

Abstract
Small organic molecules, like quaternary ammonium compounds, have long been used to probe both the permeation and gating of voltage-dependent K+ channels. For most K+ channels, intracellularly applied quaternary ammonium (QA) compounds such as tetraethylammonium (TEA) and decyltriethylammonium (C10) behave primarily as open channel blockers: they can enter the channel only when it is open, and they must dissociate before the channel can close. In some cases, it is possible to force the channel to close with a QA blocker still bound, with the result that the blocker is “trapped.” Armstrong (J. Gen. Physiol. 58:413–437) found that at very negative voltages, squid axon K+ channels exhibited a slow phase of recovery from QA blockade consistent with such trapping. In our studies on the cloned Shaker channel, we find that wild-type channels can trap neither TEA nor C10, but channels with a point mutation in S6 can trap either compound very efficiently. The trapping occurs with very little change in the energetics of channel gating, suggesting that in these channels the gate may function as a trap door or hinged lid that occludes access from the intracellular solution to the blocker site and to the narrow ion-selective pore.
Keywords: potassium channels, tetraethylammonium compounds, ion channel gating, open channel blockade, use-dependent blockade
introduction
The best studied blockers of voltage-activated K+ channels are tetraethylammonium and its derivatives, collectively called quaternary ammonium (QA)1 compounds. Armstrong (1966, 1969, 1971) pioneered the use of QA compounds for the study of K+ channel permeation and gating. He discovered that tetraethylammonium (TEA) and its QA derivatives block the ionic current only after the channel has been open. Because dissociation of the blockers was made faster by increasing the external [K+], he proposed that QA compounds bind within the pore.
For the most part, QA compounds also prevent K+ channels from closing. Armstrong (1971) found that for squid axon K+ channels, the tail currents (indicative of K+ channel closing upon repolarization) are made slower by nonyltriethylammonium (C9) blockade, and they have a rising phase (“hook”). These kinetic changes are explained by assuming that most of the channels remain open until the blocker dissociates, and then they close to the original unblocked closed state. However, the blocker's ability to prevent closing is not absolute. Armstrong observed a slow component of recovery from blockade, that was more pronounced and slower at very negative voltages. Since very negative potentials are known to favor closing of the channel, he suggested that this slow recovery from blockade was due to some of the channels closing with a QA ion trapped inside the pore.
For cloned wild-type Shaker K+ channels, QA blockers behave much as they do in the squid axon channel, except that there is no indication of trapping. Decyltriethylammonium (C10) slows the closing of the channels upon repolarization (Choi et al., 1993). Moreover, at voltages where the channel open probability is significantly lower than one, TEA and C10 show a substantially reduced apparent affinity for the channels, consistent with a scheme In which these blockers bind only to the open state (Choi et al., 1993).
We have accidentally found a point mutation, I470C, that changes the Shaker K+ channel so that it can trap TEA and C10. This mutation is in the S6 membrane-spanning region of the channel, near the 469 site previously shown to influence the binding affinity of long-chain QAs (Choi et al., 1993). Because this mutant also does not C-type inactivate, we could easily measure the voltage dependence of gating of blocked channels. This voltage dependence was not very different from the gating in the absence of blocker, suggesting that the blockers bind to a site that does not itself change very much during gating, except that the site is covered by a trap door or hinged lid when the channel closes.
materials and methods
Mutagenesis and Expression
For channel expression we used the Shaker H4 K+ channel (Kamb et al., 1988) with the Δ6-46 mutation that removes N-type inactivation (Hoshi et al., 1990). The channel cDNA was subcloned into the GW1-CMV expression vector (British Biotechnology, Oxford, UK; Choi et al., 1991). In addition, our “wild-type” channel had the following background mutations: C301S, C308S, and T449V (Holmgren et al., 1996); all references to wild-type data obtained in this paper refer to this channel. The T449V mutation reduces the amount of C-type inactivation (López-Barneo et al., 1993); with the additional I470C mutation, no inactivation at all was apparent. The mutation I470C was made by polymerase chain reaction (Ausubel et al., 1996) and was confirmed by sequencing the entire subcloned 230-bp insert between amino acids 435 and 509.
The expression system used was human embryonic 293 cell line (HEK 293; Amer. Type Culture Collection, Rockville, MD). The channel expression plasmid (20 μg) was cotransfected with the πH3-CD8 plasmid (Seed and Aruffo, 1987) which expresses the α subunit of the human CD8 lymphocyte antigen. Cells expressing the CD8 antigen were identified visually by decoration with antibody-coated beads (Jurman et al., 1994).
Solutions and Electrophysiological Recordings
The internal solution contained (mM): 160 KCl, 1 EGTA, and 10 HEPES (pH 7.4). We omitted Mg2+ from our internal solution because we found it to be a voltage-dependent open channel blocker. The external solution contained (mM): 150 NaCl, 10 KCl, 3 CaCl2, 1 MgCl2, 10 HEPES, pH 7.4.
The electrophysiological recordings were performed from inside-out excised patches (Hamill et al., 1981) using the same conditions described earlier (Holmgren et al., 1996).
Stabilization Energy Calculation
The stabilization energy associated with a blocker was calculated from the apparent equilibrium constant for gating of blocked channels, relative to the apparent equilibrium constant for gating of unblocked channels: ΔGstab = RT ln [Kg(B)/Kg]. This was calculated at the voltage for which the data for both curves were most reliable (i.e., closest to 0.5). This voltage was −50 mV for C10 and −40 mV for TEA.
results
Kinetic Properties of Blockade by C10 in the I470C Mutant
Fig. 1 shows C10 blockade of wild-type and I470C mutant Shaker K+ channels. When applied to the intracellular side of wild-type channels, C10 produces a characteristic blockade that resembles fast inactivation (Choi et al., 1993; Fig. 1, top row). The exponential decline in the current represents the association of the blocker with the channels, which begins only after the channels start to open (Armstrong, 1971; Choi et al., 1993). Although there was 78% blockade by the end of the 60-ms pulse, there was complete recovery in the 10-s interval between the first and second pulses in the presence of blocker, as seen from the identical appearance of the two pulses. This is consistent with the previous finding that Shaker channels close only after C10 dissociates from the channel, so that the closed channels are not blocked.
Figure 1.

The Shaker I470C mutant shows use-dependent blockade by C10. Voltage clamp currents from wild-type and I470C mutant Shaker K+ channels, in response to depolarization from −90 mV to 0 mV. Pulses were separated by 10-sec intervals at −90 mV; the inside-out patch was perfused with 8 μM blocker beginning immediately after the control pulse. Steady-state control current was 650 pA (wild-type) and 1020 pA (470C).
The C10 blockade of the mutant I470C channels was quite different. There were some quantitative changes: the affinity was slightly higher and the rate of binding was slightly slower. The main change, though, was qualitative. There was almost no recovery from blockade between the first and second pulses in the presence of C10, though the steady-state level at the end of the two pulses is the same. This is a strong form of use dependence: there is little or no block when the channel is maintained in the closed state (before the first pulse), but once the channel is opened, the block becomes very severe and remains so.
Could this simply be due to very tight binding of C10 to the mutant? We measured the kinetics of C10 interaction with the open state by analyzing the rate and extent of the decay in the current at different concentrations of blocker (Murrell-Lagnado and Aldrich, 1993; Holmgren et al., 1996). As for the wild-type channels, C10 blockade of the mutant had bimolecular association kinetics: the on-rate was proportional to the concentration and the off-rate was independent of concentration (Fig. 2). At 0 mV, the association rate was 1.3 × 107 M−1 s−1 (compared to 3 × 107 M−1 s−1 for the wild type), and the dissociation rate was 6.9 s−1 (compared to 60 s−1 for the wild type). Clearly, these changes are not enough to account for the different behavior of the mutant. In particular, dissociation of the blocker between depolarizing pulses must be much slower than the ∼7 s−1 dissociation from the open state.
Figure 2.

Kinetics of C10 blockade of I470C mutant channels. The kinetics were determined from the relaxation time course (τ) and extent (f = Isteady-state/Ictrl), measured in the first pulse following application of blocker. We computed kassoc as (1 − f)/τ, and kdissoc as f/τ. Each point shows the mean and standard error of determinations on at least three patches. Fit lines have values for kassoc = (1.34 ± 0.03) × 107 M−1 s−1, kdissoc = 6.9 ± 0.2 s−1.
Trapping and Untrapping of C10
We considered the possibility that in the mutant channels, the activation gates might close in the presence of a blocking C10 molecule and trap the C10 in the channel. To test for trapping of the blocker in the closed state, we performed experiments like the one illustrated in Fig. 3. In this particular example, the patch was perfused with C10 and channels were activated by a 240-ms depolarization to 0 mV, which allowed roughly 75% of the channels to be blocked. Subsequently, we closed the channels at −90 mV, removed the blocker, and waited for 5 min at a holding potential of −90 mV. After waiting, we applied a long test pulse to 0 mV to assess the remaining blockade. At the beginning of this test pulse only 25% of the channels were rapidly activated. The remaining 75% were still blocked at the beginning of the test pulse, but they proceeded to recover with the characteristic open channel dissociation rate of roughly 6–7 s−1. In experiments performed with different concentrations of C10, we found that the magnitude of the fast component of the test pulse was always the same as the preceding level of steady-state block (data not shown). These results are consistent with complete trapping of blocker at −90 mV. When the experiment of Fig. 3 is done on wild-type channels, no trapping is seen (i.e., the test pulse is identical to the control; data not shown).
Figure 3.

Trapping of C10 in I470C mutant channels. Blocker was applied ∼25 s before the second depolarizing pulse and washed out immediately afterwards. All depolarizing pulses are to 0 mV from a holding potential of −90 mV. Steady-state current in the first pulse was 250 pA.
A simple kinetic model that would explain our results is: 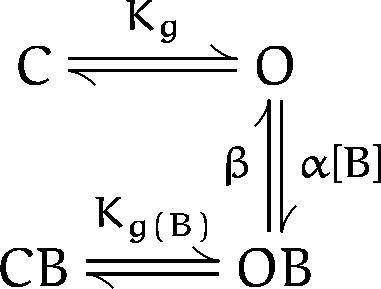
where C, O, OB, and CB represent different states of the channel: closed, open, open-blocked, and closed-blocked, respectively; Kg and Kg(B) are the effective equilibrium constants for gating in the absence or presence of blocker; B is the blocker (C10 in this case); and α and β are the association and dissociation rate constants, respectively. We assume that the C O and the CB OB transitions occur much faster than the blocking and unblocking reactions. Therefore, when the membrane was repolarized to −90 mV in the presence of C10 (Fig. 3), all of the channels closed: the 75% in the OB state closed to the CB state, while the 25% remaining in the O state closed to the C state. After removing the blocker and reopening the channels, the fast component comes from channels that open rapidly from C to O, while the slow recovery corresponds to channels that move rapidly from CB to OB but then recover slowly to the O state. In support of the assumption that the CB to OB transition is fast compared with blocker dissociation (OB to O), we find that the time course of the slow recovery matches the open state dissociation rate measured in Fig. 2.
In this model, because the blocker can dissociate only when the gate is open (i.e., from the OB state), the rate of recovery from block (βapp) should be proportional to the open probability of the blocked channels:
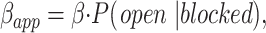 |
where P(open|blocked) is the conditional probability that a channel is open given that it is blocked (equal to P OB/(P OB + P CB)). This allows us to measure the voltage-dependent gating of blocked channels by measuring βapp at different voltages (Miller et al., 1987). We measured βapp in two ways, depending on the voltage. At more positive voltages where we could measure a significant K current, we monitored the dissociation continuously from the rise of the current after removing blocker, as in the third pulse of Fig 3. For more negative voltages, we first removed the blocker (maintaining the voltage at −90 mV to prevent escape) and then held at the test voltage and applied very brief pulses to +60 mV to monitor the time course of recovery from blockade (Fig. 4).
Figure 4.

Method for measuring the apparent dissociation rate (βapp) of C10 at more negative voltages. After C10 was applied and washed out, the voltage was held at the test potential, with brief (1 ms) pulses to +60 mV to monitor recovery (voltage and perfusion protocol shown in bottom panel). The 1-ms pulses were followed by hyperpolarization to −120 mV for 4 ms, in order to close the channels quickly. The dotted line shows the steady-state level of blockade achieved during the prepulse. Rates obtained by this method were corrected for recovery during the test pulses by subtracting the rate measured at −90 mV (middle panel).
The voltage dependence of βapp is plotted in Fig. 5, together with the relative open probability of unblocked channels as measured from tail currents. The solid line through the values of βapp was obtained by fitting the data to a Boltzmann function. The midpoint (V 1/2) of the CB OB transition was ∼10 mV more negative than the V 1/2 of the C O transition. After Miller et al. (1987), we define a “stabilization energy” for the blocker effect on gating, equal to RT ln [Kg(B)/Kg]. For C10 we found that the blocker stabilized the open state by a rather small energy of ∼1.2 kcal/mol.
Figure 5.

Voltage dependence of C10 escape from trapping. The apparent dissociation rate (βapp) of C10 was measured from the rate of current increase, as in the final pulse of Fig. 3, or from test pulses applied at various times after blocker washout, as in Fig. 4. For comparison, the normalized conductance-voltage (g-V) relationship is shown, from measurements of initial tail currents at −50 mV after 20-ms pulses to various voltages. The horizontal error bar shows the standard deviation of the midpoint of the g-V, measured from five different cells. All individual points give the mean and standard error of at least three determinations. The βapp data were fit by the function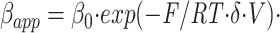
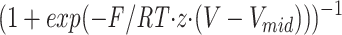 with values of β0 = 6.3 ± 0.1 s−1; z = 4.9 ± 0.5; δ = 0.15 ± 0.02; and Vm
id = −58 ± 0.8 mV. The normalized g-V was fit by the function
with values of β0 = 6.3 ± 0.1 s−1; z = 4.9 ± 0.5; δ = 0.15 ± 0.02; and Vm
id = −58 ± 0.8 mV. The normalized g-V was fit by the function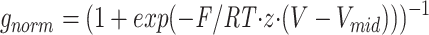 with values of z = 5.1 ± 0.1 and Vmid = −48 ± 0.2 mV.
with values of z = 5.1 ± 0.1 and Vmid = −48 ± 0.2 mV.
Because C10 is positively charged, its intrinsic dissociation rate (β) should depend on voltage if the binding site is within the membrane field. This voltage dependence of β was apparent in our data as the shallow decline in dissociation rate at positive voltages. For this reason, the data were actually fit using the product of the Boltzmann function for gating and an exponential function that describes the intrinsic voltage dependence of C10 dissociation. From the fit, the estimate of the effective electrical distance for the dissociation rate was 15% of the membrane field, which seems reasonable given that on wild-type Shaker K+ channels, C10 binds at a site ∼20% into the membrane field from the intracellular side (data not shown; similar to Choi et al., 1993).2
Trapping and Untrapping of TEA
The characteristics of blockade by TEA were also altered in the mutant and showed evidence of trapping. TEA blockade of wild-type channels is fast enough to appear as an instantaneous reduction in current (Fig. 6 A, top), but in the mutant, it is slow enough to produce a relaxation after channel opening like that seen for C10 (Fig. 6 A, bottom). The kinetics of the relaxation were consistent with a bimolecular association reaction (Fig. 6 B).
Figure 6.

TEA blockade of the I470C mutant. (A) Use-dependent blockade by TEA, measured as in Fig. 1. Wild-type with 0.6 mM TEA, and mutant with 0.5 mM TEA. Steady-state control current was 600 pA (wild-type) and 1700 pA (470C). (B) Kinetics of TEA blockade of mutant channels, measured from the relaxation, as in Fig. 2. Each point is the mean and range of two determinations. Fit lines have values for kassoc = (8.3 ± 0.1) × 105 M−1 s−1, kdissoc = 124 ± 4 s−1.
As with C10, there was a substantial difference in peak current between the first and second pulse in the presence of TEA (Fig. 6 A), consistent with the possibility that the channels could trap TEA. As seen in Fig. 7, trapping of TEA at −90 mV was very efficient. Even 5 min after removal of TEA, significant blockade remained. There is a small fast rising phase of the current, corresponding to unblocked channels, but ∼75% of the current appears with a slower time constant corresponding to blocker dissociation.
Figure 7.

Trapping of TEA in I470C mutant channels. TEA (0.2 mM) was applied about 25 s before the second depolarizing pulse, and washed out immediately afterwards. All depolarizing pulses are to +80 mV from a holding potential of −90 mV; the large depolarization improves the kinetic separation between channel opening and recovery from blockade. Steady-state blockade in the middle pulse is ∼28% of the control current. The recovery in the final pulse was well fit by the normalized two exponential function I(t) = 1 − A 1 · exp(−t/τ1) − (1 − A 1) · exp(−t/τ2), with values A 1 = 0.24 ± 0.01, τ1 = 0.48 (constrained to the rising phase of the control pulse), and τ2 = 3.8 ± 0.03. Steady-state current in the first pulse was 775 pA.
Systematic study of the voltage dependence of the recovery from TEA blockade showed that TEA produces a slight equilibrium stabilization of the closed state. This was determined using the same approach described above for C10. We measured the voltage dependence of recovery for TEA (βapp) as an indication of the gating of blocked channels (Fig. 8). This was shifted to the right by ∼12 mV compared with the voltage dependence of unblocked channel gating. This effect corresponds to a stabilization energy of ∼1.6 kcal/mol, with TEA favoring the closed state. No intrinsic voltage dependence of TEA dissociation was apparent in the voltage range explored.
Figure 8.

Voltage dependence of TEA escape from trapping. The apparent dissociation rate (βapp) of TEA was measured as for Fig. 5, except that for more negative voltages, a true two-pulse protocol, like in Fig. 7, was used. The g-V (same data as in Fig. 4) is shown for comparison. There was apparently no intrinsic voltage dependence for TEA dissociation; the dissociation rate data were fit by a Boltzmann function with z = 6.9 ± 1.5 and Vmid = −37 ± 1 mV.
discussion
Trapping of TEA and C10 by the I470C Mutant Channel
We found that in the I470C mutant of the Shaker K+ channel, TEA and C10 blocked current with slightly slower kinetics and slightly higher affinity than in the wild-type channels. Surprisingly, we observed a use- dependent blockade that arose from the trapping of the QAs when the channels were closed. Three features of the I470C mutant channel facilitated the study of this phenomenon: (1) high level expression of the channel, which permitted us to record macroscopic currents from inside-out excised patches that could be rapidly perfused; (2) complete trapping of the blockers in the closed channels, which simplified the kinetic scheme; and (3) the complete absence of C-type inactivation, which greatly simplified the experimental protocols needed to estimate the untrapping rates.
We found that TEA and C10 were trapped in the channels with little energetic perturbation of the C O transition. A molecule of C10 bound to the channel stabilized the open conformation by ∼1.2 kcal/mol, while a TEA molecule stabilized the closed conformation by ∼1.6 kcal/mol. These small and opposite effects of trapped organic blockers on the energetics of gating lead us to believe that gating mostly changes the accessibility of the blocker binding site, with very little change of the site itself. In other words, we suspect (as originally pictured by Armstrong in 1971) that the blockers bind in a relatively static intracellular vestibule that leads to the narrow ion-selective part of the pore, and that the activation gate acts like a trap door to prevent access between this vestibule and the intracellular solution (Fig. 9 A). Trapping can occur because behind the closed gate there is a cavity, which is probably filled with water in the normal closed state but can accommodate a blocker molecule. Structural studies of a mutagenized model protein (cytochrome c peroxidase) show an artificially enlarged, water-filled cavity at the active site (Fitzgerald et al., 1996). Access to this cavity is controlled by a hinged lid that can close and trap small ligands, although large ligands show a “foot in the door” effect and hold the lid open.
Figure 9.

Cartoon of two types of gating motions. (A) Trap door motion, in which a lid closes over a static binding site. A cavity in the closed state accommodates the blocker. (B) Collapsing motion, in which the binding site normally disappears in the closed state. Closing in the presence of a blocker requires distortion of the normal closed conformation in order to accommodate the blocker.
An alternative model, that trapping occurs when the closing channel “squeezes tightly” or collapses around the blocker (Fig. 9 B), seems compatible with Ba2+ trapping seen in other channels (Miller et al., 1987; Grissmer and Cahalan, 1989), because Ba2+ has the approximate dimensions of K+. It seems less likely for the trapping of relatively large organic blockers like TEA and C10, particularly because we observe little or no energetic penalty for closing in the presence of a blocker. Nevertheless we cannot completely rule this model out. The net energetic effect of a bound blocker on channel gating is necessarily the sum of unfavorable and favorable effects on the conformational change. That is, a blocker might sterically hinder the movement of the protein to the normal closed position and force it to adopt an energetically less favorable conformation; on the other hand, that less favorable closed conformation might have a better interaction with the blocker than the open state, which would tend to stabilize the modified closed state. Therefore, even though the net effect of a blocker on closing is very small (on the order of a single hydrogen bond), we cannot be completely confident that there is no change in the binding site upon closing.
Comparison with Trapping in Other Channels
In the squid axon delayed rectifier K+ channel, Armstrong (1971) found that a long-chain QA compound, C9, could be trapped at very negative voltages. Even at these voltages trapping was very inefficient, suggesting that C9 made it much more difficult for the channel to close (unlike the case studied here). Smaller blockers may be trapped more efficiently in the squid channel: John Clay (1995) found that internally applied TEA did not change the time course of channel closing in squid axon K+ channels. This is consistent with the possibility that TEA is trapped by the channels, although there is no evidence for trapping because TEA kinetics are so fast.
Although trapping was discovered in K+ channels using quaternary ammonium derivatives, the process is not exclusive to QA compounds or to K+ channels. Trapping has been observed in Na+ channels for local anesthetics (Strichartz, 1973; Hille, 1977; Ragsdale et al., 1994; Qu et al., 1995) and for disopyramide analogues (in rabbit myocytes; Carmeliet, 1988). Nicotinic acetylcholine–activated channels at the neuromuscular junction can trap chlorisondamine (Neely and Lingle, 1986). In various voltage-activated K+ channels, trapping has been shown for antiarrhythmic drugs (in rabbit myocytes; Carmeliet, 1992, 1993) and for aminopyridine compounds (Wagoner and Oxford, 1990; Choquet and Korn, 1992; Kirsch and Drewe, 1993). Divalent ions are also known to be trapped inside K+ channels (Eaton and Brodwick, 1980; Armstrong et al., 1982; Miller, 1987; Miller et al., 1987; Grissmer and Cahalan, 1989).
Ba2+ ions can block squid axon delayed rectifier K+ channels from either the extracellular or the intracellular side (Eaton and Brodwick, 1980; Armstrong and Taylor, 1980; Armstrong et al., 1982), and Ba2+ seems to reach the same site regardless of which side it is applied to. Opening the activation gates apparently increases access of Ba2+ to the pore from the intracellular side, while extracellular Ba2+ can enter the channel even in the closed state (Armstrong et al., 1982). This result argues that the activation gate is only at the intracellular side (but compare Miller et al., 1987 and Grissmer and Cahalan, 1989, both of whom found that Ba2+ could not enter from the external side in the closed state and therefore concluded that the pore is gated at both ends).
The most detailed kinetic study of trapping is for Ba2+ ions in Ca2+-activated K+ channels (Miller, 1987; Miller et al., 1987; Neyton and Pelleschi, 1991). Miller and colleagues (1987) found that internally applied Ba2+ could be trapped by closing of the channels. They measured the probability of Ba2+ escape from a single channel as a function of the holding voltage, and thus inferred (as we have here) the open probability of a blocked channel. They found that a bound Ba2+ ion stabilized the open conformation by ∼1.5 kcal/mol. This effect of Ba2+ is highly dependent on the identity and concentration of the monovalent ion bathing the extracellular face of the channel (Neyton and Pelleschi, 1991). We have not extensively studied the effect of external monovalents on the TEA and C10 effects on the Shaker I470C mutant channels, but we did not observe a difference between 10 mM external K+ and zero external K+.
Other Forms of Use-dependent Blockade in Shaker K+ Channels
Baukrowitz and Yellen (1996) showed that C10 produces a use-dependent blockade in Shaker channels that was very dependent on external [K+]. In that case, use dependence does not occur through blocker trapping, but rather by a different mechanism in which the blocker promotes the intrinsic C-type inactivation gating process. Because C-type inactivation and recovery is slow compared with blockade, both the peak and steady-state current in each pulse get progressively smaller. For the channels in this study, C-type inactivation was reduced by the 449V mutation and further reduced by the I470C mutation, and it plays no part in the use dependence observed here, which arises from trapping.
Mechanism of the Mutation's Effect
We do not know why the I470C mutation permits the Shaker K+ channel to trap QA blockers. Perhaps the mutation of four amino acids in the pore (one per subunit) from isoleucine to cysteine, which has a smaller side chain, makes extra room that allows the blocker to be retained in the closed state without interfering with the closing of the door. We wondered whether this change might also allow the blockers to bind more deeply in the channel than they ordinarily do, but we found no evidence for a change in the voltage dependence of blocker binding (data not shown).
Pharmacological Implications
In any case, the fact that a point mutation can change this channel from one that does not trap blockers to one that traps them effortlessly is quite remarkable. It suggests that the difference between channels that do or do not trap blockers is not a major architectural difference, but instead it requires only minor remodeling. Furthermore, it suggests that there are likely to be substantial differences between channel subtypes in their ability to trap blockers; even within a channel subtype, there could be significant species differences. Because blocker trapping has important consequences for the use-dependent behavior of therapeutic channel blockers, it may be important to determine this property for the precise human molecular target of a drug, rather than using an animal homologue.
Acknowledgments
We thank Mark Jurman for preparing the mutant and for a steady supply of transfected cells, and Dr. Yi Liu for helpful discussions.
This work was supported by a Muscular Dystrophy Association postdoctoral fellowship (to M. Holmgren) and by National Institutes of Health grant NS29693 (to G. Yellen).
Footnotes
The effective electrical distance for the dissociation rate corresponds to the difference in electrical potential between the C10 binding site and the position of the transition state for binding/unbinding of C10. This should be smaller than the effective electrical distance for equilibrium binding of C10, which corresponds to the difference in potential between the binding site and the intracellular solution.
Abbreviations used in this paper: C9, nonyltriethylammonium; C10, decyltriethylammonium; QA, quaternary ammonium; TEA, tetraeth-ylammonium.
references
- Armstrong CM. Time course of TEA+-induced anomalous rectification in squid giant axons. J Gen Physiol. 1966;50:491–503. doi: 10.1085/jgp.50.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM. Inactivation of the potassium conductance and related phenomena caused by quaternary ammonium ion injection in squid axons. J Gen Physiol. 1969;54:553–575. doi: 10.1085/jgp.54.5.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axon. J Gen Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Swenson RP, Taylor SR. Block of squid axon channels by internally and externally applied barium ions. J Gen Physiol. 1982;80:663–682. doi: 10.1085/jgp.80.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Taylor SR. Interaction of barium ions with potassium channels in squid giant axons. Biophys J. 1980;30:473–488. doi: 10.1016/S0006-3495(80)85108-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel, F.M., R. Brent, R.E. Kingston, D.D. Moore, J.G. Seidman, J.A. Smith, and K. Struhl. 1996. Current Protocols in Molecular Biology. Wiley, New York. Section 8.5.
- Baukrowitz T, Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+channel. Science (Wash DC) 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. Activation block and trapping of penticainide, a disopyramide analogue, in the Na+channel of rabbit cardiac Purkinje fibers. Circulation Res. 1988;63:50–60. doi: 10.1161/01.res.63.1.50. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. Voltage- and time-dependent block of the delayed K+current in cardiac myocytes by dofetilide. J Pharmacol Exp Ther. 1992;262:809–817. [PubMed] [Google Scholar]
- Carmeliet E. Use-dependent block and use-dependent unblock of the delayed rectifier K+current by almokalant in rabbit ventricular myocytes. Circ Res. 1993;73:857–868. doi: 10.1161/01.res.73.5.857. [DOI] [PubMed] [Google Scholar]
- Choi KL, Aldrich RW, Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+channels. Proc Natl Acad Sci USA. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KL, Mossman C, Aubé J, Yellen G. The internal quaternary ammonium receptor site of Shaker potassium channels. Neuron. 1993;10:533–541. doi: 10.1016/0896-6273(93)90340-w. [DOI] [PubMed] [Google Scholar]
- Choquet D, Korn H. Mechanism of 4-aminopyridine action on voltage-gated potassium channels in lymphocytes. J Gen Physiol. 1992;99:217–240. doi: 10.1085/jgp.99.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay JR. Quaternary ammonium ion blockade of IKin nerve axons revisited. Open channel block vs. state independent block. J Membr Biol. 1995;147:23–34. doi: 10.1007/BF00235395. [DOI] [PubMed] [Google Scholar]
- Eaton DC, Brodwick MS. Effects of barium on the potassium conductance of squid axon. J Gen Physiol. 1980;75:727–750. doi: 10.1085/jgp.75.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald MM, Musah RA, McRee DE, Goodin DB. A ligand-gated, hinged loop rearrangement opens a channel to a buried artificial protein cavity. Nature Structural Biology. 1996;3:626–631. doi: 10.1038/nsb0796-626. [DOI] [PubMed] [Google Scholar]
- Grissmer S, Cahalan MD. Divalent ion trapping inside potassium channels of human T lymphocytes. J Gen Physiol. 1989;93:609–630. doi: 10.1085/jgp.93.4.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor interaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren M, Jurman ME, Yellen G. Structure and function of the S4-S5 loop of the Shaker K+channel examined through cysteine mutagenesis and chemical modification. J Gen Physiol. 1996;108:195–206. doi: 10.1085/jgp.108.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science (Wash DC) 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Jurman ME, Boland LM, Liu Y, Yellen G. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. BioTechniques. 1994;17:876–881. [PubMed] [Google Scholar]
- Kamb A, Tseng-Crank JCL, Tanouye MA. Multiple products of the Drosophila Shaker gene may contribute to potassium channel diversity. Neuron. 1988;1:421–430. doi: 10.1016/0896-6273(88)90192-4. [DOI] [PubMed] [Google Scholar]
- Kirsch GE, Drewe JA. Gating-dependent mechanism of 4-aminopyridine block in two related potassium channels. J Gen Physiol. 1993;102:797–816. doi: 10.1085/jgp.102.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors and Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Miller C. Trapping single ions inside single ion channels. Biophys J. 1987;52:123–126. doi: 10.1016/S0006-3495(87)83196-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, Latorre R, Reisin I. Coupling of voltage- dependent gating and Ba++ block in the high-conductance, Ca++-activated K+channel. J Gen Physiol. 1987;90:427–449. doi: 10.1085/jgp.90.3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell-Lagnado RD, Aldrich RW. Interactions of amino terminal domains of Shaker K channels with a pore blocking site studied with synthetic peptides. J Gen Physiol. 1993;102:949–975. doi: 10.1085/jgp.102.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely A, Lingle CJ. Trapping of an open-channel blocker at the frog neuromuscular acetylcholine channel. Biophys J. 1986;50:981–986. doi: 10.1016/S0006-3495(86)83538-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyton J, Pelleschi M. Multi-ion occupancy alters gating in high-conductance, Ca2+-activated K+channels. J Gen Physiol. 1991;97:641–665. doi: 10.1085/jgp.97.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu YS, Rogers J, Tanada T, Scheuer T, Catterall WA. Molecular determinants of drug access to the receptor site for antiarrhythmic drugs in the cardiac Na+channel. Proc Natl Acad Sci USA. 1995;92:11839–11843. doi: 10.1073/pnas.92.25.11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+channels by local anesthetics. Science (Wash DC) 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Seed B, Aruffo A. Molecular cloning of the CD2 antigen, the T-cell erythrocyte receptor, by a rapid immunoselection procedure. Proc Natl Acad Sci USA. 1987;84:3365–3369. doi: 10.1073/pnas.84.10.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strichartz GR. The inhibition of sodium currents in myelinated nerve by quaternary derivatives of lidocaine. J Gen Physiol. 1973;62:37–57. doi: 10.1085/jgp.62.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagoner PK, Oxford GS. Aminopyridines block an inactivating potassium current having slow recovery kinetics. Biophys J. 1990;58:1481–1489. doi: 10.1016/S0006-3495(90)82493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]