

Abstract
β-arrestin2 and its ubiquitination play crucial roles in both internalization and signaling of seven-transmembrane receptors (7TMRs). To understand the connection between ubiquitination and β-arrestin’s endocytic and signaling functions, we generated a β-arrestin2 mutant that is defective in ubiquitination (β-arrestin20K), by mutating all the ubiquitin acceptor lysines to arginines and compared its properties with the wild type and a stably ubiquitinated β-arrestin2-Ub chimera. In vitro translated β-arrestin2 and β-arrestin20K displayed equivalent binding to recombinant β2 adrenergic receptor (β2AR) reconstituted in vesicles, while β-arrestin2-Ub bound approximately four fold more. In cellular coimmunoprecipitation assays, β-arrestin20K bound non-receptor partners such as AP-2 and c-Raf and scaffolded pERK robustly, but displayed weak binding to clathrin. Moreover, β-arrestin20K was recruited only transiently to activated receptors at the membrane, did not enhance receptor internalization and decreased the amount of pERK assimilated into isolated β2AR complexes. While the wild type β-arrestin2 formed ERK signaling complexes with the β2AR at the membrane, a stably ubiquitinated β-arrestin2-Ub chimera, not only stabilized the ERK signalosomes but also led to their endosomal targeting. Interestingly, in cellular fractionation assays the ubiquitination state of β-arrestin2 favors its distribution in membrane fractions suggesting that ubiquitination increases β-arrestin’s propensity for membrane association. Our findings suggest that although β-arrestin ubiquitination is dispensable for β-arrestin’s cytosol to membrane translocation and its ‘constitutive’ interactions with some cytosolic proteins, it nevertheless is a prerequisite both for the formation of tight complexes with 7TMRs in vivo as well as for membrane compartment interactions that are crucial for downstream endocytic and signaling processes.
The multifunctional adaptor proteins β-arrestins (1 and 2) were originally identified as desensitizing molecules that prevent the coupling between seven-transmembrane receptors (7TMRs)1 and G proteins (1–3). More recently, however, it was found that β-arrestin-binding to receptors not only stops G protein mediated second messenger signaling but also engages several novel signaling pathways including Mitogen Activated Protein Kinase (MAPK) cascades (4,5). Furthermore, β-arrestins have also been shown to bind and regulate cell surface receptors other than 7TMRs and their signaling has been implicated in regulating the actin cytoskeleton, chemotaxis, antiapoptosis and metastasis (6).
β-arrestins serve as endocytic adaptors that bind clathrin and adaptin protein subunit 2 (AP-2) and facilitate receptor internalization via clathrin-coated vesicles (7–9). The differing affinity and trafficking patterns of GFP-β-arrestins induced by several 7TMRs have led to the classification of receptors into two groups, ‘Class A’ and ‘Class B’ (10). ‘Class A’ receptors (e.g. β2 adrenergic, α1b adrenergic, μ opioid, endothelin 1A and dopamine D1A receptors) show higher affinity for β-arrestin2 than β-arrestin1, and recruit GFP-β-arrestins only to the plasma membrane. ‘Class B’ receptors (e.g. vasopressin V2, angiotensin AT1a, neurotensin1, thyrotropin-releasing hormone and neurokinin NK-1 receptors) bind to both β-arrestin1 and 2 with equal affinity and cotraffic and colocalize with GFP-β-arrestin in endocytic vesicles. Thus, complexes formed between β-arrestin and ‘Class A’ receptors are transient and exist only at the membrane whereas those formed between β-arrestin and ‘Class B’ receptors are stable and persist after receptor endocytosis (10). These differential patterns of β-arrestin2 recruitment correlate with the amplitude of β-arrestin bound phosphorylated ERK1/2 (pERK). ‘Class B’ receptors such as the Angiotensin1a and the V2 vasopressin receptors activate a β-arrestin-bound pool of ERK more persistently than ‘Class A’ receptors such as the β2 adrenergic receptor (β2AR) and the α1b adrenergic receptor (11).
β-arrestins also become ubiquitinated [attachment of ubiquitin (Ub) on lysine residues] upon agonist stimulation of various 7TMRs. Upon β2AR stimulation, Mdm2, a RING type E3 ligase, ubiquitinates β-arrestin2 and this modification is required for rapid internalization of the receptor (12). The pattern of β-arrestin ubiquitination correlates with the stability of receptor-β-arrestin interaction, i.e. transient interaction (‘Class A’) is associated with transient ubiquitination, and persistent interaction (‘Class B’) with sustained ubiquitination (13,14). Exchanging the carboxyl terminal amino acid residues of these two types of receptors reverses the patterns of β-arrestin trafficking as well as the time course of ubiquitination and the extent of β-arrestin-bound ERK activation (11,13,15). Additionally, translational fusion of ubiquitin to the C-terminus of β-arrestin (β-arrestin2-Ub) leads to its cotrafficking and colocalization with the β2AR (Class A) in endocytic vesicles thus mimicking a “Class B” trafficking pattern (13).
Interestingly, specific lysine residues are targeted for modification in response to agonist-stimulation of a particular 7TMR. For example, Angiotensin1a receptor (AT1aR) -dependent sustained β-arrestin ubiquitination occurs primarily at lysines 11 and 12 in β-arrestin2 (16). Mutation of these lysines to arginine residues leads to the reversal of Angiotensin II-stimulated β-arrestin ubiquitination from a sustained to a transient pattern, with a corresponding reversal of AT1aR-β-arrestin binding from stable endosome localized complexes to transiently associated complexes seen only at the plasma membrane.
In an attempt to understand the role of ubiquitination in the regulation of the endocytic and signaling functions of β-arrestin, we generated a β-arrestin2 mutant (β-arrestin20K) that is defective in ubiquitination, by mutating all the ubiquitin acceptor lysines in β-arrestin2 to arginines, and compared it with the wild type and a stably ubiquitinated form in its ability to interact with 7TMRs and nonreceptor partners as well as its capability to facilitate receptor internalization and signaling.
EXPERIMENTAL PROCEDURES
Cell lines, reagents and plasmids
COS-7 and HEK-293 cells were obtained from American Type Culture Collection. COS-7 cells were maintained in Dulbecco’s modified Eagle’s medium (Sigma) supplemented with 10 % Fetal Bovine Serum and 1% penicillin streptomycin and transiently transfected with Lipofectamine 2000 reagent (Invitrogen). HEK-293 cells were maintained in Minimal Essential Medium supplemented with Fetal Bovine Serum and transiently transfected with FuGene 6 reagent (Roche Diagnostics). M2 anti FLAG affinity agarose beads, isoproterenol, arginine-vasopressin peptide, anti FLAG M1 and M2 antibodies, FITC-anti mouse secondary IgG and N-ethyl maleimide (NEM) were from Sigma. Ubiquitin antibody FK2 was from Biomol. Monoclonal antibody 12CA5 to HA epitope was from Roche Diagnostics. Alexa 594 and Alexa 633, conjugated secondary antibodies were from Invitrogen. HRP-conjugated secondary antibodies were from GE/Amersham Pharmacia. Detection of active ERK was with a rabbit polyclonal anti-phospho-p44/42 MAPK (Cell Signaling Technology, 1:2000 for Western blot, and 1:200 for immunostaining). Total ERK was detected with anti-MAPK 1/2 (Upstate Technology Inc, 1:10,000 dilution for Western blots). A1CT, a rabbit polyclonal antibody to β-arrestin1 C-terminus generated in the Lefkowitz Laboratory was used to detect β-arrestin isoforms.
Rat β-arrestin2/pEGFPN1 plasmid has been previously described (17). A 1500 bp DNA fragment-encoding β-arrestin2-Ub was subcloned into the KpnI and ApaI sites of pEGFPC1 vector to obtain the expression plasmid for GFP-β-arrestin2-Ub. The lysine residue at position 48 in Ub was replaced with arginine using a QuikChange® Site-Directed Mutagenesis Kit (Stratagene). GFP-β-arrestin2-Ub used in this work actually represents GFP-β-arrestin2-UbK48R.
Five rounds of mutations accomplished the construction of β-arrestin20K, each mutagenesis step targeting 5–7 lysine residues. We used the QuikChange® MultiSite-Directed Mutagenesis Kit (Stratagene) and followed manufacturer’s instructions for the design of oligos and PCR protocols. The DNA fragment encoding β-arrestin20K was later cloned into pEGFP-N1 to yield β-arrestin20K-GFP. All DNA constructs were verified by sequencing. HA-β2AR plasmid was a gift from Dr. Neil Freedman (Duke University); HA-V2R plasmid was provided by Dr. Marc Caron (Duke University). Myc-cRaf and RFP-ERK2 have been previously reported (18).
To achieve equivalent expression of β-arrestin2 WT and lysine mutants (Fig 1), we transfected cells on a 100mm dish with 1μg DNA for the WT and −7K, 3 μg DNA for −14K, −19K, −26K and 2.5μg for −31K. For the GFP-tagged plasmids (Fig 5), 1μg was used for the WT and −7K and 2μg for the rest.
Fig 1. A lysine-less β-arrestin2 (β-arrestin20K) is not ubiquitinated upon 7TMR stimulation.
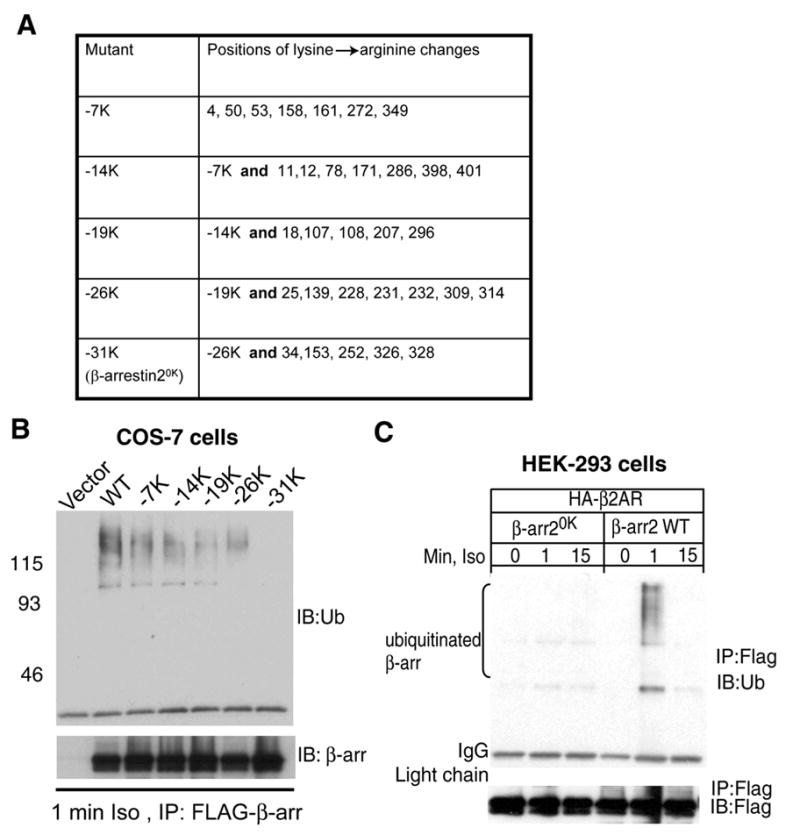
A. Left column details the total number of lysine residues mutated to arginine. The right column depicts the position of the lysine in the primary sequence of rat β-arrestin2. B. COS-7 cells were transiently transfected with vector or the indicated β-arrestin2-FLAG plasmid, stimulated with 1μM isoproterenol for 1 min. Anti-FLAG immunoprecipitates were probed for ubiquitin with FK2 antibody (top panel) and a polyclonal FLAG antibody (bottom panel). C. HEK-293 cells were transfected with HA-β2AR and either WT or β-arrestin20K containing FLAG tag. Serum-starved cells were stimulated with 1 μM isoproterenol for indicated times and FLAG immunoprecipitates were analyzed with FK2 ubiquitin antibody (top panel) and M2 FLAG antibody (bottom panel). The blots shown in B and C are from one of three separate experiments.
Fig 5. Translocation of GFP-tagged β-arrestin2 lysine mutants to activated β2ARs.
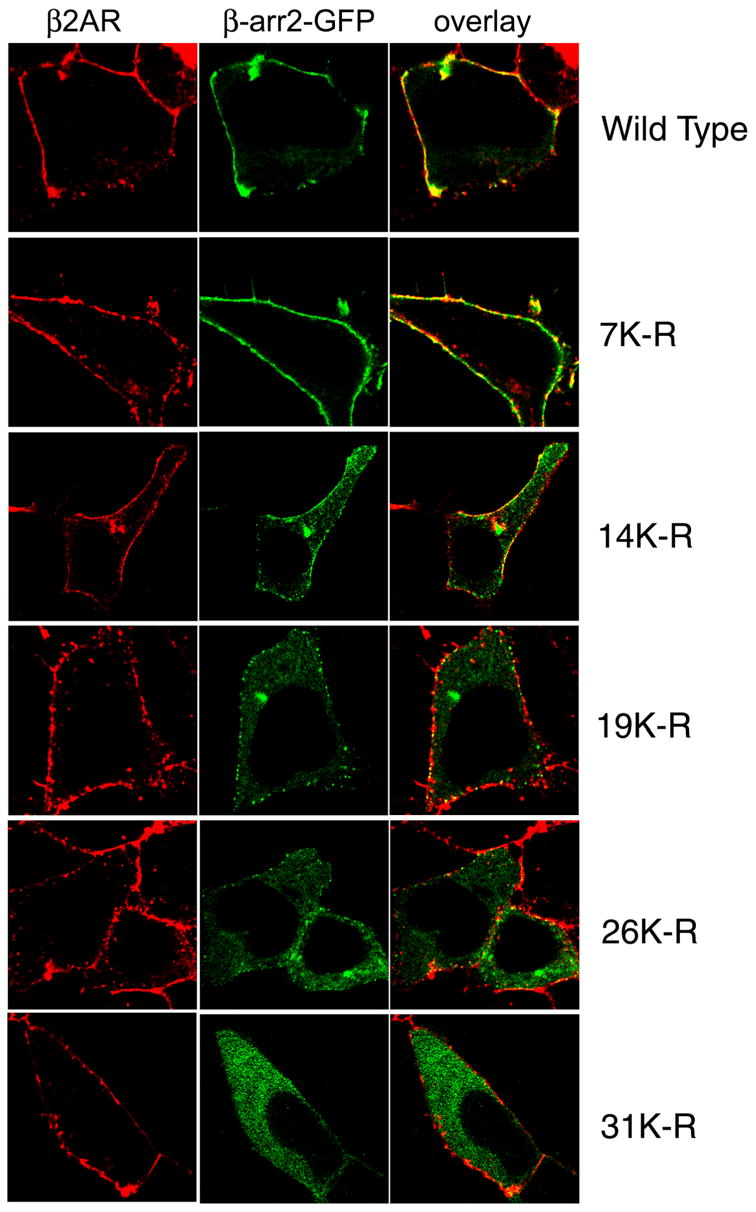
HEK-293 cells were transfected with HA-β2AR and the indicated β-arrestin2 plasmid. Cells were stimulated with isoproterenol for one minute, fixed and immunostained for the β2AR. Confocal images shown represent one of three similar experiments. β2AR detection is shown in red, β-arrestin fluorescence in green and overlay panels depict colocalization (yellow).
Immunoprecipitation and immunodetection
β-arrestin2-FLAG/pCDNA3, β-arrestin20K-FLAG/pCDNA3.1or FLAG-β-arrestin2-Ub/pCDNA3.1were used to immunoprecipitate β-arrestins. To detect active ERK in receptor immunoprecipitates, FLAG-β2AR was co-expressed with GFP-tagged β-arrestin constructs and RFP-ERK2. Cells were serum starved for 2 h (COS-7) or 4 h (HEK-293) and then stimulated or not for the indicated times with the appropriate agonists. Cells were solubilized in a lysis buffer (LB) containing 50mM HEPES (pH7.5), 0.5% NP40, 250mM NaCl, 2mM EDTA, 10% (v/v) glycerol, 1mM sodium orthovanadate, 1mM sodium fluoride, 1mM phenylmethylsulfonyl fluoride, leupeptin (5μg/ml), aprotinin (5μg/ml), pepstatin A (1μg/ml), benzaminidine (100μM) and 10 mM NEM. The use of NEM in lysis buffers in coimmunoprecipitation procedures is an important technical feature since it stabilizes ubiquitinated species by preventing their deubiquitination. Soluble extracts were mixed with FLAG M2 affinity beads and rotated at 4 °C overnight. Nonspecific binding was eliminated by repeated washes with LB and bound protein was eluted with sample buffer containing SDS. The proteins were separated on a gradient gel (4–20%, Invitrogen) and transferred to nitrocellulose membrane for Western blotting. Chemiluminescence detection was performed using SuperSignal® West Pico reagent (Pierce). pERK and β-arrestin signals were quantified by densitometry using GeneTools software.
In vitro translation of β-arrestins and β-arrestin-recombinant β2AR binding
[35S] β-arrestins were in vitro translated using a TNT® T7 Quick Coupled Transcription/Translation Systems (Invitrogen Cat.# L1170) according to the manufacture’s recommended procedure. Briefly, reactions were assembled by mixing appropriate amounts of TNT® Quick Master Mix, [35S] methionine (Amersham Biosciences Cat.# AG1094) and pCDNA3.1-β-arrestin 2 wild type, β-arrestin2-0K or β-arrestin2-Ub plasmids in 0.5 ml microcentrifuge tubes. The reactions were incubated at 30 °C for 90 minutes and the in vitro translated [35S]β-arrestins were stored at −80 °C before performing binding experiments.
To study receptor binding, the in vitro translated [35S]β-arrestins were incubated in 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA with 14.6 pmol (0.7 μg) of β2AR reconstituted in phospholipid vesicles at room temperature for one hour. Purified GRK2 (0.5 μg), 80 μM ATP, 50 μM isoproterenol or 50 μM propranolol were added to the reaction mixture, where indicated. After the incubation period, an aliquot of the reaction was set aside to determine input levels of [35S]β-arrestins and the remaining samples were diluted with ice-cold buffer and centrifuged at 85,000 rpm for 30 minutes with a bench top Optima TLX ultracentrifuge. After ultracentrifugation, the supernatants were removed and the pellets were washed with 0.5 ml of 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 2 mM EDTA. The samples were centrifuged again and the wash was repeated for five times. Finally, 30 μl of SDS-PAGE buffer were added to each sample and proteins were separated by 4–20% gel. The gels were dried and the amounts of β-arrestins bound to the β2AR were determined by autoradiography. Control experiments were performed by the same experimental procedure except that empty vesicles were used in the place of receptor containing vesicles. Bands were quantified by densitometry and the amount of each β-arrestin was normalized to its input levels.
Crosslinking
COS-7 cells were transiently transfected with FLAG-β2AR along with pEGFP or β-arrestin20K-GFP. 30 h post transfection, cells in 100-mm dishes were stimulated at 37°C in phosphate-buffered saline (PBS) containing 10 mM HEPES (pH 7.4), with vehicle or agonist. Stimulations were terminated by the addition of Dithio-bis-maleimidoethane (DTME, Pierce) to a final concentration of 2 mM, and plates were rocked for 40 min at room temperature. Cells were washed three times with PBS/HEPES to remove unreacted DTME, lysed in RIPA buffer (150 mM NaCl, 50 mM Tris, pH 8.0, 5 mM EDTA, 1% Nonidet P-40, 0.5% deoxycholate) and receptors immunoprecipitated.
Phospho-ERK time course
HEK-293 cells stably expressing the β2AR, transfected with vector, β-arrestin2-FLAG, FLAGβ-arrestin2-Ub or β-arrestin20K-FLAG on 12-well plates were starved for at least 4 h in serum-free medium prior to stimulation. After stimulation, cells were solubilized by directly adding 2X SDS-sample buffer, followed by boiling at 100 °C for 5 minutes. For each transfection, an equal portion of the cells was set aside for protein determination (modified Bradford protocol). Equal micrograms of cellular extracts were separated on 4–20% Tris-Glycine polyacrylamide gels (Invitrogen) and transferred to nitrocellulose membranes for immunoblotting. Phosphorylated ERK1/2, total ERK1/2 and β-arrestins were detected by immunoblotting with rabbit polyclonal anti-phospho-p44/42 MAPK (Cell Signaling, 1:2,000), anti-MAP kinase 1/2 (Upstate Technology Inc, 1:10,000), and anti-β-arrestin (A1CT, 1:3,000) antibodies, respectively. Chemiluminiscence detection was performed using the SuperSignal West Pico reagent (Pierce) and Phosphorylated ERK1/2 immunoblots were quantified using GeneTools software.
Confocal microscopy
HEK-293 cells have a favorable morphology such that sections of cytoplasm and nucleus can be simultaneously imaged and hence they were used in these experiments. HEK-293 cells on 10 cm dishes were transiently transfected with HA-β2AR along with β-arrestin2-GFP, GFP-β-arrestin2-Ub or βarrestin20K-GFP. Twenty-four hours post-transfection, cells were plated on collagen-coated 35-mm glass bottom plates. On the following day, cells were starved for at least 2h in serum free medium prior to stimulation. After stimulation, cells were fixed with 5% formaldehyde diluted in PBS containing Calcium and Magnesium. Fixed cells were permeabilized with 0.01% Triton in PBS containing 2% BSA for 60 minutes and incubated at room temperature with appropriate primary antibody. The secondary antibody incubations were done for one hour followed by repeated washes using PBS. Confocal images were obtained on a Zeiss LSM510 laser scanning microscope using multitrack sequential excitation (488, 568, 633 nm) and emission (515–540 nm GFP; 585–615 nm, Texas red) filter sets. Live cell GFP images were acquired using a heated (37 °C) microscope stage and collected sequentially using single line excitation (488 nm).
Receptor Internalization
FLAG or HA epitope tagged receptors expressed in HEK-293 cells in twelve-well dishes were incubated with or without agonist for 30 min in serum-free medium at 37 °C. Cell surface receptors were labeled with M1 FLAG mAb or 12CA5 mAb, and fluorescein isothiocyanate-conjugated goat antibody to mouse IgG as a secondary antibody. Receptor internalization was quantified as loss of cell surface receptors as measured by fluorescence assisted cell sorting (Flow cytometry facility, Duke University).
Subcellular Fractionation
Monolayers of COS-7 cells transfected with β-arrestin2 or β-arrestin2-Ub plasmids were gently scraped and collected in PBS containing protease inhibitors and 40 mM NaCl, subjected to two freeze-thaw cycles for lysis. Samples were centrifuged at 800 × g for 5 min to precipitate unlysed cells. The resulting supernatant was centrifuged at 100,000 × g to separate soluble and membrane components. 40 μg of each fraction was separated on SDS gels and analyzed by Western blotting.
RESULTS
A ubiquitin minus β-arrestin2 mutant
To obtain a β-arrestin2 mutant that is not ubiquitinated upon 7TMR stimulation, we made conservative changes of groups of lysines to arginines, overexpressed FLAG-tagged mutants in COS-7 cells and tested the β-arrestin precipitates for the ubiquitination signal induced by one-minute isoproterenol stimulation (Fig 1 A, B). Surprisingly, elimination of such a signal required replacement of all of β-arrestin2’s 31 lysine residues (mutant β-arrestin20K Fig 1B). When a FLAG epitope-tagged β-arrestin20K is over expressed in HEK-293 cells, no ubiquitination smear is detected upon isoproterenol stimulation (Fig 1C). Although these experiments indicate that β-arrestin20K can be expressed as a properly folded protein that is isolated and detected by the epitope tag, a concern still remains whether β-arrestin20K despite its 31 lysine to arginine changes is a bona fide form of β-arrestin.
To test whether the basic folding and binding properties of β-arrestin20K are retained, we compared the binding of in vitro translated β-arrestin2 and β-arrestin20K to purified recombinant β2AR reconstituted in vesicles. We also tested β-arrestin2-Ub for receptor binding under the same conditions. In these in vitro assays, both WT and β-arrestin20K represent non-ubiquitinated forms and only β-arrestin2-Ub chimera constitutes the ubiquitinated form. As shown in Fig 2A and B, β-arrestin20K bound to the β2AR to the same extent as β-arrestin2. However the presence of a single ubiquitin moiety increased the binding by fourfold (Fig 2A and B). These experiments suggest that while both non-ubiquitinated forms of β-arrestin2 (i.e. WT and 0K) are equipotent for β2AR binding, there is more binding between the β2AR and the ubiquitinated form (i.e. β-arrestin2-Ub). When binding was performed in the presence of isoproterenol, a small increase was observed for all three β-arrestin forms (data not shown). We hypothesized that reconstituted β2AR was already in an activated conformation due to the presence of zinterol in purification buffers. If so, inclusion of an antagonist could alter the observed binding. When β-arrestin-receptor complex formation was tested in the presence of propranolol, we found a dramatic decrease in binding for all three β-arrestin forms (Fig 2A and B), suggesting that propranolol destabilizes but does not eliminate receptor-β-arrestin binding in these experiments. Moreover, when reconstituted receptor samples were probed with a β2AR specific phosphoserine antibody (serines 355,356), a small amount of phosphorylation was detected (Fig 2C upper panel). Addition of GRK2 leads to an increase in the phosphorylation signal and isoproterenol augments it further (Fig 2C). We observed a comparable increase in binding above basal conditions for all three β-arrestin forms upon GRK2 phosphorylation and isoproterenol treatment of the reconstituted β2AR (Fig 2D). Collectively, these in vitro binding assays confirm that although ubiquitinated β-arrestin2 forms a tight complex with the β2AR, nonubiquitinated β-arrestin2 can bind reconstituted β2AR and that the protein-protein interaction domain(s) between the receptor and β-arrestin20K is mostly unperturbed.
Fig 2. Binding of in vitro translated β-arrestins to recombinant β2ARs reconstituted in phospholipid vesicles.
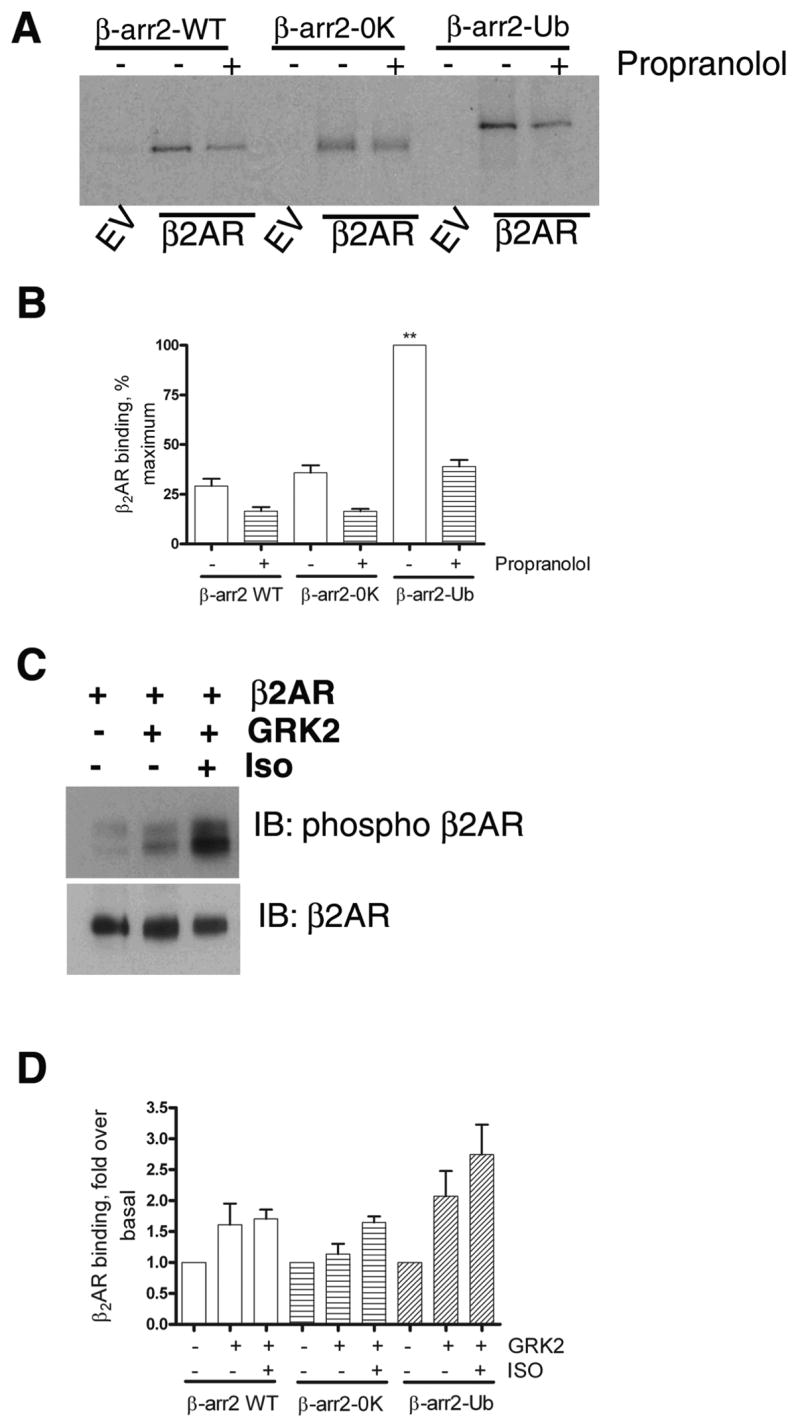
A. As described in the methods section, ‘in vitro’ translated, 35S labeled β-arrestin2, β-arrestin20K or β-arrestin2-Ub was incubated with either empty phospholipid vesicles or vesicles containing β2AR. The vesicles were then precipitated by repeated centrifugation and wash cycles. The final pellet was solubilized, separated on SDS gels and the bound β-arrestins were detected by autoradiography. In the indicated lanes, propranolol (50 μM) was included in the reaction mixture. B. Results from 3 separate binding experiments were quantified and shown as bar graphs. ** p<0.001, β-arrestin2 or β-arrestin20K versus β-arrestin2-Ub, one way ANOVA, and Tukey’s multiple comparison test. β-arrestin2 versus β-arrestin20K, no significant difference. C. Reconstituted β2AR samples were probed with a phosphoserine antibody (Serines 355, 356 within β2AR carboxyl tail) in the upper panel and with a β2AR antibody (H-20, Santacruz) in the lower panel. D. Receptor-β-arrestin binding was tested as in ‘A’. GRK2 alone or GRK2 and isoproterenol (50 μM) were added to the reaction as indicated. The bar graph is a summarization of two independent experiments performed in duplicate. For each β-arrestin form, basal binding in the absence of GRK2 and isoproterenol was assigned as 1.
To determine the isoproterenol-stimulated binding of β-arrestin20K to the β2AR in a cellular context we employed immunoprecipitation assays utilizing chemical crosslinking with a sufhydryl-reactive compound, DTME (Fig 3). We used COS-7 cells transiently transfected with FLAG-β2AR and β-arrestin20K-GFP, immunoprecipitated the receptors under nonstimulated or stimulated conditions (5 min, 1 μM isoproterenol) and detected β-arrestin20K-GFP by Western blotting (Fig 3A). β-arrestin20K-GFP binds to activated receptors with a 2–3 fold agonist-induced recruitment (Fig 3B). In similar assays, the WT and β-arrestin2-Ub were recruited 10–12 and 12–15 fold, respectively (data not shown). These experiments further suggest that β-arrestin20K-GFP albeit much weaker than the WT nevertheless binds the β2AR upon agonist stimulation. Likely, the robust association of β-arrestin20K and the β2AR does not occur in cells due to a lack of β-arrestin2 ubiquitination, which helps to stabilize the complex.
Fig 3. β-arrestin20K binding to receptors demonstrated by chemical cross-linking.
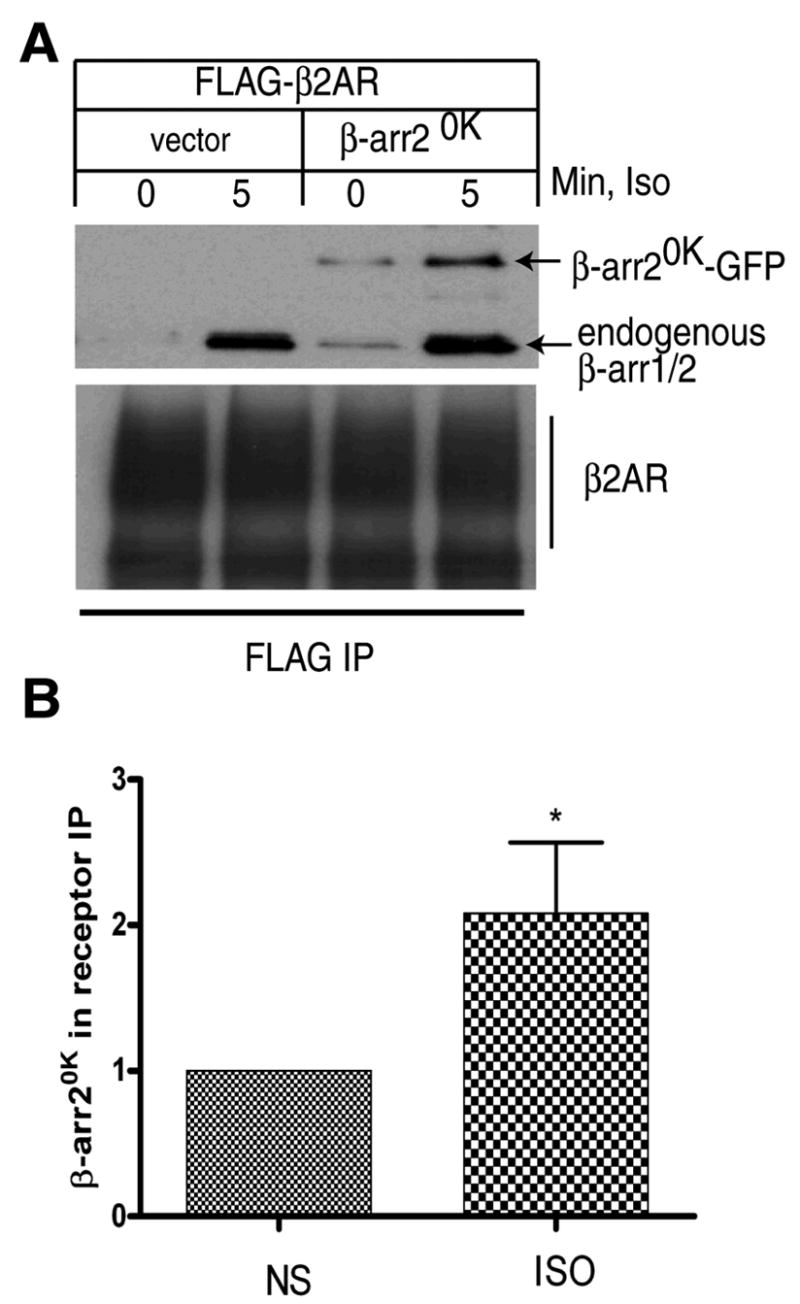
A. COS-7 cells transiently transfected with FLAG-β2AR with pEGFP vector or β-arrestin20K-GFP were stimulated with 10 μM isoproterenol for 5 min and FLAG receptors immunoprecipitated after chemical cross-linking with DTME. The IP was probed with a β-arrestin antibody (upper panel) and a receptor specific antibody, H-20 (lower panel). B. The bar graph represents quantification of β-arrestin20K in the IP from 5 independent experiments. *: p, 0.04 according to a paired t test.
We next transfected HEK-293 cells stably expressing the β2AR with either β-arrestin2-GFP, GFP-β-arrestin2-Ub (stable ubiquitination), or β-arrestin20K-GFP (no ubiquitination) and examined the translocation patterns induced by isoproterenol. All three β-arrestin variants are uniformly distributed in the cytosol prior to agonist treatment (Fig 4 A–C). Within one-minute of agonist-stimulation, both WT and GFP-β-arrestin2-Ub are recruited to the cell membrane and form distinct puncta and at 30 minutes GFP-β-arrestin2-Ub is recruited to endosomal vesicles (Fig 4B), while the WT remains at the membrane (Fig 4A). As previously shown, with a ‘Class A’ receptor a stably ubiquitinated β-arrestin traffics into endosomes while the transiently ubiquitinated WT β-arrestin dissociates and remains at the plasma membrane. On the other hand, agonist stimulation for 1 or 30 min does not lead to a major change in the intracellular distribution of β-arrestin20K-GFP (Fig 4C, center panels).
Fig 4. Recruitment of β-arrestin2, β-arrestin2-Ub and β-arrestin20K to activated 7TMRs.
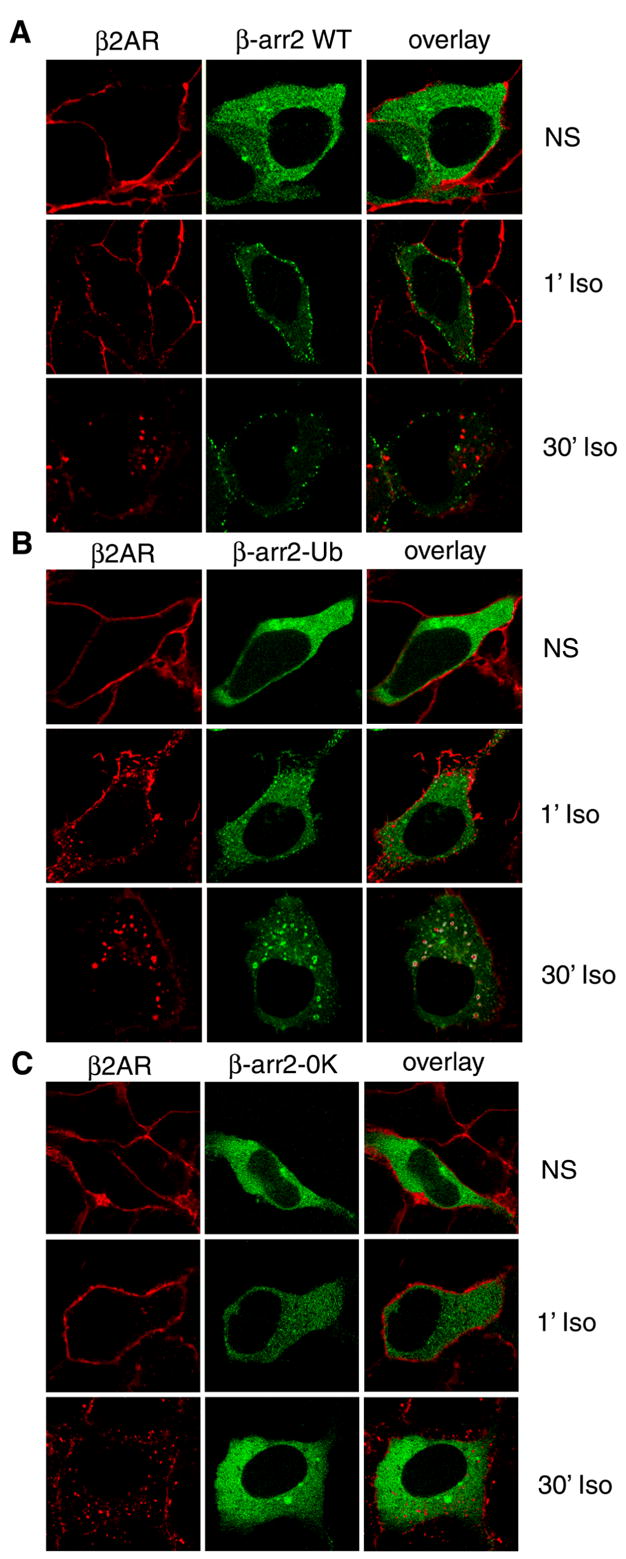
HEK-293 cells were transiently transfected with HA-β2AR and either β-arrestin2-GFP (A), GFP-β-arrestin2-Ub (B) or β-arrestin20K-GFP (C). Cells were starved for one hour in serum free media, stimulated with isoproterenol for indicated times, fixed, and immunostained for the β2AR (red). Shown are the confocal images of the receptor immunofluorescence (red) and the GFP fluorescence (green). Colocalization (yellow) of the receptor with respective β-arrestins is indicated in the overlay. These images are from one of three experiments with identical results.
To test whether the loss of translocation correlates with a loss of ubiquitination owing to cumulative lysine mutations, we examined isoproterenol-induced recruitment of all the mutants shown in Fig 1A, B. When the GFP-tagged version of each mutant was coexpressed with HA-β2AR in HEK-293 cells, we observed normal cytosolic expression under basal conditions for all the β-arrestin2 variants (supplementary Fig 1). However, upon 1 min isoproterenol stimulation, a decrease in the level of recruitment was observed correlating with the ubiquitination status of β-arrestin2 (Fig 5 middle panels and Fig 1B). Some amount of recruitment remains even when 26 lysine residues are altered. The only mutant that is totally defective in translocation is β-arrestin20K where no ubiquitination sites remain. These data suggest a strong correlation between β-arrestin ubiquitination status and its ability to bind activated receptors at the plasma membrane.
The above experiments also suggest that eliminating β-arrestin ubiquitination decreases its binding affinity for receptors in vivo and hence only unstable receptor-β-arrestin20K-GFP complexes arise at the cell membrane. In the case of ‘Class A’ receptors, such as the β2AR, although receptor-β-arrestin complexes are initially formed at the plasma membrane, these complexes are not long-lived. Thus, β-arrestin is rapidly deubiquitinated, dissociates from the receptor, and the receptor alone traffics into endosomes. This indicates that events occurring during pit/vesicle formation (e.g. β-arrestin deubiquitination) can influence the stability of β-arrestin-receptor complexes. Possibly, the lack of ubiquitin moieties on β-arrestin20K-GFP leads to its rapid disengagement from the receptor complex.
We hypothesized that β-arrestin20K binds activated receptor at the plasma membrane but that its deficiency in ubiquitination results in decreased stability of the complex as the receptor moves into pits. If this were true, then blocking the internalization of receptors should result in the retention of β-arrestin20K-receptor complexes at the plasma membrane. Indeed, when we inhibited the internalization of either the β2AR (Fig 6A) or the ‘Class B’ V2R (Fig 6B) by co-expressing dynaminK44A (a classical inhibitor of endocytosis, (19,20)) we trapped activated receptors as well as β-arrestin20K-GFP at the membrane. These experiments clearly indicate that the deficiency in ubiquitination does not inhibit translocation of cytosolic β-arrestin20K to activated receptors at the cell membrane, but rather decreases the stability of the receptor-β-arrestin complexes that are formed.
Fig 6. Agonist-stimulated translocation of β-arrestin20K.
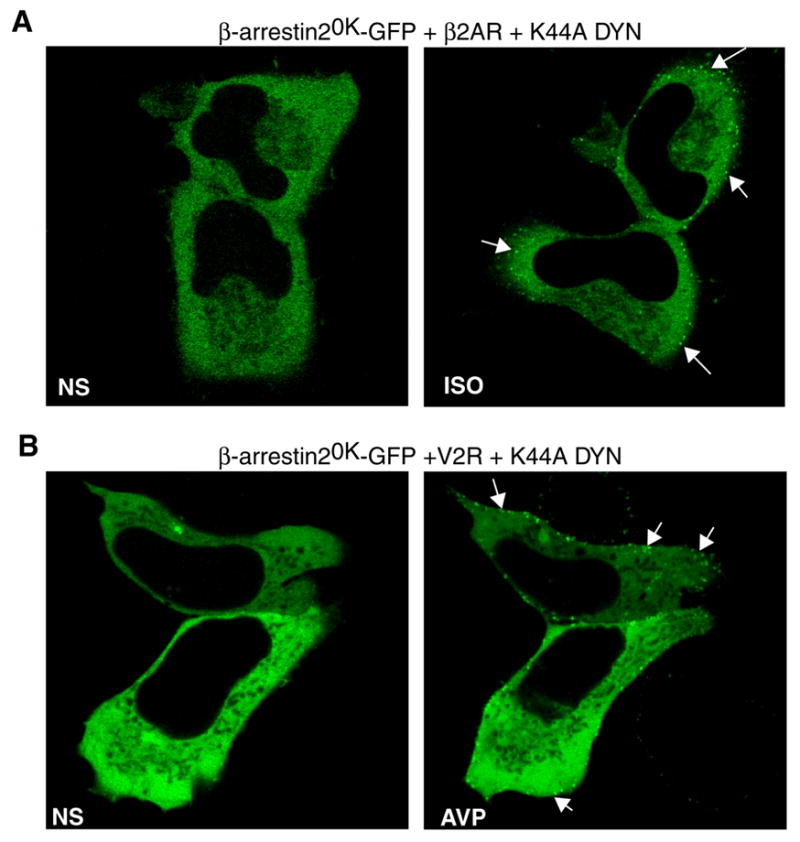
HEK-293 cells were transiently transfected with β-arrestin20K-GFP and Dynamin K44A along with the β2AR (B) or the V2R (C). Cells were starved for one hour in serum free media. The distribution of β-arrestin20K-GFP was visualized before and after treatment with 10 μM isoproterenol (B) or 1 μM AVP (C). Shown are representative confocal images of GFP-fluorescence followed in the same HEK-293 cells treated for 20 min at 37 °C.
Role of β-arrestin2 ubiquitination in receptor internalization
A characteristic feature of β-arrestin2 is its ability to augment receptor internalization upon over-expression (9). This effect is particularly striking in COS-7 cells, which express very low levels of endogenous β-arrestin2 (21). To characterize the ability of β-arrestin20K to promote receptor internalization, we over-expressed it together with HA-tagged β2AR or V2R and measured the decrease in cell surface receptors after a 30 min agonist treatment. Over expression of β-arrestin20K does not lead to any increase in receptor internalization whereas WT β-arrestin2 leads to an approximately 2.5-fold increase in both β2AR and V2R internalization (Fig 7 A, B). Similarly, over-expression of β-arrestin20K results in no change in receptor internalization in HEK-293 cells (Fig 7 C, D). Predictably, because of the unstable interaction of β-arrestin20K with activated receptors than the WT, the mutant does not have any inhibitory effect on receptor internalization in both cell types. We have previously demonstrated that the stably ubiquitinated form of β-arrestin2 (β-arrestin2-Ub) enhances receptor internalization compared to the WT (13). In contrast, β-arrestin20K, which is not ubiquitinated, forms unstable complexes with activated receptors, does not support internalization of either the β2AR or the V2R.
Fig 7. β-arrestin20K does not support receptor internalization.
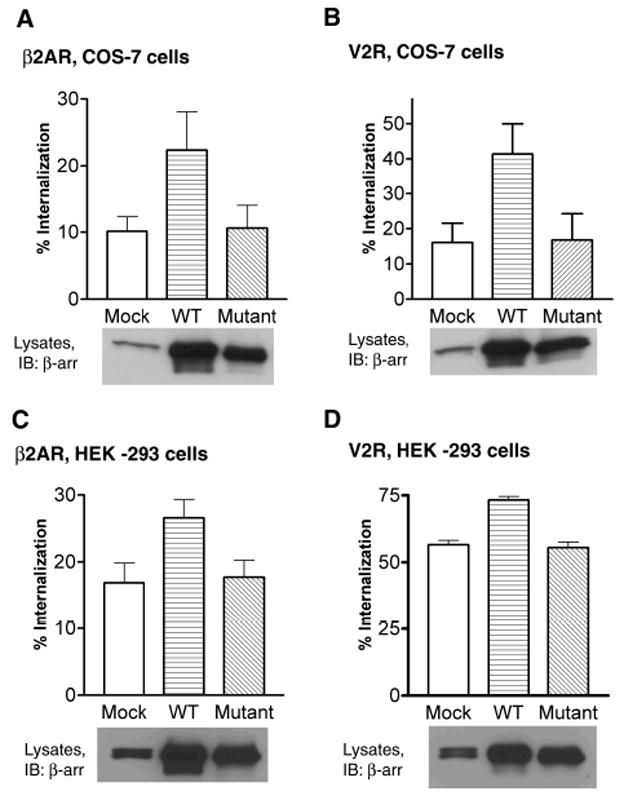
COS-7 (A, B) or HEK-293 (C, D) cells were transiently transfected with FLAG-β2AR (A, C) or HA-V2R (B, D). In each case the receptor was cotransfected with vector plasmid (Mock), β-arrestin2 (WT), or β-arrestin20K (Mutant). After serum starvation cells were treated with 10 μM isoproterenol (A, C) or 1 μM AVP (B, D) for 30 minutes at 37 °C. Cell-surface receptors before and after agonist treatment were determined by Flow cytometry. Data in (A) represent the mean ± SEM of four independent experiments done in triplicate. Data in (B and C) are the mean ± SEM of three independent experiments done in triplicate. Data in (D) represents the average of two independent experiments done in triplicate. 25 μg of lysate protein from each transfection was tested for β-arrestin2 and β-arrestin20K expression by Western blot as shown below each bar graph. A β-arrestin antibody (A1CT) was used for detection. It is to be noted that unlike HEK-293 cells that express easily detectable amounts of both β-arrestin1 and β-arrestin2 (seen as a doublet), COS cells express lesser endogenous β-arrestin2 than HEK-293 cells.
We further tested if the above lack of effect of β-arrestin20K to enhance receptor internalization was due to altered binding to endocytic proteins such as clathrin and AP-2. Clathrin binds β-arrestin directly and stoichiometrically and β-arrestin-clathrin binding is essential for receptor internalization via clathrin-coated vesicles (7). AP-2-β-arrestin interaction is required for the movement of receptors to clathrin-coated pits (8). When β-arrestin2, β-arrestin20K or β-arrestin2-Ub were immunoprecipitated from COS-7 cells transfected with HA-β2AR after 0, 1 and 10 minutes of isoproterenol treatment, an agonist-dependent increase in clathrin binding was observed for both WT and β-arrestin2-Ub but not for β-arrestin20K (Fig 8A, B). β-arrestin20K displayed only a weak interaction and a decrease in binding in the presence of isoproterenol (Fig 8A, B). For the wild type β-arrestin2, we found a five-fold increase in AP-2 binding at 1 min of agonist treatment and this binding decreased to basal levels at 10 min (Fig 8A, C). A similar time course of AP-2-β-arrestin2 interaction has been previously reported (8). Surprisingly, both β-arrestin2-Ub and β-arrestin20K displayed robust binding to AP-2 under both basal and stimulated conditions (Fig 8A and 8C). Understandably, β-arrestin20K’s weak interaction with clathrin upon isoproterenol stimulation could be a major factor in its inability to promote receptor endocytosis. Unlike the previously reported β-arrestin1319–418 which binds clathrin but lacks receptor interaction (22), β-arrestin20K did not act as an inhibitor of receptor internalization.
Fig 8. Isoproterenol stimulated clathrin and AP-2 binding to β-arrestin2, β-arrestin2-Ub and β-arrestin20K.
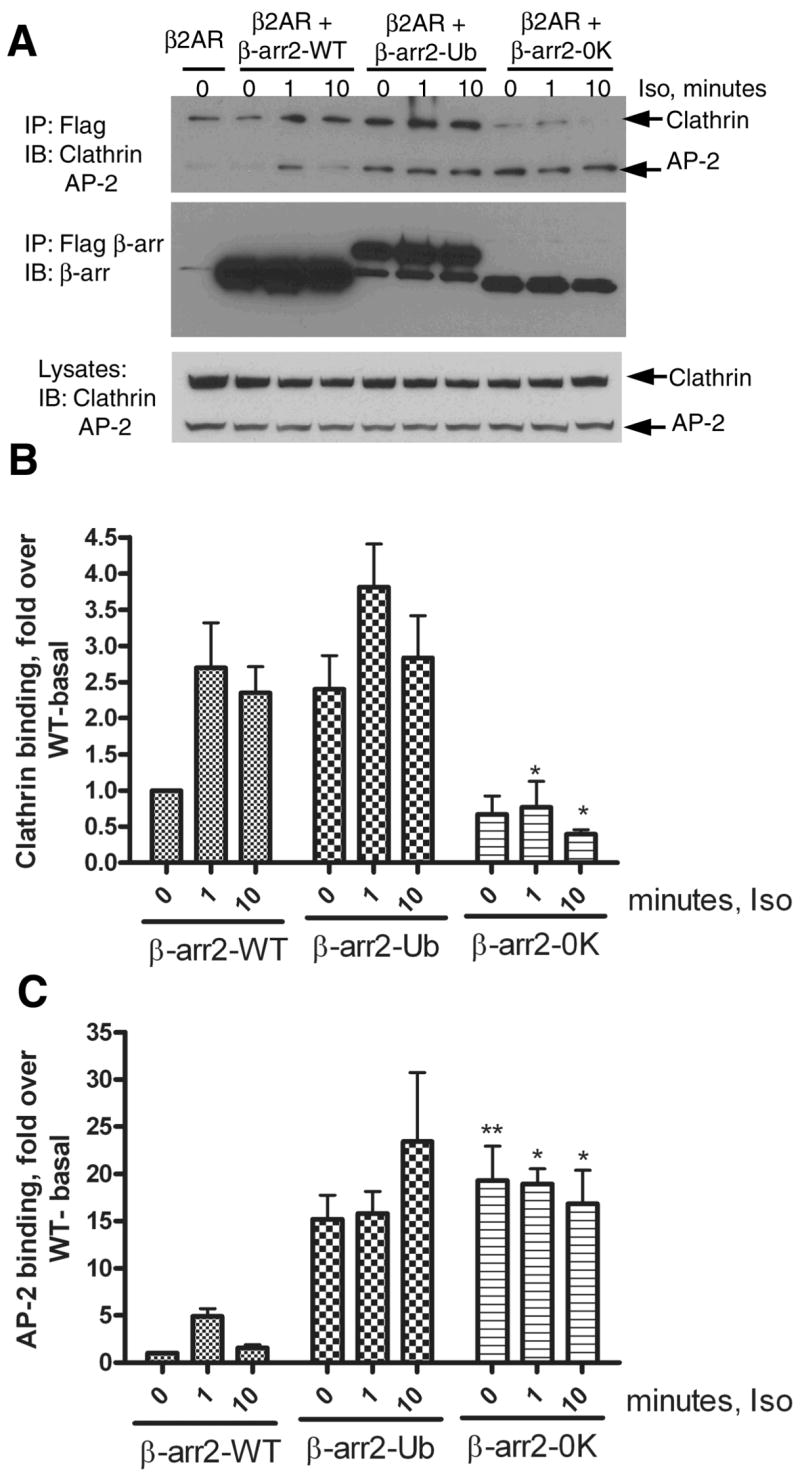
A. COS-7 cells were transiently transfected with vector or the respective FLAG-tagged β-arrestin2 plasmid and HA-β2AR. Cells were serum deprived for 1h, and stimulated for indicated times with 10 μM isoproterenol. Isolated anti-FLAG immunoprecipitates were simultaneously probed for Clathrin heavy chain and the AP-2 subunit. The bottom panel represents the endogenous levels of both these proteins in COS-7 cells as detected by the antibodies. The top panel represents the amount of Clathrin and AP-2 in β-arrestin IPs. The middle panel presents reprobing of the IP blot for β-arrestin2, β-arrestin20K and β-arrestin2-Ub with a β-arrestin antibody (A1CT). Shown are representative blots from one of four independent experiments. B. Bar graph depicts the quantification of clathrin associated with each type of β-arrestin2. Data was normalized to the amount of clathrin bound to WT under basal conditions. β-arrestin20K bound significantly lesser clathrin than the WT at 1 min and 10 min agonist treatment. *P<0.05, β-arrestin20K versus respective signal in the WT, Two way ANOVA, Bonferroni post tests. C. Bar graph shows quantification of AP-2 associated with each type of β-arrestin normalized to the levels detected in the WT immunoprecipitate under basal conditions. β-arrestin20K bound significantly higher AP2 than the WT basally and at 1 min and 10 min agonist treatment. *P<0.05, ** P<0.01 β-arrestin20K versus respective signal in the WT, Two way ANOVA, Bonferroni post tests.
Impact of ubiquitination on Raf, ERK scaffolding properties of β-arrestin2
Previous studies have shown that β-arrestin-Raf complexes are stable since their isolation is possible by gel filtration as well as by coimmunoprecipitation (18,23). We tested the interaction of the above three β-arrestin forms with the MAPKKK, cRaf (Fig 9A) and did not observe any differences between the WT and β-arrestin20K in their ability to bind myc-c-Raf1. In these assays, β-arrestin2-Ub, however, bound more c-Raf than the WT (Fig 9A). The amount of c-Raf in the immunoprecipitate normalized to total input levels was significantly higher with β-arrestin2-Ub than with the wild type as indicated by the quantification of bands from three independent experiments (data not shown).
Fig 9. Raf and ERK scaffolding properties of β-arrestin2, β-arrestin20K and β-arrestin2-Ub.
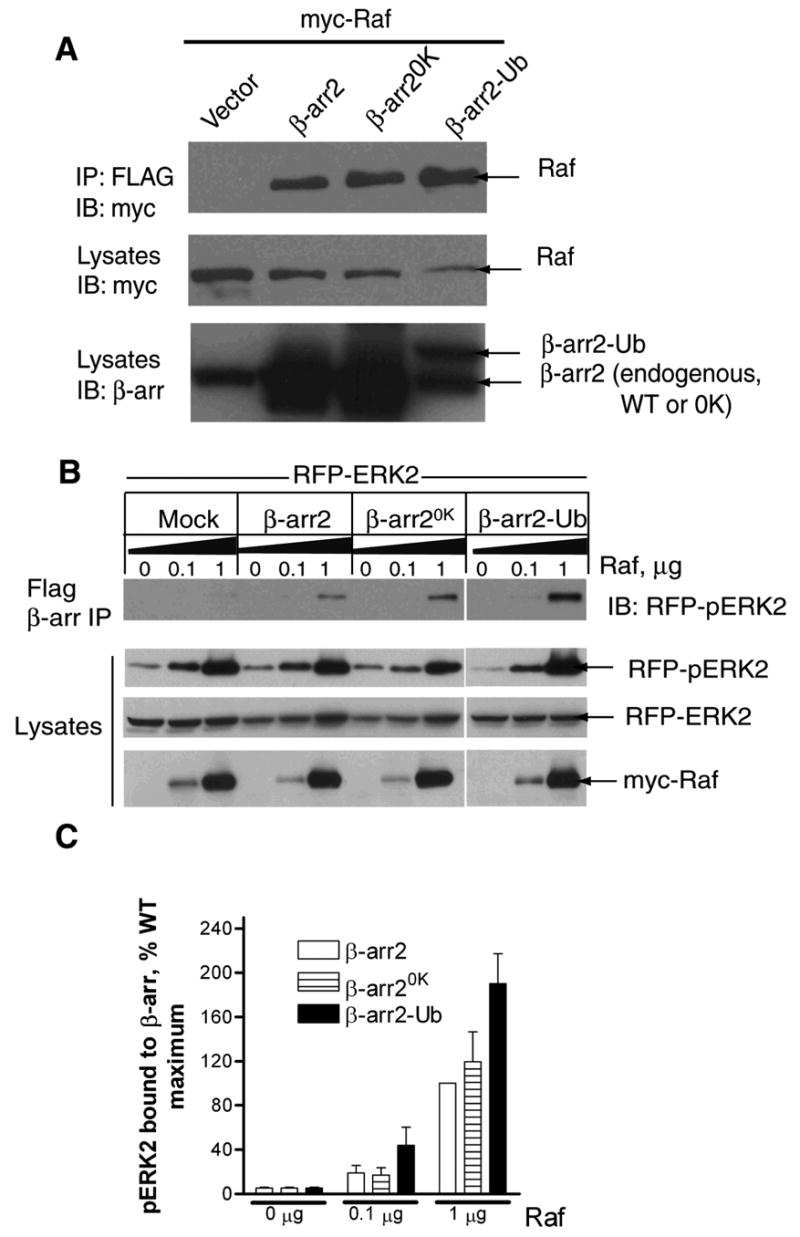
A. COS-7 cells were transfected with myc-cRaf1 along with either vector or the indicated β-arrestin2 plasmid. Anti-FLAG immunoprecipitates were probed with a monoclonal myc antibody (9E10) (top panel). The lysates were probed for the levels of transfected cRaf1 (middle panel) and β-arrestins (bottom panel) with respective antibodies. Blots are representative of four similar experiments. B. Cells were transiently transfected with RFP-ERK2 and increasing amounts of myc-cRaf1 with vector or indicated FLAG-β-arrestins. The amounts of phospho-RFP-ERK2 present in the FLAG immunoprecipitates (top panel) and lysates were determined by immunoblotting. The levels of unphosphorylated RFP-ERK2 as well as myc-cRaf1 in the same lysates are also shown. C. Bar graph depicts the amount of phospho-RFP-ERK2 in the FLAG immunoprecipitates. Data are presented in arbitrary units where the amount of maximum phospho-ERK2 present in FLAG-β-arrestin2 immunoprecipitates is defined as 100%. Data shown represent the mean ± SEM from three independent experiments.
Previous studies have also shown that by merely coexpressing β-arrestin2 with a MAPKKK (such as c-Raf, ASK-1) and a MAPK (such as ERK2, JNK3), robust activation of MAPK could be achieved (18,24,25). This property of β-arrestin2 is attributed to its capacity to simultaneously bind component enzymes of a kinase cascade thus bringing them in to proximity and allowing robust phosphorylation to occur. Accordingly, cotransfection of β-arrestin2, cRaf and GFP-ERK2 could enhance precipitation of phosphorylated ERK2 with FLAG-β-arrestin2 (18). To examine whether β-arrestin20K was capable of a similar function, we transfected COS-7 cells with either WT, β-arrestin20K or β-arrestin2-Ub along with RFP-ERK2 and increasing amounts of myc-cRaf-1. As shown in the Western blots (Fig 9B) and the bar graphs depicting quantification of pERK in β-arrestin2 precipitates (Fig 9C), β-arrestin20K could scaffold pERK to the same extent as the WT. Interestingly, β-arrestin2-Ub precipitates contained 60–80% more pERK than the WT suggesting either a greater level of kinase activation and/or a stronger interaction of β-arrestin2-Ub with pERK. All the above coimmunoprecipitation data suggest that ubiquitination is not required for β-arrestin’s interaction with c-Raf and pERK and that despite the 31 lysine mutations β-arrestin20K can interact with these β-arrestin partners.
Role of β-arrestin2 ubiquitination in the formation and subcellular targeting of receptor signalosomes
We next examined the effects of β-arrestin2, β-arrestin2-Ub, and β-arrestin20K on the assembly of receptor/β-arrestin2/ERK complexes. As depicted in Fig 10A, a significant amount of pERK was associated with β2AR immunoprecipitates from COS-7 cells upon coexpression of β-arrestin2-Ub. Lesser amounts of pERK were detected upon wild type β-arrestin2 expression, although this amount was still higher than the pERK detected with endogenous β-arrestin2 in the mock-transfected samples (Fig 10, A and B). Expression of β-arrestin20K resulted in a significant decrease in pERK in receptor complexes than what was obtained with endogenous β-arrestin2 as seen in the bar graph representing the quantification of signals from five independent experiments (Fig 10B). This decrease was not due to a decline in overall ERK activation since the level of activation in whole cell lysates was identical in all transfection conditions.
Fig. 10. Magnitude of ERK activity in receptor complexes correlates with β-arrestin ubiquitination.
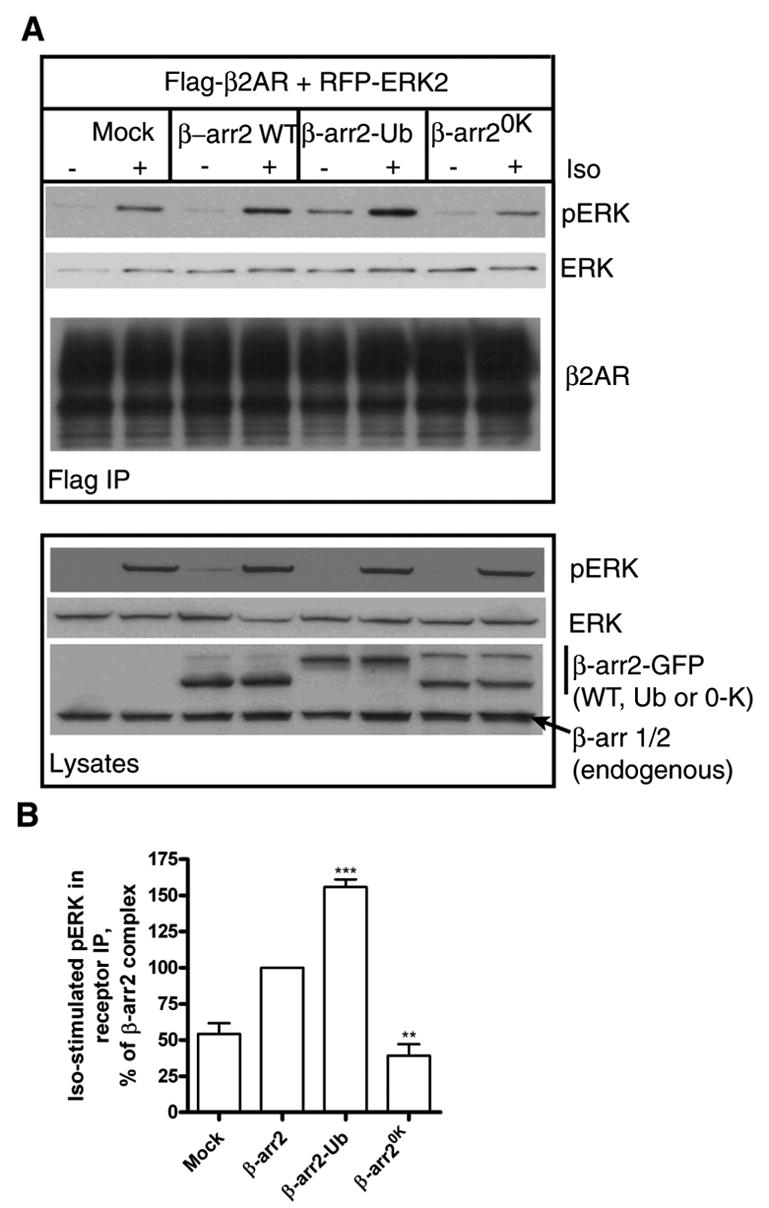
A. COS-7 cells were transfected with RFP-ERK2 over expressing FLAG-β2AR (A) along with either pEGFP vector, β-arrestin2-GFP, GFP-β-arrestin2-Ub, or β-arrestin20K-GFP. After 5 min of agonist stimulation, receptors were immunoprecipitated, separated on gels and probed for the amount of pERK content. The blots were reprobed for total ERK (second panel) followed by a second reprobe for the β2AR (third panel). The levels of RFP-pERK2, RFP-ERK2 and the different β-arrestins in the cell extracts are also shown (Lysates panel). B. Bar graphs depict the quantification of pERK coprecipitated with the agonist occupied receptor. Data represent mean value ± S.E. from five independent experiments. ***, β-arrestin2 versus β-arrestin2-Ub, p 0.0002; **, Mock versus β-arrestin20K p 0.003, as determined by paired t tests.
In general, β-arrestin-mediated ERK signals are retained in the cytosol and are prevented from entering the nucleus. To determine if β-arrestin ubiquitination plays a role in the subcellular localization of agonist-stimulated pERK, we performed confocal immunofluorescence microscopy and examined the relative distribution of agonist-activated receptors, β-arrestins and pERK. An antibody that specifically recognizes Thr202/Tyr204-phosphorylated ERK1/2 was employed to detect activated endogenous ERK. If ubiquitination of β-arrestin2 indeed plays a role in determining the spatial distribution of active ERK, then differences should be observed in the cellular distribution of pERK stimulated in the presence of β-arrestin2-Ub versus β-arrestin20K.
As depicted in Fig 11A, unstimulated cells show a uniform cytosolic distribution of β-arrestin2-GFP (green), a membrane distribution of HA-β2AR (blue) and a negligible amount of pERK (red). When the cells were stimulated for 5 minutes with isoproterenol, β-arrestin2 redistributed to the cell membrane to colocalize with the activated receptors. A robust increase in the level of pERK was observed in both cytosol and nucleus along with a clearly demarcated pERK signal on β-arrestin-studded cell membranes (2nd row, Fig 11A). After 30 minutes of isoproterenol, the β2ARs were visualized in intracellular vesicles. β-arrestins are not localized to these vesicular structures but are retained at the cell membrane. A small percentage of receptors persist at the membrane, which most likely represent recycled and/or non-internalized receptors. After 30 minutes of isoproterenol treatment, a negligible amount of pERK was detected (3rd row, Fig 11A). As a comparison, representative cells overexpressing both HA-β2AR and β-arrestin2-GFP treated with phorbol myristate acetate (PMA) are shown in the bottom row of Fig 11A. PMA stimulation leads to robust activation of ERK, which is distributed in both cytoplasm and nucleus. PMA stimulation does not lead to either β2AR internalization or β-arrestin2 translocation.
Fig 11. Subcellular distribution of 7TMR-stimulated pERK in HEK-293 cells.
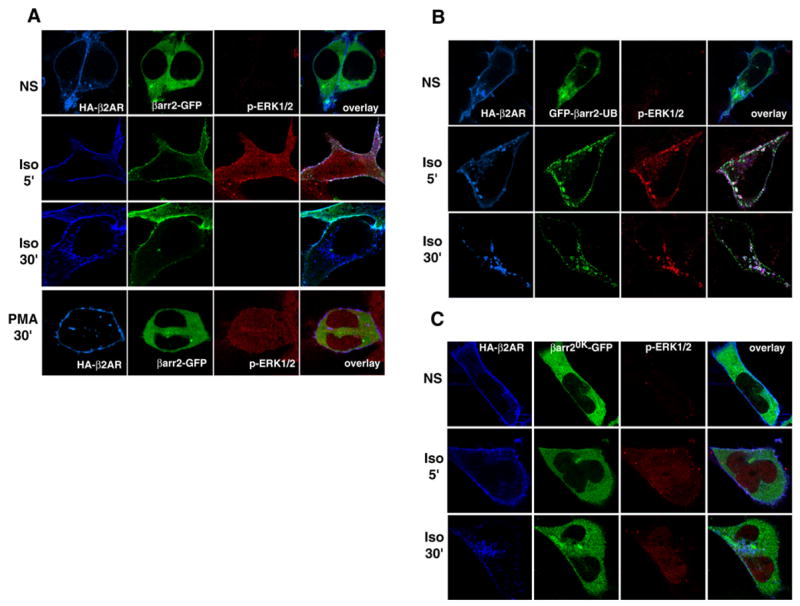
HEK-293 cells expressing HA-β2AR, along with either β-arrestin2-GFP (A), GFP-β-arrestin2-Ub (B) or β-arrestin20K-GFP (C) were stimulated with isoproterenol for 0, 5 or 30 minutes, or PMA for 30 min (A), fixed, permeabilized and labeled with a rabbit polyclonal anti-phospho-p44/42 MAPK antibody followed by Alexa633-conjugated secondary antibody. Following this receptors were labeled for HA epitope with the monoclonal antibody, 12CA5 followed by Alexa594 conjugated secondary antibody. Confocal images were collected using sequential line excitation filters (488, 568, and 633 nm) and emission filter sets at 505–550 nm for GFP detection (green), 585 nm for HA-β2AR (blue) and 650 nm for pERK (red) detection. Data represent similar results obtained from three independent experiments.
The results of similar experiments performed with GFP-β-arrestin2-Ub and HA-β2ARs are shown in Fig 11B. Under unstimulated conditions the sub-cellular distributions are identical to what is observed with the WT β-arrestin2. Quite strikingly, at 5-minute stimulation, a distinct and robust ERK activation is observed at the cell membrane coinciding with the distinct membrane recruitment of β-arrestin2-Ub. Although a majority of the cells (~80%) displayed such distribution at the cell membrane, some cells did have small vesicles in the vicinity of the cell membrane, which contained β-arrestin2-Ub, β2AR as well as pERK as shown in the figure panels (2nd row, Fig 11B). Surprisingly at the 5 minute time point, unlike the case of WT β-arrestin2 expression, little active ERK was distributed in the nucleus with β-arrestin2-Ub overexpression. We do not know the exact mechanism by which this occurs, but possibly, β-arrestin2-Ub can simultaneously promote β-arrestin-dependent cytosolic ERK and curb the G protein ERK pathway leading to lesser nuclear ERK. After 30 minutes of isoproterenol treatment, a dramatic redistribution of β-arrestin2-Ub, β2AR and pERK was seen in intracellular vesicles. These data clearly indicate that a stably ubiquitinated β-arrestin can remain associated with a ‘Class A’ receptor (i.e. β2AR) and target activated ERK to early endosomes resulting in a pool of pERK complexed with internalized receptors
In the absence of agonist, β-arrestin20K-GFP is mainly cytoplasmic, with HA-β2AR at the plasma membrane and very little active ERK (top row, Fig 11C). After 5 minutes of isoproterenol-stimulation, a robust activation of ERK occurs which is seen distributed in both cytoplasmic and nuclear compartments. However, none of this active ERK is localized with β-arrestin20K. Possibly, much of this activity is G protein mediated and is excluded from receptor complexes since less pERK is complexed with the β2AR in the presence of β-arrestin20K (see Fig 10). At 30 minutes, levels of pERK decreased but were not abolished (bottom row, Fig 11C). This situation contrasts with what is observed with the stably ubiquitinated β-arrestin2-Ub (Fig 11B), where pERK signals are stabilized and localized on endosomal vesicles at 30 min of isoproterenol stimulation. As seen in the 30 min panels of Fig 11C, β2AR internalized into endosomes which is consistent with our internalization data (Fig 7 AD), which indicate the inability of β-arrestin20K to inhibit receptor internalization.
We also determined the kinetics of ERK phosphorylation in HEK-293 cells expressing the β2AR (1 pmol per mg cellular protein) upon transfection of vector, β-arrestin2 WT, β-arrestin20K or β-arrestin2-Ub. As shown in Fig 12, expression of β-arrestin2-Ub significantly increased ERK activity at 20 min of isoproterenol treatment, β-arrestin2 led to a modest augmentation and β-arrestin20K had no effect over mock conditions (Fig 12B). Previous studies have demonstrated that, later ERK activity induced by 7TMRs is actually β-arrestin-mediated (reviewed in (26)). These results further support the idea that β-arrestin ubiquitination status underlies some aspects of β-arrestin-dependent signaling.
Fig 12. Effects of β-arrestin overexpression on the time course of isoproterenol-stimulated perk.
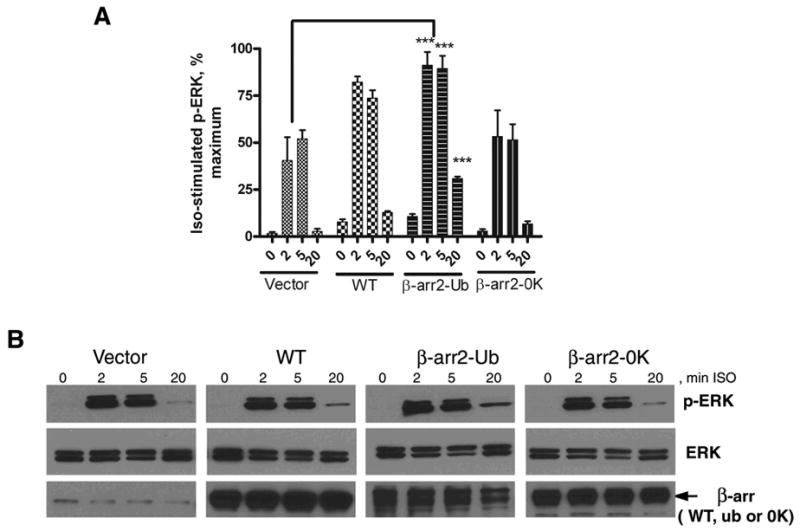
HEK-293 cells stably expressing the β2AR were transiently transfected with vector, β-arrestin2, β-arrestin2-Ub or β-arrestin20K. Monolayers of cells in 12-well dishes were stimulated with isoproterenol (10μM) for indicated times, and whole cell lysates were analyzed for ERK phosphorylation by Western blotting. Panel A shows bar graphs representing the quantification of pERK bands normalized to ERK levels obtained from 4–6 experiments. Maximal signal from an individual experiment was used as 100%. *** P<0.01, **P<0.05, Two way ANOVA, Bonferroni posttests, all compared to vector only condition. Panel B shows representative Western blots for phosphoERK in the top panels. The same blots were stripped (Restore stripping buffer, Pierce) probed for ERK1/2 levels (middle panels), and re-stripped and re-probed for β-arrestin levels (bottom panels).
Ubiquitination favors β-arrestin’s distribution in membrane compartments
The β-arrestin isoforms are mainly cytosolic proteins and are translocated to the plasma membrane upon 7TMR activation. Thus far no lipid modifications in β-arrestins favoring macromolecular membrane interactions have been identified. One well accepted mechanism that keeps them in a membrane environment is their binding to phosphorylated domains of receptors (5). Our current and previous results indicate that ubiquitination could be an important factor that determines the longevity of β-arrestin’s interactions with receptorsleading to colocalization on endosomal vesicles. Interestingly, when we analyzed the distribution of the ubiquitinated form of β-arrestin by sub-cellular fractionation, we found that the ubiquitination status of β-arrestin favors its partitioning to membrane fractions. When COS-7 cells expressing either β-arrestin2 or β-arrestin2-Ub were lysed in a detergent free low salt buffer (40 mM NaCl) and the soluble and insoluble fractions were further separated by differential centrifugation, nonubiquitinated β-arrestins were mainly cytosolic. Most of the exogenously expressed β-arrestin2 as well as YFP-β-arrestin2 were detectable in the soluble fraction (Fig 13). The YFP-β-arrestin2 band in the membrane fraction with a slightly slower mobility is unreactive to ubiquitin antibodies such as FK2, P4D1 and FK1 and its identity remains to be elucidated. On the other hand, ubiquitinated β-arrestin2 was distributed mostly in the insoluble membrane fractions (Fig 13 A and B). As seen in Fig 4B, β-arrestin2-Ub appears to be uniformly distributed in the cytosol in an undisturbed cell. Accordingly, the membrane fractionation of β-arrestin2-Ub is not due to its presence in inclusion bodies, but rather due to its affinity for membrane components. These results suggest that ubiquitination increases β-arrestin’s propensity for membrane association thus favoring β-arrestin’s prolonged localization in membrane microdomains. Although ubiquitination is dispensable for β-arrestin’s interactions with cytososlic partners, it may be necessary to facilitate the formation of functional 7TMR-β-arrestin endocytic and signaling complexes in a membrane environment.
Fig 13. Ubiquitinated β-arrestin is distributed in membrane fractions.
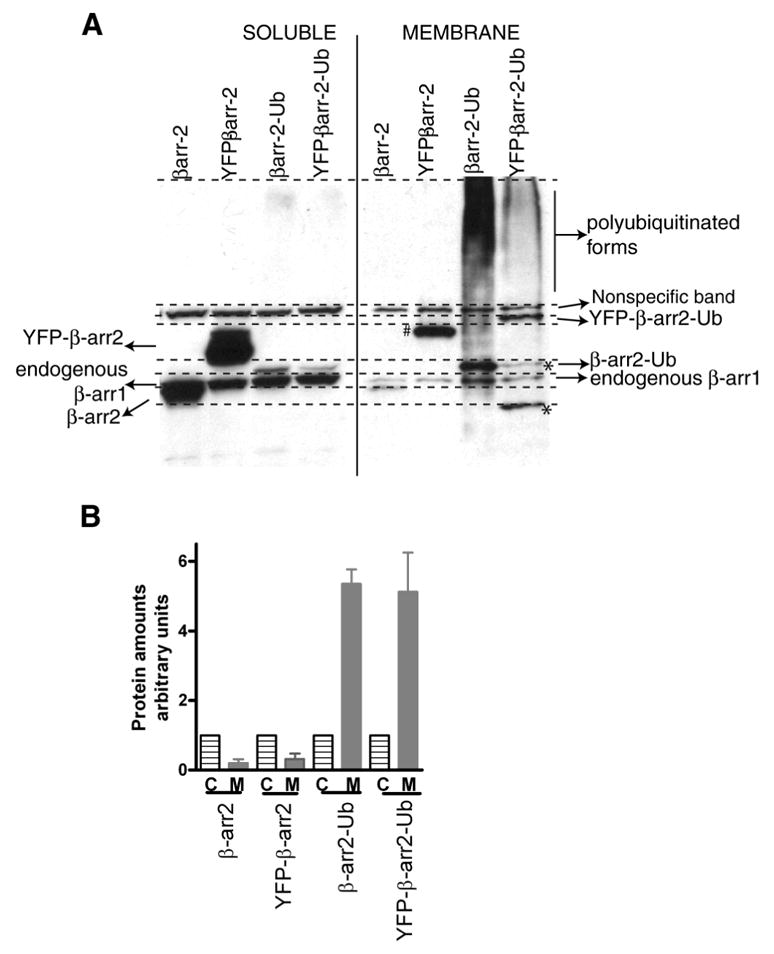
A. COS-7 cells were transfected with the indicated β-arrestin2 plasmids. 24 h post transfections, cells were collected in a detergent-free buffer, lysed by freeze thawing the samples and cytosolic and membrane fractions separated by differential centrifugation (see methods). Equal protein amount of each type of sample separated on a gradient gel was then immunoblotted with a β-arrestin antibody. The respective protein bands are indicated in the figure panels. # a modified form of YFP-β-arrestin2; * these bands are mostly due to the presence of internal methionines. B. Bar graphs depict quantification of bands representing respective populations (C, cytosolic, M, membrane) of the indicated β-arrestin types. In each case the cytosolic amount was arbitrarily assigned as ‘1’. The data represent mean ±SEM from three independent experiments.
DISCUSSION
β-arrestin1 and 2 are ubiquitously expressed proteins that function to desensitize G protein mediated signals, which arise upon the stimulation of 7TMRs (2,3). β-arrestins also bind to clathrin and AP2 and serve as endocytic adaptors for several 7TMRs to promote internalization via clathrin-coated vesicles (7–9). In addition, β-arrestin2 functions as a receptor-regulated scaffold for MAPK pathways, e.g. for JNK3 and ERK2 (18,25). Recently it has been demonstrated that β-arrestin2 can specifically initiate ERK pathways even when receptors do not couple to G proteins (17,26,27). Moreover, β-arrestin-dependent signaling plays important roles in diverse cellular processes including cell proliferation, membrane ruffling, chemotaxis as well as metastasis (28–31).
A consequence of agonist stimulation of several 7TMRs is the ubiquitination of β-arrestins, which is required for rapid receptor internalization (12). Ubiquitination is a post-translational modification that was originally described in the context of regulated destruction of many proteins by the proteasomal machinery (32). However, recent years have witnessed the discovery of a plethora of non-proteasomal roles of ubiquitin (33,34) and ubiquitination has been shown to play an important role in the lysosomal degradation of 7TMRs (35). Moreover monoubiquitination of adaptor proteins such as Eps15 and mono/multiubiquitination of cell surface receptors are implicated in endocytosis, whereas, polyubiquitination of the adaptor protein TRAF6 is suggested to be crucial for triggering NF-kappaB signaling pathways (36–38). β-arrestin functions as both an endocytic and a signaling adaptor for 7TMRs and bears a functional analogy to proteins such as Eps15 and TRAF6. Currently, the nature of β-arrestin2 ubiquitination as to being poly- or mono- ubiquitination is unknown. Nevertheless, β-arrestin ubiquitination facilitates both receptor internalization and MAPK activation.
The kinetics of β-arrestin ubiquitination and deubiquitination appear to determine the stability and duration of β-arrestin-receptor interactions, which in turn determine its trafficking pattern (13). Our studies indicate that stable β-arrestin-receptor interaction leading to cotrafficking of receptors and β-arrestins into endosomes not only results in sustained ubiquitination but also in the enhanced activation of ERK. β-arrestin ubiquitination also plays an important role in promoting receptor internalization (12,13). Are these cellular processes, namely, receptor internalization, β-arrestin trafficking, MAPK activation and β-arrestin ubiquitination independent or related events? Could β-arrestin ubiquitination serve as a locus of control for these various pathways? To understand the integration of ubiquitination in to the trafficking and signaling functions of β-arrestins, we sought to compare wild type β-arrestin2 with a non-ubiquitinated form as well as a stably ubiquitinated form. To generate β-arrestin2 totally defective in ubiquitination, we had to replace all the 31 lysines within β-arrestin2 with arginine residues. Sometimes it takes only one mutation to generate a completely misfolded protein. So it is a concern to study a protein with 31 lysine-arginine changes. However, we believe that the conservative nature of the introduced changes allowed β-arrestin20K to be functional in both in vitro assays as well as our protein-protein interaction studies. For the most part, β-arrestin20K behaved like the wild type β-arrestin2 since its binding to rβ2AR in vitro and to c-Raf and ERK was unchanged from the wild type. Interestingly, β-arrestin20K bound AP-2 much more robustly than the wild type, while its interactions with clathrin and the β2AR were impaired in vivo. Previous studies have shown that certain arginine residues in β-arrestin2 (R394 and R396) are involved in AP-2 binding (39). It is possible that by introducing 31 arginines in the place of lysines in β-arrestin20K we introduced additional AP-2 binding sites leading to a more robust interaction. While the AP-2 binding domain in the wild type β-arrestin2 is exposed only after a receptor induced conformational change, it is possible that for the β-arrestin20K some of the arginines could present an interaction domain constitutively.
Although β-arrestin20K is capable of equivalent protein-protein interactions with the β2AR as the nonubiquitinated wild type in vitro, in a cellular context it shows impairment in binding, since unlike the wild type, it can not be ubiquitinated at proper site(s). β-arrestin20K did not support internalization of either β2AR or V2R confirming that ubiquitination of β-arrestin is crucial for its role in promoting receptor endocytosis (Fig 7). Additionally, this mutant was only transiently recruited to the plasma membrane upon stimulation of the β2AR or the V2R (Fig 6). In contrast, a β-arrestin-Ub chimera which remains stably ubiquitinated, can enhance β2AR internalization (13) and is stably recruited to endosomal compartments with the β2AR (Fig 4B). Although both ubiquitinated and nonubiquitinated forms of β-arrestin can form complexes with pERK (Fig 9), only the ubiquitinated form is capable of this function in a receptor complex (Figs 10 and 11). In other words, β-arrestin ubiquitination plays a central role in stabilizing kinase activity in receptor signalosomes. However, whether β-arrestin ubiquitination acts as a “trigger mechanism” for activating the c-Raf-MEK1-ERK2 cascade remains to be determined. Thus far, TRAF6 autoubiquitination is the only example in which the adaptor protein ubiquitination initiates kinase signaling (38).
One important consequence of β-arrestin-dependent MAPK activation is the compartmentalization of the signals. Thus, AT1aR-stimulated pJNK3 scaffolded by β-arrestin2 is retained on endocytic vesicles (25). Similarly, AT1aR-stimulated ERK is concentrated on endosomes, which are also the destination for internalized receptor-β-arrestin complexes (18). As evidenced by our microscopy experiments this subcellular location of MAPKs is indeed directed by the ubiquitination status of β-arrestin2 (Fig 11 A–C). Thus, a stably ubiquitinated β-arrestin not only confers a ‘Class B’ trafficking pattern on a ‘Class A’ receptor, but also leads to changes in the compartmentalization of pERK (Fig 11B).
Our cell fractionation experiments indicate that ubiquitin moieties on β-arrestin somehow favor its membrane distribution. We currently do not know the exact mechanism by which ubiquitin chains favor membrane interactions that facilitate subsequent signalosome formation. It is possible that while β-arrestin’s concave domains interact with the phosphorylated domains of the receptor, ubiquitin chains elsewhere on β-arrestin help its retention in a membrane environment. Furthermore, β-arrestin’s ubiquitinated domains could favor a tighter interaction with c-Raf allowing robust kinase activation at the membrane. This activity is then passed down through the cascade leading to ERK phosphorylation within the β-arrestin scaffold.
The subcellular distribution of visual arrestins as well as β-arrestins is also influenced by their binding to various phosphoinositides (40,41). PIP2 and PIP3 binding facilitates plasma membrane recruitment of β-arrestins (42) while the soluble ligand IP6 regulates oligomerization of β-arrestin1 and β-arrestin2 as well as nuclear localization of β-arrestin1(43). Interestingly, mutation analyses of bovine β-arrestins as well as structural studies of β-arrestin1-IP6 co crystals indicate the presence of lysine residues within the phosphoinositide-binding domains in β-arrestin (42,43). Since our results indicate that β-arrestin ubiquitination favors its membrane interactions (Fig 13) it will be of great interest to determine if ubiquitination at distinct lysine(s) plays any role in β-arrestin oligomerization and/or modulates β-arrestin-phosphoinositide binding.
Our data suggests that β-arrestin and the ERK protein bound to it have to remain at the membrane for an optimal duration before receptor signalosomes are formed. Moreover, a subsequent stable interaction of receptor and β-arrestin is required for sustaining and targeting this activity to subcellular compartments. Interestingly, previous studies have shown that the membrane recruitment of β-arrestin2 itself can lead to ERK signaling and that a 7TMR-β-arrestin1 chimeric protein can act as a constitutive signalosome (44,45). Our findings indicate that β-arrestin ubiquitination controls not only receptor trafficking but the nature, stability and subcellular localization of active ERK signals. It seems overwhelmingly likely that this regulation also extends to the nature of the ERK substrates and hence of the cellular consequences of receptor mediated activation of ERK. Seen in this light β-arrestin ubiquitination may be viewed as the “glue” which holds the receptor “signalosome” together and directs the ultimate destination of its cellular journey. It will be of interest to determine which other signaling pathways may be regulated in this way.
Supplementary Material
Acknowledgments
We thank Vidya Venkataramanan for excellent technical help, Donna Addison and Elizabeth Hall for excellent secretarial assistance, Richard Premont for helpful suggestions, Xin Rong Jiang for purified β2AR and Darel Capel for purified GRK2. This work was supported by National Institutes of Health Grants HL080525 (to SKS), HL16037 (to RJL), and DK55524 (to LML) and the Research Service of the Department of Veterans Affairs. SKS is also supported by an American Heart Association grant 0530014N. R.J.L is an investigator with the Howard Hughes Medical Institute.
Footnotes
The abbreviations used are: 7TMR, seven transmembrane receptor; β2AR, beta2 adrenergic receptor; AP-2, adaptin protein subunit2; ERK1/2, extracellular signal regulated kinases 1 & 2; pERK, phosphorylated ERK; MAPK, mitogen activated protein kinase; Mdm2, mouse double minute2; RING, really interesting new gene, PMA, phorbol myristate acetate; Ub, ubiquitin;
References
- 1.Benovic JL, Kuhn H, Weyand I, Codina J, Caron MG, Lefkowitz RJ. Proc Natl Acad Sci U S A. 1987;84:8879–8882. doi: 10.1073/pnas.84.24.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 3.Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, Snyder SH, Caron MG, Lefkowitz RJ. J Biol Chem. 1992;267:17882–17890. [PubMed] [Google Scholar]
- 4.Lefkowitz RJ, Shenoy SK. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 5.Gurevich VV, Gurevich EV. Structure. 2003;11:1037–1042. doi: 10.1016/s0969-2126(03)00184-9. [DOI] [PubMed] [Google Scholar]
- 6.Lefkowitz RJ, Rajagopal K, Whalen EJ. Mol Cell. 2006;24:643–652. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 7.Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 8.Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG, Barak LS. Proc Natl Acad Sci U S A. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferguson SS, Downey WE, 3rd, Colapietro AM, Barak LS, Menard L, Caron MG. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- 10.Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. J Biol Chem. 2000;275:17201–17210. doi: 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- 11.Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 12.Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- 13.Shenoy SK, Lefkowitz RJ. J Biol Chem. 2003;278:14498–14506. doi: 10.1074/jbc.M209626200. [DOI] [PubMed] [Google Scholar]
- 14.Perroy J, Pontier S, Charest PG, Aubry M, Bouvier M. Nat Methods. 2004;1:203–208. doi: 10.1038/nmeth722. [DOI] [PubMed] [Google Scholar]
- 15.Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. J Biol Chem. 1999;274:32248–32257. doi: 10.1074/jbc.274.45.32248. [DOI] [PubMed] [Google Scholar]
- 16.Shenoy SK, Lefkowitz RJ. J Biol Chem. 2005;280:15315–15324. doi: 10.1074/jbc.M412418200. [DOI] [PubMed] [Google Scholar]
- 17.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. Proc Natl Acad Sci U S A. 2003;100:10782–10787. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Proc Natl Acad Sci U S A. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen MS, Obar RA, Schroeder CC, Austin TW, Poodry CA, Wadsworth SC, Vallee RB. Nature. 1991;351:583–586. doi: 10.1038/351583a0. [DOI] [PubMed] [Google Scholar]
- 20.van der Bliek AM, Redelmeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL. J Cell Biol. 1993;122:553–563. doi: 10.1083/jcb.122.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menard L, Ferguson SS, Zhang J, Lin FT, Lefkowitz RJ, Caron MG, Barak LS. Mol Pharmacol. 1997;51:800–808. [PubMed] [Google Scholar]
- 22.Krupnick JG, Santini F, Gagnon AW, Keen JH, Benovic JL. J Biol Chem. 1997;272:32507–32512. doi: 10.1074/jbc.272.51.32507. [DOI] [PubMed] [Google Scholar]
- 23.DeFea KA, Zalevsky J, Thoma MS, Dery O, Mullins RD, Bunnett NW. J Cell Biol. 2000;148:1267–1281. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller WE, McDonald PH, Cai SF, Field ME, Davis RJ, Lefkowitz RJ. J Biol Chem. 2001;276:27770–27777. doi: 10.1074/jbc.M102264200. [DOI] [PubMed] [Google Scholar]
- 25.McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ. Science. 2000;290:1574–1577. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 26.Dewire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 27.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 28.Dasgupta P, Rastogi S, Pillai S, Ordonez-Ercan D, Morris M, Haura E, Chellappan S. J Clin Invest. 2006;116:2208–2217. doi: 10.1172/JCI28164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gavard J, Gutkind JS. Nat Cell Biol. 2006;8:1223–1234. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 30.DeFea KA. Annu Rev Physiol. 2007;69:535–560. doi: 10.1146/annurev.physiol.69.022405.154804. [DOI] [PubMed] [Google Scholar]
- 31.Scott MG, Pierotti V, Storez H, Lindberg E, Thuret A, Muntaner O, Labbe-Jullie C, Pitcher JA, Marullo S. Mol Cell Biol. 2006;26:3432–3445. doi: 10.1128/MCB.26.9.3432-3445.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hershko A, Ciechanover A. Annu Rev Biochem. 1992;61:761–807. doi: 10.1146/annurev.bi.61.070192.003553. [DOI] [PubMed] [Google Scholar]
- 33.Welchman RL, Gordon C, Mayer RJ. Nat Rev Mol Cell Biol. 2005;6:599–609. doi: 10.1038/nrm1700. [DOI] [PubMed] [Google Scholar]
- 34.Mukhopadhyay D, Riezman H. Science. 2007;315:201–205. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- 35.Shenoy SK. Circ Res. 2007;100:1142–1154. doi: 10.1161/01.RES.0000261939.88744.5a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I. Nat Cell Biol. 2003;5:461–466. doi: 10.1038/ncb983. [DOI] [PubMed] [Google Scholar]
- 37.Polo S, Sigismund S, Faretta M, Guidi M, Capua MR, Bossi G, Chen H, De Camilli P, Di Fiore PP. Nature. 2002;416:451–455. doi: 10.1038/416451a. [DOI] [PubMed] [Google Scholar]
- 38.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 39.Laporte SA, Oakley RH, Holt JA, Barak LS, Caron MG. J Biol Chem. 2000;275:23120–23126. doi: 10.1074/jbc.M002581200. [DOI] [PubMed] [Google Scholar]
- 40.Moore CA, Milano SK, Benovic JL. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- 41.Lee SJ, Xu H, Kang LW, Amzel LM, Montell C. Neuron. 2003;39:121–132. doi: 10.1016/s0896-6273(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 42.Gaidarov I, Krupnick JG, Falck JR, Benovic JL, Keen JH. Embo J. 1999;18:871–881. doi: 10.1093/emboj/18.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Milano SK, Kim YM, Stefano FP, Benovic JL, Brenner C. J Biol Chem. 2006;281:9812–9823. doi: 10.1074/jbc.M512703200. [DOI] [PubMed] [Google Scholar]
- 44.Terrillon S, Bouvier M. Embo J. 2004;23:3950–3961. doi: 10.1038/sj.emboj.7600387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jafri F, El-Shewy HM, Lee MH, Kelly M, Luttrell DK, Luttrell LM. J Biol Chem. 2006;281:19346–19357. doi: 10.1074/jbc.M512643200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.