

Abstract
During the development of mammalian digits, clustered Hoxd genes are expressed following a collinear regulatory strategy, leading to both the growth of digits and their morphological identities. Because gene dosage is a key parameter in this system, we used a quantitative approach, associated with a collection of mutant stocks, to investigate the nature of the underlying regulatory mechanism(s). In parallel, we elaborated a mathematical model of quantitative collinearity, which was progressively challenged and validated by the experimental approach. This combined effort suggested a two-step mechanism, which involves initially the looping and recognition of the cluster by a complex including two enhancer sequences, followed by a second step of microscanning of genes located nearby. In this scenario, the respective rank of the genes, with respect to the 5′ extremity of the cluster, is primordial, as well as different gene-specific affinities. This model accounts for the quantitative variations observed in our many mutant strains, and reveals the molecular constraint leading to thumbness; i.e., why a morphological difference must occur between the most anterior digit and the others. We also show that the same model applies to the collinear regulation of Hox genes during the emergence of external genitalia, though with some differences likely illustrating the distinct functionalities of these structures in adults.
[Keywords: Limbs; genitals; regulatory landscapes; modeling, looping]
In vertebrates, Hox genes belonging to the HoxA and HoxD clusters are necessary for the development of the limbs (see Zakany and Duboule 2007). Genes of both clusters are expressed in a complex manner, mostly through two different phases, which depend on distinct regulatory controls (Nelson et al. 1996; Deschamps and van Nes 2005; Tarchini and Duboule 2006). During early limb budding, Hoxd gene transcription is activated in a collinear fashion, starting with Hoxd1 and progressing toward the 5′ end of the cluster (Hoxd13). This early phase of activation leads to the concomitant restriction in the expression of the most 5′-located genes (from Hoxd10 to Hoxd13) into the most posterior-distal cells of the developing limb bud. Eventually, these “posterior” Hox gene products will activate transcription of the gene sonic hedgehog (Tarchini et al. 2006), a key determinant of limb growth and polarity (Riddle et al. 1993), necessary for the second phase of limb development to occur.
This early phase of Hox genes activation corresponds to the organization and patterning of both the stylopodium (the arm) and the zygopodium (the forearm). This phase is soon followed by the morphogenesis of the most distal parts of the limbs (the autopodium; hands and feet), which is accompanied by a second wave of Hox genes activation, involving Hoxa13 as well as the four posterior most genes of the HoxD cluster (from Hoxd10 to Hoxd13). It is during this late phase of expression that the Shh signaling pathway further impacts on the shape of the expression domain, thus imposing the anterior-to-posterior (AP) polarity of our limb extremities (Drossopoulou et al. 2000). The existence of these two separate modes of Hox gene expression has been associated with the distinction between the most distal pieces of the limbs and the rest of the appendages in term of evolutionary history. In this view, the autopodium is a neomorphic structure that appeared in tetrapods, subsequent to the appearance of proximal limbs (Sordino et al. 1995), in parallel to either the de novo emergence, or the full recruitment of the appropriate regulatory sequences (see Spitz et al. 2003; Davis et al. 2007; Freitas et al. 2007; Gonzalez et al. 2007).
The nature of the genetic control underlying the early phase of Hoxd gene expression in limb buds remains to be determined. However, molecular genetic approaches in the mouse have revealed that transcriptional activation likely depends on sequences located outside from the cluster itself, on the telomeric side; i.e., 3′ from Hoxd1 (Spitz et al. 2005). In contrast, the regulatory elements that control the late phase of activation appear to locate centromeric to the cluster (5′ from Hoxd13), as judged by extensive genetic and transgenic analyses. On the one hand, both spontaneous and engineered large inversions have clearly positioned digit-specific enhancer sequences upstream of (centromeric from) the HoxD cluster (Spitz et al. 2003, 2005). On the other hand, transgenic analyses involving BAC clones as well as shorter DNA fragments have pointed to two regions of critical importance for the expression of 5′-located Hoxd genes in developing autopods.
The first of these DNA segments, located some 180 kb upstream of Hoxd13, contains several global enhancer sequences, one of which active in developing digit cells. This global control region (GCR) is conserved among all vertebrates including teleostei, even though the fish counterpart was not able to elicit a digit-specific expression when introduced into transgenic mice (Spitz et al. 2003). The second sequence (Prox) was identified subsequently, and lies between the GCR and the 5′ end of the HoxD cluster. It is also found in birds and amphibian, although not detected in teleostei genomes (Gonzalez et al. 2007). In transgenic animals, this sequence can drive expression in developing digits with a specificity slightly distinct from, and complementary to, that of the GCR. Therefore, expression of Hoxd genes during the late phase of limb development likely results from the combined action of these two regulatory sequences (Gonzalez et al. 2007). In contrast, little is known yet on the transcriptional control of Hoxa13 expression during digit morphogenesis.
In wild-type embryonic day 12.5 (E12.5) mouse embryos, the four contiguous genes Hoxd10, Hoxd11, Hoxd12, and Hoxd13 are expressed under the control of the GCR and Prox sequences. While their expression profiles in digits are virtually identical to one another, their transcriptional outputs seems to follow a collinear distribution, at least when considering steady-state levels of mRNAs (Dolle et al. 1991). Hoxd13, the gene lying at the 5′ extremity of the cluster, is transcribed with maximal efficiency, whereas Hoxd10 is only weakly expressed. Hoxd12 and Hoxd11 have intermediate amounts of transcripts, as assessed by in situ hybridization. This quantitative difference makes Hoxd13 expression detectable in presumptive cells of future digit I (the most anterior digit; the thumb in mammals), unlike the other three genes, whose transcripts are not detected in these cells, most likely due to lower expression levels. Consequently, the Hoxd13 transcript domain encompasses those of the three other genes, an observation in contrast to the general expression strategy of Hox genes and referred to as “reverse collinearity” (Nelson et al. 1996).
This difference between Hoxd13 and other Hox genes is of critical importance as it may help to individualize the morphology of digit I from those of the other digits. There is indeed an exact correlation between digit morphology and Hox gene expression, such that all reported cases where the thumb is replaced by a more “posterior” (longer) type of digits, also show a gain of posterior Hoxd genes expression in the presumptive thumb domain. Alternatively, forced expression of “posterior” Hox genes (e.g., Hoxd11) throughout the limb bud—i.e., including presumptive digit I cells—leads to the disappearance of the thumb and transformation toward a more “elongated” digit morphology, resembling posterior digits (e.g., Morgan et al. 1992). Therefore, the globally weaker dose of Hox function in presumptive digit I cells, mostly due to the nonexpression of Hoxd12, Hoxd11, and Hoxd10 there, is likely a key factor in the morphological difference that exists between this future digit and more posterior digits, which express these latter genes in addition to Hoxa13 and Hoxd13.
The nature of the molecular mechanism underlying reverse collinearity is unknown, yet it must rely on quantitative collinearity, since the Hoxd gene expressed at the highest level (Hoxd13) is the only one transcribed in presumptive digit I cells. By using a set of deletion and duplication alleles produced by targeted meiotic recombination (TAMERE) (Herault et al. 1998), we previously showed that the respective position of a gene was important, rather than the specificity of its promoter. For example, when Hoxd12 was experimentally positioned at the end of the cluster, where Hoxd13 normally stands, it was expressed with the same efficiency as Hoxd13 (Kmita et al. 2002a), including in presumptive digit I cells, where it is normally silent. Also, the introduction of supernumerary promoters at the proximity of the HoxD cluster induced regulatory reallocations in digits (Monge et al. 2003), suggesting that collinear expression of Hoxd genes in developing autopods was the result of a global regulatory equilibrium, controlled by upstream sequences and involving both the number and respective positions of target transcription units.
In this study, we investigate this collinear process by using a quantitative approach to precisely determine the impact of several mutated configurations on the relative amounts of the various Hoxd mRNAs. We use both experimental and theoretical approaches to elaborate a model of collinear regulation, after several rounds of predictions and validations, a process made possible by the large number of various mutant alleles available at this locus. We conclude that the best-fit model involves both a strong topological component, but also calls for some differences in promoter-specific responses. This model also indicates that the collinear Hoxd genes regulations underlying the development of both digits and external genitalia are very similar and likely implemented by the same elements, even though differences exist in the local control of some genes. Finally, this model accounts for the impossibility for all Hoxd genes to be expressed equally strongly in presumptive digits cells (“reverse collinearity”), and hence, it links a clustered genetic topography and the associated regulatory constraints, to a particularly important morphological output, the emergence and maintenance of thumbness.
Results
In E12.5 mouse limb buds, the four most 5′-located Hoxd genes are transcribed in developing digits, with virtually identical expression profiles (Fig. 1A). However, Hoxd13 is expressed at a much higher level and extends within the most anterior presumptive digit one (Fig. 1A, arrowhead; Kmita et al. 2002a), unlike the three other neighbor genes, whose mRNAs steady-state levels seem to decrease following their respective rank on the cluster (Fig. 1A, quantitative collinearity). This expression pattern in digits is controlled by two regulatory sequences located in cis (GCR and Prox) (Spitz et al. 2003; Gonzalez et al. 2007), which also regulate expression of the Evx2 and lunapark (Lnp) genes in the same digit domain, as a result of their location within this regulatory landscape (Fig. 1A; Spitz et al. 2003).
Figure 1.

Quantitative collinearity in developing digits. (A, top) Posterior Hoxd genes are expressed in developing digits following a gradient of transcriptional efficiency: Hoxd13 is expressed robustly, whereas more 3′-located genes display progressively weaker expression levels, up to Hoxd9, which is barely expressed in this domain. Only Hoxd13 is detected in presumptive digit I (arrowhead). (Bottom) Genomic organization of the HoxD locus and centromeric sequences. Evx2 and Lnp are coexpressed with Hoxd genes in the developing digits, under the control of two shared enhancer sequences (GCR, Prox). (B) Experimental flowchart using an in situ hybridization showing the wild-type expression of Hoxd12 at E13.5. Digits (top) and genital buds (bottom) were dissected off of either wild-type or mutant fetuses, and expression levels of all posterior Hoxd genes (from Hoxd13 to Hoxd9), Lnp, and Evx2 were quantified by real-time RT–PCR (see Fig. 2). We looked for potential similarities or differences in the regulation of Hoxd genes in digits and genitalia by comparing the detected modification occurring in both developing tissues. (C) Validation of the quantification by comparing the expression levels of Hoxd13 in the digits of either wild-type (wt) embryos or embryos heterozygous and homozygous for a deletion of the Hoxd13 locus [del(13)]. (Left) Amplification curves. (Right) Relative Hoxd13 mRNA levels. Expression levels fit exactly with gene copy number, showing the accuracy and specificity of the approach. (D) Quantitative collinearity. For all genes, transcript copy numbers in wild-type developing digits were measured by external calibration. Expression levels are shown as a proportion of Hoxd13. Hoxd13 shows the highest transcript level, and RNA copy numbers decrease progressively for more 3′-located genes, up to Hoxd9. Hoxd8 is detected near background levels. Lnp and Evx2 levels correspond to ∼10% of that of Hoxd13.
Quantitative collinearity
In order to assess the various quantities of steady-state mRNAs, and hence to establish a baseline for the relative expression levels of these genes in the wild-type condition, the most distal parts of developing E13.5 digits were dissected out, mRNA extracted and quantified by real-time RT–PCR. At the same time and for comparative purposes, the emergent genital bud was also dissected out and treated similarly (Fig. 1B), since quantitative collinearity was originally described in this developing bud (Dolle et al. 1991). The accuracy of this quantitative approach was verified using a mouse carrying a deletion of the Hoxd13 locus. In heterozygous mice, presumptive digits expressed almost exactly half the amount of the corresponding mRNAs, whereas Hoxd13 transcripts were expectedly not scored in the homozygous mutant background (Fig. 1C). This result as well as several other lines of evidence (data not shown), indicated that cross-regulatory interactions occurring between posterior Hox genes and their products, if any, were not importantly involved in the final amounts of transcripts. Consequently, this particular collinear regulation mostly, if not entirely, derives from interactions in cis.
We next quantified the relative amounts of transcripts between the various genes, such as to better visualize quantitative collinearity and to have a starting point for both the evaluation of our various mutant configurations and theoretical modeling (see below). By using comparisons with known amounts of RNA, the results depicted in Figure 1D were obtained. While they globally confirmed the existence of a collinear distribution, they revealed some unexpected aspects. Because the amount of Hoxd13 mRNAs was arguably the highest, it was arbitrarily fixed to 1, the other values being expressed in percentages of this amount. In this scale, Hoxd8 transcripts were not detected and Hoxd9 mRNAs represented <5%, which was at the limit of significance. This may reflect some weak activity, not easily detected by in situ hybridization. While Hoxd10 was clearly expressed, although at a low level, Hoxd11 and Hoxd12 had surprisingly almost identical levels of mRNAs, in the range of 35% of the Hoxd13 amount. Altogether, quantitative collinearity was confirmed, yet with no clear application to Hoxd12 and Hoxd11. Unlike what previous expression studies had suggested, the steady-state levels of both Lnp and Evx2 mRNAs were low, barely >10% of that of Hoxd13 (Fig. 1D).
Regulatory reallocations
Once these wild-type quantifications were established, we processed a set of mutant strains where both the number and respective order of the genes had been modified (Fig. 2). We first used three mutant strains where the most posterior gene Hoxd13 had been deleted, either alone (Fig. 3A, red bars), in combination with Hoxd12 (Fig. 3A, yellow bars), or together with both Hoxd12 and Hoxd11 (Fig. 3A, green bars). In all cases, quantitative collinearity was maintained, but shifted along with the number of genes deleted. For example, in the absence of the Hoxd13 locus, Hoxd12 was overexpressed by a factor of two- to threefold, to reach 80% of wild-type Hoxd13 expression level. Both Hoxd11 and Hoxd10 transcripts were also increased in amounts, although to a lesser extent. A slight increase in Hoxd9 expression was also scored, yet still below the amount of Hoxd10 mRNAs in the wild-type configuration (i.e., at the same respective positions).
Figure 2.

Stocks of mutant mice used in this study with, at the top, a drawing of the wild-type HoxD cluster. Posterior Hoxd genes are depicted by using a color code, from Hoxd13 in red, to Hoxd9 in blue. The position of conserved RXII is shown by the light-blue oval. The mutant strains used in this work are listed below. Deleted segments are shown by broken lines, whereas duplicated loci are indicated by the same color code. Breakpoints are shown as red triangles.
Figure 3.

Regulatory reallocations. Real-time PCR comparisons of gene expression levels in presumptive digits of either wild-type embryos (light-blue bars) or embryos homozygous for deletions of Hoxd loci located in 5′ of the cluster. The levels are always shown as a proportion of Hoxd13 wild-type RNA level. To better visualize the results, different scales are used for either Lnp and Evx2, or “posterior” Hoxd genes. (A) Deletions of the Hoxd13 locus, either alone [del(13), red bars] or in combination with either Hoxd12 [del(13–12), yellow bars] or Hoxd12 and Hoxd11 loci [del(13–11), green bars]. The deletions lead to up-regulations of the remaining genes. Regulatory reallocations are more important on the 3′ side (Hoxd genes) than on the 5′ side (Evx2) of the deletion. Lnp shows a significant up-regulation in the largest deletion, exclusively. In all cases, the newly positioned 5′-most Hoxd gene (rank 1) shows the strongest expression. (B) Similar genetic configurations, but combined in cis with a deletion of conserved RXII. Deleting RXII (dark-blue bars) results in both a decrease in Evx2 transcription and an overall higher expression of posterior Hoxd genes. Deleting RXII together with Hoxd13 [del(RXII)–del(13), orange bars], or Hoxd13 and Hoxd12 [del(RXII)–del(13-12), green bars] lead to partial recovery of Evx2 levels, together with an increase of the remaining Hoxd gene expression. However, quantitative collinearity is maintained in all configurations.
When all three Hoxd13 to Hoxd11 loci were removed, Hoxd10 was increased, yet not to the expected level for the “leading gene”; i.e., the gene positioned at rank number 1. In this case, a very significant increase in the amount of Hoxd9 mRNAs was detected (Fig. 3A, green bars), although also below the amount expected for a gene located at the second position with respect to the 5′ end of the cluster. Also, in all deleted configurations, Lnp was not significantly modified, except for a slight up-regulation in the del(13-11) mice. The same holds true for the regulation of Evx2, even though in this case, the magnitude of the variations was much higher. In particular, removing the Hoxd13 to Hoxd11 DNA interval induced an almost 300% increase in the transcription of Evx2, whereas the shorter deletions had a less pronounced effect, though stronger than that seen for Lnp.
From this set of experiments, we concluded that the collinear response in transcript amounts was mostly, although not entirely, dependent on the rank of the gene. A collinear response was obtained regardless of which gene was positioned first, yet the shape of the response was not identical in all cases, as clearly shown by the expression of Hoxd9 in the del(13-12) mutant limbs, which was much below the level of Hoxd10 (Fig. 3A, yellow bars), unlike the expression of Hoxd12 and Hoxd11 in the wild-type condition, where these latter two genes were at the same respective positions and are expressed in similar amounts (Fig. 3A, light-blue bars).
Conserved region XII (RXII)
RXII, a region of high DNA homology between various vertebrate species and located between Hoxd13 and Evx2 (Kmita et al. 2002b) was previously proposed, using in situ hybridization data, to help potential enhancer sequences to contact between Hoxd13 and Evx2, thereby favoring expression of Hoxd13 (Kmita et al. 2002a). By using this quantitative approach, we could not confirm this original observation, despite RXII having a genuine effect on the transcription of these target genes. Rather than equalizing transcript amounts among the various target genes, the deletion of RXII increased the transcriptional activities of Hoxd13 to Hoxd10 (Fig. 3B, cf. dark-blue and light-blue bars). When combined with the deletions of either the Hoxd13 locus, or of both the Hoxd13 and Hoxd12 loci, the same tendency was observed and quantitative collinearity was maintained (Fig. 3B, orange and green bars)—more obviously, in fact, than upon the mere deletion of RXII. In this latter case, expression of Hoxd11 was not significantly different from that of Hoxd12 when using the Student’s t-test (Fig. 3B, dark-blue bars).
Interestingly, the major modification caused by deletion of RXII was scored on Evx2 regulation, whose transcript level dropped down to 50%. As observed also in the presence of RXII (Fig. 3A), the deletion of RXII in combination with either Hoxd13 or Hoxd13 and Hoxd12 lead to an increase in the transcription of Evx2. In this case, however, the increase was much less pronounced than in the presence of RXII, and the level of Evx2 mRNAs eventually reached the wild-type level (Fig. 3B, green bar), whereas a robust increase was seen with the same deletion but in the presence of RXII (Fig. 3A, green bar). Altogether, the gain of expression observed on Hox genes upon deletion of RXII was clearly more important than the concomitant loss of Evx2 expression, indicating that the increase was not due to a mere redistribution of regulations.
Modeling quantitative collinearity
As different mechanisms could underlie both the collinear regulation in the wild-type locus and its observed variations in our mutant configurations, we tried to elaborate a theoretical model that would best fit this initial set of experimental data. We considered two major classes of mechanisms: those relying on a scanning process, and those involving direct initial contacts between the enhancers and the target DNA region(s), through the formation of loops (e.g., see Bulger and Groudine 1999; Tolhuis et al. 2002). Based on the first set of experimental data mentioned above, two important general observations were taken into account: First, while the rank of the gene with respect to the 5′ end of the cluster is a key parameter for the expression level, it is not the only element to consider. For example, regardless of which gene was positioned at the place of Hoxd13 (rank I), this gene was highly expressed. The absolute levels of expression were nevertheless significantly different from gene to gene (Fig. 4A). Generally, genes located 3′ of the breakpoint were not as highly expressed as was predicted by their new position. For example, in del(13), Hoxd12 reached only 80% of the Hoxd13 wild-type level, despite its first position within the cluster (Fig. 4A). However, no simple rule could be drawn, as shown by Hoxd11 expression in the same deletion, which in contrast was 1.4-fold that of the Hoxd12 wild-type level (Fig. 4A). Also, Evx2 expression increased strongly together with the size of the deletions, even though the respective position of this gene remained unchanged (Fig. 3A). These observations suggested that slightly different, gene-specific affinities were also involved.
Figure 4.

Modeling quantitative collinearity. (A) Expression levels of Hoxd genes as a function of their relative position with respect to the 5′ extemity of the cluster (their “rank”), in the various genetic configurations. While gene rank has a major impact on expression level, different genes are not expressed at the same absolute level whenever positioned at the same rank. (B) The total transcriptional activity at the locus (i.e., the sum of the expression levels of all Hoxd genes active in developing digits) correlates with the number of transcription units: Deletions or duplications decrease or increase the total transcription, respectively. The wild-type configuration is shown with a light-blue bar. Configurations associated with a deletion of RXII are excluded from this analysis. (C) A two-step model. The digit enhancers (GCR, Prox) first contact the Evx2-Hoxd13 intergenic region. They may subsequently activate the transcription of Lnp, Evx2, or Hoxd genes, with different probabilities. Hoxd genes are activated in a polar fashion, the enhancers scanning (or sensing) the complex in a 5′–3′ direction. This model accounts for most of the observed features and generated several predictions that were subsequently assayed (see the text).
Second, the overall transcriptional activity of the locus, as measured by cumulative amounts of transcripts, was function of the number of transcription units present, such that the deletion of one or several genes led to a corresponding overall decrease of mRNAs produced at the locus (with the exception of the codeletion of RXII, as described above). This observation was subsequently confirmed by increasing the number of genes, through various duplications (see below), leading to a quasilinear relationship between gene number and total mRNAs levels (Fig. 4B). This unexpected observation indicated that the regulatory system is not saturated in the wild-type condition, and that regulatory reallocations, following genomic modifications, do not simply reflect the redistribution of the same general regulatory potential among a reduced or increased number of target units.
After considering various model outlines, a particular class of models was found to fit the above-mentioned constraints, while not requiring an undue number of estimated parameters. These models assume a two-step process: In the first phase, a complex involving regulatory sequences (GCR, Prox), together with their bound factors, will interact with the Evx2-Hoxd13 intergenic region (Fig. 4C). The probability that the GCR/Prox complex will bind the locus is proportional to the number of promoters present at the locus.
Once this interaction has occurred, various probabilities exist to activate either Lnp, Evx2, or Hoxd genes. These probabilities depend on the affinities of the various promoters and the probability that the GCR/Prox complex did not stop previously at upstream promoters. We thus assumed that the five Hox genes promoters are not equivalent in their capacity to attract and fix the GCR/Prox complex. This second step could occur via a “microscanning” process, over the ∼40-kb large DNA interval containing from Hoxd13 to Hoxd10. Since all promoters are at the vicinity of the enhancers complex, this latter can discriminate and select in function of the respective affinities. In this class of models, the maximum number of estimated parameters is nine (see Materials and Methods). Two parameters define the regression between the total number of transcripts and the number of promoters at the locus, and seven parameters at most are required to account for the affinities of the seven loci considered (Lnp, Evx2, and Hoxd13 to Hoxd9). Such models successfully passed the test of the three observed features (Fig. 5): (1) the decrease in total transcripts amount together with promoter number, (2) the modulation of ranking by promoter efficiency to determine the expression levels, and (3) the up-regulation of Evx2 whenever Hox genes are deleted. After definition of the various parameters, this model, which includes components of both scanning and looping mechanisms, fit the first set of data much better than any other model relying exclusively on either a general scanning process or gene-specific affinities (data not shown).
Figure 5.

Predicted versus observed expression levels. Comparison between expression levels predicted by the model (yellow bars) and observed values in either the wild-type (light-blue bars) or mutant configurations (red bars). The various configurations are depicted below each graph. (A) Wild-type situation. (B–D) Deletions including Hoxd13. The wild-type levels are shown on each panel for comparison. The model accurately reproduces the observed values in wild-type and mutant configurations, all Hoxd gene predictions lying within two standard deviations of the observed values. The up-regulation of Evx2, although predicted, is slightly underestimated by the model. The blue oval between Hoxd13 and Evx2 indicates the presence of RXII.
The application of this model generated a set of values, which were then compared with both the wild-type condition and the three deleted configurations described above. In the wild-type condition, a very good match was obtained between the predicted and the observed values (Fig. 5A). When either the Hoxd13 locus, both Hoxd13 and Hoxd12 or the Hoxd13 to Hoxd11 interval were deleted from the model, the predicted values for reallocations also matched the observed data set within error bars (Fig. 5B–D, cf. red and yellow bars), with the exception, perhaps, of the gain of Evx2 expression, which was slightly but systematically underestimated by the model. Once established, the model was used to make a number of predictions, which were verified by using the appropriate mutant strains. In turn, the new results were not only used to test the resilience of the model, but also to further constrain the parameter values, progressively, through several rounds of adjustment and experimental validation. The values (predictions) of the model given in the figures below, for comparison with the experimental data sets, are extracted from the final version of the model.
Duplication alleles
This model made clear predictions regarding the effects of either gene duplications, or internal deletions. We experimentally challenged these predictions, starting with three duplications obtained via our TAMERE system (Herault et al. 1998). The first duplication was that of the Hoxd13 locus alone (Fig. 6A), the second was a duplication of the entire Hoxd13 to Hoxd11 interval (Fig. 6B), and the third allele was an “internal” duplication of the Hoxd12 to Hoxd11 locus (Fig. 6C). Generally, the predictions of the model turned out to be rather precisely validated by the experiments. In particular, the duplication of the Hoxd13 locus, leading to two copies of Hoxd13 positioned at ranks 1 and 2 did not elicit a double amount of Hoxd13 mRNAs, emphasizing again the prime importance of the promoters’ rank over the promoters’ affinity. In this same duplication, however, the impact on Evx2 transcription was once again not faithfully predicted, the observed down-regulation being more important than anticipated (Fig. 6A).
Figure 6.

Duplication alleles. Observed (red) and predicted (yellow) gene expression levels in three duplications. Wild-type levels (light blue) are shown on each panel for comparison. (A) dup(13). (B) dup(13-11). (C) dup(12-11). In all cases, the expression of duplicated loci was less than twice the wild-type level, and genes located 3′ of the duplication were down-regulated, highlighting the importance of gene rank. Hoxd13 expression remained unaffected by the duplication of Hoxd12 and Hoxd11, whereas duplications including Hoxd13 lead to a decrease in Evx2 transcription. The predictions of the model matched the observed values, with the exception of Evx2 down-regulation in dup(13) and dup(13-11), underestimated by the model. Blue ovals are as in Figure 5.
Also, in the duplication of Hoxd13 to Hoxd11, the predicted level for Hoxd11 mRNAs at first appeared too high when compared with the experimental data (Fig. 6B), yet the Student’s t-test indicated that this difference was not significant. Beside this point, the observed matching was good, as was the case for the internal duplication of both the Hoxd12 and Hoxd11 loci, where the amount of Hoxd13 transcripts remained expectedly stable. In this latter duplication, a robust increase in Hoxd11 mRNAs was scored, as predicted by the model. This increase would not be expected based on a “rank-only” model, since after duplication, one copy of Hoxd11 is now in the respective position of Hoxd9; i.e., in a virtually “silent” position with respect to digit enhancers (see above). This increase thus illustrates the “affinity component” of the system (as predicted by the model), Hoxd11 performing better than Hoxd9 when placed at the same genomic position. In all duplicated configurations, the observed decrease in the amount of Hoxd10 mRNAs matched the predictions.
Interestingly, in contrast to what was observed with the various deletions, the overall level of Hoxd gene transcripts increased together with the importance of the duplications. For instance, a mild increase in total transcription was detected with the duplication of Hoxd13, whereas a more robust increase was scored upon duplication of the Hoxd13 to Hoxd11 interval (addition of three transcription units). As mentioned earlier, this relationship between the number of genes and the total amount of transcripts was nearly linear (Fig. 4B).
Internal deletions
The predictions of the model concerning a set of five internal deletions—i.e., deletions that did not remove Hoxd13 from its first rank—were also challenged experimentally (Fig. 7). In all these deletions, the observed levels of Hoxd13 mRNAs were stable, as predicted, except for the case of the del(12-11) where an ∼20% increase was detected. Therefore, the general decrease in the amount of Hoxd genes’ transcripts did not lead to an increase in Hoxd13 transcription. Likewise, neither Evx2 nor Lnp showed significant variations in their transcriptions, in agreement again with the model.
Figure 7.

Deletions excluding Hoxd13. Observed and predicted gene expression levels in five “internal deletions.” Colors and schemes are as in Figure 6. (A) del(12). (B) del(12-11). (C) del(12-10). (D) del(11). (E) del(11-10). All deletions lead to increased expression of 3′-located genes, whereas genes located 5′ of the deletions were less affected. The observed levels are in good agreement with predicted values, with the exception of the strong up-regulation of Hoxd9 in del(11-10), not accounted for by the model. Blue ovals are as for Figure 5.
The effects of these deletions were thus scored mostly on the 3′ side of the breakpoints. Here again, the observed transcript levels were in good agreement with the predictions. For example, Hoxd10 transcription was only moderately increased in the absence of the Hoxd12 locus (hence, at respective rank 3) (Fig. 7A), whereas a robust increase was scored for the same gene when located at respective rank 2, after deletion of both Hoxd12 and Hoxd11 (Fig. 7B), thus emphasizing the importance of the rank. Interestingly, when Hoxd12, Hoxd11 and Hoxd10 were deleted (Fig. 7C), and hence, Hoxd9 was now neighboring Hoxd13 in rank 2, this latter gene was not activated at a very high absolute level, showing once again that the overall transcriptional activity distributed over the cluster was not simply reallocated among the number of resident promoters.
While the deletion of the Hoxd11 locus generated data in good agreement with the predictions (Fig. 7D), the internal deletion of both Hoxd11 and Hoxd10 generated the only truly aberrant measure of the whole experimental series. This internal deletion indeed induced a very robust increase of Hoxd9 transcription, reaching the level of endogenous Hoxd12 (Fig. 7E). Interestingly, the same 3′ breakpoint was used in the del(12-10) configuration, which did not lead to a similar increase (Fig. 7; cf. red bars in C and E).
Regulation in the genital eminence
During the emergence and further development of the external genital organs (the future penis and clitoris), the same Hoxd genes are expressed with a similar quantitative collinear distribution of transcripts (Dolle et al. 1991). This observation, as well as subsequent genetic data, have highlighted the developmental similarities between distal limb buds and genital buds (Kondo et al. 1997; Cobb and Duboule 2005), and suggested that Hoxd gene expression in both structures could be controlled by the same regulatory circuitry (Dolle et al. 1991; e.g., see Cohn 2004; Suzuki et al. 2004). We thus investigated whether the model could also be applied to both wild-type and mutant situations in the developing external genital organs. However, when the parameters of the model were adapted after considering the entire series of alleles, the global fit with the experimental data set was not optimal. This could mean that either the structure of the model was different between developing limbs and genitals, or alternatively, some classes of genetic configurations (5′ deletions, duplications, internal deletions) could not be accounted for by the actual model; hence, their consideration to adjust the parameters would decrease the general fit.
Consequently, we tried to exclude specific classes of genetic rearrangements in the adjustment of parameters, by keeping the same model structure. Interestingly, when we ignored the data set obtained with the various “internal deletions” (not containing Hoxd13) to adjust the various parameters, we obtained an excellent fit with the data sets derived from both 5′ deletions (including Hoxd13) and duplications (Fig. 8). As for the digits, the match with the wild-type condition was very good (Fig. 8A) and the same (almost linear) relationship was observed between the overall level of transcription and the absolute number of transcription units present in this DNA interval (Fig. 8F). The match between the model and either two 5′ deletions [del(13) and del(13-12)] (Fig. 8B,C), or two duplications [dup(13-11) and dup(12-11)] (Fig. 8D,E) was very good, indicating that the regulatory mechanisms at work in both developing digits and external genitalia were most likely the same.
Figure 8.

Hoxd gene regulation in the developing genital tubercle. The same model structure was applied to investigate Hoxd gene regulation in the genital eminence. The various parameters of the model were optimized using expression values observed in the genital bud in all configurations, except for internal deletions (see the text). (A) In the wild-type situation, quantitative collinearity in the genital bud was similar to what was observed in presumptive digits. (B–E) The 5′ deletions and duplications induced comparable regulatory reallocations in the genital bud, as compared with digits. (B) del(13). (C) del(13-12). (D) dup(13-11). (E) dup(12-11). (F) As in the case of digits, the overall transcription level correlated with the number of transcription units at the locus. In all cases, the observed expression levels were in good agreement with the predictions of the model. (G–I) In contrast, the model could not account for the expression levels observed following internal deletions. For instance, del(12-11) (G) and del(11) (H) lead to a decreased expression of Hoxd10, and del(11-10) leads to a down-regulation of Hoxd12 (I). As in developing digits, del(11-10) elicited a strong increase in Hoxd9 transcription, which was not predicted by the model. Blue ovals are as in Figure 5.
Because internal deletions had to be ignored to adjust the parameters, their fit with the model was expectedly rather poor. For example, in the del(12-11) configuration, Hoxd10 transcripts were much less abundant than anticipated by the model (Fig. 8G, yellow bar). Also, the deletion of both Hoxd11 and Hoxd10 loci induced a decrease of Hoxd12 mRNAs, which were thus below the predicted amount (Fig. 8I). Only the deletion of the Hoxd11 locus matched fairly well the predictions of the model (Fig. 8H). Finally, and similar to what was observed in digits, the deletion of the Hoxd11 and Hoxd10 loci induced a large and unexpected gain in the transcription of Hoxd9 (Fig. 8I, red bar) that could neither be explained nor accounted for by the model.
Discussion
We used a quantitative approach to analyze the regulatory mechanism underlying collinear Hoxd gene regulation during mammalian digit development. Such an approach is necessary due to the sensitivity of various digit morphologies to both the dose and quality of Hox gene functions delivered during fetal development (Zakany et al. 1997; Zakany and Duboule 2007). Variations in this quantitative regulation have been proposed to partially account for important morphological differences between mammalian digits (e.g., Chen et al. 2005); hence, the understanding of this regulatory control may help explain what determines the various digital formulae of mammalian hands and feet.
The ontogeny of mammalian thumbness; a dosage effect linked to gene topology
In mammals, a clear difference is observed between the morphology of the most anterior digit (the thumb) and the other digits—the former generally displaying a reduced size due to the presence of two phalanges only. At the molecular level, very few genes are differentially regulated in various presumptive digit domains, and hence could determine digit identities. Among these few candidates, Hox genes are the most promising for at least three reasons. First, the inactivation of HoxA and HoxD gene functions lead to either a reduction or a complete agenesis of digits, including the thumb (Dolle et al. 1993; Fromental-Ramain et al. 1996; Kmita et al. 2005). Second, gain of expression of posterior Hoxd genes (e.g., Hoxd12 and Hoxd11) in presumptive thumb cells lead to a more elongated morphology, resembling posterior digits (Morgan et al. 1992; Zakany et al. 2004). Finally, naturally occurring mutations, which homogenize digital morphologies, are all associated with a homogenization of Hox genes expression domains.
Among Hox genes expressed in digits during development, Hoxa13, the only member of the HoxA cluster expressed there, is equally transcribed in all digit primordia, including the thumb. Therefore, this critical difference in shape ought to be determined by Hoxd genes and indeed both Hoxd10, Hoxd11, and Hoxd12 are expressed exclusively in presumptive digits 2–5, whereas excluded from digit I (the thumb). In contrast, Hoxd13, like Hoxa13, is expressed throughout all presumptive digit cells. Accordingly, mice mutant for Hoxd13 have an abnormal thumb (Dolle et al. 1993). Interestingly, however, Hoxd13 is expressed originally with the same posterior restriction than its three immediate neighbors, with an apparent peak in posterior-distal cells and an exclusion from digit I cells. Because Hoxd13 is expressed more robustly than its neighbors, transcription is nevertheless subsequently detected in presumptive thumb cells, thus encompassing all five digital primordia, leading to what was referred to as “reverse collinearity” (Nelson et al. 1996). In this view, reverse collinearity is the mere topological translation of quantitative collinearity.
Therefore, the subtle transcriptional regulation of Hoxd genes in these cells is likely a crucial parameter in the determination of thumbness, mostly by reducing the global dose of HOX products present in digit I. This dosage effect appears to be linked to gene topology, since mice deleted for the Hoxd13 locus expressed Hoxd12 in developing digit I (Kmita et al. 2002a), indicating that the gene positioned at the extremity of the cluster is always expressed with maximal efficiency, regardless of its own specific regulatory sequences. Accordingly, genes located at the second, third, fourth, and fifth ranks respond to this regulation too, yet with lower efficiencies, leading to their nontranscription in presumptive digit I cells. The resulting dose of HOXA and HOXD proteins is thus higher in developing digits 2–5 than in digit I, which will impact on the future morphologies, Hox genes being involved in the control of cell proliferation and digit patterning. This difference between digit I and the others suggests a regulatory explanation for the existence of two developmental modules underlying the evolution of anthropoid distal forelimbs (Reno et al. 2007).
In an evolutionary context, it is likely that the recruitment of this regulation was an important step to accompany either the emergence, or the expansion of digits in an ancestral tetrapod. Because of the built-in asymmetry of the system, based on gene order, we can speculate that quantitative collinearity was already at work in these ancestral autopods. Consequently, a morphological distinction between the most anterior digit (the thumb in mammals) and the others must have existed from the start. In this view, while Hox gene clustering was certainly an efficient evolutionary opportunity, it may have also importantly constrained the system to impose some heterogeneity in digital morphologies. These morphological differences may have been a basis for further adaptive solutions and selection thereof, either by further elaborating on them, or by subsequent digital deletions or reductions.
While many mammals indeed display little if any difference in the morphologies of more posterior digits (e.g., the human digits 2–5), some vertebrate species exhibit remarkably different, although neighboring, digits; for example, associated with their flying behavior. In both bat and chicken wings, digits can be variable in sizes and number of phalanges, without obvious changes in Hox gene expression. It is nonetheless possible that slight quantitative modifications be responsible for this effect (Chen et al. 2005). Alternatively, the exact timing in the expression of these Hox genes in every presumptive digit territory may subsequently affect digital morphologies. While our various analytical tools reflect a frozen situation, a more dynamic view of the system may reveal the nature of such differences. It is equally possible that the Hox system be not used after the initial morphological distinction has occurred between the most anterior digit and the others.
Modeling quantitative collinearity
Our quantitative approach, associated with the large collection of mutant strains at this locus, allowed us to elaborate a model of large-scale gene regulation that accounts for most of the observed results. In order to model this regulation, we considered some facts as established, such as the importance of gene position (the “rank”), the fact that two other genes were also affected by this regulation (Evx2 and Lnp), and the known presence and location—upstream of the gene cluster—of regulatory sequences that control gene expression in developing distal limb buds (the GCR and Prox elements) (Spitz et al. 2003; Gonzalez et al. 2007). Other parameters were readily excluded, due to their incapacity to explain the observed results. For example, differences in various mRNAs half-lives, if at all involved, would have a very minor impact. Also, models involving the scanning of the landscape by a protein complex, after initial recognition of enhancer sequences (see Blackwood and Kadonaga 1998) were rapidly discarded due to the difficulty of reconciling them with any available data set. We thus focused on models based on an initial looping, followed by the construction of a particular regulatory microarchitecture. The predictions formulated from an early model were subsequently tested by using the appropriate set of mutant strains. In turn, the new data were used to adjust the various parameters such as to improve the fit of the model. This bilateral process led to a final formulation built on nine different parameters, which could account for the 69 quantitative observations made by using 12 different mouse strains.
Surprisingly, the model helped to clarify two issues raised previously on the basis of in situ hybridization patterns. First, the gene rank alone cannot account for the observed transcriptional efficiencies. While of primary importance, this parameter is not sufficient to explain the various data sets. In fact, even wild-type absolute transcript levels indicate no real difference between Hoxd12 and Hoxd11. Second, while largely increased, the transcript levels of either Hoxd12 or Hoxd11, when placed at the first rank, never reached the wild-type level of Hoxd13 transcripts. Also, the total amounts of Hoxd13 transcripts in duplicated configurations containing two copies of this locus were the same regardless of the rank of the second Hoxd13 copy; i.e., when placed either at rank number 2 or number 4. Therefore, the importance of gene rank has to be somehow modulated by specific affinities between the enhancer complex and the various gene promoters. In the model, the introduction of parameters reflecting this previously unanticipated feature (see Kmita and Duboule 2003) was necessary to reach the requested global fit. Interestingly, the affinities derived from the model for Lnp, Evx2, and Hoxd9 are very similar to each other (see Materials and Methods), possibly reflecting a generic, baseline promoter affinity, whereas Hoxd10 to Hoxd13 promoters would have evolved more robust affinities for the enhancer complex.
The second issue concerns the DNA segment initially contacted by the enhancer complex. Our various data sets suggested a model whereby enhancers, in a first step, contact the Evx2 to Hoxd13 intergenic region via a loop. In a second step, the enhancer complex scans (senses) the immediate environment preferentially from the 5′ toward the 3′ part of the cluster, regulated by promoter-specific affinities. While anticipated, the importance of the Evx2-Hoxd13 intergenic region does not seem to rely on the presence of the highly conserved sequence RXII, unlike originally reported based on in situ hybridization patterns (Kmita et al. 2002a). In this latter case, expression of genes located at ranks number 2 and 3 were also detected in presumptive cells of digit I, which was interpreted as a break in the collinear response. Our quantification of transcripts derived from mice deleted for this sequence XII indeed revealed that quantitative collinearity was maintained despite a generally elevated level of Hoxd gene expression, which likely explains their transcription in presumptive digit I cells.
In support of this explanation, the deletion used by Kmita et al. (2002a) removed both the RXII and the Hoxd13 locus. Our analysis of this genetic condition revealed a very robust transcriptional increase (>100%) of the remaining three genes, explaining their expression in presumptive digit I cells and, hence, the misleading impression that all patterns were alike in these embryos. In the absence of RXII, Evx2 transcription was down-regulated, which suggests that this sequence may be part of the Evx2-specific transcriptional requirements (in the promoter region or in the 5′ untranslated region). However, the effects observed upon Hoxd gene transcription cannot be explained solely on this basis, and call for a specific function for RXII. For example, this conserved DNA sequence may act as a switch for the direction of scanning, once enhancers have recognized the Hoxd13-Evx2 intergenic region. The factors that favor the contact between the GCR/Prox enhancers and this intergenic region remain to be characterized.
Finally, the model integrated an interesting phenomenon observed with both deleted and duplicated configurations; i.e., the fact that the total amount of transcripts produced in digit cells by all genes present in the landscape is a function of the number of genes. This has several implications regarding the transcriptional mechanism at work, as it means that the system is not saturated, and adding more transcription units will increase the global output of the system. This suggests that the wild-type situation likely corresponds to a particular equilibrium, as imposed by the regulatory architecture of the locus. Likewise, the deletion of several promoters will not merely reallocate the regulatory potential over the remaining units, but instead, will produce a novel equilibrium that will use less of the global transcriptional potential available. One possibility is that such equilibrium imposes a number of constraints to the system—for example, in the spatial organization—preventing situations in which any number of genes would use their full regulatory potential.
While in good agreement with the vast majority of the data sets obtained experimentally, our model showed some minor and acceptable discrepancies with the reality (for example, the underestimation of Evx2 down-regulation in Hoxd13 duplication), as well as one major problem that remains unsolved; i.e., the behavior of Hoxd9 in the internal deletion del(10-11). In this case, an almost 20-fold increase was scored in the expression of Hoxd9, whereas the model predicted only a slight increase (Fig. 7E). This particular result is the only one suggesting the manifestation of a new phenomenon, such as the emergence of a novel, increased affinity for Hoxd9 following the fusion with the 3′ part of the Hoxd12 locus. The validation of this hypothesis would require yet another set of genetic modifications, not readily available.
The digits versus genitals connection
Because of similar Hox expression dynamics during the development of both digits and external genitalia, it was proposed that these structures share both ontogenetic principles and a phylogenetic history (Dolle et al. 1991; Kondo et al. 1997). Since then, the conservation of developmental principles during the emergence and growth of these various buds has been largely documented, notably through the presence and function of the same key regulatory molecules and signaling pathways (for reviews, see Cohn 2004; Suzuki et al. 2004). In fact, quantitative collinearity was originally described in the growing genital eminence (Dolle et al. 1991), and we thus looked at whether the model constructed for the digits could equally account for the results obtained with developing external genitalia.
Clearly, the same type of model could be applied to the results obtained with RNA extracted from developing genital buds. In particular, the model had a very good fit for the duplicated configurations and the deletions including Hoxd13 (5′ deletions). Also, the quasilinear relationship between the total transcriptional readout of the system and the number of transcription units was also observed in genitalia. Altogether, these results suggested that the regulatory strategy leading to quantitative collinearity was identical in both digits and genitalia, and that these various structures not only use the same genes, but also the same regulatory circuitry.
However, the consideration of all experimental data, as obtained from genital bud RNAs, did not fit well, at first, with the model elaborated for digit RNAs, even after adaptation of some parameter values. Interestingly, the set of internal deletions—i.e., those deletions excluding Hoxd13—was systematically at odds with the model. Whenever these deletions were ignored and parameters values adapted accordingly, a close to perfect match with the model was obtained. This observation ought to be interpreted in two different contexts. First, Hox gene function and regulation in these structures must be compared at slightly different developmental time points (see Cobb and Duboule 2005), as the distal part of the limb buds should be compared with the distal part of the developing genitalia (the future penis, in males, or clitoris, in females) and not with more proximal parts of the bud, the labioscrotal, and preputial swellings, giving rise to either the scrotum, the labia, or the prepuce. Because there was no clear morphological landmark to dissociate these parts at our dissection time point, unlike in subsequent developmental stages, it is possible that a contamination of more proximal cells induced a bias in the results.
Another, nonexclusive explanation is that additional regulatory sequences evolved within the gene cluster (e.g., see Gould et al. 1997) to fine-tune or accompany the evolution of the external genital apparatus, on top of pre-existing mechanisms. Internal deletions may have a particularly pronounced impact on the function of these genital-specific sequences, thus leading to results that cannot possibly be accounted for by the model. Such targeted differences in the regulation of these genes may not be so surprising, given the end result that is achieved in both cases. Indeed, in the external genitals, the adaptive value may reside in the elongation of the future organ. In digits, however, the function of these genes is not restricted to the mere growth of the structures but also, as documented above, to their patterning. These slightly different and sex-dependent contexts ultimately had their own adaptive values, and it is perhaps not surprising if each structure evolved some additional regulation, on top of a shared and potentially ancestral regulatory circuitry.
Materials and methods
Stocks of mice
The various mutant strains used in this study were described previously (Kmita et al. 2002a, b; Tarchini and Duboule 2006; Di-Poi et al. 2007). Besides the deletion of RXII, all mutant alleles were produced by TAMERE (Herault et al. 1998). Genotyping of mice and embryos was performed using Southern blot and PCR analysis, according to standard procedures. For duplicated alleles, a real-time PCR strategy was used to quantify copy numbers of the duplicated segments, in order to discriminate between heterozygous and homozygous mutant embryos (Supplemental Material).
RNA samples and real-time RT–PCR
For each mutant line, heterozygous mice were crossed to obtain wild-type, heterozygous, and homozygous mutant embryos. Presumptive digits and genital tubercles were dissected from E13.5 embryos and stored in RNA later reagent (Qiagen), until genotyped. RNA was isolated from individual embryo samples using the RNeasy microkit (Qiagen) after disruption and homogenization with a Polytron device (Kinematica). Single-stranded cDNA was synthesized using SuperScript II RT (Invitrogen)
Real-time PCR primers and TaqMan probes were designed using Primer Express 2.0 software (Applied Biosystems), and PCR efficiencies were measured using serial dilutions of cDNA. cDNAs were PCR-amplified in a 7900HT SDS system (Applied Biosystems). Results were comparable when using either TaqMan or SYBR green strategies. Specificity of the SYBR green reactions was determined by examination of product melting curves. Relative quantities (RQ) were calculated from the threshold cycle (Ct) values with the formula RQ = (1 + E)−Ct, where E is the PCR efficiency calculated from standard curves. A mean quantity was calculated from triplicate reactions for each sample and normalized to two or three similarly measured quantities of housekeeping genes (Rps9, Tbp, and Tubb). Sequences of primers and probes used in this assay are listed in Supplemental Table S1.
For the determination of absolute transcripts levels, external calibration was performed using a standard consisting of known amounts of the various mRNAs. Sense RNA was produced by in vitro transcription of full-length or partial cDNA clones (Supplemental Material) using T7 or SP6 RNA polymerases (Promega). Standard curves were obtained by serial dilution of known amounts of these RNAs, and reverse transcription with Drosophila L2 cell total RNA as a carrier. Absolute quantification was performed by parallel real-time PCR amplification of digit and genital bud total cDNA together with this standard curve. Similar results were obtained using serial dilutions of a BAC clone covering the locus as a standard (data not shown).
Looping and tracking model in digits
A preliminary analysis of the data revealed two striking features of this system: First, the total amount of transcripts—i.e., the sum of absolute levels of expression for all genes in a given configuration—strongly correlated with the number of promoters present at the locus (Fig. 4B). The Pearson coefficient is 0.93 in digits for the eleven configurations [excluding del(10-11), see the text]. Second, the five Hoxd genes showed a constant pattern where, in any given pair, the 5′-located gene is more robustly expressed than its 3′-located neighbor. However, rank alone is not a good predictor of absolute levels of expression (see the text). Therefore, we designed a two-step model assuming the existence of seven different promoter efficiencies (Elnp, Eevx, E13 to E9), which would determine a probability for the GCR/Prox transcription complex to interact with any of these promoters. In the first step, the GCR/Prox elements loop over and bind the locus somewhere between Evx2 and Hoxd13 (Fig. 4C). The probability of this event is assumed to be proportional to the number of promoters present at the locus. This will determine the overall level of transcription; i.e., the total number of transcripts from all the genes present. In a second step, the frequency f of interaction between the GCR/Prox complex and either Lnp, Evx2, or Hoxd13, is proportional to the “efficiency” E of their respective promoters. A potential interaction with any downstream-located Hoxd gene is processive and will depend on (1) the intrinsic efficiency of its promoter and (2) the cumulative effect of previous interactions with all upstream-located Hoxd promoters. Thus, the theoretical frequency fi of interaction of GCR/Prox with a Hoxd promoter i is
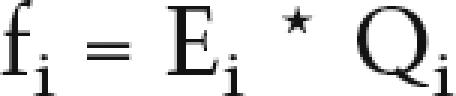 |
where Qi is the probability that the GCR did not stop upstream of i in the sweep, computable from the Ei; i.e., Q12 = (1-E13); Q11 = [1 − E13 − E12 * (1 − E13)] and so forth. Relative activities may then be obtained by
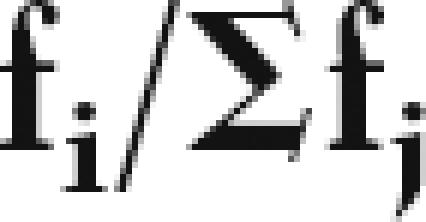 |
where Σfj is the sum over the seven genes. Absolute levels of activity (ai) are computed by applying this ratio to the predicted total number of transcripts for a given configuration (TTc), as derived from the linear regression
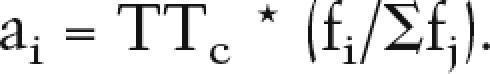 |
These absolute levels of activity are to be compared with the observed absolute levels of expression. The best fit between this model and the data sets was looked for by using an optimum set of nine parameters: two parameters for the linear regression to account for the total number of transcripts, and seven parameters to account for the “efficiencies” of the promoters. The seven latter parameters were derived from minimizing the sum of weighted least squares:
 |
where aip and aio are the predicted and observed absolute levels of expression, respectively, Vi is the variance of measurements for the gene i, and ΣΣ is the sum over all configurations and all genes. All computations were done using Matlab 7.0.4 (The MathWorks) software.
Among the 69 data points, one observation, Hoxd9 in the del(10-11) configuration, significantly deteriorated the goodness-of-fit of both the linear regression and the least-square fit: The weighted sum of least squares was almost halved when Hoxd9 in del(10-11) was excluded. Not taking this observation into account in the fit (see the text), we obtained the following optimum model efficiencies:
Efficiencies Ei in digits
Lnp 0.035
Evx2 0.031
Hoxd13 0.347
Hoxd12 0.198
Hoxd11 0.210
Hoxd10 0.107
Hoxd9 0.033
The total number of transcripts (TTc) was derived from the following regression line, with R2 = 0.93 (Fig. 4B):
The corresponding predicted absolute levels for each gene in the 12 configurations are given in Supplemental Table S7 and compared with observed values (Figs. 5–7).
Model in genitals
The same model was used to predict absolute transcripts levels in genitals. As discussed in the text, internal deletions were ignored when minimizing the sum of weighted least squares. The optimum model efficiencies, the regression equation for the total number of transcripts, and the corresponding predicted absolute levels are given in Supplemental Table S8 and compared with observed values (Fig. 8).
Acknowledgments
We thank J. Zakany, M. Kmita, and B. Tarchini for sharing mice, and M. Docquier and P. Descombes, from the NCCR genomic platform, for their help and advice. This work was supported by funds from the canton de Genève, the Louis-Jeantet and Claraz foundations, the Swiss National Research Fund, the National Center for Competence in Research (NCCR) “Frontiers in Genetics,” and the EU programs “Cells into Organs” and “Crescendo” to D.D.
Footnotes
Supplemental material is available at http://www.genesdev.org.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.1631708
References
- Blackwood E.M., Kadonaga J.T. Going the distance: A current view of enhancer action. Science. 1998;281:60–63. doi: 10.1126/science.281.5373.60. [DOI] [PubMed] [Google Scholar]
- Bulger M., Groudine M. Looping versus linking: Toward a model for long-distance gene activation. Genes & Dev. 1999;13:2465–2477. doi: 10.1101/gad.13.19.2465. [DOI] [PubMed] [Google Scholar]
- Chen C.H., Cretekos C.J., Rasweiler J.J.T., Behringer R.R. Hoxd13 expression in the developing limbs of the short-tailed fruit bat, Carollia perspicillata. Evol. Dev. 2005;7:130–141. doi: 10.1111/j.1525-142X.2005.05015.x. [DOI] [PubMed] [Google Scholar]
- Cobb J., Duboule D. Comparative analysis of genes downstream of the Hoxd cluster in developing digits and external genitalia. Development. 2005;132:3055–3067. doi: 10.1242/dev.01885. [DOI] [PubMed] [Google Scholar]
- Cohn M.J. Developmental genetics of the external genitalia. Adv. Exp. Med. Biol. 2004;545:149–157. doi: 10.1007/978-1-4419-8995-6_9. [DOI] [PubMed] [Google Scholar]
- Davis M.C., Dahn R.D., Shubin N.H. An autopodial-like pattern of Hox expression in the fins of a basal actinopterygian fish. Nature. 2007;447:473–476. doi: 10.1038/nature05838. [DOI] [PubMed] [Google Scholar]
- Deschamps J., van Nes J. Developmental regulation of the Hox genes during axial morphogenesis in the mouse. Development. 2005;132:2931–2942. doi: 10.1242/dev.01897. [DOI] [PubMed] [Google Scholar]
- Di-Poi N., Zakany J., Duboule D. Distinct roles and regulations for posterior and anterior Hoxd genes in metanephric kidney development. PLoS Genet. 2007;3:e232. doi: 10.1371/journal.pgen.0030232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolle P., Izpisua-Belmonte J.C., Brown J.M., Tickle C., Duboule D. HOX-4 genes and the morphogenesis of mammalian genitalia. Genes & Dev. 1991;5:1767–1776. doi: 10.1101/gad.5.10.1767. [DOI] [PubMed] [Google Scholar]
- Dolle P., Dierich A., LeMeur M., Schimmang T., Schuhbaur B., Chambon P., Duboule D. Disruption of the Hoxd-13 gene induces localized heterochrony leading to mice with neotenic limbs. Cell. 1993;75:431–441. doi: 10.1016/0092-8674(93)90378-4. [DOI] [PubMed] [Google Scholar]
- Drossopoulou G., Lewis K.E., Sanz-Ezquerro J.J., Nikbakht N., McMahon A.P., Hofmann C., Tickle C. A model for anteroposterior patterning of the vertebrate limb based on sequential long- and short-range Shh signalling and Bmp signalling. Development. 2000;127:1337–1348. doi: 10.1242/dev.127.7.1337. [DOI] [PubMed] [Google Scholar]
- Freitas R., Zhang G., Cohn M.J.2007Biphasic Hoxd gene expression in shark paired fins reveals an ancient origin of the distal limb domain PLoS ONE 2 :e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromental-Ramain C., Warot X., Messadecq N., LeMeur M., Dolle P., Chambon P. Hoxa-13 and Hoxd-13 play a crucial role in the patterning of the limb autopod. Development. 1996;122:2997–3011. doi: 10.1242/dev.122.10.2997. [DOI] [PubMed] [Google Scholar]
- Gonzalez F., Duboule D., Spitz F. Transgenic analysis of Hoxd gene regulation during digit development. Dev. Biol. 2007;306:847–859. doi: 10.1016/j.ydbio.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Gould A., Morrison A., Sproat G., White R.A., Krumlauf R. Positive cross-regulation and enghancer sharing: Two mechanisms for specifying overlapping Hox expression domains. Genes & Dev. 1997;11:900–913. doi: 10.1101/gad.11.7.900. [DOI] [PubMed] [Google Scholar]
- Herault Y., Rassoulzadegan M., Cuzin F., Duboule D. Engineering chromosomes in mice through targeted meiotic recombination (TAMERE) Nat. Genet. 1998;20:381–384. doi: 10.1038/3861. [DOI] [PubMed] [Google Scholar]
- Kmita M., Duboule D. Organizing axes in time and space; 25 years of colinear tinkering. Science. 2003;301:331–333. doi: 10.1126/science.1085753. [DOI] [PubMed] [Google Scholar]
- Kmita M., Fraudeau N., Herault Y., Duboule D. Serial deletions and duplications suggest a mechanism for the collinearity of Hoxd genes in limbs. Nature. 2002a;420:145–150. doi: 10.1038/nature01189. [DOI] [PubMed] [Google Scholar]
- Kmita M., Tarchini B., Duboule D., Herault Y. Evolutionary conserved sequences are required for the insulation of the vertebrate Hoxd complex in neural cells. Development. 2002b;129:5521–5528. doi: 10.1242/dev.00151. [DOI] [PubMed] [Google Scholar]
- Kmita M., Tarchini B., Zakany J., Logan M., Tabin C.J., Duboule D. Early developmental arrest of mammalian limbs lacking HoxA/HoxD gene function. Nature. 2005;435:1113–1116. doi: 10.1038/nature03648. [DOI] [PubMed] [Google Scholar]
- Kondo T., Zakany J., Innis J.W., Duboule D. Of fingers, toes and penises. Nature. 1997;390:29. doi: 10.1038/36234. [DOI] [PubMed] [Google Scholar]
- Monge I., Kondo T., Duboule D. An enhancer-titration effect induces digit-specific regulatory alleles of the HoxD cluster. Dev. Biol. 2003;256:212–220. doi: 10.1016/s0012-1606(02)00136-7. [DOI] [PubMed] [Google Scholar]
- Morgan B.A., Izpisúa-Belmonte J.-C., Duboule D., Tabin C.J. Targeted misexpression of Hox4.6 in the avian limb bud causes apparent homeotic transformations. Nature. 1992;358:236–239. doi: 10.1038/358236a0. [DOI] [PubMed] [Google Scholar]
- Nelson C.E., Morgan B.A., Burke A.C., Laufer E., DiMambro E., Murtaugh L.C., Gonzales E., Tessarollo L., Parada L.F., Tabin C. Analysis of Hox gene expression in the chick limb bud. Development. 1996;122:1449–1466. doi: 10.1242/dev.122.5.1449. [DOI] [PubMed] [Google Scholar]
- Reno P.L., McCollum M.A., Cohn M.J., Meindl R.S., Hamrick M., Lovejoy C.O. Patterns of correlation and covariation of anthropoid distal forelimb segments correspond to Hoxd expression territories. J. Exp. Zool. Part B: Mol. Dev. Evol. 2007 doi: 10.1002/jez.b.21207. [DOI] [PubMed] [Google Scholar]
- Riddle R.D., Johnson R.L., Laufer E., Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75:1401–1416. doi: 10.1016/0092-8674(93)90626-2. [DOI] [PubMed] [Google Scholar]
- Sordino P., van der Hoeven F., Duboule D. Hox gene expression in teleost fins and the origin of vertebrate digits. Nature. 1995;375:678–681. doi: 10.1038/375678a0. [DOI] [PubMed] [Google Scholar]
- Spitz F., Gonzalez F., Duboule D. A global control region defines a chromosomal regulatory landscape containing the HoxD cluster. Cell. 2003;113:405–417. doi: 10.1016/s0092-8674(03)00310-6. [DOI] [PubMed] [Google Scholar]
- Spitz F., Herkenne C., Morris M.A., Duboule D. Inversion-induced disruption of the Hoxd cluster leads to the partition of regulatory landscapes. Nat. Genet. 2005;37:889–893. doi: 10.1038/ng1597. [DOI] [PubMed] [Google Scholar]
- Suzuki K., Shiota K., Zhang Y., Lei L., Yamada G. Development of the mouse external genitalia: Unique model of organogenesis. Adv. Exp. Med. Biol. 2004;545:159–172. doi: 10.1007/978-1-4419-8995-6_10. [DOI] [PubMed] [Google Scholar]
- Tarchini B., Duboule D. Control of Hoxd genes’ collinearity during early limb development. Dev. Cell. 2006;10:93–103. doi: 10.1016/j.devcel.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Tarchini B., Duboule D., Kmita M. Regulatory constraints in the evolution of the tetrapod limb anterior–posterior polarity. Nature. 2006;443:985–988. doi: 10.1038/nature05247. [DOI] [PubMed] [Google Scholar]
- Tolhuis B., Palstra R.J., Splinter E., Grosveld F., de Laat W. Looping and interaction between hypersensitive sites in the active β-globin locus. Mol. Cell. 2002;10:1453–1465. doi: 10.1016/s1097-2765(02)00781-5. [DOI] [PubMed] [Google Scholar]
- Zakany J., Duboule D. The role of Hox genes during vertebrate limb development. Curr. Opin. Genet. Dev. 2007;17:359–366. doi: 10.1016/j.gde.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Zakany J., Fromental-Ramain C., Warot X., Duboule D. Regulation of number and size of digits by posterior Hox genes: A dose-dependent mechanism with potential evolutionary implications. Proc. Natl. Acad. Sci. 1997;94:13695–13700. doi: 10.1073/pnas.94.25.13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakany J., Kmita M., Duboule D. A dual role for Hox genes in limb anterior–posterior asymmetry. Science. 2004;304:1669–1672. doi: 10.1126/science.1096049. [DOI] [PubMed] [Google Scholar]