

Abstract
The physiological ligands for Na,K-ATPase (the Na,K-pump) are ions, and electrostatic forces, that could be revealed by their ionic strength dependence, are therefore expected to be important for their reaction with the enzyme. We found that the affinities for ADP3−, eosin2−, p-nitrophenylphosphate, and Vmax for Na,K-ATPase and K+-activated p-nitrophenylphosphatase activity, were all decreased by increasing salt concentration and by specific anions. Equilibrium binding of ADP was measured at 0–0.5 M of NaCl, Na2SO4, and NaNO3 and in 0.1 M Na-acetate, NaSCN, and NaClO4. The apparent affinity for ADP decreased up to 30 times. At equal ionic strength, I, the ranking of the salt effect was NaCl ≈ Na2SO4 ≈ Na-acetate < NaNO3 < NaSCN < NaClO4. We treated the influence of NaCl and Na2SO4 on K diss for E·ADP as a “pure” ionic strength effect. It is quantitatively simulated by a model where the binding site and ADP are point charges, and where their activity coefficients are related to I by the limiting law of Debye and Hückel. The estimated net charge at the binding site of the enzyme was about +1. Eosin binding followed the same model. The NO3 − effect was compatible with competitive binding of NO3 − and ADP in addition to the general I-effect. K diss for E·NO3 was ∼32 mM. Analysis of Vmax/K m for Na,K-ATPase and K+-p-nitrophenylphosphatase activity shows that electrostatic forces are important for the binding of p-nitrophenylphosphate but not for the catalytic effect of ATP on the low affinity site. The net charge at the p-nitrophenylphosphate-binding site was also about +1. The results reported here indicate that the reversible interactions between ions and Na,K-ATPase can be grouped according to either simple Debye-Hückel behavior or to specific anion or cation interactions with the enzyme.
Keywords: Debye-Hückel theory, eosin binding, K+-phosphatase activity, protein electrostatics, nucleotide binding
introduction
Na,K-ATPase, identical to the Na,K-pump, catalyzes an Na+ + K+-activated hydrolysis of ATP to ADP and inorganic phosphate, Pi. In plasma membranes this is manifested in the coupling of the chemical hydrolysis of ATP to the vectorial transport of Na+ out of, and K+ into the cell.
The present paper focuses on the importance of electrostatic effects for Na,K-ATPase, as manifested by the enzyme's response to changes in the salt concentration and to the anion composition of the medium surrounding the membrane bound enzyme. Usually experiments with Na,K-ATPase are performed using chloride salts of the cations Na+, K+ and Mg2+, and care is generally taken to keep the ionic strength constant as it is well known that changes in this parameter affects most of the biochemical reactions of the system. Given the ionic nature of the transport ligands Na+ and K+, the substrates Mg2+ and ATP4− and the products ADP3− and HPO4 2−/H2PO4 − it is highly probable that electrostatic phenomena play a significant role in the Na,K-ATPase reaction. Generally, electrostatic effects in proteins are important for structure as well as stability and biological function, and, especially in enzymes, they play a decisive role in substrate recognition and transition-state stabilization (Warshel and Russell, 1984; Matthew, 1985; Allewell and Oberoi, 1991). An important manifestation of electrostatic interactions is the ionic strength dependence (García-Moreno E, 1994), and by analogy with a large variety of other systems (see discussion for examples of enzymes; Record et al., 1978; McLaughlin, 1989; Green and Andersen, 1991) this can be exploited to shed light on structure and reaction mechanism. Surprisingly, however, no systematic investigation of ionic strength effects on Na,K-ATPase has been reported.
With regard to the mechanism of possible specific effects of anions (other than the substrate, the products and their analogues) only little attention has been given to this subject. Recently it has been found (Post and Suzuki, 1991; Klodos et al., 1994) that the type of anion in the medium was a determinant of the steady-state value of the ratio between the two phosphorylated intermediates, E1P/E2P, during turnover. It was noted that the anions ranked like the Hofmeister lyotropic series but no further interpretation of the results was presented.
The present investigation of the ionic strength and anion dependence of the reactions of Na,K-ATPase was prompted by the observation that substitution of NO3 − for Cl− resulted in a dramatic decrease in the affinity of Na,K-ATPase for ADP and ATP at the substrate site (Rossi and Nørby, 1993). Presumably the binding of the negatively charged ADP and ATP is assisted by attraction to positive charges at or around the binding site, and the inhibitory effect of NO3 − could be a result of a neutralization of these charges. Characterization of the ionic strength dependence and the special inhibitory effect of NO3 − (and other anions) could be useful in evaluating the events in both substrate binding and turnover.
Here we report first a detailed characterization of anion and ionic strength dependence of the equilibrium binding of ADP (and some eosin analogues) to the high affinity substrate site of Na,K-ATPase, and secondly we describe the effects of anions on the overall Na,K-ATPase as well as the K+-activated p-nitrophenylphosphatase (K+-pNPPase)1 reactions. It was found that the binding affinity decreased with increasing ionic strength and was further decreased by replacement of Cl− with NO3 −, SCN−, and ClO4 −, indicative of both unspecific electrostatic interactions and specific site-binding of anions. In the absence of information about the steric structure of the binding site and the electrostatic charges of Na,K-ATPase we have evaluated the ionic strength dependence of the ADP-binding by the simplest possible model. This considers the electrostatic properties of the binding site as those of a point charge, and we found that the interaction of this point charge with the charges on ADP or eosin could be quantified by the limiting law of Debye and Hückel (1923). The charge on the high-affinity site of Na,K-ATPase was calculated to +1. The Na,K-ATPase and K+-pNPPase activities were also inhibited by increasing ionic strength and specific anions. Quantitative evaluation by conventional kinetics and transition-state theory of the K+-pNPPase results was also compatible with a charge of +1 on the pNPP binding site, whereas there seemed to be little electrostatic interaction at the low affinity ATP-binding site.
materials and methods
Enzyme Preparations
Two types of membrane-bound Na,K-ATPase preparation from pig kidney outer medulla were used: EI, “zonal enzyme,” Vmax for Na,K-ATPase activity = 30–32 U · (mg protein)−1, was prepared according to Jørgensen (1974) by selective extraction of plasma membranes with SDS (sodiumdodecylsulphate) in the presence of ATP, followed by isopycnic zonal centrifugation. EII, “purified enzyme,” Vmax for Na,K-ATPase = 20–24 U · (mg protein)−1, also was prepared using the procedures of Jørgensen (1974) but without zonal centrifugation and with the SDS-activation modifications described by Jensen et al. (1984). Both preparations were stored at −20°C in 17.6 mM imidazole, 0.625 mM EDTA, and 250 mM sucrose, titrated to pH 7.4 with HCl, with 1.3 (EI) and 4 (EII) mg protein · ml−1, which is equal to an ADP-binding site concentration of about 4.3 μM (EI) and 10 μM (EII). The two preparations behaved identically and their properties were equal to those published earlier: The turnover, calculated from the Bmax of ADP-binding (e.g., see Table I) and Vmax, is close to 9,000 min−1, which is the same as that of the best preparations of pig kidney enzyme as well as that found for less purified preparations (Jørgensen, 1974, 1988). Also, the K diss for ADP-binding is 0.27 μM at basal conditions (see Table I, experiment 1A at I = 78 mM) which agrees well with the value of 0.22–0.25 μM obtained under similar conditions (Jensen et al., 1984). Furthermore, the response of the two preparations to changing ionic strength and [NO3 −] is the same (see Fig. 5).
Table I.
Effect of Ionic Strength and Anions on Kdiss for ADP-binding to Na,K-ATPase
| NaCl experiments | Na2SO4 experiments | NaNO3 experiments | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exp. | [Cl−] | I | Bmax | K diss | Exp. | [SO4 2−] | I | Bmax | K diss | Exp. | [NO3 −] | I | Bmax | K diss | ||||||||||||||
| mM | mM | μM | μM | mM | mM | μM | μM | mM | mM | μM | μM | |||||||||||||||||
| A1 | 19 | 78 | 1.17 ± 0.02 | 0.270 ± 0.004 | B1 | 6 | 78 | 1.70 ± 0.04 | 0.434 ± 0.010 | C1 | 19 | 78 | 1.80 ± 0.02 | 0.423 ± 0.004 | ||||||||||||||
| A2 | 119 | 178 | 1.20 ± 0.01 | 0.649 ± 0.006 | B2 | 6 | 78 | 1.73 ± 0.02 | 0.379 ± 0.003 | C2 | 24 | 83 | 1.83 ± 0.03 | 0.481 ± 0.007 | ||||||||||||||
| A3 | 219 | 278 | 1.20 ± 0.02 | 1.606 ± 0.012 | B3 | 36 | 168 | 1.71 ± 0.02 | 0.870 ± 0.009 | C3 | 49 | 108 | 1.80 ± 0.03 | 0.939 ± 0.011 | ||||||||||||||
| A4 | 319 | 378 | 1.13 ± 0.11 | 3.347 ± 0.054 | B4 | 66 | 258 | 1.77 ± 0.03 | 1.756 ± 0.017 | C4 | 79 | 138 | 1.84 ± 0.04 | 1.875 ± 0.025 | ||||||||||||||
| A5 | 394 | 453 | 1.29 ± 0.09 | 4.955 ± 0.089 | B5 | 96 | 348 | 1.72 ± 0.06 | 2.833 ± 0.040 | C5 | 79 | 138 | 1.84 ± 0.01 | 1.848 ± 0.011 | ||||||||||||||
| A6 | 469 | 528 | 1.20 ± 0.11 | 7.111 ± 0.124 | B6 | 126 | 438 | 1.96 ± 0.10 | 4.735 ± 0.086 | C6 | 109 | 168 | 1.78 ± 0.06 | 3.233 ± 0.050 | ||||||||||||||
| A7 | 519 | 578 | 0.98 ± 0.09 | 9.119 ± 0.181 | B7 | 156 | 528 | 1.77 ± 0.08 | 6.243 ± 0.061 | C7 | 139 | 198 | 1.80 ± 0.06 | 4.945 ± 0.059 | ||||||||||||||
| C8 | 169 | 228 | 1.82 ± 0.08 | 7.512 ± 0.079 | ||||||||||||||||||||||||
Values obtained by nonlinear regression are ± SE. Bmax and K diss (the latter calculated by nonlinear regression using a common Bmax in each series) were obtained from the data in Fig. 1 to which the numbers of the experiments also refer. The ionic strength before addition of the Na+ salts (experiments A1, B1, B2, and C1) was calculated as described under materials and methods.
Figure 5.

The effect of NO3 − on the binding of ADP, analyzed according to the model in Fig. 4. K A and K diss are the apparent EADP-dissociation constants in the absence and presence of NO3 −, respectively. Two series of data are shown: (•) are data taken from Table I, where K diss in NO3 − is divided by a corresponding (same I) K A-value in Cl− medium. In these experiments I varies from 78 to 228 mM. The open circles (○) are from a set of experiments (not shown, see text) where I was constant = 178 mM, where [Cl−] + [NO3 −] = 119 mM, and where [NO3 −] was varied from 0 to 100 mM. The line is drawn through (0,1) with a slope ± SE of 0.0309 ± 0.0006 mM−1. From the analysis in Fig. 4, this corresponds to a dissociation constant, K N, for ENO3 equal to ≃32 mM.
Protein was measured (Lowry et al., 1951) with a standard of BSA.
Equilibrium ADP-Binding
These measurements were performed as described previously (Nørby and Jensen, 1988; Jensen, 1992). The binding assays in Cl− were done by mixing 4 ml enzyme-stock suspension with 5.7 ml of either H2O or NaCl of the appropriate concentration, 1.2 ml buffer (88 mM imidazole, 1,250 mM sucrose, and 3.125 mM EDTA, titrated to pH 7.4 with HCl) and 0.865 ml 150 mM EDTA/imidazole, pH 7.4. Each assay consisted of 1,300 μl of the mixture, 195–0 μl 150 mM Tris/HCl, pH = 6.3, and 5–200 μl 14C-ADP (e.g., 40 μM) in 150 mM Tris/HCl, pH = 6.3.
After mixing, several 75-μl aliquots from each assay were transferred to counting vials for determination of total (T) radioactivity, and 1 ml was transferred to a cold centrifuge tube and centrifuged at 0–2°C for 60 min at 100,000 g. The tubes were cautiously removed, and after transfer of the supernatant to another tube, several 75-μl aliquots of this supernatant were pipetted to counting vials to determine unbound radioactivity (F). Bound radioactive ADP (B) was determined as B = T − F.
For binding experiments in NO3 − or SO4 2−, 20 ml enzyme-stock suspension in Cl− buffer was diluted with 80 ml ice-cold ISE buffer (17.6 mM imidazole, 250 mM sucrose, 0.625 mM EDTA titrated to pH 7.4 with HNO3 or H2SO4), centrifuged 60 min at 60,000 g at 2°C, and the pellet was resuspended in 35 ml of the appropriate ISE buffer corresponding to an ADP-binding capacity of ∼6.5 μM in the suspension. 4 ml of this suspension was mixed on ice with 5.7 ml of either H2O, NaNO3, or Na2SO4 of the appropriate concentration, 1.2 ml 5× concentrated ISE buffer with NO3 − or SO4 2−, and 0.865 ml 150 mM EDTA/imidazole, pH = 7.4. Each assay consisted of 1,300 μl of this mixture, 195–0 μl 150 mM Tris/HNO3 or 100 mM Tris/H2SO4 buffer, pH = 6.3, and 5–200 μl 14C- ADP (e.g., 40 μM) in 150 mM Tris/HNO3 or 100 mM Tris/H2SO4 buffer. Determination of bound and free ADP was done as described for the experiments in Cl−.
The binding experiments were performed at 0–2°C and the pH was 7.66–7.71 (Cl− experiments), 7.69–7.74 (NO3 −), and 7.65–7.82 (SO4 2−).
Calculation of Ionic Strength, I, for the ADP-Binding Experiments
The ionic strength is I = 1/2 Σ c i z i 2, where c i and z i are the concentration and valence of the ith ion in the solution (Robinson and Stokes, 1970). To calculate I for the ADP-binding assay solutions, it is necessary to have appropriate pKa-values for imidazoleH+, H2EDTA2−, and TrisH+. The pKa-values depend on the ionic strength of the solution, but since the buffers are only dominating at relatively low ionic strength (before the addition of Na+ salts), we have chosen constants corresponding to I around 0.1 M. Thus, the following pKa values should be acceptable approximations: imidazoleH+, pKa = 7.5 (Perrin, 1965); H2EDTA2−, pKa = 6.5 (Kortüm et al., 1961); TrisH+, pKa = 8.8 (Perrin, 1965). From the pH ≃ 7.7 and the total concentrations of these three compounds, the approximate concentrations of the different charged species can be calculated: Common to all experiments: [imidazoleH+] = 32.6 mM, [HEDTA3−] = 9.4 mM, [H2EDTA2−] = 0.6 mM. In Cl− and NO3 − experiments: [TrisH+] = 18.5 mM, and in SO4 2− assays: [TrisH+] = 12.3 mM. This gives a “basal” ionic strength = 78 mM in the binding experiments, and the concentration of Cl−, NO3 −, and SO4 2− is 19, 19, and 6 mM, respectively.
Na,K-ATPase Activity
This activity was measured at 37°C at pH = 7.4 in 30 mM histidine/HCl buffer, with 10 pmol enzyme · ml−1, 3 mM ATP, and 4 mM MgCl2 (if not otherwise indicated), and the concentration and type of Na+ and K+ salts indicated in the figures. The basal ionic strength was ∼42 mM. The assay was started by addition of enzyme, stopped after 3 min with ice-cold TCA (trichloroacetic acid; final concentration 5%), and the amount of liberated phosphate was determined (Esmann, 1988). Blanks contained 1 mM ouabain, which was added before the enzyme.
K+-dependent p-Nitrophenylphosphatase Activity
This catalytic activity was measured at 37°C and pH = 7.4 in 30 mM histidine/HCl buffer with 10 pmol enzyme · ml−1, 10 mM Na2pNPP (if not otherwise indicated), 20 mM MgCl2 and the concentration and type of K+ salts indicated in the figures. The basal ionic strength was ∼90 mM (including 10 mM Na2pNPP). Reaction time was 3 min; ice-cold TCA (final concentration 5%) was added, and the reaction product p -nitrophenol determined as described (Esmann, 1988). The blank was a reagent-blank without enzyme.
Fluorescence Measurement of Eosin- and 6-Carboxyeosin Binding
The fluorescence of eosin or 6-carboxyeosin was measured at 20°C at pH = 7.0 with a Perkin-Elmer MPF 44A spectrofluorometer with two monochromators (Perkin-Elmer Cetus Instruments, Emeryville, CA) (Esmann, 1992). The excitation was at 530 nm, and the emission was monitored at 560 nm, both slits being 10 nm. The cuvette contained 0.2 nmol enzyme · ml−1, 10 mM histidine/HCl buffer, 1 mM CDTA (trans-1,2-cyclohexylenedinitrilotetraacetic acid)/2- amino-2-methyl-1,3-propanediol, 20 mM NaCl, 0.5 μM eosin or 6-carboxyeosin and the Na+ salts indicated in Fig. 11. The basal ionic strength was ∼27 mM.
Figure 11.

Titration of eosin (▪, •, ▴) and 6-carboxyeosin (□, ○, ▵) fluorescence with NaCl (▪, □), NaNO3 (•, ○), or NaClO4 (▴, ▵). Na,K-ATPase, 0.08 mg · ml−1, in 10 mM histidine/HCl-buffer (pH = 7.0), 1 mM CDTA, 20 mM NaCl, 0.5 μM eosin, or 6-carboxyeosin was titrated with additional Na+ salts (the basal ionic strength was ≃27 mM). The fluorescence in the presence of 0.1 mM ADP (where all bound eosin or 6-carboxyeosin is displaced from the enzyme [Esmann, 1991; 1992]) is set to 100. Note that the fluorescence-data points have not been corrected for the dilution caused by the addition of Na+ salts (initial vol 2 ml, final vol 2.5 ml). The temperature was 20°C. The lines through the Cl− data are calculated (also taking dilution effects into account) using the following assumptions: K
diss = [E]·[eo]/[E·eo] and log(K
diss/ K
0) = −z
E·z
eo
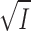 (cf. Eq. 8); [E] + [E·eo] = 0.2 μM; [eo] + [E·eo] = 0.5 μM. The fluorescence quantum yield for E·eo is about five times that of free eo and is assumed to be independent of the ionic strength, and K
0 is taken as 0.2 μM for eosin, and 0.1 μM for carboxyeosin (see Esmann and Fedosova, 1997). The curves correspond to z
E·z
eo = −1.5 (eosin) or −1.7 (carboxyeosin).
(cf. Eq. 8); [E] + [E·eo] = 0.2 μM; [eo] + [E·eo] = 0.5 μM. The fluorescence quantum yield for E·eo is about five times that of free eo and is assumed to be independent of the ionic strength, and K
0 is taken as 0.2 μM for eosin, and 0.1 μM for carboxyeosin (see Esmann and Fedosova, 1997). The curves correspond to z
E·z
eo = −1.5 (eosin) or −1.7 (carboxyeosin).
Reagents and Processing of Data
The reagents were of analytical grade. The various Na+ and K+ salts, H4EDTA (titriplex II), CDTA, Na2pNPP, and histidine were from Merck (Darmstadt, Germany); 2-amino-2-methyl-1,3-propanediol was from Fluka Chemie AG (Buchs, Switzerland); Tris-base and imidazole were from Sigma Chemical Co. (St. Louis, MO); sucrose was from British Drug House (Poole, UK); Na2ATP was from Boehringer Mannheim Biochemicals (Indianapolis, IN); ouabain was from Serva Biochemicals (Paramus, NJ); eosin was from Hopkin and Williams (Chadwell Heath, UK), and 6-carboxyeosin was custom-synthesized by Molecular Probes, Inc. (Eugene, OR). 14C-ADP from NEN (Boston, MA) was chromatographed on DEAE Sephadex (Pharmacia Fine Chemicals, Uppsala, Sweden) and converted to Tris salt in 150 mM Tris/HCl or HNO3 buffer or 100 mM Tris/H2SO4 buffer, pH 6.3 as described (Nørby and Jensen, 1971). The 14C-ADP concentration was 40 μM and the specific activity was about 1.4 × 106 Bq · μmol−1.
Curvefitting and presentation of data was done with the Origin software, Microcal, Amherst, MA.
Theory
The influence of ionic strength, I, on the equilibrium dissociation constant.
The dissociation of ADP from EADP (E is Na,K-ATPase)
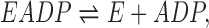 |
is characterized by the measured (apparent) equilibrium constant, K diss:
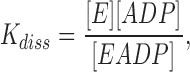 |
1 |
calculated using the concentrations of unbound ADP, [ADP], bound ADP, [EADP], and nonoccupied enzyme binding sites, [E], in moles · liter−1.
According to experiment (and theory, see below), K diss may vary with the ionic strength, I, and this dependence can be evaluated by the introduction of the relation (Tanford, 1961; Robinson and Stokes, 1970)
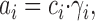 |
2 |
where a i is the I-independent activity, c i the molar concentration, and γi the molar activity coefficient. By combining Eqs. 1 and 2 we get
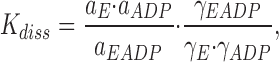 |
3 |
or
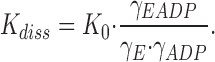 |
4 |
Per definition (Johnson, 1960; Kirkwood and Oppenheim, 1961), the thermodynamic equilibrium constant, K 0, is thus independent of the ionic strength, and any influence of I on K diss stems from the dependence of the ratio of activity coefficients on I.
Debye and Hückel (1923) derived a relationship between the individual activity coefficients for an ion, γi, the ionic strength, I, the ionic charge, z i, and a number of physical constants and parameters, here condensed in the terms A and B (for details and assumptions in the derivation see Tanford, 1961, and Robinson and Stokes, 1970):2
 |
5 |
where a is the distance (in cm) of closest approach of the ions. In water at 25°C, A is 0.508 (l · mol−1)1/2 and B is 0.329 × 108 cm−1 · (l · mol−1)1/2. A and B are quite independent of temperature. Eq. 5 is only theoretically valid for dilute solutions, the upper limit for I being about 0.1 M, or somewhat higher if a is considered an adjustable parameter (Kielland, 1937; Robinson and Stokes, 1970). Various empirical extensions of Eq. 5 have been introduced (e.g., Kielland, 1937; Edsall and Wyman, 1958; Johnson, 1960; Robinson and Stokes, 1970), and some of these equations have proven valid for simple electrolytes at I up to 2 M (Robinson and Stokes, 1970).
If we in Eq. 5 set a to 3 × 10−8 cm, the denominator becomes 1 +  , and for I < 0.01 M a good approximation for log γ is:
, and for I < 0.01 M a good approximation for log γ is:
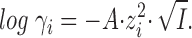 |
6 |
This is the so-called “limiting law” of Debye and Hückel.
An expression describing the effect of ionic strength on the dissociation constant is now obtained by insertion of Eq. 6 into the logarithmically transformed Eq. 4
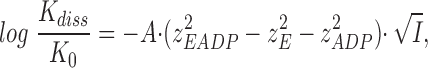 |
7 |
and if we furthermore assume that z EADP = z E + z ADP, and set A = 0.5 (l · mol−1)1/2, see above, Eq. 7 reduces to
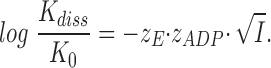 |
8 |
A similar equation is used to describe eosin-binding to Na,K-ATPase.
The effect of ionic strength on enzyme reactions.
We have not found any general description or evaluation of this subject in the literature, although special features like substrate- and ligand-binding (including protons) as influenced by electrolyte concentration have been measured and treated theoretically (see discussion).
In the present work we have determined Vmax and K m (K 0.5) for ATP or pNPP for the Na,K-ATPase and K+-pNPPase activity in media of different ionic strength. To evaluate the observed changes in Vmax and K m we develop a simple model based on conventional enzyme kinetics (Cleland, 1963; Segel, 1975; Palmer, 1991) and transition-state theory for chemical and enzymatic reactions (Kraut, 1988). Kinetics of single-substrate, S, enzyme catalyzed reactions (or multiple substrate reactions where all but one substrate are saturating) follow the Michaelis-Menten equation
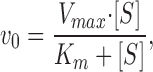 |
9 |
where v0 is the initial rate (mol · liter−1 · s−1) of formation of product, Vmax is v0 for [S] → Θ, and K m is a constant. Both Vmax and K m can be determined from measurements of v0 as a function of [S], e.g., Figs. 7 and 9.
Figure 7.

Na,K-ATPase activity as a function of [ATP] at, from top to bottom, 0.2 M (▪),0.4 M (□), 0.6 M (▪), or 0.75 M NaCl (□). The Mg2+-concentration was always [ATP] + 1 mM, and conditions were otherwise as described in materials and methods and Fig. 6, i.e., 20 mM KCl, t = 37°C, pH = 7.4. Activity = 100 equals 21 U · mg−1 like in Fig. 6. Data are average of two to four experiments and the relative SE was <6%. The lines are rectangular hyperbolae fitted to the data (Theory, Eq. 9), and on the inset the values derived for K m (•) and Vmax (▪) are shown (included are data from an experiment at 0.16 M NaCl).
Figure 9.

The K+-pNPPase activity as a function of [Na2pNPP] at, from top to bottom, 0.05 M (▪), 0.125 M (□), 0.2 M (▪), 0.3 M (□), or 0.4 M KCl (▪). Conditions were otherwise as in Fig. 8 and activity = 100 corresponds to 5 U · mg−1. Data are average of two to four experiments, and the error bars give the SE. Since [Na+] increases with [Na2pNPP] the inhibition seen at high [Na2pNPP] for the experiment at 0.05 M KCl is expected (Skou, 1974). A simple model taking this Na+/K+ competition into account:
ENa2⇌E⇌EK2⇌EK2pNPP → products,
was therefore used to determine the K m values (•) and Vmax values (▪) shown on the inset (included here are the derived data from two experiments at 0.5 and 0.6 M). The lines on the main panel are nonlinear, least-squares fits of the above model to the data. We chose K 0.5 = 2 mM for K+ (cf. Beaugé et al., 1984) and kept the ratio K 0.5,K/K 0.5,Na constant. Also, see the text.
We shall use a simple transition-state model as basis for the evaluation of the effect of ionic strength on the enzyme reactions:
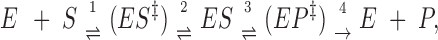 |
10 |
where E is the enzyme, S = substrate, P = product, ES is the (stable) enzyme-substrate complex and (ES‡) and (EP‡) are transition-state complexes. Since [P] = 0, reaction 4 is considered irreversible. The total concentration of enzyme is
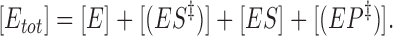 |
11 |
In transition-state theory (e.g., Segel, 1975; Kreevoy and Truhlar, 1986) the transition state is an activated complex through which the reactants must pass before proceeding to form products. Transition-state theory is based on two assumptions: equilibrium between the transition-state and the reactants, and the “dynamic bottleneck assumption” meaning that the reaction rate, v0, is controlled by decomposition of the activated transition-state complex (Segel, 1975; Kreevoy and Truhlar, 1986; Kraut, 1988).
In accordance with Eq. 10 we have
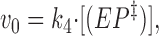 |
12 |
where k 4 is independent of I, whereas [(EP‡)] need not be. Expressed in terms of the conventional (possibly I-dependent) equilibrium constants corresponding to Eq. 10
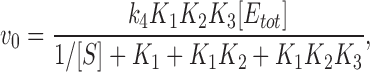 |
13 |
from which, when [S] → Θ
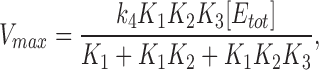 |
14 |
and K m, see also Eq. 9,
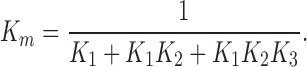 |
15 |
As seen from Eqs. 14 and 15 and as pointed out by Plesner (1986), Vmax and K m in general are complicated functions containing all the rate constants in the reaction mechanism, whereas the ratio, Vmax/K m, between these parameters is simpler “. . . and contain(s) only rate constants characterizing a) intermediates directly interacting with the appropriate substrate, b) the complex of the enzyme with the substrate in question or c) intermediates reversibly connected to them, if any . . .” When we form this ratio from Eqs. 14 and 15, we get
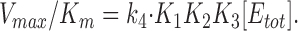 |
16 |
Now, since
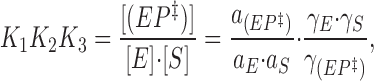 |
17 |
we obtain
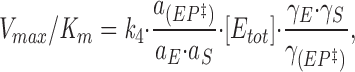 |
18 |
which converts to
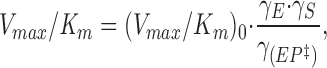 |
19 |
where (Vmax/K m)0 is the “thermodynamic ratio,” independent of I.
The calculated values for (Vmax/K m) are evaluated using the same mathematical procedure as outlined for the dissociation constant K diss (Eqs. 6–8). We assume that
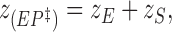 |
20 |
and apply the limiting law of Debye and Hückel, to get
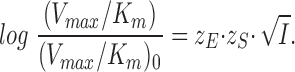 |
21 |
This means that, like the equilibrium measurements (Eq. 8), the kinetic measurements may be used for estimation of the charge at the site where the substrate (or ligand) interacts with the enzyme.
results
Equilibrium Binding of ADP: The Effect of Ionic Strength and Type of Anion on the Dissociation Constant, Kdiss
The equilibrium binding of ADP to pig kidney Na,K-ATPase was characterized in three series of experiments where the ionic strength, I, was varied by addition of NaCl, Na2SO4, or NaNO3. The binding isotherms are shown in Fig. 1, where ADP-binding to the enzyme, E, is described by a model in which E has only one ADP-binding site. The anion concentrations, the ionic strength, and the values for Bmax and K diss derived from Fig.1 are given in Table I, which shows that the binding capacity, Bmax (= [EADP]max) is independent of I. The only parameter that varies between the curves is the dissociation constant, K diss, for EADP. For all three salts K diss increases dramatically with increasing I.
Figure 1.



Binding isotherms at 0°C and pH 7.7 for binding of ADP to Na,K-ATPase in (A) NaCl-, (B) Na2SO4-, and (C) NaNO3-solutions of increasing concentration. In each panel, the anion concentration (and thereby the ionic strength) increases from the top-curve (1) to the bottom-curve (7 or 8), see Table I. In panel A is also shown a binding isotherm in 100 mM Na-acetate (□). The concentrations of bound and free ADP are those in the assay solutions, so that the Bmax value in each series depends upon the (constant) concentration of enzyme-preparation used for the assays of that series—for further details see materials and methods. The relative error on F due to pipetting and counting is ±2% and that on B is about ±3% (see also the double determinations at the highest B values). The curves are the best fit by nonlinear regression (data weighted by B−2, see Jensen et al., 1984) to a one-site model: B = Bmax · F/(K diss + F), and the values for Bmax and K diss derived from these data are given in Table I.
A few ADP-binding experiments, in which 100 mM of some other Na+ salts were added to the basal medium, are shown as Scatchard plots in Fig. 2. Compared to Cl−, NO3 − decreases the binding affinity and the decrease is even more pronounced with SCN− and ClO4 − whereas 100 mM Na-acetate has the same effect on K diss as 100 mM NaCl (Fig. 2). For further illustration, the complete isotherm for 100 mM acetate is shown in Fig. 1 A.
Figure 2.

Normalized Scatchard plots of ADP-binding to Na,K-ATPase in media with 19 mM Cl− + 100 mM Na+ salts of the anions indicated. The ionic strength, I, was 178 mM in all experiments. The complete isotherms for Cl− and CH3COO− (acetate) are given in Fig. 1 A. In these experiments the same enzyme concentration was used throughout. The calculated values for K diss (μM) ± SE are: 0.640 ± 0.007 (Cl−); 0.635 ± 0.004 (acetate); 3.24 ± 0.05 (NO3 −); 18.3 ± 0.2 (SCN−); 38.6 ± 0.70 (ClO4 −).
The relationship between K diss for EADP and the ionic strength is illustrated in Table I and Fig. 3. In Cl−- and SO4 2−-media, K diss at a given ionic strength is about the same. It increases from 0.3 to 7 μM when I is increased from 0.075 to 0.52 M, whereas in NaNO3 the same rise in K diss requires only addition of ∼150 mM NaNO3. Since no salt has less effect on K diss than NaCl, Na2SO4, or Na-acetate when I is considered the independent variable, we ascribe the influence of these salts on K diss to a “pure” ionic strength effect. Consequently, the response in the other salt media is considered a resultant of an ionic strength effect plus a specific interaction of the particular anion (in the binding experiments the cation is always Na+) with the enzyme protein.
Figure 3.

The values for K
diss for the experiments in Cl− (▪) or SO4
2− (▴), at varying ionic strength, I, are taken from Table I, and here plotted as log(K
diss/μM) versus 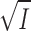 in order to investigate the Debye-Hückel relation (see Theory, Eq. 8 and the text). The lines were calculated by nonweighted, linear regression. The ordinate intercept corresponds to the thermodynamic equilibrium constant, K
0 ± SE = 0.070 ± 0.005 μM (in SO4
2−) and 0.031 ± 0.003 μM (in Cl−). The slope ± SE of the lines is 2.72 ± 0.07 M−1/2 (in SO4
2−) and 3.25 ± 0.07 M−1/2 (in Cl−).
in order to investigate the Debye-Hückel relation (see Theory, Eq. 8 and the text). The lines were calculated by nonweighted, linear regression. The ordinate intercept corresponds to the thermodynamic equilibrium constant, K
0 ± SE = 0.070 ± 0.005 μM (in SO4
2−) and 0.031 ± 0.003 μM (in Cl−). The slope ± SE of the lines is 2.72 ± 0.07 M−1/2 (in SO4
2−) and 3.25 ± 0.07 M−1/2 (in Cl−).
Influence of I on Kdiss of EADP
The dissociation constants for EADP in Table I are apparent dissociation constants calculated using the concentrations of the species involved, cf. Eq. 1 in the Theory section. In that section we have evaluated the possible effects of ionic strength on K
diss using the Debye- Hückel theory for interaction between ions in solution (Eqs. 1–8). As mentioned above, we regard the effects of [NaCl] and [Na2SO4] on K
diss to be due solely to a change in I, and in Fig. 3 we have explored this relationship by plotting log K
diss for the NaCl- and Na2SO4- experiments as a function of 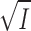 . It appears that straight lines fit the data perfectly. Interpretation of these data according to Eq. 8 determines the thermodynamic equilibrium constant K
0 to be between 3 and 7 × 10−8 M. The slope of the lines has the value of 2.72 and 3.25 (both with a SE of ±0.07), which is equal to −z
E · z
ADP. Since z
ADP is −3 this leads to a value for the charge of E (the enzyme site), z
E = +0.91 (SO4
2− data) or +1.08 (Cl− data), both with SE = ±0.02.
. It appears that straight lines fit the data perfectly. Interpretation of these data according to Eq. 8 determines the thermodynamic equilibrium constant K
0 to be between 3 and 7 × 10−8 M. The slope of the lines has the value of 2.72 and 3.25 (both with a SE of ±0.07), which is equal to −z
E · z
ADP. Since z
ADP is −3 this leads to a value for the charge of E (the enzyme site), z
E = +0.91 (SO4
2− data) or +1.08 (Cl− data), both with SE = ±0.02.
The Effect of NO3 − on ADP-binding
Assuming that the influence of the ionic strength per se is described by the NaCl- and Na2SO4-experiments, we have analyzed the more pronounced effect of NaNO3 on K diss (Table I) using the simple model shown in Fig. 4. If the simultaneous occupancy with NO3 − and ADP is prohibited, i.e., if ENO3ADP does not exist, the model corresponds to competitive inhibition of ADP-binding by NO3 −.
Figure 4.

A simple model for the effect of NO3 − on ADP-binding. K A and K diss are the apparent EADP-dissociation constants in the absence and presence of NO3 −.
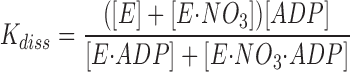
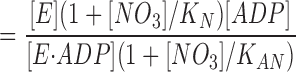 ,
,
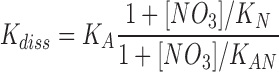 .
.
If K AN ≫ [NO3], then: K diss/K A = 1 + [NO3] /K N.
K A, the dissociation constant for EADP in the absence of NO3 −, varies with I. This variation contributes to the variation of K diss in the presence of NO3 −, and we therefore use the ratio K diss/K A, where K diss and K A are measured at the same ionic strength, to illustrate the specific effect of NO3 −. Fig. 5 is a plot of K diss/K A for two series of experiments: One in which the data were calculated (filled circles) from Table I and where I varies from about 78 to 228 mM, and another (open circles) where the ionic strength was kept constant at 178 mM (binding isotherms not shown).
The data in Fig. 5 fall on a straight line with a slope of 0.031 mM−1. For these data to be described by the simple model in Fig. 4, K AN must be much larger than the highest value of [NO3 −] used in these experiments, i.e., K AN ≫ 150 mM. In this case we can obtain a value for the dissociation constant of ENO3, K N, which is the reciprocal slope: K N ≃ 32 mM. Using this value for K N we can estimate a value for K AN (at I ≃ 0.178 M) from the relation K N · K NA = K A · K AN by insertion of K A = 6 × 10−7 M (Table I) and K NA = 10−4 M (this is a minimum estimate of the EADP dissociation constant made at high [NO3 −]; Rossi and Nørby, 1993). The result is that K AN ≃ 5 M. These considerations do not exclude simultaneous binding of NO3 − and ADP to the enzyme, but they strongly suggest that the affinity of EADP for NO3 − is manyfold smaller than the affinity of E for NO3 −, indicating that NO3 −-binding is close to being competitive.
The Influence of Different Anions and of the Ionic Strength on the Na,K-ATPase Activity and the K+-pNPPase Activity of Na,K-ATPase
Na,K-ATPase activity.
The effect of anions and salt concentration on the rate of ATP-hydrolysis was characterized in several series of experiments. In one, the results of which are given in Fig. 6 with closed symbols, different Na+ salts were added to a medium with constant [KCl] = 20 mM, 3 mM ATP and 4 mM MgCl2. With all salts there is an increase in the activity at low salt concentration (due to the activation by Na+) followed by a decline in rate of hydrolysis with increasing salt concentration. With NaNO3 and NaClO4 the inhibition is more pronounced than with NaCl, as was the case when ADP-binding was measured. The Na2SO4-curve follows the NaCl curve when [Na+] is the independent variable, whereas when the activity is plotted versus I (not shown), the inhibitory effect of Na2SO4 is 10–30% less than that of NaCl. Experiments with Na-acetate up to 800 mM (not shown) gave the same results as with NaCl.
Figure 6.

Na,K-ATPase activity with 3 mM ATP, 4 mM MgCl2, 20 mM KCl (closed symbols), and the concentrations of NaCl (▪), Na2SO4 (▴), NaNO3 (•), and NaClO4 (♦) shown. Also shown (□) is the Na,K-ATPase activity with 3 mM ATP, 4 mM MgCl2, and with NaCl + KCl added in a constant molar ratio = 8. The ionic strength, I, was 0.042 M before addition of Na+ and K+ salts, see materials and methods. The temperature was 37°C, pH was 7.4, and activity = 100 equals 21 U · mg−1. Data are average of three to four experiments and the relative SE was <6%.
The reversibility of the salt effect was demonstrated by first suspending the enzyme in 2 M NaCl or 0.5 M NaNO3 and thereafter removing the salt by centrifugation. After resuspension of the pellet in the normal storage medium, the Na,K-ATPase activity was the same as before the salt treatment.
A second series of Na,K-ATPase measurements addressed the problem of whether increasing the concentration of Na+ (relative to [K+]) is a decisive factor in the decline in activity. Hypothetically this could occur because of competition between Na+ and K+ for the activating, extracellular K+ site. We therefore varied the salt concentration like in the first series, but kept the ratio [NaCl]/[KCl] = 8 (the ratio at optimal activity, i.e., 160 mM NaCl/20 mM KCl). From Fig. 6 (open symbols) it is seen that at any salt concentration (Cl− salts) above I ≃ 0.2 M, the Na,K-ATPase activity is the same whether the [NaCl]/[KCl] ratio is constant = 8 or whether it increases from 8 at optimal activity to 40 at 0.8 M salt. It therefore appears that specific, competitive inhibition by Na+ can be ruled out in the experiments of Figs. 6 and 7.
To explore the possibility that a drastic increase in K m for ATP with increasing salt concentration could contribute to the observed fall in activity, K m for ATP for the Na,K- ATPase activity was measured with 20 mM KCl, [Mgtot] = [ATP] + 1 mM and 0.16–0.75 M NaCl (Fig. 7). As seen from the inset on Fig.7 there is a slight decrease in K m with increasing I, so lack of ATP cannot contribute to the fall in activity (Fig. 6, and Vmax on the inset of Fig. 7). Similar experiments in 0.2 M NaClO4 (not shown) likewise revealed no effect on activity of an increase in [ATP] from 3 to 10 mM.
According to the model in the Theory section regarding the effect of ionic strength on enzyme kinetics, it might be possible, from the slope of a plot of log(Vmax/ K
m) versus 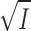 (Eq. 21), to estimate the charge of the low-affinity ATP-binding site (z
E). Such a plot, using the values from the inset of Fig. 7, is shown on Fig. 10. The value of the slope, which is equal to z
E·z
ATP is determined by linear regression to −0.50 ± 0.05. Since z
ATP is −4, z
E becomes +0.13 ± 0.01.
(Eq. 21), to estimate the charge of the low-affinity ATP-binding site (z
E). Such a plot, using the values from the inset of Fig. 7, is shown on Fig. 10. The value of the slope, which is equal to z
E·z
ATP is determined by linear regression to −0.50 ± 0.05. Since z
ATP is −4, z
E becomes +0.13 ± 0.01.
Figure 10.

Quantitative characterization of the effect of ionic strength on Na,K-ATPase activity (□), data directly from Fig. 7, and on pNPPase activity in KCl (▪), data directly from Fig. 9. Also shown are data from pNPPase measurements in K2SO4- solutions (▴). For the latter, K
m was taken from Fig. 9, and Vmax was then calculated from the v0 values in Fig. 8 using the Michaelis-Menten equation (Theory, Eq. 9). Following the model for the effect of I on enzyme reactions outlined in the Theory section, resulting in Eq. 21, the calculated log(Vmax/K
m) are plotted versus 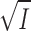 . The slopes ± SE (M−1/2) obtained from linear regression are: Na,K-ATPase (□) = −0.50 ± 0.05; pNPPase in KCl (▪) = −2.11 ± 0.08; and pNPPase in K2SO4 (▴) = −1.68 ± 0.06.
. The slopes ± SE (M−1/2) obtained from linear regression are: Na,K-ATPase (□) = −0.50 ± 0.05; pNPPase in KCl (▪) = −2.11 ± 0.08; and pNPPase in K2SO4 (▴) = −1.68 ± 0.06.
K+-activated pNPPase activity.
Fig. 8 shows the value for this activity as a function of the concentration of different K+ salts. After the initial activation by K+, further addition of salt in all cases leads to a lower pNPPase activity. In contrast to what is seen with ADP binding and Na,K-ATPase activity, NO3 − does not have any special effect. The similarity between the effect of KCl and KNO3 is also found when the activity is measured as a function of I at a higher [pNPP] = 25 mM (not shown). When the SO4 2− data are related to the ionic strength (rather than to [K+]), the apparent special effect of SO4 2− also disappears.
Figure 8.

K+-p-nitrophenylphosphatase (K+-pNPPase) activity of Na,K-ATPase measured with 10 mM Na2pNPP, 20 mM MgCl2, and the concentrations of KCl (▪), KNO3 (•), K2SO4 (▴), and KClO4 (♦) shown. The temperature was 37°C, pH was 7.4, and activity = 100 corresponds to 5 U · mg−1. The data are plotted versus the K+-concentration. The ionic strength, I, before addition of K+ salts was 0.090 M, see materials and methods. In each particular experiment with Cl−, NO3 −, or ClO4 −, I (M) is thus [K+] + 0.09. For the SO4 2−-data, I = 1.5 · [K+] + 0.09. Data are average of three to four experiments and the relative SE was <66%.
Under the conditions of the assay for the data of Fig. 8 ([pNPP] = 10 mM) the enzyme is only 70–80% saturated with substrate when the salt concentration is 0.05 M or lower, since K m for pNPP is 2–3 mM at low ionic strength (Skou, 1974; Campos et al., 1988; Fig. 9 in this paper). To investigate whether the inhibition of the K+-pNPPase activity at high ionic strength could result from an increase in K m for pNPP (substrate desaturation), we measured the pNPPase activity as a function of [pNPP] in 0.05–0.6 M KCl. It was found (Fig. 9, inset) that K m increased up to 10–12 mM with increasing [KCl]. The consequence is, that in 0.5 M KCl, Vmax would be twice the activity measured with 10 mM pNPP (Fig. 8). Substrate desaturation therefore contributes significantly to the decrease in activity with increasing [KCl]. The special ClO4 − effect (relative to that of Cl−) is not an effect on K m: in an experiment (not shown) similar to that in Fig. 9, with varying [pNPP] and with 0.125 M Cl− or 0.125 M ClO4 − we found K 0.5 to be similar, namely about 4.5 mM in 0.125 M Cl− and 5.5 mM in 0.125 M ClO4 −.
Like for the Na,K-ATPase activity, the effect of I on the K+-pNPPase activity was quantified using the ratio Vmax/K
m as outlined in the Theory section (Eq. 21). For the experiments with KCl, the set of 7 directly determined K
m and Vmax values shown on the inset of Fig. 9 was used. In the case of the K2SO4 experiments, the K
m values of Fig. 9 and the v0 data from Fig. 8 were used to calculate Vmax (Eq.9). On Fig. 10, log(Vmax/K
m) for K+-pNPPase is plotted as a function of 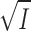 . According to Eq. 21 the slope of such a plot is the product of the charges at the binding site and of the substrate, z
E·z
pNPP. The values for the slope ± SE are −1.68 ± 0.06 (in SO4
2−) and −2.11 ± 0.08 (in Cl−) which, since z
pNPP = −2, estimates z
E to +0.84 ± 0.03 (in SO4
2−) and +1.06 ± 0.04 (Cl−), i.e., the same values as obtained in the ADP-binding experiments.
. According to Eq. 21 the slope of such a plot is the product of the charges at the binding site and of the substrate, z
E·z
pNPP. The values for the slope ± SE are −1.68 ± 0.06 (in SO4
2−) and −2.11 ± 0.08 (in Cl−) which, since z
pNPP = −2, estimates z
E to +0.84 ± 0.03 (in SO4
2−) and +1.06 ± 0.04 (Cl−), i.e., the same values as obtained in the ADP-binding experiments.
Binding of Eosin and 6-Carboxyeosin to Na,K-ATPase: The Effect of Increasing Concentrations of NaCl, NaNO3 and NaClO4
When the enzyme has bound Na+, eosin or 6-carboxy-eosin can bind to the substrate site of Na,K-ATPase with an affinity comparable to that of ATP. The bound species have a higher fluorescence than the free (Skou and Esmann, 1981). We have exploited this to investigate whether the results obtained with equilibrium binding of ADP (Fig. 1, Table I) could be replicated with these “substrate analogues.” Fig. 11 shows that the equilibrium fluorescence of eosin and 6-carboxyeosin decrease when the ionic strength is increased by any of the monovalent salts NaCl, NaNO3, or NaClO4. The fluorescence level of unbound eosin or carboxyeosin (experimentally the fluorescence after addition of excess ADP) is taken as “100.” If, as assumed for the ADP-binding experiments, the effect of increasing [NaCl] is a pure ionic strength effect, the fluorescence of 6-carboxyeosin (charge −3) would be expected to decrease faster with increasing [NaCl] than the fluorescence of eosin (charge −2). This is what we observe. As with the ADP-binding (Figs. 1, 2 and Table I) NO3 − and ClO4 − have specific inhibitory effects relative to Cl− on the binding of the eosins.
The curves fitted to the Cl− data in Fig. 11 are based on the same equations as used in the ADP-binding experiments (Theory, Eqs. 4, 6 and 8), i.e., we assume that K
diss for the process E·eo E + eo is a simple function of the activity coefficients, γ, for E (enzyme), eo (eosin or carboxyeosin) and the E·eo complex, and that γ is related to I by the limiting law of Debye and Hückel, i.e., log(K
diss/K
0) = −z
E·z
eo
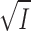 . Assuming that the fluorescence yield of bound dye, independently of I, is about five times that of the free (Esmann and Fedosova, 1997), we obtain a value for the charge product z
E·z
eo ≃ −1.5. Since z
eo = −2, this corresponds to z
E ≃ +0.75. For carboxyeosin the charge product is ≃ −1.7, which with a net carboxyeosin charge of −3 corresponds to z
E ≃ +0.6. Both calculated values for z
E are somewhat lower than the value of +0.9 to +1.1 obtained from the ionic strength dependence of ADP-binding and pNPP-binding in the K+-pNPPase activity.
. Assuming that the fluorescence yield of bound dye, independently of I, is about five times that of the free (Esmann and Fedosova, 1997), we obtain a value for the charge product z
E·z
eo ≃ −1.5. Since z
eo = −2, this corresponds to z
E ≃ +0.75. For carboxyeosin the charge product is ≃ −1.7, which with a net carboxyeosin charge of −3 corresponds to z
E ≃ +0.6. Both calculated values for z
E are somewhat lower than the value of +0.9 to +1.1 obtained from the ionic strength dependence of ADP-binding and pNPP-binding in the K+-pNPPase activity.
discussion
The present work concerns the influence of various salts on equilibrium binding of ligands (ADP, eosin and 6-carboxyeosin) to Na,K-ATPase as well as on two hydrolytic activities of the enzyme, the Na,K-ATPase activity and the K+-pNPPase activity. We observed a dramatic decrease in affinity for the ligands as well as an inhibition of both of the enzymatic activities with increasing salt concentration. In a theoretical paper Record et al. (1978) have listed five potential origins for such salt effects: (1) differential cation binding; (2) differential anion binding.; (3) differential hydration (at high electrolyte concentration); (4) differential screening (Debye-Hückel) effects of electrolyte on the macroion charges, reflected in a variation of the macromolecular activity coefficient; (5) effects of electrolyte on the activity coefficient of the ligand. Of these items, No. 3 will not be discussed in any detail since we have no relevant results in this work. The concept of “Hofmeister series” of anions will be briefly discussed later.
The salts used in this work are Na+ salts or K+ salts. These cations are the natural activators of the enzyme, and they influence the nucleotide binding profoundly. The enzyme binds ATP, ADP and eosin with only low affinity at very low ionic strength (no Na+ or “Na+-like” buffers), or in the presence of K+ salts (Skou and Esmann, 1981; Glynn and Richards, 1982; Jensen and Ottolenghi, 1983b ). When the ionic strength, I, is increased somewhat either by buffers or Na+ salts, the enzyme system changes to a Na+-form (E1- form) that displays a high affinity (e.g., K diss < 1 μM at I < 0.1 M) for ATP, ADP and the eosins (Nørby and Jensen, 1971; Hegyvary and Post, 1971; Jørgensen, 1975; Skou and Esmann, 1981). This effect of increasing I, as well as the specific effects of Na+ and K+, all saturate at relatively low salt concentrations. There is no experimental evidence that increasing the Na+ concentration changes the enzyme from a high-affinity nucleotide-binding E1-form to a low-affinity form (E2). Likewise, results with the pNPPase reaction at higher K+ concentrations do not reflect a conformational change from the E2-form to the E1-form. Thus, as will be substantiated below, we believe that we can rule out differential cation binding (item No. 1 of the list above) as a cause for the observed inhibitions by increasing salt concentration.
It is possible that effects of increasing [salt] are mediated by differential anion binding (item No. 2 of Record et al.'s list). In this regard our experiments seem to fall into two groups. At equal ionic strength, the results with Cl−, SO4 2− and acetate are comparable in that they show the smallest inhibition of ADP- and eosin-binding (Figs. 1, A and B, 2, and 11), Na,K-ATPase activity (Fig. 6) and pNPPase activity (Fig. 8), whereas the NO3 −, SCN−, and ClO4 − salts show progressively larger effects, the lack of a special inhibition of K+-pNPPase by NO3 − being an exception. We therefore hypothesize that the observations with Cl−, SO4 2−, and acetate salts are related to electrostatic events alone, and that the more pronounced responses obtained with the other anions reflect additional specific anion-binding, and perhaps other properties of strong electrolytes.
Ionic Strength, I, and Equilibrium, High-Affinity Binding of ADP, Eosin, and 6-Carboxyeosin to the Substrate Site of Na,K-ATPase
Increasing the concentrations of NaCl, Na2SO4, and Na-acetate led to a marked fall in the affinity of Na,K-ATPase for ADP, eosin and 6-carboxyeosin (Figs. 1, A and B, 3, 11 and Table I). Comparison of the results for ADP (charge −3) and eosin (charge −2) following the procedure outlined in the Theory section, Eqs. 1–8, shows that the larger the charge on the binding ligand, the more pronounced is the effect of the salt concentration on binding. This supports the theory that electrostatic interactions are important in this process.
There are few published data that allow a judgement of the influence of I on the binding of ligands to the substrate site of Na,K-ATPase, but they all show the same qualitative trend as here. Nørby and Jensen (1974) reported that increasing I from 0.08 to 0.12 M with Tris-buffer increased K diss for EATP by a factor of 1.5, and the same can be deduced from comparison of the results in Ottolenghi and Jensen (1983) and Jensen and Ottolenghi (1983a) where I was changed with NaCl. A very pronounced inhibition of eosin binding by increased concentrations of choline-Cl or NaCl up to 0.45 M was demonstrated by Skou and Esmann (1980, 1981). Like our experiments with NaCl and Na2SO4, where [Na+] in the latter is only two-thirds of that in NaCl at equal ionic strength, these studies support that the important parameter is the ionic strength rather than the type of cation used.
The high-affinity ATP-binding site is located in the large cytoplasmic loop of the α-subunit, presumably in a polar milieu, and several charged amino acid residues (see reviews by Kaplan, 1991; Lingrel and Kuntzweiler, 1994) have been implicated as part of this site. The picture in Kaplan (1991) for example shows 7 lysines (2 of which may not be essential for ATP-interactions [Lingrel and Kuntzweiler, 1994]), 1 arginine, and 3 aspartates. The importance of a net positive charge for substrate binding was already suggested by the pH profile of K diss for EATP (Hegyvary and Post, 1971): K diss was low and constant for pH = 5 to 7, but it increased sharply when pH was raised from 7.5 to 9. This must reflect the removal of a positive charge or the creation of a negative charge. The importance of electrostatic interaction for ATP-binding is likewise illustrated by the finding that substitution of Lys480 to Arg or Ala had little effect on K diss but in the mutant Lys480Glu the affinity was reduced ∼10-fold (Wang and Farley, 1992). The negatively charged Asp369 is a crucial residue at the catalytic site of the ATPase, since this is the residue phosphorylated by the γ-phosphate of ATP during turnover. During binding of the negatively charged ATP this residue must exert a repulsive force, and this is convincingly demonstrated in a recent work (Pedersen et al., 1996) where substitution of Asp369 with uncharged Asn or Ala resulted in a 20–30-fold increase in the affinity for ATP at the high-affinity site. At the same time the catalytic activity was completely lost.
In the evaluation of the effect of ionic strength on ADP and eosin binding we model the substrate site as a point charge and use the Debye-Hückel limiting law for the relation between the activity coefficient and ionic strength (Eq. 6). The validity of these assumptions is discussed in the next section. The analysis (Figs. 3 and 11) leads to the estimate that the net charge of the high-affinity binding site (the substrate site of Na,K-ATPase) is +0.9 to +1.1 (ADP-binding experiments) and about +0.7 (eosin and carboxyeosin experiments).
The Applicability of the “Point Charge” Assumption for the Binding Site and the Limiting Law of Debye-Hückel to Evaluate the Ionic Strength Effects
There are two main principles in the derivation of models for the electrostatics of proteins: the macroscopic, continuum model, and the microscopic, all atoms model (Warshel and Russell, 1984; Matthew, 1985; Allewell and Oberoi, 1991; Nakamura, 1996). The microscopic model depend on x-ray structural information. Unfortunately no x-ray data are available for Na,K-ATPase, and we have therefore used the macroscopic approach.
In both types of model it is essential to know the roles of the different charges on the protein. For the macroscopic model, the two extremes are: does the charge on the whole protein or the charge only in the microenvironment where the reaction takes place (the binding site or cleft) determine the direction and magnitude of the salt effects? The latter view, which we here call the “point charge assumption for the binding site,” has considerable support in previous studies in which it was demonstrated that the local charges at the site of reaction are much more important than distant charged groups, the effect of which furthermore dramatically decrease with increasing I (Alberty and Hammes, 1958; Hammes and Alberty, 1959; Snyder et al., 1981; Loewenthal et al., 1993). The selective importance of local electrostatic attraction is also underscored for superoxide dismutase by Getzoff et al. (1992). The net charge of this enzyme is negative like that of the substrate O2·− but site directed mutagenesis and ionic strength effects convincingly demonstrate that it is the charge at the substrate site that decides substrate recognition and attraction.
In the analysis of the electrostatic interaction between the charge of the substrate site and the charge of the substrate, we have applied the limiting law of Debye-Hückel (Eq. 6). Theoretically this equation is only valid in very dilute solutions and the good correlations between our data and Eqs. 8 and 21 derived on the basis of the limiting law (see Figs. 3 and 10) are therefore perhaps surprising. A survey of the literature reveals, however, that this finding is by no means unique. One prominent example is the usefulness of the limiting law of Debye-Hückel in relating the calculated effective dielectric constant, Deff, in the active site cleft of subtilisin to the ionic strength of the bulk medium (Russell et al., 1987). Deff was calculated by assuming two point-charges separated by a certain distance, and the simple relationship ln Deff = ln Dwater + α · 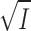 was found to be valid over the entire range of [KCl] from 0.1 to 1 M.
was found to be valid over the entire range of [KCl] from 0.1 to 1 M.
Studies of the effects of ionic strength on enzyme reactions have often concentrated on the rate of binding of the substrate. The I-dependence of this reaction can also be found to follow the law of Debye and Hückel (Eq. 5) or the limiting law (Eq. 6), far beyond the upper limit of their theoretical validity (the Cu,Zn superoxide dismutase reaction, see Argese et al. [1987], and Getzoff et al. [1992], binding of ATP to myosin subfragment 1, Johnson and Taylor [1978], binding of butyrylthiocholine to butyrylcholinesterase, Masson et al. [1996], hydrogen exchange of various peptide groups in bovine trypsin inhibitor, Christoffersen et al. [1996]). Likewise at NaCl concentrations up to 1 M, Snyder et al. (1981) found that the ionic strength dependence of the bimolecular rate constant for reaction of negative disulfide substrates with cysteines in naturally occurring proteins fits with that described by the Debye-Hückel law.
Ionic Strength and the Kinetics of the Na,K-ATPase and K+-pNPPase Activity
The maximal rate of hydrolysis of ATP as well as pNPP, i.e., the rate at saturating substrate and activating ligand concentrations, decreased with increasing ionic strength. For the Na,K-ATPase activity the decrease was roughly the same whether Cl−, SO4 2− or acetate− was the anion (Fig. 6), and the relationship between Vmax and I was also the same whether I was increased with NaCl ([KCl] fixed at 20 mM) or with NaCl + KCl in a constant ratio [NaCl]/[KCl] = 8 (Fig. 6). This supports the hypothesis that it is an ionic strength effect rather than a competitive binding of Na+ to activating K+-sites. For K+-pNPPase the I- dependence of v0 (with 10 mM pNPP) was the same in KCl, K2SO4 and KNO3 (Fig. 8), emphasizing the importance of I rather than specific anions also here. According to the transition-state model in the Theory section (Eq. 14), there are no simple explanations for the dependence of Vmax on ionic strength.
We did, however, exploit the kinetic measurements using the expression Vmax/K m that characterizes the reaction of the enzyme with the substrate (Theory, Eqs. 18–21). The ionic strength dependence of this ratio (Fig. 10) revealed that there is a significant electrostatic interaction involved in the binding of pNPP2− to the substrate site. The net charge at this enzyme site for pNPP was calculated to be between +0.8 and +1.1, i.e., close to the values found for the high-affinity ADP- and eosin-binding site. In contrast, the low affinity binding of ATP4−, which has a decisive rate-determining role in the Na,K-ATPase activity, was much less I dependent (the estimated net charge at the enzyme site was +0.13). If the low-affinity ATP binding is in the same topological region as the high-affinity binding, then this region must undergo a drastic electrostatic rearrangement (e.g., a deprotonation), when it changes from the high-affinity to the low-affinity conformation. Likewise, we may suggest, as did also Beaugé et al. (1984), that the pNPP-binding site is different from the low-affinity ATP site. The enzyme is known to be phosphorylated by ATP at Asp369 when in the high affinity form, and it is also phosphorylated by pNPP when Na+ is present, presumably at the same Asp369-residue (Yamazaki et al., 1994). If pNPP binds at the same place during both the Na+,K+-pNPPase and K+-pNPPase reaction, this is likely to be at the high affinity ATP binding site in the same effective “electrostatic conformation.” The difference in NO3 −-sensitivity between the equilibrium binding of ADP and pNPP in the pNPPase reaction could be due to the fact that ADP has 3 but pNPP only 2 negative charges.
Our results are comparable to the few observations on the effect of ionic strength on the hydrolytic activities of Na,K-ATPase that have been published. In the absence of K+, the Na+-ATPase activity was clearly inhibited by both [NaCl] > 0.2 M and by Tris-Cl (Nørby et al., 1983), whereas if I was kept constant with Tris-Cl + NaCl, only activation by Na+ was seen. Likewise, in experiments with a constant ratio of [Na+]/[K+] = 6.5, the Na,K- ATPase activity increased with the (Na++K+) salt concentration when I was kept constant with Tris-Cl, but without Tris-Cl there was inhibition above 0.2 M salt (Skou, 1979). There is thus an inhibitory effect on the Na,K-ATPase activity of NaCl, NaCl + KCl, or Tris-Cl (provided that there is a certain minimum concentration of the activating ions). The pNPPase activity was inhibited by increasing ionic strength, irrespective whether I was increased by KCl (Koyal et al., 1971; Skou, 1974) or choline-Cl (Skou, 1974). All these observations again support our assumption that we are dealing with effects of ionic strength and not of specific cations.
The Effect of Special Anions on the Properties of the Na,K-pump
Among the anions examined in this work, NO3 − and ClO4 − have special inhibitory effects on equilibrium binding of ADP (Table I, Fig. 2), eosin and carboxyeosin (Fig. 11), and on Na,K-ATPase activity under Vmax conditions (Fig. 6), whereas only ClO4 − showed specific inhibition of the K+-pNPPase activity (Fig. 8). ADP-binding is also specifically inhibited by SCN− (Fig. 2).
The effect of NO3 − on ADP binding is described in detail (Figs. 1 C, and 5, Table I) and in the concentration range studied, it is found to be compatible with a simple model (Fig. 4) involving competitive binding between ADP and NO3 − with a dissociation constant for E·NO3 of 32 mM (Fig. 5). According to this model, NO3 − may also bind to EADP but with a very low affinity, K diss for EADP·NO3 probably being larger than 5 M. We do not know whether SCN− and ClO4 − act on ADP and eosin binding by a similar mechanism. Since we have obtained evidence for the binding of NO3 − to the substrate site of Na,K-ATPase, it is relevant to mention that inhibition of creatine kinase by NO3 − is proposed to be caused by a formation of a dead-end complex creatine-enzyme-MgADP-NO3 (review by Watts, 1973). K diss for NO3 − is <1 mM. Further molecular details are not known, but it seems that the NO3 − binding observed in this work is different (K diss ≃ 32 mM for ENO3 and > 5 M for EADP·NO3) also because it is competitive to ADP-binding and not synergistic. It is also of particular interest that some H+-translocating Mg-ATPases from tissues of higher plants are inhibited by NO3 − with a K 0.5 of 10–50 mM, i.e., comparable to the K diss found here (Sze, 1985; Jacoby, 1987; Blumwald et al., 1987). The mechanism for inhibition is not known. There are several papers dealing with anion inhibition of ATPases from other tissues, but a discussion of those is beyond the scope of this article. We should point out though, that in the light of the results presented here and the findings just discussed, the statement that inhibition by “KNO3 and KSCN . . . appear to be more specific for the V- than the P- and F- type ATPases” (Pedersen and Carafoli, 1987) may need revision.
None of the anions investigated in this work seem to interfere with the low-affinity ATP-binding site involved in Na,K-ATPase activity. Perhaps this reflects that electrostatic interactions are not especially important at this site (see above). The lack of specific interference of NO3 − with the K+-pNPPase activity shows that at least one anion sensitive step is different between the two hydrolytic processes of the enzyme.
Regarding Vmax for Na,K-ATPase activity (Figs. 6 and 7) and K+-pNPPase activity (Figs. 8 and 9), we have no explanation for either the special effect of the anions or for the effect of increasing I, but these results show that other steps in the reaction mechanism than the binding of substrate and ligands are sensitive to anions and ionic strength. According to recent studies by Post and Suzuki (1991) and by Klodos and colleagues (Klodos and Forbush, 1991; Klodos, 1991; Klodos et al., 1994) both anions and salt concentration as such have profound influence on the formation and dephosphorylation of the phosphorylated intermediates, EP, in the Na+-ATPase reaction. The model proposed for the results of changing salt (NaCl) concentrations does not use the concept of ionic strength effects on the protein but rather a “control of enzyme conformations by changes in separate unmixed phases of the lipid of the membrane” (Klodos et al., 1994). The effect of the anions was ranked according to their ability to influence the ratio between ADP- and K+-sensitive phosphoenzyme, E1P/E2P, and the rate of dephosphorylation of EP, and it was found that the ranking conformed with the Hofmeister series for anions (see below and the review by Collins and Washabaugh, 1985). This suggested that water structure could be an important parameter in these enzymatic phenomena (although it has been claimed [p. 277 in Edsall and Wyman, 1958] “that the significance of the series is quite unrelated to colloidal chemistry in any special sense. It is rather at general function of the size and hydration of ions”).
The effects of anions reported in the present work do not rank systematically in the Hofmeister series, although this does not exclude that the lyotropic properties could be influential especially perhaps at higher salt concentrations (Leberman, 1991; Parsegian, 1995; Leberman and Soper, 1995). For the anions used here, the Hofmeister series is (Collins and Washabaugh, 1985; Leberman, 1991): SO4 2− > acetate > Cl− > NO3 − > ClO4 − > SCN−. The ranking is to be understood in such a way that to obtain a given result (originally the precipitation of eggwhite) a higher equivalent concentration of SO4 2− (e.g., 0.5 M Na2SO4 is 1 eq · l−1 of SO4 2−) than acetate is required, etc. For comparison, in the phenomena investigated here, the rankings are: ADP-binding, acetate = Cl− > SO4 2− > NO3 − > SCN− > ClO4 −; Na,K-ATPase activity, acetate = Cl− = SO4 2− > NO3 − > ClO4 −; K+-pNPPase activity, acetate > Cl− = NO3 − > SO4 2− > ClO4 −.
Pertinent to the reaction of anions with the Na,K-pump are also the three papers (Dissing and Hoffman, 1990; Marín and Hoffman, 1994a , b ) describing a coefflux of Na+ and SO4 2− or HPO4 2− (Pi) in red blood cells. The transport is ouabain inhibitable and intimately connected to the so-called uncoupled Na+ efflux through the Na,K-pump. The mechanism is unknown and the anion effects observed in this paper do not seem to be related to this peculiar mode of operation of the pump.
Concluding Remarks
The results presented in this paper show a considerable influence of salt-concentration and special anions on Na,K-ATPase. The quantitative treatment of ligand binding to the substrate site, and the discussion of previous theoretical and experimental work show the versatility of regarding the unspecific salt effect as an effect of ionic strength. This, and the specific interactions with the anions NO3 −, SCN−, and ClO4 −, signifies the importance of electrostatic interactions between substrate and enzyme. Similar measurements of the Na,K-ATPase and the K+-pNPPase activity reveal that also other steps in the reaction mechanism are dependent on such interactions.
Acknowledgments
This work was supported by EU Contract CI1*CT93-0048 and HFSP-grant RG-511/95 M.
Footnotes
The expert technical assistance of Edith Bjørn Møller, Birthe Bjerring Jensen, and Angielina Damgaard is gratefully acknowledged, and we are thankful to our colleague Dr. Jørgen Jensen for his collaboration in the early phases of this study. We also thank professors emeriti Jacqueline A. Reynolds and Charles Tanford, Easingwold, UK, for helpful suggestions, and Dr. Igor W. Plesner, University of Aarhus, for discussions, on kinetic and thermodynamic aspects of ionic strength effects. We are grateful to Dr. Haruki Nakamura, Biomolecular Engineering Research Institute, Osaka, Japan, for sending us a preprint of his review in advance of its publication.
Abbreviations used in this paper: CDTA, trans-1,2-cyclohexylenedinitrilotetraacetic acid; K+-pNPPase, K+-activated p-nitrophenylphosphatase; pNPP, p-nitrophenylphosphate.
In the Debye-Hückel expression, Eq. 5
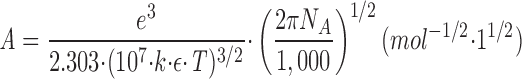 ,
, 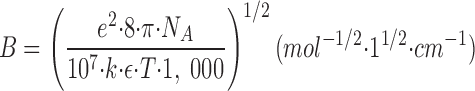 , where e (the protonic charge) = 4.8 × 10−10 (electrostatic U), N
A (Avogadros number) = 6.023 × 1023 (mol−1), k (Boltzmann's constant) = 1.38 × 10−23 (Joule · K
−1), ε (the dielectric constant of water) = 78.5 at 25°C, and T (the temperature in K). In the temperature range 0–40°C the product εT decreases by less than 5%, so A and B are quite independent of temperature.
, where e (the protonic charge) = 4.8 × 10−10 (electrostatic U), N
A (Avogadros number) = 6.023 × 1023 (mol−1), k (Boltzmann's constant) = 1.38 × 10−23 (Joule · K
−1), ε (the dielectric constant of water) = 78.5 at 25°C, and T (the temperature in K). In the temperature range 0–40°C the product εT decreases by less than 5%, so A and B are quite independent of temperature.
references
- Alberty RA, Hammes GG. Application of the theory of diffusion-controlled reactions to enzyme kinetics. J Phys Chem. 1958;62:154–159. [Google Scholar]
- Allewell NM, Oberoi H. Electrostatic effects in protein folding, stability, and function. Methods Enzymol. 1991;202:3–19. doi: 10.1016/0076-6879(91)02003-r. [DOI] [PubMed] [Google Scholar]
- Argese E, Viglino P, Rotilio G, Scarpa M, Rigo A. Electrostatic control of the rate-determining step of the copper, zinc superoxide dismutase catalytic reaction. Biochemistry. 1987;26:3224–3228. doi: 10.1021/bi00385a043. [DOI] [PubMed] [Google Scholar]
- Beaugé L, Berberián G, Campos M. Potassium-p-nitrophenyl phosphate interactions with (Na++K+)-ATPase. Their relevance to phosphatase activity. Biochim Biophys Acta. 1984;773:157–164. doi: 10.1016/0005-2736(84)90560-1. [DOI] [PubMed] [Google Scholar]
- Blumwald E, Rea PA, Poole RA. Preparation of tonoplast vesicles: application to H+-coupled secondary transport in plant vacuoles. Methods Enzymol. 1987;148:115–123. [Google Scholar]
- Campos M, Berberián G, Beaugé L. Phosphatase activity of Na+/K+-ATPase. Enzyme conformations from ligand interactions and Rb occlusion experiments. Biochim Biophys Acta. 1988;940:43–50. doi: 10.1016/0005-2736(88)90006-5. [DOI] [PubMed] [Google Scholar]
- Christoffersen M, Bolvig S, Tüchsen E. Salt effects on the amide hydrogen exchange of bovine pancreatic trypsin inhibitor. Biochemistry. 1996;35:2309–2315. doi: 10.1021/bi951711q. [DOI] [PubMed] [Google Scholar]
- Cleland WW. The kinetics of enzyme-catalyzed reactions with two or more substrates or products. III. Prediction of initial velocity and inhibition patterns by inspection. Biochim Biophys Acta. 1963;67:188–196. doi: 10.1016/0006-3002(63)91816-x. [DOI] [PubMed] [Google Scholar]
- Collins KD, Washabaugh MW. The Hofmeister effect and the behaviour of water at interphases. Q Rev Biophys. 1985;18:323–422. doi: 10.1017/s0033583500005369. [DOI] [PubMed] [Google Scholar]
- Debye P, Hückel E. Zur Theorie der Elektrolyte. I. Gefrierpunktsniedrigung und verwandte Erscheinungen. Physik Z. 1923;24:185–208. [Google Scholar]
- Dissing S, Hoffman JF. Anion-coupled Na efflux mediated by the human red cell Na/K pump. J Gen Physiol. 1990;96:167–193. doi: 10.1085/jgp.96.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edsall, J.T., and J. Wyman. 1958. Biophysical Chemistry. Vol. 1. Academic Press, New York. pp. 699.
- Esmann M. ATPase and phosphatase activity of Na+,K+-ATPase: molar and specific activity, protein determination. Methods Enzymol. 1988;156:105–115. doi: 10.1016/0076-6879(88)56013-5. [DOI] [PubMed] [Google Scholar]
- Esmann M. Conformational transitions of detergent-solubilized Na,K-ATPase are conveniently monitored by the fluorescent probe 6-carboxy-eosin. Biochem Biophys Res Commun. 1991;174:63–70. doi: 10.1016/0006-291x(91)90485-p. [DOI] [PubMed] [Google Scholar]
- Esmann M. Determination of rates of nucleotide binding and dissociation from Na,K-ATPase. Biochim Biophys Acta. 1992;1110:20–28. doi: 10.1016/0005-2736(92)90289-x. [DOI] [PubMed] [Google Scholar]
- Esmann, M., and N.U. Fedosova. 1997. Eosin as a probe for conformational transitions and nucleotide binding in Na,K-ATPase. Ann. NY Acad. Sci. In press. [DOI] [PubMed]
- García-Moreno E B. Estimating binding constants for site-specific interactions between monovalent ions and proteins. Methods Enzymol. 1994;240:645–667. doi: 10.1016/s0076-6879(94)40067-9. [DOI] [PubMed] [Google Scholar]
- Getzoff ED, Cabelli DE, Fisher CL, Parge HE, Viezzoli MS, Banci L, Hallewell RA. Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature (Lond) 1992;358:347–351. doi: 10.1038/358347a0. [DOI] [PubMed] [Google Scholar]
- Glynn IM, Richards DE. Occlusion of rubidium by the sodium-potassium pump: its implications for the mechanism of potassium transport. J Physiol (Lond) 1982;303:17–43. doi: 10.1113/jphysiol.1982.sp014326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green WN, Andersen OS. Surface charge and ion channel function. Annu Rev Physiol. 1991;53:341–359. doi: 10.1146/annurev.ph.53.030191.002013. [DOI] [PubMed] [Google Scholar]
- Hammes GG, Alberty RA. The influence of net protein charge on the rate of formation of enzyme-substrate complexes. J Phys Chem. 1959;63:274–279. [Google Scholar]
- Hegyvary C, Post RL. Binding of adenosine triphosphate to sodium and potassium ion-stimulated adenosine triphosphatase. J Biol Chem. 1971;246:5234–5240. [PubMed] [Google Scholar]
- Jacoby B. Characterization of tonoplast enzyme activities and transport. Methods Enzymol. 1987;148:105–114. [Google Scholar]
- Jensen J. Heterogeneity of pig kidney Na,K-ATPase as indicated by ADP- and ouabain-binding stoichiometry. Biochim Biophys Acta. 1992;1110:81–87. doi: 10.1016/0005-2736(92)90297-y. [DOI] [PubMed] [Google Scholar]
- Jensen J, Nørby JG, Ottolenghi P. Binding of sodium and potassium to the sodium pump of pig kidney evaluated from nucleotide-binding behaviour. J Physiol (Lond) 1984;346:219–241. doi: 10.1113/jphysiol.1984.sp015018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J, Ottolenghi P. ATP binding to solubilized (Na++K+)-ATPase. The abolition of subunit-subunit interaction and the maximum weight of the nucleotide binding unit. Biochim Biophys Acta. 1983a;731:282–289. doi: 10.1016/0005-2736(83)90020-2. [DOI] [PubMed] [Google Scholar]
- Jensen J, Ottolenghi P. Binding of Rb+and ADP to a potassium-like form of Na,K-ATPase. Curr Top Membr Transp. 1983b;19:223–227. [Google Scholar]
- Johnson KA, Taylor EW. Intermediate states of subfragment 1 and actosubfragment 1 ATPase: reevaluation of mechanism. Biochemistry. 1978;17:3432–3442. doi: 10.1021/bi00610a002. [DOI] [PubMed] [Google Scholar]
- Johnson, M.J. 1960. Enzymic equilibria and thermodynamics. In The Enzymes, Vol. 3B, second edition. P.D. Boyer, H. Lardy, and K. Myrbäck, editors. Academic Press, New York/London. 407–441.
- Jørgensen PL. Purification and characterization of (Na++K+)-ATPase. III. Purification from the outer medulla of mammalian kidney after selective removal of membrane components by sodium dodecylsulphate. Biochim Biophys Acta. 1974;356:36–52. doi: 10.1016/0005-2736(74)90292-2. [DOI] [PubMed] [Google Scholar]
- Jørgensen PL. Purification and characterization of (Na+,K+)-ATPase. V. Conformational changes in the enzyme. Transitions between the Na-form and the K-form studied with tryptic digestion as a tool. Biochim Biophys Acta. 1975;401:399–415. doi: 10.1016/0005-2736(75)90239-4. [DOI] [PubMed] [Google Scholar]
- Jørgensen PL. Purification of Na+,K+-ATPase: enzyme sources, preparative problems, and preparation from mammalian kidney. Methods Enzymol. 1988;156:29–43. doi: 10.1016/0076-6879(88)56005-6. [DOI] [PubMed] [Google Scholar]
- Kaplan, J.H. 1991. Localization of ligand binding from studies of chemical modification. In The Sodium Pump: Structure, Mechanism and Regulation. J.H. Kaplan and P. De Weer, editors. Society of General Physiologists series, Vol. 46. The Rockefeller University Press, New York. 117–128. [PubMed]
- Kielland J. Individual activity coefficients of ions in aqueous solutions. J Am Chem Soc. 1937;59:1675–1678. [Google Scholar]
- Kirkwood, G., and I. Oppenheim. 1961. Chemical Thermodynamics. McGraw-Hill Book Co., New York/Toronto/London. pp. 261.
- Klodos, I. 1991. Effect of lyotropic anions on the dephosphorylation of Na,K-ATPase phosphointermediates. In The Sodium Pump, Recent Developments. J.H. Kaplan and P. De Weer, editors. Society of General Physiologists series, Vol. 46. The Rockefeller University Press, New York. 333–337.
- Klodos, I., and B. Forbush III. 1991. Transient kinetics of dephosphorylation of Na,K-ATPase after dilution of NaCl. In The Sodium Pump, Recent Developments. J.H. Kaplan and P. De Weer, editors. Society of General Physiologists series, Vol. 46. The Rockefeller University Press, New York. 327–331.
- Klodos I, Post RL, Forbush B., III Kinetic heterogeneity of phosphoenzyme of Na,K-ATPase modeled by unmixed phases. Competence of the phosphointermediate. J Biol Chem. 1994;269:1734–1743. [PubMed] [Google Scholar]
- Kortüm, G., W. Vogel, and K. Andrussow. 1961. Dissociation Constants of Organic Acids in Aqueous Solution. (IUPAC) Butterworths, London. pp. 536.
- Koyal D, Rao SN, Askari A. Studies on the partial reactions catalyzed by the (Na++K+)-activated ATPase. I. Effects of simple anions and nucleoside triphosphates on the alkali-cation specificity of the p-nitrophenylphosphatase. Biochim Biophys Acta. 1971;225:11–19. doi: 10.1016/0005-2736(71)90278-1. [DOI] [PubMed] [Google Scholar]
- Kraut J. How do enzymes work? . Science (Wash DC) 1988;242:533–540. doi: 10.1126/science.3051385. [DOI] [PubMed] [Google Scholar]
- Kreevoy, M.M, and D.G. Truhlar. 1986. Transition state theory. In Investigations of Rates and Mechanisms of Reactions. Part I. fourth edition. C. Bernasconi, editor. John Wiley and Sons, New York. 14–95.
- Leberman R. The Hofmeister series and ionic strength. FEBS Lett. 1991;284:293–294. doi: 10.1016/0014-5793(91)80707-a. [DOI] [PubMed] [Google Scholar]
- Leberman R, Soper AK. Effect of high salt concentrations on water structure. Nature (Lond) 1995;378:364–366. doi: 10.1038/378364a0. [DOI] [PubMed] [Google Scholar]
- Lingrel JB, Kuntzweiler T. Na,K-ATPase. J Biol Chem. 1994;269:19659–19662. [PubMed] [Google Scholar]
- Loewenthal R, Sancho J, Reinikainen T, Fersht AR. Long-range surface charge-charge interactions in proteins. Comparison of experimental results with calculations from a theoretical method. J Mol Biol. 1993;232:574–583. doi: 10.1006/jmbi.1993.1412. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Marín R, Hoffman JF. Phosphate from the phosphointermediate (EP) of the human red blood cell Na/K pump is coeffluxed with Na, in the absence of external K. J Gen Physiol. 1994a;104:1–32. doi: 10.1085/jgp.104.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín R, Hoffman JF. ADP+orthophosphate (Pi) stimulates an Na/K pump-mediated coefflux of Piand Na in human red blood cell ghosts. J Gen Physiol. 1994b;104:33–55. doi: 10.1085/jgp.104.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson P, Froment M-T, Bartels CF, Lockridge O. Asp70 in the peripheral anionic site of human butyrylcholineesterase. Eur J Biochem. 1996;235:36–48. doi: 10.1111/j.1432-1033.1996.00036.x. [DOI] [PubMed] [Google Scholar]
- Matthew JB. Electrostatic effects in proteins. Annu Rev Biophys Biophys Chem. 1985;14:387–417. doi: 10.1146/annurev.bb.14.060185.002131. [DOI] [PubMed] [Google Scholar]
- McLaughlin S. The electrostatic properties of membranes. Annu Rev Biophys Biophys Chem. 1989;18:113–136. doi: 10.1146/annurev.bb.18.060189.000553. [DOI] [PubMed] [Google Scholar]
- Nakamura H. Roles of electrostatic interactions in proteins. Q Rev Biophys. 1996;29:1–90. doi: 10.1017/s0033583500005746. [DOI] [PubMed] [Google Scholar]
- Nørby JG, Jensen J. Binding of ATP to brain microsomal ATPase. Determination of the ATP-binding capacity and the dissociation constant of the enzyme-ATP complex as a function of K+concentration. Biochim Biophys Acta. 1971;233:104–116. doi: 10.1016/0005-2736(71)90362-2. [DOI] [PubMed] [Google Scholar]
- Nørby JG, Jensen J. Binding of ATP to Na+,K+-ATPase. Ann NY Acad Sci. 1974;242:158–167. doi: 10.1111/j.1749-6632.1974.tb19088.x. [DOI] [PubMed] [Google Scholar]
- Nørby JG, Jensen J. Measurement of binding of ATP and ADP to Na+,K+-ATPase. Methods Enzymol. 1988;156:191–201. doi: 10.1016/0076-6879(88)56021-4. [DOI] [PubMed] [Google Scholar]
- Nørby JG, Klodos I, Christiansen NO. Kinetics of the Na-ATPase activity by the Na,K-pump. Interactions of the phosphorylated intermediates with Na+, Tris+, and K+ . J Gen Physiol. 1983;82:725–759. doi: 10.1085/jgp.82.6.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottolenghi P, Jensen J. The K+-induced apparent heterogeneity of high-affinity nucleotide binding-sites in (Na++K+)- ATPase can only be due to the oligomeric structure of the enzyme. Biochim Biophys Acta. 1983;727:89–100. doi: 10.1016/0005-2736(83)90372-3. [DOI] [PubMed] [Google Scholar]
- Palmer, T. 1991. Understanding Enzymes, third ed. Ellis Horwood, New York. pp. 399.
- Parsegian VA. Hopes for Hofmeister. Nature (Lond) 1995;378:335–336. [Google Scholar]
- Pedersen PL, Carafoli E. Ion motive ATPases. I. Ubiquity, properties, and significance to cell function. TIBS. 1987;12:146–150. [Google Scholar]
- Pedersen PA, Rasmussen JH, Jørgensen PL. Expression in high yield of pig α1β1 Na,K-ATPase and inactive mutants D369N and D807N in Saccharomyces cerevisiae. . J Biol Chem. 1996;271:2514–2522. doi: 10.1074/jbc.271.5.2514. [DOI] [PubMed] [Google Scholar]
- Perrin, D.D. 1965. Dissociation Constants of Organic Bases in Aqueous Solution. (IUPAC) Butterworths, London. pp. 473.
- Plesner I. The apparent Kmis a misleading kinetic indicator. Biochem J. 1986;239:175–178. doi: 10.1042/bj2390175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post, R.L., and K. Suzuki. 1991. A Hofmeister effect on the phosphoenzyme of Na,K-ATPase. In The Sodium Pump: Structure, Mechanism, and Regulation. J.H. Kaplan and P. De Weer, editors. Society of General Physiologists series, Vol. 46. The Rockefeller University Press, New York. 201–209. [PubMed]
- Record MT, Jr, Anderson CF, Lohman TM. Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: the roles of ion association or release, screening, and ion effects on water activity. Q Rev Biophys. 1978;11:103–178. doi: 10.1017/s003358350000202x. [DOI] [PubMed] [Google Scholar]
- Robinson, R.A., and R.H. Stokes. 1970. Electrolyte Solutions, second edition. Butterworth, London. pp. 571.
- Rossi RC, Nørby JG. Kinetics of K+-stimulated dephosphorylation and simultaneous K+ occlusion by Na,K-ATPase, studied with the K+ congener Tl+. The possibility of differences between the first turnover and steady state. J Biol Chem. 1993;268:12579–12590. [PubMed] [Google Scholar]
- Russell AJ, Thomas PG, Fersht AR. Electrostatic effects on modification of charged groups in the active site cleft of subtilisin by protein engineering. J Mol Biol. 1987;193:803–813. doi: 10.1016/0022-2836(87)90360-3. [DOI] [PubMed] [Google Scholar]
- Segel, I.H. 1975. Enzyme Kinetics. Behaviour and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. John Wiley and Sons, New York. pp. 957.
- Skou JC. Effect of ATP on the intermediary steps of the reaction of the (Na++K+)- dependent enzyme system. III. Effect on the p-nitrophenylphosphatase activity of the system. Biochim Biophys Acta. 1974;339:258–273. [PubMed] [Google Scholar]
- Skou JC. Effects of ATP on the intermediary steps of the reaction of the (Na++K+)- ATPase. IV. Effect of ATP on K0.5 for Na+and on hydrolysis at different pH and temperature. Biochim Biophys Acta. 1979;567:421–435. doi: 10.1016/0005-2744(79)90128-1. [DOI] [PubMed] [Google Scholar]
- Skou JC, Esmann M. Effects of ATP and protons on the Na:K selectivity of the (Na++K+)-ATPase studied by ligand effects on intrinsic and extrinsic fluorescence. Biochim Biophys Acta. 1980;601:386–402. doi: 10.1016/0005-2736(80)90543-x. [DOI] [PubMed] [Google Scholar]
- Skou JC, Esmann M. Eosin, a fluorescent probe of ATP binding to the (Na++K+)-ATPase. Biochim Biophys Acta. 1981;647:232–240. doi: 10.1016/0005-2736(81)90251-0. [DOI] [PubMed] [Google Scholar]
- Snyder GH, Cennerazzo MJ, Karalis AJ, Field D. Electrostatic influence of local cysteine environments on disulfide exchange kinetics. Biochemistry. 1981;20:6509–6519. doi: 10.1021/bi00526a001. [DOI] [PubMed] [Google Scholar]
- Sze H. H+-translocating ATPases: advances using membrane vesicles. Annu Rev Plant Physiol. 1985;36:175–208. [Google Scholar]
- Tanford, C. 1961. Physical Chemistry of Macromolecules. John Wiley and Sons, Inc., New York/London/Sidney. pp. 710.
- Wang K, Farley RA. Lysine 480 is not an essential residue for ATP binding or hydrolysis by Na,K-ATPase. J Biol Chem. 1992;267:3577–3580. [PubMed] [Google Scholar]
- Warshel A, Russell ST. Calculation of electrostatic interactions in biological systems and in solutions. Q Rev Biophys. 1984;17:283–422. doi: 10.1017/s0033583500005333. [DOI] [PubMed] [Google Scholar]
- Watts, D.C. 1973. Creatine kinase (adenosine 5'-triphosphate-creatine phosphotransferase). In The Enzymes, Vol. IIIA, third edition. P.D. Boyer, editor. Academic Press, London. 383–455.
- Yamazaki A, Kaya S, Tsuda T, Araki Y, Hayashi Y, Taniguchi K. An extra phosphorylation of Na+,K+-ATPase by paranitrophenylphosphate (pNPP): evidence for the oligomeric nature of the enzyme. J Biochem (Tokyo) 1994;116:1360–1369. doi: 10.1093/oxfordjournals.jbchem.a124688. [DOI] [PubMed] [Google Scholar]