

Abstract
We have investigated the interaction of charybdotoxin (CTX) with Shaker K channels. We substituted a histidine residue for the wild-type phenylalanine (at position 425) in an inactivation-removed channel. The nature of the imidazole ring of the histidine provides the ability to change the charge on this amino acid side chain with solution hydrogen ion concentration. Wild-type, recombinant CTX blocked wild-type Shaker channels in a bimolecular fashion with a half-blocking concentration (K d) of 650 nM (at a membrane potential of 0 mV). The F425H mutant channels were much more sensitive to CTX block with an apparent K d (at pH 7.0) of 75 nM. Block of F425H but not wild-type channels was strongly pH sensitive. A pH change from 7 to 5.5 rendered the F425H channels >200-fold less sensitive to CTX. The pH dependence of CTX block was steeper than expected for inhibition produced by H+ ions binding to identical, independent sites. The data were consistent with H+ ions interacting with subunits of the channel homotetrameric structure. The in situ pK for the imidazole group on the histidine at channel position 425 was determined to be near 6.4 and the dissociation constant for binding of toxin to the unprotonated channel was near 50 nM. We estimate that the binding of a H+ ion to each subunit adds 0.8 kcal/mol or more of interaction energy with CTX. We used mutant toxins to test electrostatic and steric interactions between specific CTX residues and channel position 425. Our results are consistent with a model in which protons on F425H channel subunits interact with three positive charges on CTX at an effective distance 6–7 Å from this channel position.
Keywords: recombinant toxin, ion channels, voltage clamp, Xenopus oocyte
introduction
The loop of amino acids between the fifth and sixth membrane-spanning domains of voltage-gated K+ channels is the locus for many of the permeation and pharmacological properties of proteins. This area contains the highly conserved P-region that controls much of the ion selectivity process (Yool and Schwarz, 1991; Heginbotham et al., 1992; De Biasi et al., 1993) and the receptor for scorpion α-K-toxins.
The α-K-toxins, including charybdotoxin (CTX),1 bind with high affinity to these channels and block ion movements through the pore (Miller, 1995). The toxin binding site is located at the outer entrance to the channel pore and is formed by two amino acid segments, including outer parts of the P-region and flanking regions extending toward the fifth and sixth membrane-spanning domains.
Several studies have exploited the known tertiary structure of the scorpion α-K-toxins to probe the architecture of voltage-gated K+ channels. These studies have revealed the location of the ion permeation pathway within the channel primary structure (MacKinnon and Miller, 1989; MacKinnon et al., 1990) and the homotetrameric nature of the channels in heterologous expression systems (MacKinnon, 1991). Recombinant toxins have been used to map the toxin:channel interaction surface (Stampe et al., 1994) and to identify strongly interacting pairs of toxin:channel amino acids (Hidalgo and MacKinnon, 1995; Aiyar et al., 1995; Naranjo and Miller, 1996). Additional, similar efforts have led to the identification of electrostatic interactions between specific charged amino acids on the toxin and channel (Stocker and Miller, 1994; Naini and Miller, 1996).
There is a large range in the sensitivity of different K+ channels to scorpion toxins (Miller, 1995). One important determinant of channel sensitivity has been shown to be the residue at Shaker position 425 (Goldstein and Miller, 1992). The replacement of the wild-type phenylalanine with a glycine at this position produces a 2,000-fold increase in channel sensitivity to CTX. To further examine the interaction of CTX with Shaker K+ channels, we introduced a histidine at position 425 and probed this interaction with external H+ ions.
We found that at pH 7 the F425H mutant was almost 10-fold more sensitive to CTX block than wild-type channels. Block of wild-type channels was only weakly pH dependent, and then only at pH levels <∼5.5. In contrast, block of F425H channels was especially pH sensitive over the range of pH 6–7. F425H channels at pH 5.5 were at least 200-fold less sensitive to CTX than at pH 7. The pH dependence of CTX block was steeper than expected for inhibition produced by H+ ions binding to identical, independent sites, but was consistent with H+ ions interacting with CTX by binding to subunits within the channel homotetrameric structure. The pH sensitivity of F425H channels was moderately reduced after neutralization of toxin lysines (K11 and K31) known to electrostatically interact with channel locations near 425. We also tested a steric mechanism for the pH-induced inhibition by replacing the threonines at toxin positions 8 and 9 (which may sterically interact with channel position 425) with smaller serine and glycine residues. This double mutant toxin remained sensitive to the protonation of the histidine at position 425. These results provide some constraints on the location of channel position 425 with respect to the toxin molecule and help refine the picture of the mechanism of toxin:channel interaction.
methods
Molecular Biological Methods
In this work, the wild-type channel was the inactivation-deletion version of Shaker B, ShB Δ6-46 (a gift from Dr. Richard Aldrich's laboratory, Stanford University, Stanford, CA). The channel cDNA was subcloned into the pBluescript II KS (−) vector (Stratagene Inc., La Jolla, CA). The F425H mutation introduced into the ShB Δ6-46 clone was carried out using a two-step PCR protocol and the resulting mutant clone was analyzed by DNA sequencing.
Recombinant Charybdotoxin
Some of the important amino acids for the interaction of CTX and the Shaker K+ channel are illustrated in Fig. 1. This figure reflects, in large part, the results and conclusion of Goldstein et al. (1994). The outline of a single CTX molecule is shown occupying one of four identical orientations with the channel homotetramer. Toxin lysine 27 is approximately centered over the channel pore opening. Lysines 11 and 31 have been shown to electrostatically interact with channel location 427 (Naini and Miller, 1996) on channels with an F425G mutation. Goldstein et al. (1994) suggest that toxin threonines 8 and 9 interact with the phenylalanine ring at ShB location 425 as indicated in the figure. We examined the actions of three CTX mutants on ShB and F425H channels: K11Q, K31Q, and the double mutant T8ST9G. The locations of these amino acids on the CTX molecule are indicated in Fig. 1 with italics type.
Figure 1.

A model for the topology of the CTX:Shaker K channel interaction. The location of the putative channel position 425 on each of the subunits of the homotetrameric protein are indicated by the solid circles. The approximate extent of the phenylalanine side chain at these locations is indicated by the dashed semi circle. The CTX profile seen from above is also indicated with toxin lysine 27 centered in the channel. Toxin amino acids mutated in this study are shown in italics in approximately their physical locations. This picture is derived in large part from the results of Goldstein et al. (1994) and was adapted from Fig. 6 of that work.
The variants of CTX were produced by expressing a cleavable fusion protein in Escherichia coli. Sequence-specific proteases were used to cleave the fusion protein from the toxin. The recombinant CTX was purified using standard biochemical methods, oxidized to form the disulfide bonds, and the NH2-terminal end was cyclized to form pyroglutamate. For details, see Park et al. (1991) and Stampe et al. (1994).
Oocyte Isolation and Microinjection
Frogs, Xenopus laevis, were maintained as described by Goldin (1992). Isolated ovarian lobes were rinsed with Ca-free OR-2 solution (82.5 mM NaCl, 2.5 mM KCl, 1 mM MgCl, and 5 mM HEPES, pH 7.6 with NaOH), and then defolliculated by incubation for 60–90 min with 2 mg/ml collagenase Type IA (Sigma Chemical Co., St. Louis, Mo). Cleaned, healthy oocytes were transferred and maintained for 2 h in ND-96 solution (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl, 1 mM MgCl, 5 mM HEPES, and 2.5 mM Na-pyruvate, pH 7.6 with NaOH) before injection. Injected oocytes were transferred to multi-well tissue culture plates and incubated at 16°C in ND-96 solution supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin.
Electrophysiological Recordings
Potassium channel currents were assayed electrophysiologically 2–3 d after injection. Electrophysiological recordings were done at room temperature (22–24°C) using a custom-built, two-electrode voltage clamp apparatus. Electrodes were made of 1 BBL glass with filament (1.5 mm o.d.) from World Precision Instruments, Inc. (Sarasota, FL). The resistance of the electrodes was between 0.3 and 0.5 MΩ when filled with a 3-M KCl solution. Voltage clamp pulses of 20- or 80-ms duration were used, but all determinations of toxin block were done with currents obtained at 20 ms, which minimizes the effects of voltage-dependent unblock (e.g., see Fig. 7 and the associated discussion). Data acquisition was performed using a 12-bit analog/digital converter controlled by a personal computer. Current records were filtered at 5 kHz. No compensation for series resistance was made. Currents were obtained before, during, and after the application of various concentrations of recombinant CTX. Experiments in which the currents after CTX did not recover to at least 90% of control values were rejected.
Figure 7.

pH dependence of T8ST9G CTX block of F425H channels. Currents before, during, and after application of 0.25 μM T8ST9G CTX recorded at 0, 20, and 40 mV are shown at pH 5 (top) and 7 (bottom).
The composition of the external solution used for electrophysiological recordings was (mM): 100 NaCl, 2 KCl, 1 MgCl, and 1.8 CaCl2. Solutions of pH 5.0, 5.5, and 6.0 were buffered with 10 mM MES; solutions of pH 6.5, 7.0, and 8.0 were buffered with 10 mM MOPS, HEPES, and EPPS, respectively. Toxin was added to the external solution with 30 μg/ml BSA.
results
CTX Sensitivity of Wild-Type and F425H Mutant Channels
The dose-dependent block of channel current at 0 mV and at pH 7.0 by recombinant, wild-type CTX is illustrated in Fig. 2, •. The ordinate in the figure represents the fraction of the current inhibited by the indicated toxin concentration. The inhibition of channel current by CTX was well-described by a 1:1 toxin:channel interaction (Fig. 2, line) with a half-blocking concentration (K d) of 650 nM. Under identical conditions, the F425H mutant channels (Fig. 2, ○) were substantially more sensitive to CTX and best described with a half-blocking concentration of 75 nM.
Figure 2.

Block of wild-type and mutant Shaker K channels by CTX at pH 7.0. Ordinate: amount of current inhibited by CTX expressed as a fraction of the average of current before and after toxin application. Currents measured at the end of 20-ms pulses to a potential of 0 mV. Data from wild-type (•) and F425H (○) channels are illustrated with standard error limits. The solid curve is the fit to the experimental data described by the relation: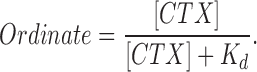 K
d values obtained from these fits are indicated. The number of determinations (if greater than three) are given next to the data points.
K
d values obtained from these fits are indicated. The number of determinations (if greater than three) are given next to the data points.
pH Sensitivity of CTX Block
The F425H mutation in Shaker channels allowed us to use H+ ions to probe the interaction of CTX with the toxin receptor on the channel surface. The protonation state of the imidazole group of the histidine placed at position 425 is expected to change over the pH range from 5 to 8. Consequently, we investigated the pH dependence of CTX block of the F425H mutant channels over this pH range. As the external pH was reduced, channel gating was shifted to more depolarized potentials. Consequently, for these experiments it was necessary to determine CTX block at more depolarized potentials, and we used a value of +40 mV. Fig. 3 shows CTX block of F425H channels at pH 7.0 (○) and 5.5 (•) at this potential. The block at pH 7.0 was consistent with a K d value of 91 nM. This is slightly larger than the 75 nM value at 0 mV of Fig. 2 and is generally consistent with a small voltage dependence of the CTX off rate (Goldstein and Miller, 1993, and see Fig. 7). At an external pH of 5.5, the channel was almost insensitive to CTX. The very small block at pH 5.5 seen in Fig. 3 is consistent with a K d value of ∼21 μM (more than 200-fold higher than at pH 7.0).
Figure 3.

CTX block of F425H channels at pH 7 and 5.5. Ordinate values as described in Fig. 2. Data at pH 7 (○) and 5.5 (•) are illustrated. Currents were measured at the end of 20-ms pulses to +40 mV. Solid lines are as in Fig. 2, with indicated K d values. The number of determinations (if greater than three) are given next to the data points.
Fig. 4 contains data showing that the large effect of low pH on CTX block of the F425H channels was not due to titration of residues on the channel other than at 425 or on the CTX molecule. Fig. 4 A shows that there was little difference in the dose-dependent block of wild-type channels at pH 7.0 (•) and 5.5 (○)—quite different from the results with F425H channels illustrated in Fig. 3. Fig. 4 B shows that block of wild-type channels by 0.5 μM CTX was almost independent of pH over the range 8.0–5.5, but there was some decrease in the amount of block at pH 5.
Figure 4.

pH dependence of CTX block of wild-type channels. (A) Block of current at the indicated CTX concentration at pH 7 (•) and 5.5 (○). Data at pH 7 are the averages of 3–10 observations, except for a single determination at 5 μM. Solid line as in Fig. 2 with indicated K d value. (B) pH dependence of block by 0.5 μM CTX. Data are the averages of 5–10 measurements; standard error limits omitted if smaller than symbol. Line was fit to data of Eq. 1 with indicated parameters.
To quantitatively describe the pH-induced inhibition of toxin block, we considered that H+ ions could protonate the channel and in so doing inhibit CTX binding. In this competitive inhibition model, the fraction of channels blocked is given by:
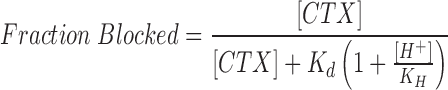 |
1 |
where K d is the dissociation constant for CTX interaction with the unprotonated channel, and K H is the dissociation constant for channel protonation.
Eq. 1 provides a reasonable description of the H+ ion titration of CTX block of wild-type channels as seen in Fig. 4 B (line). The K d value obtained from the fit of Eq. 1 to these data is 0.86 μM, generally consistent with that obtained from the complete CTX dose–response relation of Fig. 4 A. The H+ ion inhibition of CTX block of wild-type channels is consistent with a pK H value of 5.4. Thus, there are no chemical groups on the channel or on CTX that are important for toxin binding that are protonated at pH levels of 6.0 or above. The small effect on channel block seen at pH 5.0 consistent with a pK value of 5.4 suggests that some channel or toxin residue is protonated at pH values of 5.5 or lower.
The pH dependence of block of F425H mutant channels produced by 100 nM CTX is shown in Fig. 5 A. At pH 7.0, this amount of toxin blocked more of the mutant channel current than 0.5 μM did of wild-type channels (see Fig. 2). Thus, replacing the wild-type channel phenylalanine at position 425 with a histidine produced a large increase in channel affinity for CTX. Moreover, CTX block of F425H channels was much more pH sensitive than block of wild-type channels, especially over the pH range of 6 to 7. The solid line in Fig. 5 A is the best fit of Eq. 1 to these data with K d and pK H values of 42 nM and 7.2, respectively.
Figure 5.

pH dependence of CTX block of F425H channels. Currents measured at +40 mV. The CTX concentration used was 100 nM. Data are the averages of 4–7 observations and indicated next to the mean values. SEM limits as shown. (A) Solid line: fit of Eq. 1 to data with K d and pK H values of 42 nM and 7.2, respectively. Dotted line: see text. Dashed line: fit of Eq. 2 to data with K d and pK H values of 48 nM and 5.9, respectively. (B) Data as in A. Lines: fits of Eq. 3 to data. See text for details.
While Eq. 1 provides a qualitative description of the data of Fig. 5 A (solid line), it is apparent that the CTX block of F425H channels was more sensitive to external H+ ions than could be accounted for by this equation, the derivation of which considers all the H+ ion binding sites to be identical and independent and ignores the homotetrameric organization of the channels as expressed in Xenopus oocytes (MacKinnon, 1991).
Implications of Homotetrameric Channel Structure
The homotetrameric structure of K+ channels provides four overlapping receptors for a single CTX molecule (Stampe et al., 1994). Thus, protonation of the histidine in one channel subunit could reduce toxin affinity simply by reducing the number of available sites by 25%, protonating a second subunit would reduce the affinity by a factor of two, etc. The fraction of channels blocked by a fixed CTX concentration as a function of H+ ion concentration is equal to:
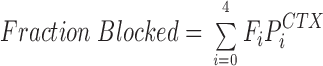 |
where Fi is the fraction of channels with i subunits protonated and is given by
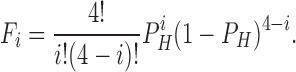 |
P H represents the probability of protonating one site:
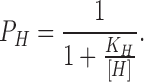 |
P i CTX is the probability of CTX binding to a channel with i of four subunits protonated and for the analysis here:
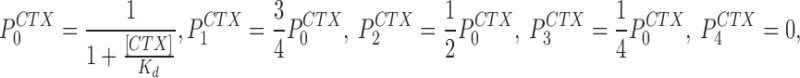 |
and represents the probability of protonating one site.
The dotted line in Fig. 5 A is the best fit of this set of equations to the data and provides a poor representation of the results. Thus, protonating each subunit in the channel does more to inhibit CTX block than simply reducing the number of overlapping receptors.
The limiting case of a larger effect of subunit protonation is to consider that the protonation of any one of the four histidine residues is sufficient to entirely inhibit toxin block. With this assumption, and again taking into account the different combinations of each protonated state, the fraction of channels blocked by CTX is:
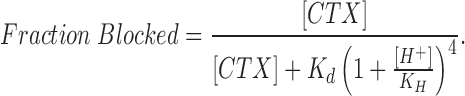 |
2 |
Thus, owing to the 4th power relation in Eq. 2, CTX block of homotetrameric channels is expected to be rather more sensitive to H+ ions than the prediction expressed by Eq. 1. The dashed line in Fig. 5 A is the fit of Eq. 2 to the data (with K d and pK H values of 48 nM and 5.9, respectively) and more closely accounts for the steep dependence of CTX block on H+ ions than does Eq. 1.
While Eq. 2 provides a good description of the experimental data, it is unlikely that a single protonated subunit would be sufficient to entirely inhibit toxin block. MacKinnon (1991) and Naranjo and Miller (1996) showed that channels with a single unfavorable subunit remain reasonably toxin sensitive. We thus considered a more general and, likely, realistic model that considers that the protonation of each subunit adds a constant, unfavorable energy for the interaction with CTX. The fraction of channels blocked is then given by:
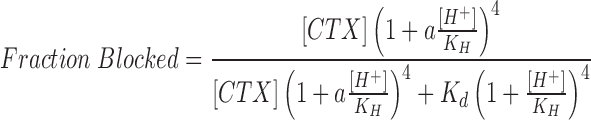 |
3 |
where a represents the fractional change in the CTX dissociation constant that occurs for each subunit that is protonated and is less than unity for an unfavorable interaction. In this model, protons may bind to channels with CTX but do so with an a-fold change in binding affinity; there is no interaction assumed between protons. The factor a can be related to the change in free energy caused by subunit protonation: ΔΔG0 = −RT lna.
We fit Eq. 3 to the pH dependence of CTX block of F425H channels with values of free energy change of 0.42, 0.66, 0.83, and 1.0 kcal/mol representing reductions of binding affinity by factors of 2, 3, 4, and 5, respectively. The best-fit results to the data are illustrated in Fig. 5 B. The effects of this interaction energy are seen mostly at low pH where multiple subunits become protonated. An interaction energy of only 0.42 kcal/ mol appears inconsistent with the data but energies above 0.66 kcal/mol provide a reasonable description of the results, especially since the block of F425H channels at pH 5 and, to a lesser extent, pH 6 may be underestimated by the small pH dependence present even in wild-type channels (see Fig. 4 B). The CTX dissociation constant and pK values associated with the fits with energies of 0.83 and 1.0 kcal/mol were essentially the same with values of ∼50 nM and 6.4, respectively.
pH Sensitivity of Charge Neutralizing CTX Mutants
CTX is a rather basic protein with several charged lysine residues, at least two of which, K11 and K31, have been shown to electrostatically interact with channel location 427 (Naini and Miller, 1996, and see methods). Given the likely close proximity of positions 425 and 427 on the channel surface, a possible mechanism for H+ ion-induced reduction in CTX affinity would be the electrostatic repulsion between the protonated imidazole ring at channel position 425 and K31 and K11 on CTX. We tested this possibility by examining the pH dependence of block of wild-type and F425H channels by mutant K31Q CTX. These results are illustrated in Fig. 6.
Figure 6.

pH dependence of mutant CTX block of wild-type and F425H channels. K31Q block of wild-type (○) and F425H (•) channels and K11Q block of F425H channels (▪). Toxin concentrations of 0.5 μM; currents measured at +40 mV. Dotted line: fit of Eq. 1 to data with K31Q CTX and wild-type channels with K d and pK H values of 0.78 μM and 5.4, respectively. Solid line: see discussion. Data values are averages of 3–4 (○), 4–5 (▪), or 5–6 (•); measurements shown with SEM limits.
As we found for the interaction of wild-type CTX with wild-type channels, a reduction in extracellular pH from 7.0 to 6.0 had little effect on block of wild-type channels by K31Q CTX (Fig. 6, ○), but block was reduced at pH 5.0. We fit Eq. 1 to these data (Fig. 6, dotted line) and obtained estimates for K d and pKi values of 0.78 μM and 5.4, respectively. These values are quite similar to the values of 0.86 μM and 5.4 for the interaction of wild-type channels with wild-type toxin (see Fig. 4 B).
Block of F425H channels by K31Q CTX was pH dependent but appeared to be somewhat less pH sensitive than block by wild-type toxin. The data of Fig. 5 A show that at pH 7 100 nM wild-type toxin blocked ∼55% of the F425H channel current, but only ∼5% at pH 6. K31Q CTX (500 nM, Fig. 6, •) blocked a similar fraction of current at pH 7 but, in contrast to wild-type toxin, blocked ∼30% at pH 6. This reduced pH sensitivity may reflect a reduced interaction energy between protonated subunits and the reduced valence of the K31Q mutant CTX (see discussion). We also examined F425H channel block by the CTX mutant K11Q. The inhibition produced by 0.5 μM K11Q (Fig. 6, ▪) was quite similar in magnitude and pH dependence to that produced by K31Q CTX.
Test of Steric Hindrance at Channel Position 425
Goldstein et al. (1994) suggest that some of the 2,000-fold increase in CTX sensitivity of F425G channels is due to the ability of the toxin to move deeper into the channel vestibule owing to the elimination of the steric interaction of toxin threonines 8 and 9 and the phenol ring on the phenylalanines at channel position 425 (see Fig. 1). So another possible mechanism for the inhibition of CTX block of F425H channels at low pH might be the slightly increased size of the protonated imidazole ring. To test this possibility, we investigated block by a toxin of reduced size at positions 8 and 9 by the double mutation T8ST9G.
An example of the actions of T8ST9G CTX with F425H channels is illustrated in Fig. 7. Fig. 7 (top) shows currents recorded before, during, and after application of 0.25 μM T8ST9G CTX at pH 5, indicating little or no block of protonated channels by this double mutant toxin. As for wild-type CTX, unprotonated channels are quite sensitive to T8ST9G CTX as illustrated in Fig. 7 (bottom). At a membrane voltage of 40 mV (and at 20 ms), the average fractional block of F425H channels at pH 7 by 0.25 μM T8ST9G CTX was 0.86 ± 0.01 (n = 3). The small, slow increase in the current during the 80-ms pulse to +40 mV at pH 7 in the presence of toxin is due to unblock of channels at this positive potential (Goldstein and Miller, 1993).
discussion
One of the results of this study was that there appeared not to be any chemical groups important for the interaction of wild-type CTX with wild-type Shaker K+ channels that are protonated over the pH range from 8 to near 5.5. Toxin binding was inhibited at lower pH values, consistent with a pK near 5.4, which may suggest the involvement of a carboxyl group in the interaction of the toxin with the channel. Aside from the carboxy terminus, CTX has only a single carboxyl group on the glutamate at position 12. Since this amino acid is not important for CTX block (Goldstein et al., 1994), the inhibition of block of wild-type channels at low pH is not likely due to the carboxyl group at this position. Neutralizing mutants of the aspartate at channel position 431 produce large changes in the interaction of CTX with F425G channels and in interaction of the closely related agitoxin 2 with wild-type Shaker channels (Hidalgo and MacKinnon, 1995). Thus, a candidate for the inhibition of CTX block of wild-type channels at very low pH is the carboxyl side chain of D431.
The replacement of the phenylalanine in the wild-type channel at position 425 with a histidine makes the toxin:channel interaction strongly pH dependent over the range from 7 to 6. The pH sensitivity was significantly larger than that expected from classical competitive inhibition between identical H+ ion binding sites and CTX (Fig. 5 A, solid line). This result is not surprising since the binding sites are not identical; the homotetrameric channel structure causes these sites to be arranged in groups of four. One consequence of this arrangement is a high order dependence of CTX block on subunit protonation as seen by comparing Eqs. 2 and 3 with Eq. 1. It is this high-order sensitivity that causes even relatively small interaction energies per subunit to produce pH-dependent CTX block that is indistinguishable from that of the case where even a single protonated subunit totally inhibited CTX block (see Fig. 5, A and B). Since MacKinnon (1991) and Naranjo and Miller (1996) showed that channels with a single unfavorable subunit remain reasonably toxin sensitive, we think it unlikely that the protonation of a single histidine at channel position 425 renders the channel completely resistant to CTX. The analysis associated with Fig. 5 B suggests the minimum interaction energy between a single protonated subunit and CTX to be near 0.83 kcal/mol.
The results illustrated in Fig. 7 indicate that this interaction energy is not likely to be steric in nature. Goldstein et al. (1994) suggest that the 2,000-fold increase in channel affinity for CTX produced by F425G mutation is a result of the bulky aromatic side chain ring of the phenylalanine at position 425 making unfavorable contact with the toxin threonines at positions 8 and 9. According to this picture, the F425G mutation removes the interaction with T8 and T9 and allows the toxin to settle closer to the channel surface in a more favorable binding location. Fig. 7 shows that a strong pH dependence of block was present for CTX reduced in size at positions 8 and 9. From experiments like that illustrated in Fig. 7, we estimate the K d for F425H channel block (at pH 7.0 and at +40 mV) by T8S/T9G CTX to be near 40 nM—similar to the value for the interaction of wild-type toxin with these mutant channels. Thus, block of F425H channels is not sensitive to the size of the amino acids at toxin positions 8 and 9 and the H+ ion-induced reduction of block is not likely a result of steric effects.
Since there are considerable electrostatic interactions between scorpion α-K-toxins and Shaker channels involving, among others, the nearby channel location 427 (e.g., Hidalgo and MacKinnon, 1995; Naini and Miller, 1996), it seems likely that the inhibition of CTX block of F425H channels at low pH is electrostatic in nature. The data of Fig. 6 show that any such electrostatic interaction does not reside entirely in interactions with toxin lysines 11 or 31. These residues may be involved (see below), but each must interact sufficiently weakly with channel subunits at position 425 that much of the pH dependence of block is preserved.
The fitting of Eq. 3 to the data (Fig. 5 B) indicate that our results can be accounted for if the protonation of each subunit reduces CTX affinity by a factor of 3 to 5. If this energy is electrostatic in nature, constraints may be placed on the distances between positive charges on the toxin molecule and channel position 425 (Stocker and Miller, 1994). For the discussion that follows, we consider that one proton at channel position 425 reduces CTX block by a factor of 4, equivalent to an interaction energy of ∼0.83 kcal/mol (see Fig. 5 B).
As discussed in Stocker and Miller (1994), an approximation of the magnitude of the electrostatic energy between a charge on a protein and a test charge of valence z a distance r into the aqueous solution from the protein–water interface is provided by:
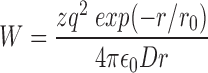 |
where q is the electronic charge, D the dielectric constant, and r 0 the Debye length.
CTX is a very basic protein with several lysine and arginine residues including, among others, the vital K27, and also K11 and K31, known to interact with channel position 427. Thus, for the purpose of this calculation, we consider the proton at channel position 425 to interact with a toxin with an effective valence of 3. The interaction energy of 0.83 kcal/mol then places channel position 425 a distance of 6–7 Å from (the center of) these charges on CTX, which is generally consistent with the picture illustrated in Fig. 1.
As a test of this analysis, consider that the only effect of the K31Q and K11Q mutations was to reduce the effective valence of CTX from 3 to 2. This would reduce the interaction energy to 2/3 of 0.83 or 0.55 kcal/mol. Eq. 3 (with K d for CTX of 50 nM and pK of 6.4, as obtained from the analysis of Fig. 5 B) can then be used to predict the pH dependence for block by these charge-reduced toxins. The solid line in Fig. 6 is the prediction from this equation and accounts rather well for the amount of block by each toxin at pH 7 and 6. More block is predicted at pH 5 than is actually observed, but this may be due to inhibition of block at low pH even with wild-type channels (e.g. Fig. 6, ○).
From the results and analyses of this study, we conclude that the affinity of CTX for Shaker K channels depends on the character as well as size of the amino acid side chain at position 425. Replacing the wild-type phenylalanine at this position with the very small (and polar) glycine increases CTX affinity by several orders of magnitude (Goldstein et al., 1994). We found that replacing the wild-type phenylalanine with a similar sized but more polar histidine residue produced a 10- to 12-fold increase in CTX affinity. Protonating the imidazole ring of the introduced histidine at this position substantially inhibited block by CTX, most likely through an electrostatic mechanism that may involve at least the toxin lysines at positions 11 and 31. Furthermore, the magnitude and pH dependence of block of F425H channel by neutralizing mutants of each of these lysines were very similar, suggesting that channel position 425 is approximately equidistant from toxin locations 11 and 31.
These conclusions are summarized in Fig. 1, in which we suggest that Shaker 425 has the position (relative to CTX) indicated by the open circle. This location is approximately equidistant from K11 and K31 and is ∼6–7 Å from the center of the triangle formed by K11, K31, and K27, which is not in the plane of the figure since K27 likely sits lower in the channel than K11 and K31 (Goldstein et al., 1994). This location for 425 is also consistent with our finding that size-reducing mutations at T8 and T9 have little effect on block of F425H channels. All our results are consistent with this physical picture and with protons at position 425 interacting electrostatically with three CTX positive charges that include K11, K31, and some other as yet unidentified charge. We speculate that this could be K27, but it may be difficult to test as charge neutralizing mutants at this position decrease the toxin affinity by more than three orders of magnitude.
Acknowledgments
We thank Jill Thompson for technical help with these experiments and for critically reading the manuscript. We are grateful to Chris Miller for keeping us from sliding down the slippery slope of scientific simplification.
This work was supported by National Institutes of Health grant NS-14138 and by a grant from the Fondo de Apoyo a la Investigation (FAI-UASLP) to P. Perez-Cornejo.
Abbreviation used in this paper
- CTX
charybdotoxin
references
- Aiyar J, Withka JM, Rizzi JP, Singleton DH, Andrews GC, Lin W, Boyd J, Hanson D, Simon M, Dethlefs B, et al. Topology of the pore-forming region of a K+channel revealed by the NMR-derived structures of scorpion toxins. Neuron. 1995;15:1169–1181. doi: 10.1016/0896-6273(95)90104-3. [DOI] [PubMed] [Google Scholar]
- De Biasi M, Drewe JA, Kirsch GE, Brown AM. Histidine substitution identifies a surface position and confers Cs+ selectivity on a K+pore. Biophys J. 1993;65:1235–1242. doi: 10.1016/S0006-3495(93)81154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL. Maintenance of Xenopus laevisand oocyte injection. Methods Enzymol. 1992;207:266–279. doi: 10.1016/0076-6879(92)07017-i. [DOI] [PubMed] [Google Scholar]
- Goldstein SAN, Miller C. A point mutation in a Shaker K+channel changes its charybdotoxin binding site from low to high affinity. Biophys J. 1992;62:5–7. doi: 10.1016/S0006-3495(92)81760-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein SAN, Miller C. Mechanism of charybdotoxin block of a voltage-gated K+channel. Biophys J. 1993;65:1613–1619. doi: 10.1016/S0006-3495(93)81200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein SAN, Pheasant DJ, Miller C. The charybdotoxin receptor of a Shaker K+channel: peptide and channel residues mediating molecular recognition. Neuron. 1994;12:1377–1388. doi: 10.1016/0896-6273(94)90452-9. [DOI] [PubMed] [Google Scholar]
- Heginbotham L, Abramson T, MacKinnon R. A functional connection between the pores of distantly related ion channels as revealed by mutant K+channels. Science. 1992;258:1152–1155. doi: 10.1126/science.1279807. [DOI] [PubMed] [Google Scholar]
- Hidalgo P, MacKinnon R. Revealing the architecture of a K+channel pore through mutant cycles with a peptide inhibitor. Science. 1995;268:307–310. doi: 10.1126/science.7716527. [DOI] [PubMed] [Google Scholar]
- MacKinnon R. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature. 1991;350:232–235. doi: 10.1038/350232a0. [DOI] [PubMed] [Google Scholar]
- MacKinnon R, Miller C. Mutant potassium channels with altered binding of charybdotoxin, a pore-blocking peptide inhibitor. Science. 1989;245:1382–1385. doi: 10.1126/science.2476850. [DOI] [PubMed] [Google Scholar]
- MacKinnon R, Heginbotham L, Abramson T. Mapping the receptor site for charybdotoxin, a pore-blocking potassium channel inhibitor. Neuron. 1990;5:767–771. doi: 10.1016/0896-6273(90)90335-d. [DOI] [PubMed] [Google Scholar]
- Miller C. The charybdotoxin family of K+channel-blocking peptides. Neuron. 1995;15:5–10. doi: 10.1016/0896-6273(95)90057-8. [DOI] [PubMed] [Google Scholar]
- Naini AA, Miller C. A symmetry-driven search for electrostatic interaction partners in charybdotoxin and a voltage-gated K+channel. Biochemistry. 1996;35:6181–6187. doi: 10.1021/bi960067s. [DOI] [PubMed] [Google Scholar]
- Naranjo D, Miller C. A strongly interacting pair of residues on the contact surface of charybdotoxin and a Shaker K+channel. Neuron. 1996;16:123–130. doi: 10.1016/s0896-6273(00)80029-x. [DOI] [PubMed] [Google Scholar]
- Park C-S, Hausdorff SF, Miller C. Design, synthesis, and functional expression of a gene for charybdotoxin, a peptide blocker of K+channels. Proc Natl Acad Sci USA. 1991;88:2046–2050. doi: 10.1073/pnas.88.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampe P, Kolmakova-Partensky L, Miller C. Intimations of K+channel structure from a complete functional map of the molecular surface of charybdotoxin. Biochemistry. 1994;33:443–450. doi: 10.1021/bi00168a008. [DOI] [PubMed] [Google Scholar]
- Stocker M, Miller C. Electrostatic distance geometry in a K channel vestibule. Proc Natl Acad Sci USA. 1994;91:9509–9513. doi: 10.1073/pnas.91.20.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yool AJ, Schwarz TL. Alteration of ionic selectivity of a K+channel by mutation of the H5 region. Nature. 1991;349:700–704. doi: 10.1038/349700a0. [DOI] [PubMed] [Google Scholar]