

Abstract
We used patch clamp techniques to study the inhibitory effects of pentobarbital and barbital on nicotinic acetylcholine receptor channels from BC3H-1 cells. Single channel recording from outside-out patches reveals that both drugs cause acetylcholine-activated channel events to occur in bursts. The mean duration of gaps within bursts is 2 ms for 0.1 mM pentobarbital and 0.05 ms for 1 mM barbital. In addition, 1 mM barbital reduces the apparent single channel current by 15%. Both barbiturates decrease the duration of openings within a burst but have only a small effect on the burst duration. Macroscopic currents were activated by rapid perfusion of 300 μM acetylcholine to outside-out patches. The concentration dependence of peak current inhibition was fit with a Hill function; for pentobarbital, K i = 32 μM, n = 1.09; for barbital, K i = 1900 μM, n = 1.24. Inhibition is voltage independent. The kinetics of inhibition by pentobarbital are at least 30 times faster than inhibition by barbital (3 ms vs. <0.1 ms at the K i). Pentobarbital binds ≥10-fold more tightly to open channels than to closed channels; we could not determine whether the binding of barbital is state dependent. Experiments performed with both barbiturates reveal that they do not compete for a single binding site on the acetylcholine receptor channel protein, but the binding of one barbiturate destabilizes the binding of the other. These results support a kinetic model in which barbiturates bind to both open and closed states of the AChR and block the flow of ions through the channel. An additional, lower-affinity binding site for pentobarbital may explain the effects seen at >100 μM pentobarbital.
Keywords: pentobarbital, barbital, anesthetic, patch clamp, ion channels
introduction
Barbiturates have several therapeutic effects on humans including light sleep (at low doses), deep coma (at high doses), amnesia, muscle relaxation, protection against cerebral ischemia, and reversal of seizures (Fragen, 1994). Thus, these drugs probably have multiple effects on the central and peripheral nervous systems. Ion channels are among the possible targets of barbiturates. Barbiturates are known to affect many types of ion channels including GABAA receptor channels (Tanelian et al., 1993), some (ffrench-Mullen et al., 1993) but not all (Hall et al., 1994) calcium channels, sodium channels (Frenkel et al., 1990; Barann et al., 1993), glutamate receptor channels (Marszalec and Narahashi, 1993), 5-HT3 receptor channels (Barann et al., 1993), and muscle-type nicotinic acetylcholine receptor (AChR)1 channels (deArmendi et al., 1993).
Inhibition of AChR by barbiturates has been studied with electrophysiological (Lee-Son et al., 1975; Gage and McKinnon, 1985; Jacobson et al., 1991; Yost and Dodson, 1993), flux (Firestone et al., 1986b ; Roth et al., 1989; deArmendi et al., 1993), and binding (Dodson et al., 1990) techniques. Several effects have been observed (Firestone et al., 1986b , 1994), but the dominant effect is a direct inhibitory action of the drug on the open state of the channel. The potency of barbiturates for inhibiting the channel is related to but not completely determined by lipid solubility (deArmendi et al., 1993). A study of the single channel kinetics in the presence of pentobarbital (PB) allowed Gage and McKinnon (1985) to dismiss a simple, sequential open channel blocking mechanism for PB action. They suggested that the mechanism might be allosteric in that the binding of one molecule of PB to the open channel protein induces a conformational change to a new closed state of the channel. In this scenario, the binding of PB is not concomitant with inhibition.
Here, we use several patch clamp recording protocols to study the effects of PB and barbital (Barb) on nicotinic AChRs in outside-out patches from BC3H-1 cells. We examine single channel kinetics, the equilibrium and kinetic properties of macroscopic currents, and interactions between PB and Barb. We conclude that inhibition of the AChR by barbiturates is temporally coincident with binding of the drug to a site on the channel protein. This can be described by a model in which barbiturates bind to both the open and closed states of the channel. PB shows a strong preference for binding to the open state. The binding sites for PB and Barb do not coincide but are probably close to each other. There is evidence for an additional, lower affinity binding site for PB.
materials and methods
BC3H-1 cells that express the α2βγδ-type nicotinic AChR were cultured as described previously (Sine and Steinbach, 1984). To prepare cells for patch clamp recording, the culture medium was replaced with an “extracellular” solution (ECS) containing (in mM): NaCl (150), KCl (5.6), CaCl2 (1.8), MgCl2 (1.0), and HEPES (10), pH 7.3. Patch pipettes were filled with a solution consisting of (in mM) KCl (140), EGTA (5), MgCl2 (5), and HEPES (10), pH 7.3, and had resistances of 3–6 MΩ. An outside-out patch (Hamill et al., 1981) with a seal resistance of 10 GΩ or greater was obtained from a cell and moved into position at the outflow of a perfusion system. The perfusion system consisted of solution reservoirs, manual switching valves, and a V-shaped piece of plastic tubing inserted into the culture dish (Liu and Dilger, 1991). For single channel current measurements, the manual valves were used to switch from drug-free to drug-containing solutions containing 0.2 μM ACh. For macroscopic current measurements, the perfusion system also contained a solenoid-driven pinch valve. One arm of the “V” contained ECS without agonist (normal solution); the other arm contained ECS with 300 μM ACh (test solution). In the resting position of the pinch valve, normal solution perfused the patch. The pinch valve was then triggered to stop the flow of normal solution and start the flow of test solution. After the patch was exposed to test solution for 0.2 s, the pinch valve was returned to its resting position for several seconds. In this way, we treated the patch to a series of timed exposures to agonist-containing solution while minimizing the desensitizing effects of prolonged exposure to high concentrations of ACh. The perfusion system allows for a rapid (0.1–1 ms) exchange of the solution bathing the patch.
On the day of the experiment, we prepared a stock solution of 20 mM Barb or 1 mM PB (Sigma Chemical Co., St. Louis, MO) in ECS. The 20 mM Barb solution had a pH of 8.3; this was titrated to pH 7.3 with concentrated HCl. The stock solution was then diluted with ECS to obtain the desired concentration of barbiturate. The solution was transferred to a perfusion reservoir, a plastic intravenous drip bag.
The currents flowing during exposure of the patch to ACh were measured with a patch clamp amplifier (EPC-7; List Electronic, Darmstadt, Germany), filtered at 3 kHz (−3 db frequency, 8-pole Bessel filter, 902LPF; Frequency Devices, Haverhill, MA), digitized and stored on the hard disk of a laboratory computer (PDP-11-73; Digital Equipment Corp., Maynard, MA). Data analysis was performed off-line with the aid of our own computer programs. Experiments were performed at room temperature (20– 23°C). Results are expressed as means ± SD.
For macroscopic currents, the first step was to record current responses (at −50 mV) during 200-ms applications (at 5-s intervals and sampled at 100–200 μs per point) of ECS containing 300 μM ACh, a concentration that opens ∼95% of the AChR channels from BC3H-1 cells (Dilger and Brett, 1990). This current served as a reference point for estimating the number of channels in the patch. We returned to this test solution frequently during the life of the patch to quantify any loss of channel activity. Both the normal and test perfusion solutions were then switched to barbiturate-containing solutions by means of manual valves. Responses of the patch to applications of 300 μM ACh during continuous exposure to barbiturate were recorded. The drug-free solutions were then re-introduced, and recovery currents were measured. This protocol was continued with other barbiturate concentrations until the demise of the seal or a large loss of channel activity (anywhere from 10 min to 3 h). Data were accepted when the drug-free currents obtained before and after exposure to drug had not changed by >10%. For some experiments (see Figs. 10 and 11), only the test solution contained barbiturate. Using this protocol, a simultaneous jump in the concentration of both ACh and barbiturate was made. The resulting current provides information about the kinetics of drug inhibition.
Figure 10.

High time-resolution traces showing the onset of macroscopic current inhibition by 100 μM PB. The trace labeled “control” was obtained in the absence of PB. The trace labeled “equilibrium” was obtained when both normal (agonist-free) and test (agonist-containing) solutions contained 100 μM PB. The trace labeled “onset” was obtained when only the test solution contained PB. The close similarity between the “equilibrium” and “onset” traces indicates that PB does not interact strongly with closed channels. 300 μM ACh, −50 mV.
Figure 11.

Very high time-resolution traces showing the onset of macroscopic current inhibition by 5 mM Barb. Currents evoked by 10 mM ACh at an applied potential of +50 mV. See legend to Fig. 10 for definitions of labels. The kinetics of current inhibition by Barb are much faster than those of PB. It is not possible to determine whether Barb interacts with closed channels.
The ensemble mean current was calculated from between 10 and 60 individual current traces. Mean currents were fit to a 1- or 2-exponential function.
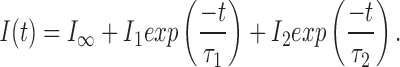 |
1 |
The time constant of the 1-exponential fit and the slower time constant of the 2-exponential fit, τ1, measures the current decay due to desensitization (Dilger and Liu, 1992). In the presence of PB, the current contained an additional fast component, τ2. This represents the time course of inhibition by PB (see results). Fractional inhibition of the peak mean current, I p, the maximum inward current obtained after perfusing ACh, was calculated as the ratio of the current in the presence of drug, I p′ to the current in the absence of drug, I p. For PB experiments, I p′ was obtained from the amplitude of the slow component of the decay, I p′ = I Θ + I 2 (see results).
Single channel recordings were made while the patch was exposed to ECS + 0.2 μM ACh at a patch potential of −100 mV. Data was digitized in 5-s segments at a rate of 50 μs per point. 3–10 data segments were collected (enough to obtain 200–1,000 single channel events, depending on the channel activity in the patch). Data collection was repeated with ECS + ACh + barbiturate and then again with ECS + ACh (recovery). Data were accepted if, after analysis, we found that the channel kinetics during recovery were within 20% of the values obtained during the initial data collection segments.
Single channel analysis consisted of identifying opening and closing transitions, obtaining the distribution of open, closed, and burst durations, and fitting the distributions (expressed as the number of events vs. log-binned duration, 10 bins per decade) to 1- and 2-exponential probability distribution functions by finding the maximum log-likelihood using a simplex algorithm. The single-exponential fit was considered adequate when the fractional amplitude of one of the components of the 2-exponential fit was <0.1. The definition of bursts was based on the distribution of short (gap) and long closures (Colquhoun and Sakmann, 1985). Mean gap and open durations and the number of openings per burst were corrected for undetected events using equations derived for a two-state mechanism (Colquhoun and Hawkes, 1995a ). This approximation is probably adequate because of the large time separation between brief and long closed durations and our observation that the open time histogram in the presence of barbiturate usually has only one component. Mean single channel amplitudes were calculated by taking the average of the amplitudes of those openings lasting >0.25 ms; these are not attenuated by the 3 kHz low-pass filter.
results
The Effects of Barbital and Pentobarbital on AChR Single Channel Currents
The effects of Barb and PB on single AChR channels are illustrated in Fig. 1. Inward single channel currents at −100 mV were activated by a low concentration of ACh (0.2 μM). Under control conditions (Fig. 1, left), channel activity consists of −4 pA openings lasting an average of 4 ms and separated by tens of milliseconds. Occasionally, a brief closing transition is seen. Both 100 μM PB and 500 μM Barb (Fig. 1, right) induce a bursting pattern of channel activity. The closures within a burst are much longer for PB than for Barb. The Barb-induced closures are so brief that the single channel amplitude appears to be attenuated by ∼8%. The attenuation is more pronounced at 1,000 μM Barb (see Fig. 5 D).
Figure 1.

Single AChR channels in control and in the presence of 100 μM PB or 500 μM Barb. Channels activated by 0.2 μM ACh, −100 mV, two separate outside-out patches. Both barbiturates induce a bursting behavior of the channels but the closures within the bursts are longer for PB than for Barb. Patches L82 and L105.
Figure 5.

The Barb concentration dependence of channel kinetics and amplitudes. (A) Open and burst durations (longer components are used when two components are present). (B) The number of openings per burst. (C) The duration of gaps between bursts. (D) The single channel current amplitude. The solid lines represent the predictions of scheme i with the kinetic parameters given in Table I. 0.2 μM ACh, −100 mV.
The closed duration histograms constructed from single channel data have two components (Fig. 2). The dominant component of the control histogram occurs near 60 ms and corresponds to the time between activation of different channels in the patch. For 0.2 μM ACh, a long closed time of 60 ms indicates that there are ∼150 active channels in the patch.2 The small, brief component of the control histograms occurring at <100 μs most likely corresponds to the closing of a channel followed by the rapid reopening of the same channel. The bursting activity of the barbiturates is represented by a large number of gaps: the brief component of the closed time histogram. PB induces gaps near 2 ms; Barb induces gaps near 70 μs. Neither barbiturate has a significant effect on the long closed component.3
Figure 2.

Closed duration histograms compiled from the same two experiments exemplified in Fig. 1. The solid lines are fits to the two-exponential probability distribution function. The long component represents the closed time between openings of different channels in the patch. The brief component represents gaps within a burst of activity of a single channel. Gaps are rarely seen in control recordings but are frequently seen in the presence of a barbiturate. The vertical calibration bar represents 10 events in control (linear scale), see below for vertical scaling of barbiturate histograms. 0.2 μM ACh, −100 mV. (A) Control: 236 events, gap duration = 66 μs (fraction = 0.12); long duration 63 ms. 100 μM PB: 251 events, gap duration = 1.8 ms (0.48); long duration = 140 ms. Calibration bar = 6 events. (B) Control: 509 events: gap duration = 35 μs (0.07); long duration = 51 ms. 500 μM Barb: 632 events: gap duration = 50 μs (0.64); long duration = 66 ms, calibration bar = 50 events.
The barbiturates decrease the open duration of single AChR channels but have little effect on the burst duration (Fig. 3). Under control conditions, there are two components in the open duration histogram; a brief one at 200–500 μs comprising 20–50% of the events and a long one at 4–5 ms. Because there are very few brief closures within a burst, the control burst duration histogram is very similar to the control open duration histogram. In the presence of either 100 μM PB or 500 μM Barb, the open duration histogram collapses to a single component with a time constant of 1.2–1.4 ms. In contrast, there is very little difference between the control and barbiturate burst duration histograms both in the number and time constants of the components.
Figure 3.

Open and burst duration histograms compiled from the same two experiments exemplified in Figs. 1 and 2. The open duration histogram normally has two components. In the presence of either barbiturate, the open histogram collapses to a single component with an intermediate duration. The burst duration histograms contain two components under control and with exposure to barbiturate. There is very little difference between the control and barbiturate burst duration histograms. The vertical calibration bar represents 10 events in control (linear scale), see below for vertical scaling of barbiturate histograms. 0.2 μM ACh, −100 mV. (A) Open durations. Control: 226 events, τfast = 0.48 ms (.35), τslow = 4.9 ms. 100 μM PB: 251 events, τ = 1.3 ms, calibration bar = 14 events. (B) Open durations. Control: 304 events, τfast = 0.26 ms (.34), τslow = 4.1 ms. 500 μM Barb: 723 events, τ = 1.2 ms, calibration bar = 25 events. (C) Burst durations. Control: 212 events, 1.07 openings per burst, τfast = 0.96 ms (.46), τslow = 6.3 ms. 100 μM PB: 136 events, 1.85 openings per burst, τfast = 0.21 ms (.25), τslow = 4.7 ms, calibration bar = 7 events. (D) Burst durations. Control: 294 events, 1.03 openings per burst, τfast = 0.23 ms (.33), τslow = 4.2 ms. 500 μM Barb: 380 events, 1.90 openings per burst, τfast = 0.16 ms (.28), τslow = 3.3 ms, calibration bar = 13 events.
The results from single channel experiments on 15 patches with PB and 6 patches with Barb are summarized in Figs. 4 and 5. We have plotted the concentration dependence of the open, τopen, and burst, τburst, durations (the longer component when there are two components) (Figs. 4 A and 5 A), the number of openings per burst, Nopen/burst, (Figs. 4 B and 5 B), the gap duration, τgap, (Figs. 4 C and 5 C), and current amplitude (Figs. 4 D and 5 D). Both barbiturates cause a monotonic decrease in the open duration with 50 μM PB or <200 μM Barb causing a 50% decrease in τopen. The burst duration is nearly constant except for [PB] > 100 μM. The number of openings per burst increases to 1.5 and 5 at high concentrations of PB and Barb, respectively. The duration of PB-induced gaps varies between 1 and 4 ms. The gap duration at 25 μM PB may be an underestimate because the closed time component probably consists of a mixture of PB-induced gaps and the briefer channel closures seen in control. For Barb, the gap duration is 40–60 μs. The single channel current amplitude is independent of [PB] but there is a decrease in the absolute value of the apparent current amplitude with increasing concentrations of Barb.
Figure 4.

The PB concentration dependence of channel kinetics and amplitudes. (A) Open and burst durations (longer components are used when two components are present). (B) The number of openings per burst. (C) The duration of gaps between bursts. (D) The single channel current amplitude. The solid lines represent the predictions of scheme SI with the kinetic parameters given in Table I. The dashed lines represent predictions of scheme SIII for burst duration, gap duration, and openings per burst. 0.2 μM ACh, −100 mV.
For Barb, many gaps are not detected when a filter frequency of 3 kHz and sampling time of 50 μs are used. To verify that the correction procedure for accounting for missed events provides good estimates of the mean open and gap durations, we studied six patches using a cutoff frequency of 6 kHz, a sampling time of 25 μs, and an applied voltage of −125 mV. Under these conditions, there was very little variation of the current amplitude with Barb concentration; the amplitude was 4% lower with 1 mM Barb than with control. The gap duration remained 50 μs. At the 6 kHz resolution, a greater number of gaps are resolved, but after correcting for undetected events, the values of Nopen/burst and τopen are no different than at the 3 kHz resolution. The Barb concentration dependence of the burst duration was the same for both the 3 kHz and 6 kHz data.
If we assume that the Barb-induced bursts are composed of brief openings to the fully opened state and brief closures to the fully closed state, we can use a beta function analysis of the amplitude histogram to estimate the open and closed time within bursts (Yellen, 1984). To do this, we applied a 1-kHz Gaussian digital filter to the single channel data and constructed an amplitude distribution from segments containing single bursts. This distribution is then fit to a beta function containing two parameters: the average open and closed times within bursts. For four patches with 1 mM Barb, the average open time was 150 ± 70 μs, and the average closed time was 46 ± 17 μs. This estimate of the open time is shorter than the one obtained by analyzing single channel data with 1 mM Barb (540 ± 110 μs, Fig. 5 A), but the estimate of the closed time is similar to the average gap duration from single channel analysis. This suggests that even after correcting the single channel data for unresolved events, we may overestimate the open time and underestimate the number of openings per burst.
The single channel data suggest that both PB and Barb act, at least qualitatively, as blockers of the AChR channel. In this interpretation, the two barbiturates differ in the duration of blocking events: the less potent drug, Barb, blocks for <0.1 ms, and the more potent drug, PB, blocks for ∼2 ms. In the discussion, we make a quantitative test of models in which barbiturates block both open and closed AChR channels. Before doing so, we present data from macroscopic current experiments that provide additional information about the action of barbiturates on the channel.
The Effects of Barbital and Pentobarbital on Macroscopic AChR Currents
Both Barb and PB inhibit the macroscopic currents evoked by rapid perfusion of ACh. Fig. 6 presents examples of currents activated by 300 μM ACh in control and in the continuous presence of 50 and 400 μM PB (Fig. 6 A) or 2 and 20 mM Barb (Fig. 6 B). In the control traces, the current reaches a peak within <1 ms and then decays with a time constant of 50–60 ms due to desensitization. With 2 or 20 mM Barb, the peak currents are reduced to 60 or 5% of control and desensitization occurs with the same time course as in control. It appears that Barb interacts with the channels either before they are opened by ACh or very quickly after ACh is perfused. With 50 or 400 μM PB, an initial fast decay precedes desensitization. This suggests that, unlike Barb, PB is not very effective at interacting with closed channels. We explore this in more detail below. To determine the degree of inhibition of open channels by PB, we fit the data to a 2-exponential decay and extrapolate the slow component (desensitization) to t = 0 (Fig. 6 B, dotted lines). The extrapolated peak currents are reduced to 35% (50 μM) or 4% (400 μM PB) of control.
Figure 6.


Examples of macroscopic current traces for control, PB, and Barb. Currents activated by 300 μM ACh for 200 ms (the current during removal of ACh is not shown), −50 mV. (A) Comparison of currents activated in control and in the presence of 50 and 400 μM PB in the same patch. Controls were obtained before 50 μM PB, before 400 μM PB, and after 400 μM PB; the control current decreased by 10% during this experiment. The decay in the control traces and the slow decay component in the PB traces has a time constant of 60–70 ms and is due to desensitization in the presence of ACh. The fast decay in the PB traces gives the kinetics of open channel block by PB (3.2 and 1.9 ms for 50 and 400 μM PB, respectively). (B) Comparison of currents activated in control and in the presence of 2 and 20 mM Barb in the same patch (different patch from A). Barb does not exhibit a fast decay component partly because the kinetics of inhibition by Barb are too fast to be resolved in this experiment.
The results from experiments on nine patches with Barb (relative peak currents) and six patches with PB (relative extrapolated peak currents) are summarized in Fig. 7. These data were fit to the Hill equation (Fig. 7, solid lines):
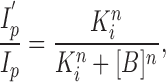 |
2 |
Figure 7.

Concentration-inhibition curves for PB and Barb obtained from measuring the extrapolated peak macroscopic currents (for PB) or peak macroscopic currents (Barb). The solid lines are fits to the Hill equation (Eq. 2); the parameters are given in the text. The dashed line is the prediction of a two-site equilibrium blocking model for PB (Eq. 9), with K 1 = 38 μM and K 2 = 460 μM. 300 μM ACh, −50 mV.
where [B] is the drug concentration, K i is the drug concentration needed for 50% inhibition (IC50), and n is the Hill coefficient. For Barb: K i = 1.9 ± 0.2 mM, n = 1.24 ± 0.07; for PB: K i = 32 ± 2 μM, n = 1.09 ± 0.06. Thus, PB is 60 times more potent than Barb at inhibiting the AChR. Because the Hill slopes are close to unity, it is possible that only one barbiturate molecule is involved in the inhibition of a channel.
The fast decay that occurs in the macroscopic currents with PB provides information about the rate of equilibration of PB with the channel. The decay is faster with higher concentrations of PB. In Fig. 6 A, the time constants are 3.2 and 1.9 ms for 50 and 400 μM PB, respectively. Fig. 8 shows that the fast decay time decreases monotonically with the concentration of PB.
Figure 8.

The PB concentration of τonset obtained from the fast decay component of macroscopic currents. The solid line is a fit of the data to Eq. 8 with f = 4.0 ± 0.6 × 106/M/s and b = 210 ± 20/s. 300 μM ACh, −50 mV.
The inhibitory effect of PB is not voltage dependent. This is illustrated in Fig. 9 in which ACh-activated currents at three different voltages, −100, −50, and +50 mV, are compared for a single patch. The traces are scaled so that the controls have the same peak current level. Neither the time constant of the fast current decay, nor the extrapolated level of the residual current is affected by voltage. There was no difference in the effect of Barb on macroscopic currents over this voltage range either (not shown).
Figure 9.

Superposition of macroscopic current traces obtained with 100 μM PB at +50, −50, and −100 mV. Traces are scaled (as noted on figure) so that the controls coincide. Neither the magnitude nor the time course of current inhibition by PB is affected by voltage. 300 μM ACh.
The macroscopic current data used for Figs. 6–9 were obtained with equilibrium drug concentrations; that is, the barbiturate was present in both the normal and test perfusion solutions. Information about the kinetics of current inhibition by the barbiturates can be obtained from experiments in which the drug is applied simultaneously with ACh. Fig. 10 shows that, for 100 μM PB, the current decay under equilibrium conditions and the current decay after rapid addition of the drug (onset) are identical. This supports the idea that PB has very little interaction with channels when they are closed; prior exposure to PB does not change the degree of inhibition either at the peak or during the decay of the current response.
Rapid addition of Barb also produces a current decay, but on a much faster time scale (Fig. 11). For this experiment, the time resolution was increased by perfusing with 10 mM ACh (this saturates the ACh binding sites within microseconds so that the 20–80% risetime of the current, 40 μs, is determined mainly by the channel opening rate [Liu and Dilger, 1991]), filtering at 15 kHz, sampling at 5 μs per point, and using +50 mV to avoid channel block by ACh. Two pieces of qualitative information about the effects of 5 mM Barb can be extracted from Fig. 11. The equilibrium trace shows that, in contrast to PB, the inhibitory effect of Barb is present at all times after perfusion with ACh. We conclude that Barb either interacts with closed channels to the same degree as it interacts with open channels or, it does not interact with closed channels but equilibrates with open channels extremely quickly, on the order of tens of microseconds or faster. The second observation, that the onset current trace exhibits a 60-μs decay, probably reflects both the binding kinetics of Barb and the time course of the Barb concentration jump. Similarly, the kinetics of recovery from block by Barb show a relaxation from the equilibration level of inhibition to control with a time course of 50 μs (not shown). The time resolution of these experiments is not sufficient to determine if this represents the kinetics of Barb dissociation from its inhibitory site or simply the diffusion of Barb away from the patch.
Interactions between Barbital and Pentobarbital
To determine whether PB and Barb inhibit the AChR channel by binding to a single site on the AChR protein, we performed experiments with both barbiturates. Fig. 12 is an example with 5 mM Barb and 100 μM PB. In the left panel, 100 μM PB decreases the extrapolated peak current to 22% of control. In the right panel, 5 mM Barb decreases the current to 34% of control (note different current scale). When both barbiturates are present, the current decreases to 40% of the 5 mM Barb current (14% of control). Thus, PB is less effective when applied in combination with Barb than when applied by itself. If the two drugs acted independently, the current would have been 22 × 34% or 7.5% of the control. If the two drugs compete for the same binding site, the predicted current is 15% of control (see discussion). The time constant of the fast decay component is 1.6 ms for PB alone and 1.8 ms for both barbiturates in combination.
Figure 12.

Evidence for competitive antagonism between 5 mM Barb and 100 μM PB. Macroscopic AChR currents from a single patch (300 μM ACh, −50 mV, continuous application of barbiturates). At left, 100 μM PB decreases the peak current (after subtraction of fast decay component) to 22% of control. At right, 5 mM Barb decreases the current to 34% of control (note different current scale). When both barbiturates are present, the current decreases to 40% of the 5 mM Barb current (14% of control). Thus, PB is less effective when given in combination with Barb than when given alone.
Inhibition curves for PB alone and PB + 5 mM Barb are shown in Fig. 13. The latter data are normalized to the relative current observed in the presence of 5 mM Barb alone, 0.30 ± 0.02 (n = 25). Fits of the data to the Hill equation (Eq. 2), give K i = 28 ± 2 μM for PB alone and K i = 53 ± 3 μM for PB + 5 mM Barb. For both data sets, the Hill coefficient was close to unity: 1.09 ± 0.07 and 0.96 ± 0.07, respectively. Thus, there is a considerable decrease in the effectiveness of PB when Barb is present. However, this decrease is not as great as would be expected if PB and Barb were competing for a single binding site (Fig. 13, dotted line ; see discussion). In the presence of 5 mM Barb, the time constant of the fast decay seen with PB is decreased at 25 and 50 μM [PB] but is unchanged at [PB] ≥ 100 μM (Fig. 14).
Figure 13.

The concentration-inhibition curves for PB alone and PB with 5 mM Barb. The data for the mixture of PB and Barb are normalized to 1.0 at [PB] = 0. The solid lines are fits to the Hill equation (Eq. 2) with K i = 28 μM, n = 1.09 (PB alone) and K i = 56 μM, n = 0.96 (PB+5 mM Barb). The dashed line is the predicted curve with the assumption that both barbiturates compete for a single binding site to produce inhibition (Eq. 9 with [Barb]/ KBarb = 2.24 and normalized to the inhibition with 5 mM Barb alone, 0.31). 300 μM ACh, −50 mV.
Figure 14.

The PB concentration of τonset obtained from the fast decay component of macroscopic currents in the absence (open symbols) and presence (closed symbols) of 5 mM Barb. The solid lines are fits of the data to Eq. 8 with f = 4.8 × 106/M/s and b = 200/s (PB alone) and f = 3.5 × 106/M/s and b = 340/s (PB + 5 mM Barb). In the presence of Barb, PB binds less quickly and dissociates more quickly. 300 μM ACh, −50 mV.
discussion
Effects Seen with One Barbiturate
The bursting effect of the barbiturates on single AChR channels suggests a model in which drug molecules bind to the AChR and block the flow of ions through the channel. This bursting cannot be explained by a model in which drug molecules bind to and block only the open state of the AChR (purely open channel block) because the expected increase in burst duration (Neher, 1983) is not seen (Figs. 4 A and 5 A). We will test the adequacy of the model shown as scheme SI (Murrell et al., 1991; Dilger et al., 1992) in which barbiturate molecules (B) can bind to both the open (O) and closed (C) conformations of the channel.
Scheme I.
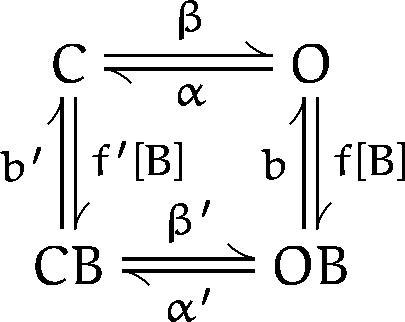
In scheme SI, the various closed states of the AChR (unliganded, singly liganded, and doubly liganded) are represented by a single state. The effective opening rate, β, depends on agonist binding, agonist concentration, and the channel opening rate. The channel closing rate is α. A barbiturate molecule may bind to either the closed state (to form CB) or open state (to form OB); the association (f and f′) and dissociation (b and b′) rates may depend on the channel conformation. The gating transition rates between drug-bound open and closed states (α′ and β′) may differ from the normal gating transition rates. In scheme SI, a single barbiturate molecule is sufficient to inhibit one channel; this is supported by the concentration-inhibition curves for PB and Barb (Fig. 7) that have Hill coefficients close to unity.
scheme SI makes quantitative predictions about the drug concentration dependence of τopen, τburst, τgap, and Nopen/burst (Dilger et al., 1992).
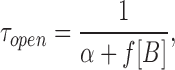 |
3 |
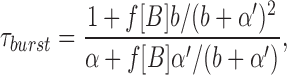 |
4 |
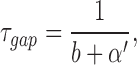 |
5 |
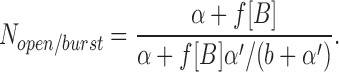 |
6 |
The relative peak current induced by rapid perfusion of saturating concentrations of ACh can also be calculated from scheme SI.
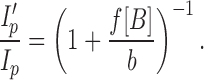 |
7 |
scheme SI predicts that the time constant of the current decay induced by a jump in drug concentration is:
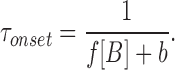 |
8 |
Note that, in scheme SI, the time constant of the macroscopic current decay is dependent on the same parameters as the kinetics of channel flickering. Hence, the decay is the multi-channel correlate of single channel flickering. Neither α nor α′ appear in the macroscopic current expressions (Eqs. 7 and 8) because, with saturating concentrations of ACh, dissociation of one molecule of ACh is quickly followed by binding of another. Under these conditions, the concept of burst loses its meaning.
The K i values (from Fig. 7) for PB (32 μM) and Barb (1.9 mM) determine the equilibrium between open and open-blocked channels (b/f) in scheme SI. These values are about twofold greater than those reported for PB and Barb inhibition of flux in Torpedo AChR (deArmendi et al., 1993). For PB, the association rates are given by fitting the concentration dependence of the single channel open time (Eq. 3; Fig. 4 A, solid line); f = 6.5 × 106/M/s. The dissociation constant can be calculated from f × b/f; b = 210/s. Very similar values for the association and dissociation constants for PB are obtained by fitting the concentration dependence of the fast decay time constant (Eq. 8, Fig. 8); f = 4.0 ± 0.6 × 106/M/s and b = 210 ± 20/s.
For Barb, analysis of the amplitude distribution suggests the open duration is more severely affected by the limited time resolution of the recording system than are the gap durations. Assuming for the moment that b >> α′, the observed gap duration gives b = 2 × 104/s (Eq. 5, Fig. 5 C). Combining this with the equilibrium dissociation constant gives f = 1 × 107/M/s. This is faster than the value obtained by fitting the Barb concentration dependence of the open duration (f = 4 × 106/M/s) but within the range obtained from analysis of the amplitude distribution (6–12 × 106/M/s).
Estimates for the remaining undetermined parameter in scheme SI, α′, can be obtained by fitting the concentration dependence of either the burst duration or the number of openings per burst. However, the burst duration may be the better measurement to fit because unresolved events will affect the number of openings per burst more than the burst duration. For PB, the burst duration is very sensitive to the value of α′; only values in the range 150–220/s provide a good description of the data at ≤100 μM PB. With α′ = 200/s, the predicted number of openings per burst do not differ very much from the observed values (Fig. 4 B, solid line). For Barb, α′ is not as well defined; values in the range 100–800/s all predict a fairly flat concentration dependence of the burst duration. With all of these values of α′, the predicted number of openings per burst is much higher than the observed values at 1,000 μM Barb: Nopen/burst = 32 with α′ = 100/s and 16 with α′ = 800/s. This range of values for α′ satisfies the assumption that b >> α′, validating the estimate of b from the gap duration. The predictions of scheme SI are shown with solid lines in Figs. 4 and 5 using the best fitting values (or intermediate values when there is a range of acceptable values) of f, b, and α′ (Table I).
Table I.
| Pentobarbital | Barbital | |||
|---|---|---|---|---|
| f (M−1s−1) | 6.5 × 106 | 1.0 × 107 | ||
| b (s−1) | 2.5 × 102 | 2.0 × 104 | ||
| α′ (s−1) | 2.0 × 102 | 4.0 × 102 | ||
| b/f (M−1) | 3.8 × 10−5 | 2.0 × 10−3 |
In their single channel study of PB on ACh receptors in denervated mouse muscle, Gage and McKinnon found quantitatively similar results on the open, gap, and burst durations. From the concentration dependence of the open duration, they calculated f = 3.4 × 106/M/s (16°C). They found a fivefold increase in the gap duration over the range of 10–500 μM PB from 1 to 5 ms. They considered this latter result as definite evidence against a sequential open channel blocking mechanism (scheme SI without the CB state) but did not explore any additional models.
A state dependence for barbiturate binding to Torpedo AChRs was observed by deArmendi et al. (1993). They found that the open state is preferred over the closed state by a factor of 4.7 (PB) and 3.2 (Barb). These values were determined by comparing the concentration of barbiturate needed to inhibit flux with the concentration needed to displace [14C]amobarbital bound to the resting receptor (Dodson et al., 1990). The ∼100 μs time resolution of our patch clamp experiments limits our ability to quantify the degree of barbiturate binding to the closed state. Fig. 10 indicates that there is no more than a 10% block of the closed channel with 100 μM PB. This implies a binding affinity to the closed state on the order of 1 mM and an open/ closed state preference of about 30-fold for PB in AChRs from BC3H-1 cells. We cannot determine the state preference of Barb for our experiments (Fig. 11).
Interactions between Barbital and Pentobarbital
The experiments illustrated in Figs. 12–14 address the question of whether PB and Barb compete for a single binding site on the AChR channel. If binding of the two drugs were absolutely competitive, the inhibition curve for PB in the presence of Barb, would be described by Eq. 9.
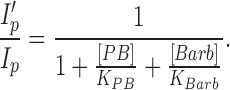 |
9 |
With 5 mM Barb, Eq. 9 predicts a 3.3-fold shift ([Barb]/KBarb) of the PB inhibition curve to a half maximum effect at 94 μM PB (Fig. 13, dashed line). The observed shift of the half maximum concentration is only 1.9-fold. Thus, the binding of PB does not exclude the binding of Barb. The binding of the two drugs is not independent either. The presence of Barb decreases the binding affinity of PB. This is also apparent from measurements of onset kinetics (Fig. 14). In the presence of Barb, PB exhibits faster kinetics. Fits of the data to Eq. 8 indicate that, in the presence of 5 mM Barb, the association rate of PB is decreased (from 4.8 ± 0.6 × 106/M/s to 3.5 ± 0.8 × 106/M/s), and the dissociation rate of PB is increased (from 200 ± 25/s to b = 340 ± 70/s). One interpretation is that the binding sites for PB and Barb are the same but when both drugs bind, they have to move to nearby, less stable positions. Alternatively, there could be two distinct binding sites for the drugs and these sites interact allosterically. Our data cannot distinguish between these two possibilities.
Allosteric Model
An alternative interpretation of the bursting behavior induced by barbiturates is to consider bursting to arise from the control burst activity at rates modified by barbiturates. scheme SII is a model that is often used to describe the normal kinetics of AChR single channels (Auerbach, 1993).
Scheme II.
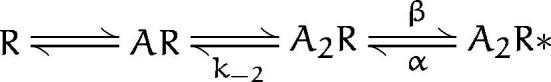
In this scheme, R represents the receptor and A represents ACh. Channel activation results from the binding to two molecules of ACh followed by a conformational change from the doubly-liganded closed state (A2R) to the open state (A2R*). At low concentrations of ACh, a burst consists of one or more transitions between A2R and A2R* terminated by the dissociation of the agonist at a rate k−2. The open time is given by 1/α, the gap duration by 1/(β + k−2) and the number of gaps per burst by β/k−2. Under control conditions, α = 0.5/ms, b ≈ k−2 ≈ 30/ms (Auerbach, 1993), so that the average burst consists of two 2-ms openings and one 20-μs gap (with the time resolution of our experiments, very few of these gaps are detected). Assume that the binding of barbiturates (with microsecond kinetics) modifies these rates to produce the observed burst kinetics. We have calculated the rates at each concentration of barbiturate. For both PB and Barb, α increases as a function of concentration and is on the order of 1/ms for 100 μM PB and 250 μM Barb. 100 μM PB decreases both β and k−2 by a factor of 100. The effects of 250 mM Barb are more moderate; β decreases by a factor of 2 and k−2 decreases by a factor of 5. The difficulty with scheme SII, however, is that it cannot account for the fast decay seen in macroscopic currents with rapid perfusion of 300 μM ACh in the presence of PB (Fig. 10). Moreover, scheme SII predicts that the onset of macroscopic currents would have an onset time (at high concentrations of ACh) of 1/(α+β), which is predicted to be 0.8 ms at 100 μM PB. Experimentally, we do not see any decrease in onset time (Fig. 10). We conclude that an allosteric model such as scheme SII is not viable explanation for the effects of barbiturates.
Extension of the Blocking Model
In scheme SI, the single channel gap duration (Eq. 5) is inversely proportional to the sum of b and α′ and is independent of the barbiturate concentration. For PB, the predicted gap duration is 2.2 ms (Fig. 4 C, solid line). The observed gap durations vary from 1.1 to 3.4 ms. The measured burst durations at high concentrations of PB also differ from the predictions of the model. One could argue that these deviations from the predicted values are unimportant because they occur at concentrations greater than three-times the K i (after all, even the archetypal AChR open channel blocker, QX-222, shows deviations from predictions at high concentrations [Neher, 1983]). However, we wanted to determine whether we could use this information to gain further insights into the mechanism of action of the barbiturates. Several pieces of evidence suggest that the observed deviations from scheme SI may be due to the binding of a second molecule of PB: (a) an increase in gap duration with [PB] is expected if the second molecule binds with low affinity and postpones the re-opening (unblocking) of the channel, (b) the interactions seen when both PB and Barb are present suggest that two barbiturate molecules may bind simultaneously, and (c) when the macroscopic current inhibition data (Fig. 7) is fit to a two-site inhibition function (Eq. 10), the two binding affinities are K 1 = 34 ± 3 μM and K 2 = 800 ± 500 μM (the dashed line in Fig. 7 is the prediction for two binding sites with affinities of 38 and 460 μM).
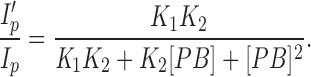 |
10 |
We then considered whether scheme SIII could be used to quantitatively predict the observed single channel gap and burst distributions. scheme SIII contains one additional state with two barbiturate molecules bound. We used Mathematica (version 2.2; Wolfram Research, Inc., Champaign, IL) to numerically evaluate the relevant matrix operations (Colquhoun and Hawkes, 1995b ) for this model.
Scheme III.
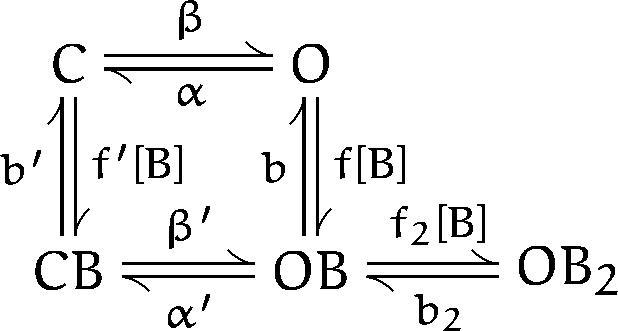
Numerical evaluation of scheme SIII requires values for nine independent constants. The channel activation rate, β, which depends on ACh binding, channel isomerization and the number of active channels, was set to 0.02/s to correspond to a typical control interburst interval of 400 ms. The previously determined values of α and f were used (Table I). The values of α′ and b were adjusted to account for the gap duration at low concentrations of PB; the best agreement was obtained with α′ = 400/s and b = 300/s. We assumed that the poor interaction of PB with closed channels results from a low association rate and a normal dissociation rate: f′ = f/10, b′ = b. Detailed balancing was used to evaluate β′. We also assumed that the binding of a second molecule of PB has a normal association rate and a fast dissociation rate: f2 = f, b2 = 12 × b (giving b2/f2 = 460 μM which is near the lower limit of the range obtained from a two-site fit). The results of the numerical evaluation are shown with dashed lines in Fig. 4 A, B, and C.4 scheme SIII quantitatively predicts the PB concentration dependence of the gap and burst durations and the number of openings per burst (the prediction for open duration is not shown because it is identical to that of scheme SI). Similar results are obtained when different assumptions are used (second binding site having a slow association rate and a normal dissociation rate: f2 = f/12, b2 = b; binding to closed channel having a normal association rate and a fast dissociation rate: f′ = f, b′ = 10*b). As might be expected, as the affinity of the second binding site is decreased, higher concentrations of PB are needed to obtain comparable changes in the predicted gap and burst durations (e.g., the predictions for b2/f2 = 920 μMat 1,000 μM PB are similar to the predictions for b2/f2 = 460 μM at 500 μM PB). The predictions of scheme SIII for macroscopic currents and kinetics do not differ significantly from the predictions of scheme SI.
Summary
The action of PB and Barb differs primarily in the dissociation rate; Barb dissociates 80 times faster than PB. This is a greater difference than would be expected if lipid solubility were the only factor that determines barbiturate potency; the octanol:buffer partition coefficients are 106 (PB) and 4.5 (Barb), giving a ratio of 23 (Firestone et al., 1986a). The same conclusion was reached, based on flux experiments with 14 barbiturates, by deArmendi et al. (1993). Our experiments suggest that the inhibitory binding site is not identical for PB and Barb. This is a plausible explanation for the poor correlation between potency and lipid solubility. Interestingly, the potency ratio for Barb and PB anesthesia in tadpoles is also large: 14.6 mM/0.16 mM = 90 (Lee-Son et al., 1975).
The kinetic experiments described here do not directly address the question of the location of the barbiturate binding site(s). The close temporal association between the duration of inhibitory events seen at the single channel level (the gap duration in Figs. 4 and 5) and the kinetics of macroscopic current inhibition after rapid perfusion of barbiturate (the onset time in Figs. 10 and 11) suggest that barbiturate binding and channel inhibition are inseparable. This favors a steric blocking mechanism over an allosteric effect. This has also been observed with other anesthetics acting on the AChR channel (Dilger et al. 1994). Allosteric mechanisms cannot be completely dismissed, though. One possibility is that the barbiturates bind and dissociate on the microsecond time scale and induce a conformational change to a new closed state of the channel. In this scenario, the transition rates between the open and new closed states determine bursting and relaxation kinetics. These rates would have to be exquisitely sensitive to the difference in chemical structure between PB and Barb to account for the 100-fold difference in the kinetic actions of these drugs.
The question of the location of the barbiturate binding site(s) might be answered more convincingly by site-directed mutagenesis experiments as has been done for open channel blockers such as QX-222 (Charnet et al., 1990). Yost and Dodson (1993) have argued that the site of action for amobarbital does not involve amino- acids at the 10′ level (near the center of the membrane) of the M2 transmembrane region of the channel. This does not, however, rule out other sites within the pore of the channel, nor does it rule out the 10′ level as being part of the binding site for other barbiturates. Inhibition of AMPA-selective glutamate receptor channels by PB is influenced by amino acids within the M2 region of the channel pore (Yamakura et al., 1995). However, so far there is no kinetic evidence that PB acts as blocker of this channel.
Acknowledgments
We thank Ms. Claire Mettewie for culturing cells.
Footnotes
This research was supported in part by a grant from the National Institute of General Medical Sciences (GM 42095); Klinik für Anästhesiologie, Universität Bonn; Department of Anesthesiology, University at Stony Brook; and an institutional grant from the School of Medicine, University at Stony Brook.
A preliminary report of these results has appeared in abstract form (Boguslavsky, R., and J.P. Dilger. 1995. Biophys. J. 68:A377).
Abbreviations used in this paper: ACh, acetylcholine; AChR, ACh receptor; Barb, barbital; PB, pentobarbital.
This estimate comes from comparison of single channel burst frequency with peak macroscopic currents measured on the same patch(Liu, Y., and J.P. Dilger, unpublished data).
For the data shown in Fig. 2 with 100 μM PB, the long closed component increased to 140 ms. However, subsequent return to control conditions showed the long closed time to be 140 ms. We assume that, in this patch, there was a rundown in channel activity between the first control and 100 μM PB runs. In patches that did not exhibit any rundown, 100 μM PB did not affect the long closed time. We did observe that 250 and 500 μM PB tended to decrease the long closed interval.
The predicted closed duration histogram for scheme SIII has four components. The longest represents the time between bursts and is not a part of the gap duration. We plotted the weighted average of the remaining three components in Fig. 4 C. We defined a burst in scheme SIII as sojourns starting in state O and ending in state C. The predicted burst duration histogram has three components but the briefest component has a negligible area. The remaining two components are separated by a factor of three or more. Because the measured burst duration histograms have two components and we have been considering the longer of the two observed components, we plotted the longest predicted burst duration component in Fig. 4 A. The predicted number of openings per burst for scheme SIII has a single component, and this is plotted in Fig. 4 B.
references
- Auerbach A. A statistical analysis of acetylcholine receptor activation in Xenopusmyocytes: stepwise versus concerted models of gating. J Physiol (Lond) 1993;461:339–378. doi: 10.1113/jphysiol.1993.sp019517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barann M, Gothert M, Fink K, Bonisch H. Inhibition by anaesthetics of 14C-guanidinium flux through the voltage-gated sodium channel and the cation channel of the 5-HT3 receptor of N1E-115 neuroblastoma cells. Naunyn-Schmiedebergs Arch Pharmacol. 1993;347:125–132. doi: 10.1007/BF00169256. [DOI] [PubMed] [Google Scholar]
- Charnet P, Labarca C, Leonard RJ, Vogelaar NJ, Czyzyk L, Gouin A, Davidson N, Lester HA. An open-channel blocker interacts with adjacent turns of α-helices in the nicotinic acetylcholine receptor. Neuron. 1990;2:87–95. doi: 10.1016/0896-6273(90)90445-l. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B. Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol (Lond) 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun, D., and A.G. Hawkes. 1995a The principles of the stochastic interpretation of ion-channel mechanisms. In Single Channel Recording. B. Sakmann and E. Neher, editors. Plenum Press, New York. 397–482.
- Colquhoun, D., and A.G. Hawkes. 1995b A Q-matrix cookbook. In Single Channel Recording. B. Sakmann and E. Neher, editors. Plenum Press, New York. 589–633.
- deArmendi AJ, Tonner PH, Bugge B, Miller KW. Barbiturate action is dependent on the conformational state of the acetylcholine receptor. Anesthesiology. 1993;79:1033–1041. doi: 10.1097/00000542-199311000-00022. [DOI] [PubMed] [Google Scholar]
- Dilger JP, Brett RS. Direct measurement of the concentration- and time-dependent open probability of the nicotinic acetylcholine receptor channel. Biophys J. 1990;57:723–731. doi: 10.1016/S0006-3495(90)82593-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilger JP, Liu Y. Desensitization of acetylcholine receptors in BC3H-1 cells. Pflüg Arch. 1992;420:479–485. doi: 10.1007/BF00374622. [DOI] [PubMed] [Google Scholar]
- Dilger JP, Brett RS, Lesko L. Effects of isoflurane on acetylcholine receptor channels. 1. Single-channel currents. Mol Pharmacol. 1992;41:127–133. [PubMed] [Google Scholar]
- Dilger JP, Vidal AM, Mody HI, Liu Y. Evidence for direct actions of general anesthetics on an ion channel protein: a new look at a unified mechanism of action. Anesthesiology. 1994;81:431–442. doi: 10.1097/00000542-199408000-00022. [DOI] [PubMed] [Google Scholar]
- Dodson BA, Urh RR, Miller KW. Relative potencies for barbiturate binding to the Torpedoacetylcholine receptor. Br J Pharmacol. 1990;101:710–714. doi: 10.1111/j.1476-5381.1990.tb14145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ffrench-Mullen, J.M.H., J.L. Barker, and M.A. Rogawski. Calcium current block by (−)-pentobarbital, phenobarbital, and CHEB but not (+)-pentobarbital in acutely isolated hippocampal CA1 neurons: comparison with effects on GABA-activated Cl−current. J Neurosci. 1993;13:3211–3221. doi: 10.1523/JNEUROSCI.13-08-03211.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestone, L.L., J.C. Miller, and K.W. Miller. 1986a Tables of physical and pharmacological properties of anesthetics. In Molecular and Cellular Mechanisms of Anesthetics. S.H. Roth and K.W. Miller, editors. Plenum Publishing Corp., New York. 267–277.
- Firestone LL, Sauter JF, Braswell LM, Miller KW. Actions of general anesthetics on acetylcholine receptor-rich membranes from Torpedo californica. . Anesthesiology. 1986b;64:694–702. doi: 10.1097/00000542-198606000-00004. [DOI] [PubMed] [Google Scholar]
- Firestone LL, Alifimoff JK, Miller KW. Does general anesthetic-induced desensitization of the Torpedo acetylcholine receptor correlate with lipid disordering? . Mol Pharmacol. 1994;46:508–515. [PubMed] [Google Scholar]
- Fragen, R.J. 1994. Clinical pharmacology and applications of intravenous anesthetic induction agents. In The Pharmacologic Basis of Anesthesiology. T.A. Bowdle, A. Horita, and E.D. Kharasch, editors. Churchill Livingstone, New York. 319–336.
- Frenkel C, Duch DS, Urban BW. Molecular actions of pentobarbital isomers on sodium channels from human brain cortex. Anesthesiology. 1990;72:640–649. doi: 10.1097/00000542-199004000-00012. [DOI] [PubMed] [Google Scholar]
- Gage PW, McKinnon D. Effects of pentobarbitone on acetylcholine-activated channels in mammalian muscle. Br J Pharmacol. 1985;85:229–235. doi: 10.1111/j.1476-5381.1985.tb08851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AC, Lieb WR, Franks NP. Insensitivity of P-type calcium channels to inhalational and intravenous general anesthetics. Anesthesiology. 1994;81:117–123. doi: 10.1097/00000542-199407000-00017. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell free membrane patches. Pflüg Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Jacobson I, Pocock G, Richards CD. Effects of pentobarbitone on the properties of nicotinic channels of chromaffin cells. Eur J Pharmacol. 1991;202:331–339. doi: 10.1016/0014-2999(91)90275-u. [DOI] [PubMed] [Google Scholar]
- Lee-Son S, Waud BE, Waud DR. A comparison of the potencies of a series of barbiturates at the neuromuscular junction and on the central nervous system. J Pharmacol Exp Therap. 1975;195:251–256. [PubMed] [Google Scholar]
- Liu Y, Dilger JP. Opening rate of acetylcholine receptor channels. Biophys J. 1991;60:424–432. doi: 10.1016/S0006-3495(91)82068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalec W, Narahashi T. Use-dependent pentobarbital block of kainate and quisqualate currents. Brain Res. 1993;608:7–15. doi: 10.1016/0006-8993(93)90766-g. [DOI] [PubMed] [Google Scholar]
- Murrell RD, Braun MS, Haydon DA. Actions of n-alcohols on nicotinic acetylcholine receptor channels in cultured rat myotubes. J Physiol (Lond) 1991;437:431–448. doi: 10.1113/jphysiol.1991.sp018604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. The charge carried by single-channel currents of rat cultured muscle cells in the presence of local anaesthetics. J Physiol (Lond) 1983;339:663–678. doi: 10.1113/jphysiol.1983.sp014741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S, Forman SA, Braswell LM, Miller KW. Actions of pentobarbital enantiomers on nicotinic cholinergic receptors. Mol Pharmacol. 1989;36:874–880. [PubMed] [Google Scholar]
- Sine SM, Steinbach JH. Activation of a nicotinic acetylcholine receptor. Biophys J. 1984;45:175–185. doi: 10.1016/S0006-3495(84)84146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanelian DL, Kosek P, Mody I, MacIver MB. The role of the GABAAreceptor/chloride channel complex in anesthesia. Anesthesiology. 1993;78:757–776. doi: 10.1097/00000542-199304000-00020. [DOI] [PubMed] [Google Scholar]
- Yamakura T, Sakimura K, Mishina M, Shimoji K. The sensitivity of AMPA-selective glutamate receptor channels to pentobarbital is determined by a single amino acid residue of the α2 subunit. FEBS Lett. 1995;374:412–414. doi: 10.1016/0014-5793(95)01163-9. [DOI] [PubMed] [Google Scholar]
- Yellen G. Ionic permeation and blockade in Ca2+-activated K+channels of bovine chromaffin cells. J Gen Physiol. 1984;84:157–186. doi: 10.1085/jgp.84.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost CS, Dodson BA. Inhibition of the nicotinic acetylcholine receptor by barbiturates and by procaine: do they act at different sites? . Cell Mol Neurobiol. 1993;13:159–172. doi: 10.1007/BF00735372. [DOI] [PMC free article] [PubMed] [Google Scholar]