation in Barrett’s esophagus and associated adenocarcinoma. Cancer Res. 2000;60:5021–5026. [PubMed] [Google Scholar]
- Kang GH, Lee S, Kim WH, Lee HW, Kim JC, Rhyu MG, Ro JY. Epstein-barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am J Pathol. 2002;160:787–794. doi: 10.1016/S0002-9440(10)64901-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JO, Kwun HJ, Jung JK, Choi KH, Min DS, Jang KL. Hepatitis B virus X protein represses E-cadherin expression via activation of DNA methyltransferase 1. Oncogene. 2005;24:6617–6625. doi: 10.1038/sj.onc.1208827. [DOI] [PubMed] [Google Scholar]
- Goffin J, Eisenhauer E. DNA methyltransferase inhibitors—state of the art. Ann Oncol. 2002;13:1699–1716. doi: 10.1093/annonc/mdf314. [DOI] [PubMed] [Google Scholar]
- Issa JP, Gharibyan V, Cortes J, Jelinek J, Morris G, Verstovsek S, Talpaz M, Garcia-Manero G, Kantarjian HM. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol. 2005;23:3948–3956. doi: 10.1200/JCO.2005.11.981. [DOI] [PubMed] [Google Scholar]
- Laird C, Jaffe E, Karpen G, Lamb M, Nelson R. Fragile sites in human chromosomes as regions of late-replicating DNA. Trends Genet. 1987;3:274–281. [Google Scholar]
- Selig S, Ariel M, Goitein R, Marcus M, Cedar H. Regulation of mouse satellite DNA replication time. EMBO J. 1988;7:419–426. doi: 10.1002/j.1460-2075.1988.tb02829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisler LE, Torti D, Boutros PC, Watson J, Chan C, Winegarden N, Takahashi M, Yau P, Huang TH, Farnham PJ, Jurisica I, Woodgett JR, Bremner R, Penn LZ, Der SD. CpG Island microarray probe sequences derived from a physical library are representative of CpG Islands annotated on the human genome. Nucleic Acids Res. 2005;33:2952–2961. doi: 10.1093/nar/gki582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew CT, Li HM, Lo KW, Leow CK, Chan JY, Hin LY, Lau WY, Lai PB, Lim BK, Huang J, Leung WT, Wu S, Lee JC. High frequency of p16INK4A gene alterations in hepatocellular carcinoma. Oncogene. 1999;18:789–795. doi: 10.1038/sj.onc.1202359. [DOI] [PubMed] [Google Scholar]
- Hu M, Yao J, Cai L, Bachman KE, van den Brule F, Velculescu V, Polyak K. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37:899–905. doi: 10.1038/ng1596. [DOI] [PubMed] [Google Scholar]
- Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]