

Abstract
Normal activation–inactivation coupling in sodium channels insures that inactivation is slow at small but rapid at large depolarizations. M1651Q/M1652Q substitutions in the cytoplasmic loop connecting the fourth and fifth transmembrane segments of Domain 4 (S4–S5/D4) of the human heart sodium channel subtype 1 (hH1) affect the kinetics and voltage dependence of inactivation (Tang, L., R.G. Kallen, and R. Horn. 1996. J. Gen. Physiol. 108:89–104.). We now show that glutamine substitutions NH2-terminal to the methionines (L1646, L1647, F1648, A1649, L1650) also influence the kinetics and voltage dependence of inactivation compared with the wild-type channel. In contrast, mutations at the COOH-terminal end of the S4–S5/D4 segment (L1654, P1655, A1656) are without significant effect. Strikingly, the A1649Q mutation renders the current decay time constants virtually voltage independent and decreases the voltage dependences of steady state inactivation and the time constants for the recovery from inactivation. Single-channel measurements show that at negative voltages latency times to first opening are shorter and less voltage dependent in A1649Q than in wild-type channels; peak open probabilities are significantly smaller and the mean open times are shorter. This indicates that the rate constants for inactivation and, probably, activation are increased at negative voltages by the A1649Q mutation reminiscent of Y1494Q/ Y1495Q mutations in the cytoplasmic loop between the third and fourth domains (O'Leary, M.E., L.Q. Chen, R.G. Kallen, and R. Horn. 1995. J. Gen. Physiol. 106:641–658.). Other substitutions, A1649S and A1649V, decrease but fail to eliminate the voltage dependence of time constants for inactivation, suggesting that the decreased hydrophobicity of glutamine at either residues A1649 or Y1494Y1495 may disrupt a linkage between S4–S5/D4 and the interdomain 3–4 loop interfering with normal activation–inactivation coupling.
Keywords: sodium channels, activation–inactivation coupling, gating, S4–S5 segment
introduction
The membrane action potential in nerve, striated muscle, and other excitable cells is initiated through Na+ influx via voltage-sensitive Na+ channels. Depolarization from the resting potential triggers activation (opening) of the Na+ channels. After a few milliseconds, if the depolarization is maintained, the channels enter a stable, nonconducting, inactivated state. After membrane repolarization, the Na+ channels return to a closed resting state capable of being activated once again. Under whole cell voltage-clamp conditions, both the activation and inactivation of Na+ currents are voltage dependent (Armstrong, 1981; Bezanilla, 1985; Patlak, 1991; Keynes, 1994; Sigworth, 1994). The precise control of the activation and inactivation processes insures that the action potential can be triggered reliably, that the depolarization is transient, and that the channels will appropriately recover from inactivation so that the membrane is returned to its original state of excitability. The molecular details of inactivation and its coupling to activation are yet poorly understood.
Activation derives its voltage dependence from the ability of buried charges in the channel protein, the highly charged S4 segments, to function as the voltage sensor by moving in response to changes in the membrane potential (Stuhmer et al., 1989; Auld et al., 1990; Liman et al., 1991; Lopez et al., 1991; McCormack et al., 1991; Logothetis et al., 1993; Fleig et al., 1994; Perozo et al., 1994; Sigworth, 1994; Papazian et al., 1995; Larsson et al., 1996; Yang et al., 1996). Every third amino acid in the S4 transmembrane segments is positively charged (either arginine or lysine) with intervening hydrophobic residues (Kallen et al., 1993). It is generally believed that inactivation derives most of its voltage dependence from being coupled to activation. According to this view, depolarization causes voltage-dependent activation gates to open, and the rate of inactivation increases as a consequence of conformation changes in the channel protein associated with the activation process, perhaps by organizing a portion of the protein as an inactivation particle receptor (IPR)1 into which the ball, in the ball and chain model, can swing to block the channel (Armstrong and Bezanilla, 1977; Armstrong, 1981; Aldrich et al., 1983; Zagotta et al., 1990; Patlak, 1991; Keynes, 1994; Sigworth, 1994; O'Leary et al., 1995; Chen et al., 1996).
Much evidence from the effects on inactivation of a variety of protein reagents, including proteases, kinases, and antibodies (Rojas and Armstrong, 1971; Eaton et al., 1978; Oxford et al., 1978), and mutagenesis studies (Vassilev et al., 1988) supports the view that one cytoplasmic region of the sodium channel, the isoleucine, phenylalanine, methionine (IFM) triad in the interdomain (ID) 3–4 loop, acts as the inactivation particle or ball to block the pore in the inactivated state when it is bound to the IPR. Substitution by other amino acids in this triad, particularly for the phenylalanine, but not at 11 lysines elsewhere in ID3-4, abolishes fast inactivation (Moorman et al., 1990; West et al., 1992; Hartmann et al., 1994; Chahine et al., 1997). However, mutations at two adjacent tyrosines (Y1494Q/ Y1495Q), seven residues downstream of IFM, affect the voltage dependence of both activation and inactivation, suggesting that they play an important role in the coupling between activation and inactivation (O'Leary et al., 1995; Kellenberger et al., 1997). Activation and inactivation are also coupled in K+ channels since mutations in the NH2 terminus, where the inactivation ball is located, can also affect activation (VanDongen et al., 1990; Schonherr and Heinemann, 1996; Marten and Hoshi, 1997; Terlau et al., 1997) (compare Hoshi et al., 1990). Furthermore, the activation voltage sensors of the sodium channel, the S4 segments, appear, at least for Domain 4 (D4), to play an important and unique role in inactivation since S4D4 mutations cause a slowing and decreased voltage dependence of inactivation (Chahine et al., 1994; Bennett et al., 1995; Chen et al., 1996; Lerche et al., 1996). Currently, it appears widely accepted that the IPR is mainly formed from portions of the S4–S5 segments in both potassium and sodium channels (Isacoff et al., 1991; Depp and Goldin, 1996; Holmgren et al., 1996; Kontis et al., 1997; Lerche et al., 1997; Smith and Goldin, 1997).
Besides a binding reaction between the inactivation particle and its receptor, which depends upon protein conformation changes induced by voltage sensors in the activation pathway, additional intramolecular interactions associated with the inactivation process have been proposed. One of these may be visualized as an interaction physically coupling the ID3-4 segment to the core of the channel protein in a way that allows the voltage-sensing activation elements to have knowledge of the state of the inactivation particle and vice-versa (Keynes, 1992; O'Leary et al., 1995). In this paper, we consider the core of the channel to be that part of the protein that is not part of the ID3-4 loop. We refer to this postulated additional physical interaction as the inactivation–activation linkage (IAL) and hypothesize that the protein components involved are centered on or near the Y1494Y1495 dyad in ID3-4 and a cognate site located in S4–S5/D4. We and others have begun to explore this region of sodium channels by mutagenesis (Depp and Goldin, 1996; McPhee et al., 1996; Lerche et al., 1997). For example, our mutations at a pair of adjacent conserved methionines (M1651Q/M1652Q) in the S4–S5 linker of Domain 4 revealed effects on the rate and voltage dependence of channel inactivation (Tang et al., 1996) and pointed to a possible contribution of these residues to the structure of the IPR. We have extended our investigation of the role of amino acids in the S4–S5 linker of Domain 4 and report that mutations at several sites closer to the S4D4 transmembrane segment also affect activation and/or inactivation and, in the case of the A1649Q channel, detail mechanistic aspects of these effects with single-channel experiments.
methods
Mutagenesis
Site-directed substitutions were carried out using the Altered Sites-II in vitro mutagenesis system according to the manufacturer's instructions (Promega Corp., Madison, WI). Briefly, a single-stranded template was prepared from JM109 cells with R408 helper phage. The synthetic antisense mutagenic oligonucleotide containing desired mutations (underlined) were as follows: L1646Q/L1647Q: 5′-CATCATGAGGGCAAACTGCTGCGTGCGGATCCCCTTG-3′; F1648Q/A1649Q: 5′-GGACATCATGAGCTGCTGGAGCAGCGTGCGGAT-3′; F1648Q: 5′-GGACATCATGAGGGCCTGGAGCAGCGTGCGGAT-3′; A1649Q: 5′-GGACATCATGAGCTGAAAGAGCAGCGTGCGGAT-3′; A1649V: 5′-AGGCAG-GGACATCATCAGGACAAAGAGCAGCGTGCG-3′; A1649S: 5′-AGG-CAGGGACATCATCAGGGAAAAGAGCAGCGTGCG-3′; L1650Q: 5′-AGGCAGGGACATCATCTGGGCAAAGAGCAGCAGCG -3′ ; M1651Q/M1652Q: 5′-GAGGGCAGGCAGGGACTGCTGGAGGGC-AAAGAGCAG-3′; L1654Q: 5′-GTTGAAGAGGGCAGGCTGGGACATCATGAGGGCA-3′; P1655Q/A1656Q: 5′-GATGTTGAAGAGCTGCTGCAGGGACATCATGAG-3′.
The oligonucleotides were annealed to the single-stranded DNA template and the reaction mixture, after polymerization and ligation, was transformed into ES 1301 mut S cells. The plasmid DNA was isolated and transformed into JM109 cells. The colonies were screened and mutations were verified by dideoxynucleotide sequencing. pCDNA-1 vector (Invitrogen Corp., San Diego, CA) was used to drive the expression of human heart sodium channel subtype 1 (hH1) in the tsA 201 cell line.
Na+ Channel Expression in tsA201 Cells
Cells of the human embryonic kidney tsA201 cell line were maintained in DMEM (GIBCO-BRL, Life Technologies, Inc., Gaithersburg, MD) containing 10% fetal bovine serum, 2 mM L-glutamine, and 1% penicillin-streptomycin in equilibrium with 5% CO2 in a humidified incubator. DNA encoding sodium channels, purified by exchange columns (QIAGEN Inc., Chatsworth, CA), was transiently transfected into tsA201 cells by standard calcium phosphate methods. Note that cells were cotransfected with 10 μg each of cDNAs encoding Na+ channels, either wild type (WT) or mutant, with DNA expressing a surface marker, pHookTM-1 (Invitrogen Corp.) (Margolskee et al., 1993). Both cDNAs were added to 0.5 ml of 255 mM CaCl2. The cDNA-CaCl2 mixture was added dropwise to 0.5 ml of 2× HEPES solution containing (mM): 274 NaCl, 40 HEPES, 12 dextrose, 10 KCl, 1.4 Na2HPO4, at pH 7.05. This mixture was incubated for 20 min at room temperature, and then added dropwise to a 100-mm petri dish of cultured tsA201 cells ∼30–55% confluent in DMEM. The next day, cells were split, transferred to 35-mm petri dishes, and cultured overnight. Cells were used for recording 2–3 d after transfection. Before patch recording, transfected cells were incubated with Capture-Tec™ magnetic beads containing antibodies directed against the expressed membrane peptide encoded by pHookTM-1 in DPBS (GIBCO-BRL, Life Technologies, Inc.) for 30 min at 37°C, 5% CO2, and washed three times with external electrophysiological recording solution (see below). More than 90% of the bead-decorated cells expressed Na+ currents.
Electrophysiology and Data Analysis
Standard whole-cell voltage-clamp recordings were carried out as described (Hamill et al., 1981; Tang et al., 1996). Sylgard-coated, fire-polished pipettes (8161; Dow Corning Corp., Midland, MI) were used. Currents were recorded and filtered at 5 kHz with an Axopatch 200A (Axon Instruments, Inc., Burlingame, CA) or a Warner PC501 patch clamp amplifier (Warner Instruments, Hamden, CT) and a digital interface (Scientific Solutions, Solon, OH, or Axon Instruments, Inc.). Cells were dialyzed at least 10 min before recording data. The whole cell current recordings were obtained at room temperature (21–23°C) with bath solution containing (mM): 155 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2, and 10 Na-HEPES, pH 7.4. Patch electrodes contained (mM): 35 NaCl, 105 CsF, 10 EGTA, and 10 Cs-HEPES, pH 7.4. Whole cell data were analyzed by a combination of pCLAMP programs, Microsoft Excel, and Origin (MicroCal, Northampton, MA). When quantifying the voltage sensitivity of various parameters, we used a Boltzmann function, I/I max = 1/{1 + exp[(V − V 1/2)Z m F/RT]}, where I is the current at each voltage, I max is the maximum current, V 1/2 is the half-maximal voltage, and Z m is the apparent gating valence in equivalent electronic charges (e o). F, R, and T have their usual meanings. Single channel recordings were carried out using a cell-attached configuration with the following bath solution containing (mM): 100 K methyl sulfate, 55 KCl, 1.5 CaCl2, 1 MgCl2, 10 K-HEPES, pH 7.4, at room temperature (O'Leary et al., 1995). The pipette solution contained (mM): 155 NaCl, 10 tetraethylammonium Cl, 2 KCl, 1.5 CaCl2, 1 MgCl2, 10 Na-HEPES, pH 7.3. Data were sampled at 20 kHz, filtered at 5 kHz, and idealized using TRANSIT (Dr. A. VanDongen, Duke University, Durham, NC). Single channel data were further analyzed using FORTRAN programs written by Dr. R. Horn (Jefferson Medical College, Philadelphia, PA). Data are expressed as mean ± SEM. All comparisons were performed by Student's unpaired t test and expressed as probability of significance values (P).
results
Glutamine-Scanning Mutations in the S4–S5/D4 Segment Alter the Time Constants and Voltage-dependence of Fast Inactivation
To identify residues that are important for channel gating, each amino acid in S4–S5/D4 has been mutated to glutamine singly or in pairs (with the exception of S1653). The amino acid sequence of the cytoplasmic loop of the S4–S5 segment in Domain 4 shows the relationship of the present mutations to the previously studied M1651Q/M1652Q substitutions in the primary sequence of the channel (Fig. 1 A). The mutant and WT channels were transiently expressed in the tsA201 cell line. Families of whole-cell sodium channel currents from WT and mutant channels show a typical pattern of rapid voltage-dependent activation followed by a slower and complete inactivation (Fig. 1 B). The decay of current after depolarization is well-fitted by a single exponential function at all voltages tested. The time constants of current decay (τh) for WT channels are characterized by a relatively strong voltage dependence with slower inactivation rates at relatively negative potentials (−55 to 0 mV) in contrast to little voltage dependence with faster inactivation rates at more positive potentials (0 to +75 mV, Fig. 2 A). The strongly voltage-dependent range coincides with the voltage range in which Na+ channel open probability changes (see Fig. 4 A, −60 to −20 mV). Mutations located NH2 terminal to the highly conserved S1653, including the pair of conserved methionines (L1646, L1647, F1648, A1649, L1650, M1651, M1652), are shown in solid symbols (Fig. 2 A) and are associated with a variable degree of slowing of the kinetics of inactivation (larger τh values at voltages more positive than −25 mV) when compared with WT channels (Fig. 2 A). The current decay phenotypes for mutant channels that vary from WT fall into two major categories: (a) the voltage dependence of τh for the mutant parallels that of WT channel (Fig. 2 A), but the time constants of inactivation are larger than WT at all voltages measured (L1646Q/L1647Q, F1648Q, and M1651Q/M1652Q); and (b) the voltage dependence of τh is clearly less than that of the WT channel (Fig. 2, A and C) with the time constants of inactivation smaller than those for WT channels at negative voltages but larger than WT at positive voltages (L1650Q, F1648Q/ A1649Q, A1649Q, and A1649V). The mutations with the least voltage dependence of τh are those involving the replacement of the conserved A1649 residue by glutamine (F1648Q/A1649Q and A1649Q), rendering the inactivation time constants virtually voltage independent in contrast to the >10-fold decrease in τh for WT channels over the voltage range −55 to +75 mV (Figs. 1 B and 2 C), a range in which channel activation is strongly voltage dependent (see Fig. 4 A). The net effect of the mutant curves crossing that of the WT channel (Fig. 2 C) is that the channels containing the A1649Q mutation inactivate approximately fourfold more rapidly than WT channel at −55 mV and approximately threefold more slowly at +75 mV. Since the voltage dependence of the τh values of the F1648Q mutant channel resembles that of WT more than that of A1649Q-containing channels (Fig. 2 A), we conclude that the change of the amino acid residue at A1649 is responsible for the unusual voltage-independent inactivation and that the A1649 site (and, to a lesser extent, the adjacent position L1650) is normally involved in the coupling between activation and inactivation. We therefore chose to focus on the mutations that result in the most extreme phenotypes, those containing A1649Q mutations, for more detailed analysis including single channel studies. It is of interest that the τh values at depolarizations more positive than −55 mV are considerably larger in magnitude for F1648Q than those of WT, and yet combining the two mutations, F1648Q and A1649Q, does not lead to additive effects (Fig. 2 A).
Figure 1.
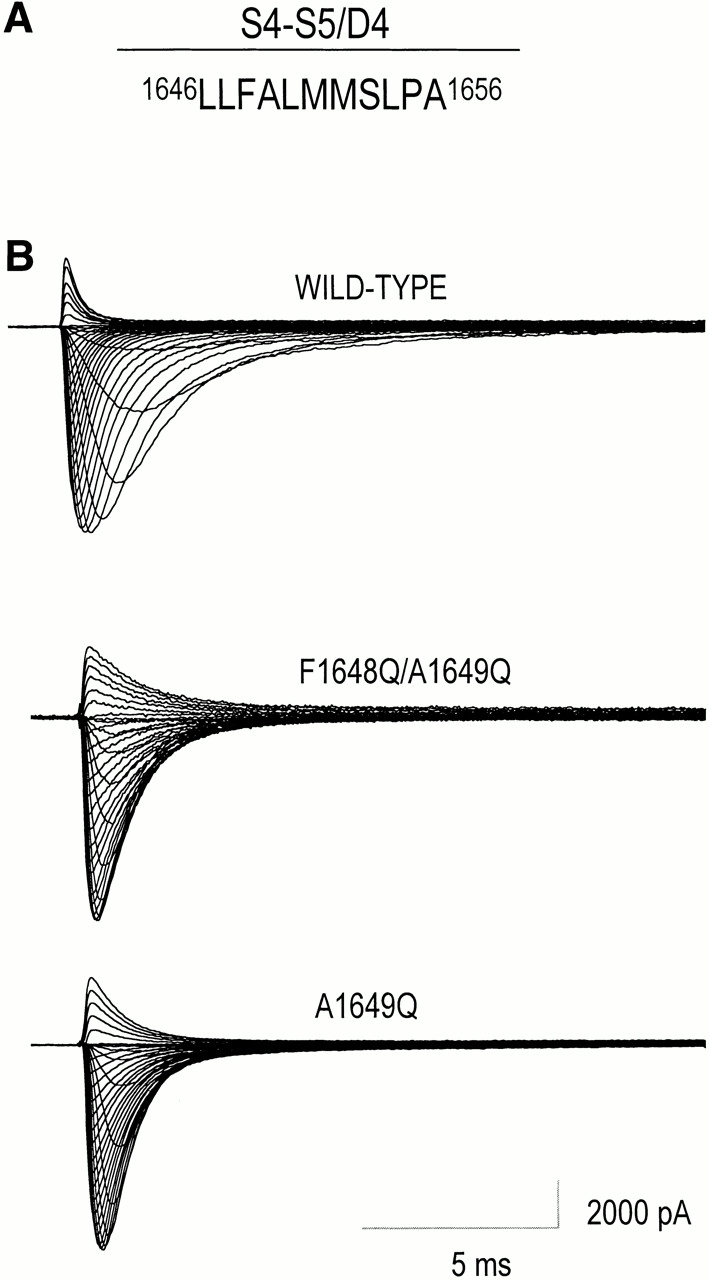
Sodium currents of wild-type hH1 and two mutant channels, F1648Q/A1649Q and A1649Q. (A) Diagram of the amino acids in the S4–S5/D4 loop of hH1that were replaced either singly or doubly by glutamine residues (S1653 was not examined). (B) Effects of F1648Q/A1649Q and A1649Q mutations in S4–S5/D4 segment of hH1 on families of Na+ currents. (top) WT human heart sodium channel subtype 1. (middle and bottom) F1648Q/A1649Q and A1649Q mutant channels, respectively, expressed transiently in tsA201 cells and activated by depolarization in 5-mV increments from −80 to +75 mV; holding potential, −120 mV.
Figure 2.

Effects of mutations of S4–S5/D4 on Na+ current inactivation. The decay of current after a depolarization is well-fitted by a single exponential function at all voltages. (A) Plot of time constant of inactivation (τh) vs. voltage for WT and a series of channels with glutamine scanning mutations through the NH2-terminal portion of S4–S5/ D4 loop. (B) τh values vs. voltage for WT and a series of channels with glutamine scanning mutations through the COOH-terminal portion of S4–S5/D4 loop. (C) τh values vs. voltage for WT and a series of channels with mutations (Ser, Val, and Gln) at A1649.
Figure 4.

Effects of mutations on activation. (A) Normalized peak G-V relationship from current–voltage (I-V) curves (not shown) for WT, F1648Q/ A1649Q, and A1649Q channels. Currents were elicited by 45-ms pulses to a range of test potentials as indicated from a holding potential of −120 mV. Each test current (I) was normalized by maximal peak current. Standard error bars are generally smaller than the symbols. Normalized G-V relationship for I-V curves, determined as I/(45 mV − V t), where V t is the test potential and 45 mV is the reversal potential in this study. Normalized conductance was fitted to a Boltzmann relationship and midpoints and slopes were determined from the fitted G-V curves (Table I). (B) Half-time to peak current, t 1/2, vs. test voltage. (C) Activation time course of channels containing the A1649Q and F1648Q/ A1649Q mutations and WT on expanded scale at −40 mV.
Because glutamine substitution for alanine changes both polarity and molecular size, we explored the effects of replacements of alanine by either serine (A1649S) or valine (A1649V). Relative to alanine, the A1649V mutation conserves hydrophobicity but changes size, while replacement of alanine by serine would primarily alter hydrophobicity. The behaviors of the A1649S and A1649V mutations are similar to that of A1649Q (Fig. 2 C) in that the voltage dependence of the time constants of inactivation are decreased and the curves for these mutants also cross that of the WT channel but, in contrast to the phenotype of A1649Q, the voltage dependence is not completely eliminated. The magnitude of deviation from WT phenotype is A1649Q > A1649S > A1649V, suggesting that normal function is associated with a hydrophobic side chain, but this remains to be proven.
Mutations located COOH terminal to the highly conserved S1653, namely L1654 and P1655/A1656, are without significant effect either on the magnitude or voltage dependence of inactivation time constants (Fig. 2 B, ○ and □).
Steady state inactivation was determined by measuring the availability of channels to be activated, that is, to produce currents after a 500-ms prepulse at various conditioning voltages. Prepulse durations of 200 ms gave identical results, supporting the view that the h∞•-V curve reflects primarily, or only, fast inactivation (Featherstone et al., 1996). All channels containing substitutions at A1649 (F1648Q/A1649Q, A1649Q, A1649V, or A1649S) manifest significantly decreased voltage dependencies of steady state inactivation compared with WT (P < 0.001), equivalent to losses of 1.49–1.83 electronic changes (e o), although the individual mutant channels are not significantly different between themselves (Fig. 3 A and Table I). In addition, the A1649-substituted channels tend to show hyperpolarizing shifts in steady state inactivation, indicating that the inactivated state is relatively more stable relative to noninactivated states in these mutants compared with the WT channel (Fig. 3 A and Table I).
Figure 3.
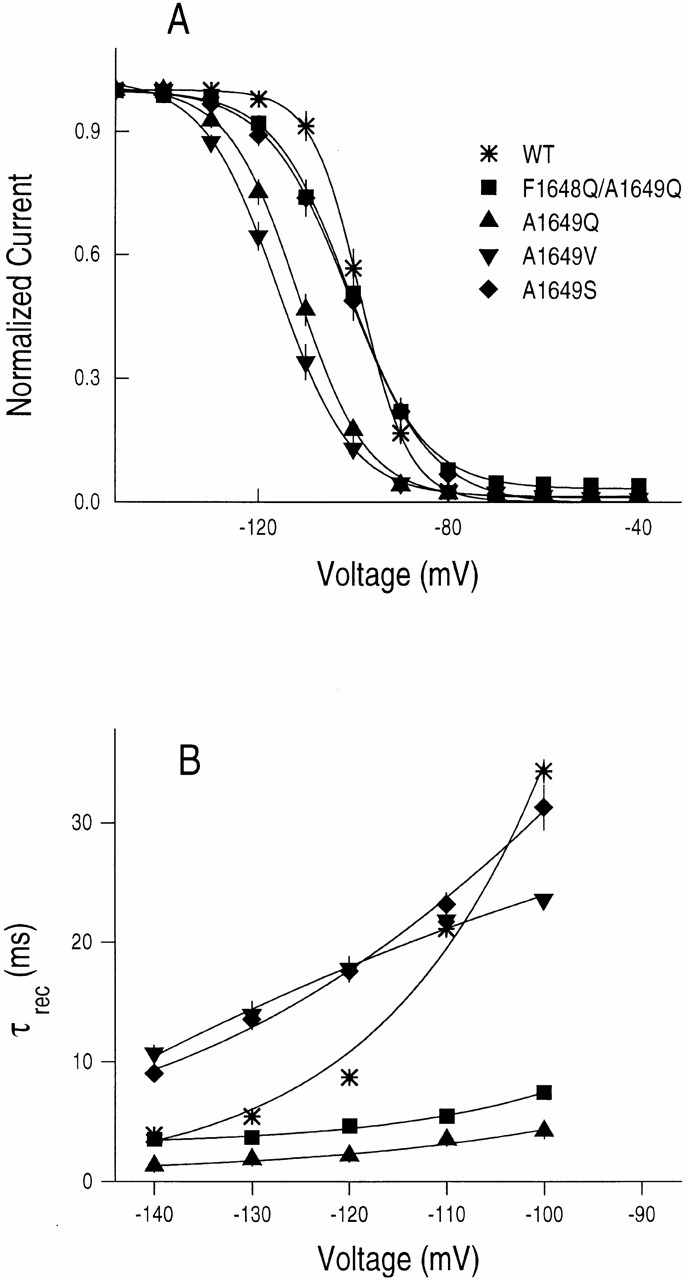
(A) Steady state inactivation for WT, F1648Q/A1649Q, A1649S, A1649V, and A1649Q channels. Currents were measured at a test potential of −30 mV after 500-ms conditioning pulses at indicated voltages from a holding potential of −120 mV. Each peak current was normalized by maximal peak current and fit by the Boltzmann equation I/I max = 1/{1 + exp[(V − V 1/2)Z m F/RT]}, where I is the current at each voltage, I max is the maximum current, V 1/2 is the half-maximal voltage and Z m is the apparent gating valence in equivalent electronic charges (e o) (Table I). (B) Time constants for recovery from inactivation (τrec). Inactivation elicited by 15-ms prepulses to 0 mV from a holding potential of −120 mV, followed by a given recovery potential for a variable time interval, and tested at 0 mV. The normalized peak currents of the test pulse were plotted as a function of time (t) between the pulses. Such plots were fit with single exponential functions to determine the recovery time constant (τrec) at each potential, and τrec values are from I test /I prepulse = 1 − exp(t/τrec) plotted as a function of recovery potential.
Table I.
Voltage-dependent Characteristics of Activation and Inactivation
| Activation | Inactivation | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Channels | Midpoint | Slope | Midpoint | Slope | n | |||||
| mV | eo | mV | eo | |||||||
| WT | −49.9 ± 0.5 | 2.45 ± 0.11 | −98.5 ± 0.1 | 4.60 ± 0.10 | 7 | |||||
| L1646Q/L1647Q | −43.0 ± 0.5 | 3.48 ± 0.20 | −78.7 ± 0.3 | 3.26 ± 0.11 | 5 | |||||
| F1648Q/A1649Q | −40.7 ± 2.3 | 2.52 ± 0.28 | −101.0 ± 0.5 | 3.11 ± 0.17 | 6 | |||||
| F1648Q | −51.2 ± 0.5 | 3.00 ± 0.16 | −82.1 ± 0.3 | 3.43 ± 0.11 | 5 | |||||
| A1649Q | −39.4 ± 2.5 | 2.35 ± 0.27 | −111.7 ± 0.3 | 3.30 ± 0.14 | 4 | |||||
| A1649S | −57.2 ± 1.7 | 3.12 ± 0.33 | −100.8 ± 0.3 | 2.77 ± 0.20 | 6 | |||||
| A1649V | −53.7 ± 2.5 | 2.72 ± 0.20 | −115.9 ± 0.5 | 3.06 ± 0.10 | 7 | |||||
| L1650Q | −51.0 ± 0.5 | 2.79 ± 0.10 | −97.5 ± 0.3 | 3.10 ± 0.10 | 6 | |||||
| L1654Q | −46.3 ± 1.4 | 3.47 ± 0.40 | −89.4 ± 0.3 | 2.70 ± 0.09 | 5 | |||||
| P1655Q/L1656Q | −50.8 ± 1.3 | 2.53 ± 0.51 | −108.2 ± 0.3 | 2.83 ± 0.10 | 2 | |||||
Activation and inactivation data were obtained from Boltzmann fits. M1651Q/M1652Q and M1651A/M1652A mutant channels do not show effects on the slopes of activation and inactivation (Tang et al., 1996). n, number of cells. Data are expressed as mean ± SEM.
Since steady state inactivation is determined by the rate constants for entry into and exit from inactivated states, we investigated the time constant for recovery from inactivation (τrec) for channel mutations at the A1649 site. Values of τrec furnish information on the rate of leaving inactivated states at negative voltages. The up to 10-fold lower τrec values for the F1648Q/A1649Q and A1649Q amino acid replacements than WT (Fig. 3 B), and the largely unchanged (F1648Q/A1649Q) or slightly left-shifted (A1649Q) steady state inactivation curves (Fig. 3 A), indicate that the net rates of entry into an inactivated state must be 10-fold greater in the mutant channels in the voltage range in which steady state inactivation changes (−130 to −80 mV). Estimates of rate constants for entry into the inactivated state, from time constants for recovery from inactivation (Fig. 3 B) (assuming that entry and exit from the inactivated state behaves like a two-state first order reaction; Hodgkin and Huxley, 1952) and the steady state probability of inactivation (Fig. 3 A), are consistent with the suggestion that entry into the inactivated state for the A1649Q channel is more rapid than WT channels in this voltage range (see also O'Leary et al., 1995). The reduction in voltage dependences of the rates of inactivation and recovery from inactivation for substitutions at A1649 are expected to contribute to the decreased slope of the steady state inactivation curve (Fig. 3 A) and provide strong support for the view that the A1649 site contributes to the coupling of activation and inactivation.
The reversal potential (E rev) and the current amplitude values (Table II) for single channels are unchanged by mutation (E rev values range from +44.2 to +47.6 mV), indicating that these mutations do not affect permeation and ion selectivity.
Table II.
Data of Single Channel Analysis
| V | WT | A1649Q | R | P | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| mV | ||||||||||
| Peak P open | −20 | 0.30 ± 0.03 | 0.22 ± 0.02 | 0.73 | <0.05 | |||||
| −40 | 0.18 ± 0.02 | 0.13 ± 0.02 | 0.72 | <0.05 | ||||||
| Median first latency (ms) | −20 | 0.79 ± 0.12 | 0.77 ± 0.06 | 0.97 | NS | |||||
| −40 | 1.79 ± 0.30 | 0.93 ± 0.08 | 0.52 | <0.05 | ||||||
| τh (ms) | −20 | 0.71 ± 0.07 | 0.72 ± 0.04 | 1.01 | NS | |||||
| −40 | 2.77 ± 0.67 | 0.80 ± 0.14 | 0.23 | <0.01 | ||||||
| τopen (ms) | −20 | 0.25 ± 0.03 | 0.15 ± 0.02 | 0.60 | <0.01 | |||||
| −40 | 0.23 ± 0.03 | 0.16 ± 0.02 | 0.63 | <0.05 | ||||||
| Current amplitude (pA) | −20 | 1.80 ± 0.03 | 1.81 ± 0.03 | 1.0 | NS | |||||
| −40 | 1.93 ± 0.03 | 2.00 ± 0.02 | 1.05 | NS |
These data derive from nine and six patches in WT and A1649Q, respectively. Total sweeps are 1,336 (−20 mV) and 1,593 (−40 mV) for WT, 992 (−20 mV), and 1,042 (−40 mV) for A1649Q. R, ratio = A1649Q/WT. Data are expressed as mean ± SEM.
The A1649Q Mutation Alters the Rate of Activation of Macroscopic Current
Although early formulations of sodium channel gating assumed that activation and inactivation are independent, with each having its own intrinsic voltage dependence, it is presently accepted that the processes are coupled such that inactivation derives most of its voltage dependence from being coupled to activation. Mutations that alter the coupling between activation and inactivation might, therefore, be expected to have additional effects on activation (O'Leary et al., 1995). We have examined this possibility in both whole-cell and single-channel configurations.
The normalized conductance–voltage (G-V) relationships, derived from the peak current–voltage relationship for activation of whole-cell Na+ currents, show differences for all mutations at A1649 from the curve of WT channels (Fig. 4 A and Table I). We will discuss mutations with rightward shifts in midpoint potentials first and those with leftward shifts second. The G-V relationships, when fitted to Boltzmann equations, show 9.2- and 10.5-mV depolarizing shifts, respectively, for the F1648Q/A1649Q (P < 0.01) and A1649Q (P < 0.01) mutations without any significant alterations in the slopes (P > 0.05) (Fig. 4 A and Table I). Thus, for these mutants, the open state is destabilized relative to nonopen states reflecting either decreased net rates of activation and/or increased net rates of inactivation/deactivation, which could reduce the peak conductance (O'Leary, et al., 1995). Consistent with this requirement, the τh values for inactivation in the voltage range −55 to −25 mV are smaller for F1648Q/A1649Q and A1649Q mutants than for WT channels.
To examine the kinetics of activation, the half-times of current increase (t 1/2) were estimated and plotted against test potentials from −55 to +75 mV (Fig. 4 B). Clearly, the macroscopic net rates of activation appear faster and less voltage dependent for both the F1648Q/ A1649Q and A1649Q mutants than WT channels at voltages more negative than −25 mV (Figs. 4, B and C). However, for a simple two-state model (C → O), the right-shifted G-V curve (Fig. 4 A) is inconsistent with what appears to be a left-shifted t 1/2 vs. voltage curve for the mutant channels relative to WT (Fig. 4 B), unless inactivation/deactivation are disproportionately speeded up. Since activation is not first-order but is clearly multi-step (note the lag in the time course of channel opening; Fig. 4 C), it may be that neither the G-V nor the t 1/2-V plots capture the full details of the entire process.
The second group consists of mutant channels with hyperpolarizing shifts of 7.3 and 3.8 mV in the midpoints of normalized G-V relationships for the A1649S (P < 0.01) and A1649V (P > 0.05) mutations with slight increases in slopes (P > 0.05) relative to WT channel (Table I). Thus, for these mutants, the open state is stabilized relative to nonopen states, reflecting increased net rates of activation and/or decreased net rates of inactivation/deactivation, which could increase peak conductance (O'Leary et al., 1995). Since inactivation rate constants for these mutations are increased at voltages more negative than −40 mV (Fig. 2 C), one might expect to find disproportionately increased net rates of activation in future studies.
Correlations of Shifts in Midpoints of Steady State Activation (G-V) and Inactivation (h∞•-V) Due to Mutations
Shifts in midpoints of activation (G-V) and inactivation (h∞•-V) due to the introduction of mutations into channel proteins are often difficult to rationalize in structural terms (Chen et al., 1996; Fleig et al., 1994; Kontis et al., 1997) and such seems to be the case in the series of mutations reported in this paper. Attempts to correlate midpoint shifts of activation or inactivation with position of the mutation in the primary sequence looking for a gradation of effects at sites progressively more distant from S4 or a pattern compatible with an α-helical conformation of the S4–S5/D4 loop failed. In addition, shifts in steady state activation do not correlate in magnitude or direction with the shifts in steady state inactivation. In terms of voltage sensitivity, all but two of the h∞•-V slopes are significantly smaller (by ≥1 e o unit) than that of the WT channel (the exceptions are M1651Q/M1652Q and M1651A/M1652A; Table I). In contrast, while many of the G-V slope factors values are at or just below that of the WT channel, only the L1646Q/L1647Q, L1654Q, M1651Q/M1652Q, and M1651A/M1652A mutation slopes are ≥1 e o unit larger than that of the nonmutated channel (Table I). This suggests that mutations in S4–S5/D4 have greater effects on the voltage sensitivity of steady state inactivation than on the voltage sensitivity of steady state activation. This may be related to the greater effect of mutations at S4D4 cationic sites than at similar sites in Domains 1–3 on τh values and their voltage dependencies (Chen et al., 1996).
Single Channel Properties of the A1649Q Mutant
One of the remarkable features of the A1649Q mutant channels is that the τh values are constant over the voltage range −55 to +75 mV (Fig. 2 C), a range in which channel activation is strongly voltage dependent (Fig. 4 A). Previous studies have shown that τh is largely determined by the first-latency values at moderate depolarizations, that is, the time to the first opening of a single Na+ channel after depolarization (Aldrich et al., 1983). During this time period, the Na+ channel transits through multiple closed states in the activation pathway (Armstrong and Bezanilla, 1973; Armstrong, 1981). Thus, the voltage-independent time constant of current decay obtained from whole-cell recordings in the A1649Q channel would be expected to be associated with a reduced voltage dependence of the latency to first opening in single channel records of the mutant compared with WT (see below).
Cell-attached patches containing either WT or A1649Q mutant channels expressed in tsA201 cells were depolarized from a holding potential of −120 mV to test potentials of −40 and −20 mV, respectively, for 40 ms (Fig. 5). In response to depolarization to −40 and −20 mV, both WT and A1649Q channels generally open once, occasionally twice, at the beginning of the depolarization. The first-latency times, corrected for the number of channels and displayed as a cumulative distribution function for WT and A1649Q channels, show a significantly decreased maximum probability of opening for A1649Q (n = 6), compared with WT (n = 9), at −40 mV (29 ± 1.2 vs. 51 ± 4.7%) and −20 mV (42 ± 1.2 vs. 61 ± 1.1%) (Fig. 6, C and D). This is consistent with the shift in the midpoints of the whole-cell G-V curves to depolarizing potentials for mutant channels (Fig. 4 A and Table I). In addition, the relative increase in cumulative open probability from −40 to −20 mV is similar for WT and A1649Q channels, which is consistent with parallel slopes of the whole-cell G-V curves of mutant and WT channels (Fig. 4 A and Table I).
Figure 5.

Recordings from cell-attached patches for WT and A1649Q channels. Sweep examples were selected from one-channel patches depolarized to test potentials of −40 and −20 mV from a holding potential of −120 mV for 40 ms.
Figure 6.

Analysis of single-channel properties. (A and B) Ensemble average open probability for WT and A1649Q channel patches, respectively, at −20 and −40 mV. (C and D) First latency distributions for the WT and A1649Q channel patches, respectively, corrected for the number of channels. Number of sweeps and median first latency values are contained in Table II.
Upon depolarization to −40 and −20 mV, in contrast to WT channels, the first latency times for A1649Q channels are shorter, particularly at −40 mV, and the voltage dependence of the first latency times over this voltage range is less in the A1649Q mutant compared with WT channel (Figs. 5 and 6, C and D). In WT channels, the median first latency time decreased 2.3-fold between −40 and −20 mV compared with 1.2-fold for A1649Q (Fig. 6, C and D, and Table II). Thus, the single channel results for A1649Q are consistent with the reduced t 1/2 values and lesser voltage dependencies observed earlier for the macroscopic kinetics of activation relative to WT channel (Fig. 4 B). The ensemble average of A1649Q shows a narrower P open distribution and a significantly decreased peak open probability compared with the WT channel at test potentials of −40 and −20 mV (Fig. 6, A and B, and Table II), which is consistent with the shift of midpoints of the mutant G-V curves in the depolarized direction. The decreased open probability in A1649Q suggests a decreased transition from closed to open states in the activation pathway and an increased inactivation rate from closed states at negative potentials. The amplified transition rate from the closed to inactivated state was suggested previously by the macroscopic steady state inactivation curve shifts in the hyperpolarizing direction for the A1649Q channel (Fig. 3 A).
The macroscopic time constants of current decay (τh), which are contributed to by activation, deactivation, and inactivation to various extents at different voltages, can be derived from the ensemble-averaged single-channel currents. The τh values from ensembles show a smaller voltage dependence at test potentials of −40 and −20 mV for the A1649Q channel than for WT (Fig. 6, A and B, and Table II), consistent with the behavior observed with whole-cell recordings (Fig. 2 C).
Because channels are increasingly likely to enter the inactivated state from the open state after activation as the depolarization voltages become more positive (Armstrong, 1981; Aldrich et al., 1983), the single channel open times become increasingly a reflection of the transition rate from the open to the inactivated state due to the progressively smaller contribution from deactivation. Mutations that slow the entry into the inactivated state from the open state are expected to increase single channel mean open times. Histograms of the distributions of the channel open times for WT and A1649Q channels at test potentials of −40 and −20 mV have been fitted by a single exponential (Fig. 7, dashed lines). The good fit to a single exponential suggests that each channel experiences a single open state that is short lived. The time constant of these fits (τopen) corresponds to the mean open time and the shorter mean open time for the A1649Q mutation at each of the voltages tested, relative to those of the WT channel (Table II), which suggests that the transition to the inactivated state from the open state is accelerated in the mutated channel. In WT and A1649Q channels, the open times (τopen) were not significantly voltage dependent at test potentials from −20 to −40 mV, consistent with previous studies that showed that the transition rate from the open state to the inactivated state is relatively voltage independent (Armstrong, 1981; Aldrich et al., 1983; Zagotta and Aldrich, 1990). This increased rate of inactivation (τh values from the ensemble of single channel experiments) combined with a smaller voltage dependency of first latency values could explain the fast and less voltage-dependent inactivation observed at negative potentials in the A1649Q channel (Fig. 2 C).
Figure 7.

Analysis of single channel open times. Open-time histograms are displayed for WT and A1649Q at −40 and −20 mV. The open times derive from fits to a single exponential shown as dashed lines. These histograms are one example of nine and six patches for WT and A1649Q, respectively. The time constants are 0.228 ± 0.013, 0.313 ± 0.017, 0.116 ± 0.014, and 0.126 ± 0.031 ms for A–D, respectively.
discussion
The most striking result is the similarity between the effects of the Y1494Q/Y1495Q mutations (O'Leary et al., 1995), COOH terminal to the IF1486M triplet in the ID3-4 segment, and the A1649Q mutation positioned in the S4–S5/D4 loop. It is unusual for a mutation to abolish the voltage dependence of τh values. In the case of the Y1494Q/Y1495Q mutation, a compelling case has been made that the substitutions increase the rate of activation and inactivation, in the latter case from both open and closed states (O'Leary et al., 1995).
We will discuss our experimental results in terms of a widely accepted kinetic gating model of voltage-dependent sodium channels in which the protein undergoes voltage-driven conformational changes as it transits through multiple closed states (Cn) into either open state (O) or inactivated state (I) (Scheme I). This scheme includes the possibility that, at depolarizing potentials, the channels can traverse a pathway to the inactivated state directly from either closed or open states, and that inactivation by such routes is irreversible over the course of a few milliseconds (Aldrich et al., 1983; Patlak, 1991).
According to some models, the S4 segment is an α-helix that experiences a large, helical screw movement normal to the lipid bilayer to transfer the necessary gating charge (Catterall, 1986; Durell and Guy, 1992). If the S4 segment moves in this fashion, then the S3–S4 and S4–S5 segments, tethered to the NH2- and COOH-terminal ends of S4, respectively, must be able to accommodate S4 displacement. The S3–S4 loop in the Shaker potassium channel appears unlikely to participate in a large conformational change during activation (Mathur et al., 1997). In contrast, the importance of the S4–S5/D4 segment in sodium channel inactivation is made obvious by altered electrophysiological parameters due to mutations at the majority of the sites in this loop. Substitution of glutamine at 7 of 10 positions, especially those in the portion of the segment bordering S4D4 (positions 1646–1652), results in significantly altered time constants of inactivation (Fig. 2). Mutations at sites 1654–1656, most distant from the S4 segment, showed little difference in τh values from the WT channel. We believe that the movement of S4D4 requires changes in the conformation or position of the NH2-terminal portion of the S4–S5/D4 segment, making this region sensitive to mutation. Consistent with our findings, the F1473S substitution in human skeletal muscle sodium channel (analogous to F1648 in hH1) is associated with slowing of fast inactivation and, apparently, is responsible for one form of Paramyotonia Congenita in humans (Fleischhauer et al., 1996).
We and others have shown that mutations in several channel regions decrease the voltage dependence of inactivation time constants or their magnitude. First, recent studies support the view that the S4 of D4, in conjunction with acting as a voltage sensor (Yang and Horn, 1995; Yang et al., 1996), is uniquely involved in the coupling between activation and inactivation (Chen et al., 1996). Thus, charge-neutralizing or -reversing substitutions at the outermost and third from outermost cationic positions in S4 segments of each domain of human heart sodium channel revealed that mutations only in D4 decrease the voltage dependence of the time constants for inactivation. Second, a pair of tyrosine residues, Y1494Q/Y1495Q, in the ID3-4 linker of hH1 is required for normal coupling between activation voltage sensors and the inactivation gate, because mutations at this site alter the macroscopic rates and decrease the voltage dependence of both activation and inactivation. These observations led to consideration of a molecular link between the core of the channel and the inactivation gate and a proposal that it is the S4–S5/D4 loop that provides the IAL (O'Leary et al., 1995; Chen et al., 1996). Third, the effects of mutations at a pair of methionine residues in the cytoplasmic loop S4–S5/D4 affect the voltage dependence and rate of inactivation (Tang et al., 1996).
The present study of the cytoplasmic loop of S4–S5/ D4 has identified a single alanine (A1649) that, when mutated to serine, valine, or glutamine, decreases or completely removes the voltage dependence of inactivation time constants. This loss of voltage dependence of inactivation appears to be unique for the A1649 site because no other amino acid replacement in this region that was tested has this effect (Fig. 2). The rates of inactivation are faster than WT at voltages more negative than −25 mV, but slightly slower than WT at depolarizations more positive than −25 mV. It appears as if the A1649Q substitution diminishes or eliminates an “encumbrance” to inactivation that is present in the WT channel, one that prevents the WT channel from prematurely entering an inactivated state at negative potentials.
The A1649Q Mutation in the S4–S5/D4 Loop Affects Activation
There is no precedent for mutations in cytoplasmic loops of sodium channels increasing the rate of activation other than the Y1494Q/Y1495Q mutations. On the contrary, only a single mutation has been reported to affect activation and in this case (equivalent to R1512E in the ID3-4 segment of hH1) the mutation in the rat brain isotype III channel causes a slowing of activation (Moorman et al., 1990). The smaller median first-latency values in A1649Q are consistent with the smaller t 1/2 values for activation, which would suggest that a mutation in a cytoplasmic loop has, unexpectedly, a pronounced effect on activation, reminiscent of the Y1494Q/Y1495Q mutation.
The increased macroscopic rates of inactivation at negative potentials can be rationalized in either of two ways or a combination of both. First, inactivation rate constants from closed states are increased in the mutant channel at negative voltages, which is consistent with the observed reduction of the peak open probability and cumulative open probability values (Fig. 6 and Table II). Second, the activation rate constants are increased. An accelerated transition along the activation pathway, associated with the shorter first latency times manifest by the mutant, could cause a more rapid inactivation at very negative voltages, due to the coupling of activation and inactivation. However, increased activation rate constants predict an increase in the peak open probability, while increased inactivation rate constants are expected to be associated with a decrease in the peak open probability in the mutant. Peak open probability estimates contain some uncertainty due to patch-to-patch variability and the number of channels in the patch. Therefore, we tentatively suggest that the large change in inactivation rate constants necessary to account for the effect of the mutation on first latency times cannot quantitatively be accommodated by the observed modest reduction of P open values in the A1649Q mutant unless activation (closed-to-open state) rate constants at voltages negative to −25 mV are also increased (see Y1494Q/Y1495Q; O'Leary et al., 1995).
The A1649Q Mutation in the S4–S5/D4 Loop Affects Inactivation
Inactivation per se is generally believed to have little intrinsic voltage dependence and most of its apparent voltage dependence derives from inactivation being coupled to activation, which is highly voltage dependent in sodium channels (Armstrong and Bezanilla, 1977; Armstrong, 1981; Aldrich et al., 1983; Zagotta and Aldrich, 1990; Patlak, 1991; Keynes, 1994; Sigworth, 1994; O'Leary et al., 1995; Chen et al., 1996). In this formulation, because inactivation of the WT Na+ channel is slower at negative potentials at which the channel activates more slowly and less completely, a substantial shift in the voltage dependence of channel activation toward more positive membrane potentials in a mutant channel would be expected to cause a corresponding slowing of inactivation if channels remain normally coupled (McPhee et al., 1996). Clearly, electrophysiological measurements of the A1649Q mutation (Figs. 2 C and 4 A) are not in accord with this expectation. The A1649Q mutation produces shifts in the midpoint of the steady state activation curve to more positive voltages but is associated with a speeding up of fast inactivation at negative potentials (≤−25 mV). We believe that this result is because the A1649Q channel has lost a component of coupling between activation and inactivation normally present in WT channels, with the result that changes in G-V curves have become largely independent of those in τh vs. V plots.
Our analysis has been further refined with single-channel studies. The ensemble-averaged currents show, as in whole-cell recordings, that τh values for current decay are less voltage dependent and smaller in the A1649Q channel at negative potentials in the range studied, from −40 to −20 mV (Fig. 6, A and B, and Table II). An explanation for these effects is that after a depolarization, a mutant channel can diverge from the activation pathway by inactivating more readily than WT from a closed state. As a result of an increase in the rate of closed-channel inactivation (overshadowing any increase in activation rate constants), there is a decrease in channel open probability. Thus, the first latency times decrease in the mutant because the channels that open and contribute to the first latency distribution must do so quickly before inactivating from closed states. In addition, a faster rate of inactivation from a closed state is expected to result in lower voltage dependence of the first latency times compared with WT channels (O'Leary et al., 1995). The decrease in magnitude and voltage dependence of first latency times in A1649Q channels causes τh to be smaller and less voltage dependent at voltages more negative than −25 mV. Finally, if WT inactivation is coupled to a slow conformational transition (e.g., a closed–closed transition in the activation pathway), this transition is either faster in A1649Q and no longer rate limiting, or the link between this slow transition and inactivation is broken in the A1649Q channel (O'Leary et al., 1995).
The time constant of the open channel inactivation also depends on the mean open time, τopen, since the activated channel primarily enters the inactivated state (rather than returning to a closed state by deactivation) at strongly depolarizing voltages (Armstrong, 1981; Aldrich et al., 1983; McPhee et al., 1995). Single-channel data show that there is a reduced length of time that a channel is open in the case of the A1649Q mutant at −40 and −20 mV compared with the WT channel, a result that is consistent with the smaller τh value measurements from macroscopic current analysis in this voltage range.
The most striking finding in this paper is the remarkable similarity in the inactivation phenotypes of the A1649Q and Y1494Q/Y1495Q mutations. The virtual elimination of the voltage dependence of τh values, an unusual effect, by mutations in two different regions of the channel, the S4–S5/D4 segment and the ID3-4 loop, suggests either that these cytoplasmic regions interact directly or, a less attractive possibility, that they individually interact with another region. We postulate that an inactivation–activation linkage, the IAL, exists by virtue of a physical interaction between the A1649 and Y1494 Y1495 regions of their respective segments, which establishes a direct link between the ID3-4 loop and the core of the channel protein via the S4–S5/D4 segment. This would explain the importance of the S4–S5/D4 segment in inactivation and might be the mechanism by which, in WT channels, the linked activation and inactivation gates have knowledge of each other's state. This linkage could be sensitive to changes in conformation of S4–S5/D4 (an unwinding of a coil or helix or a small translocation coordinated with the movement of S4) with subsequent indirect effects on the functional roles of adjacent regions. The net result is a change in the position of the ID3-4 loop, with its inactivation particle, relative to that of the core protein containing the IPR. We leave open the question of whether the IAL exists as a static, permanent noncovalent bonding interaction or whether the association is transient, occurring only at certain times during channel function. In any event, we propose that any changes in conformation, movements, and associations play two roles precisely controlling inactivation: first, contributing to the creation of a high affinity IPR, and second, establishing the proper relationship between the position of the ID3-4 loop and the core of the channel protein via the IAL. Constraints imposed by the IPR affinity and the positioning of the inactivation particle by the IAL ensure that inactivation will be slow at small depolarizations and rapid at large depolarizations in WT channels; these effects undoubtedly also contribute to the stability of the inactivation particle with its receptor. Thus, they become factors in determining the completeness of inactivation (i.e., whether there exists a residual current) and the kinetics of recovery from inactivation further contributing to the control of excitability.
Scheme I.

Acknowledgments
We thank Dr. R. Horn for conversations during this work and comments on the manuscript and the reviewers for helpful suggestions. Dr. L.-Q. Chen contributed molecular biological expertise.
Supported by grants to R.G. Kallen from the National Institutes of Health (AR-41,762), the American Heart Association (National and Southeastern Pennsylvania Affiliate), the University of Pennsylvania Research Foundation, the Department of Biochemistry and Biophysics of the University of Pennsylvania School of Medicine, and the Muscular Dystrophy Association, and to S.J. Wieland from the Muscular Dystrophy Association.
Abbreviations used in this paper
- G-V
conductance–voltage
- hH1
human heart Na+ channel subtype 1
- IAL
inactivation–activation linkage
- ID
interdomain
- IPR
inactivation particle receptor
- S
Q, and V, amino acids serine, glutamine, and valine
- WT
wild type
references
- Aldrich RW, Corey DP, Stevens CF. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 1983;306:436–441. doi: 10.1038/306436a0. [DOI] [PubMed] [Google Scholar]
- Armstrong CM. Sodium channels and gating currents. Physiol Rev. 1981;61:644–683. doi: 10.1152/physrev.1981.61.3.644. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F. Currents related to movement of the gating particles of the sodium channels. Nature. 1973;242:459–461. doi: 10.1038/242459a0. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld VJ, Goldin AL, Krafte DS, Catterall WA, Lester HA, Davidson N, Dunn RJ. A neutral amino acid change in segment IIS4 dramatically alters the gating properties of the voltage-dependent sodium channel. Proc Natl Acad Sci USA. 1990;87:323–327. doi: 10.1073/pnas.87.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N, George AJ. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- Bezanilla F. Gating of sodium and potassium channels. J Membr Biol. 1985;88:97–111. doi: 10.1007/BF01868424. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Molecular properties of voltage-sensitive sodium channels. Annu Rev Biochem. 1986;55:953–985. doi: 10.1146/annurev.bi.55.070186.004513. [DOI] [PubMed] [Google Scholar]
- Chahine M, Deschenes I, Trottier E, Chen L-Q, Kallen RG. Restoration of fast inactivation in an inactivation-defective human heart Na+channel by the cysteine-modifying reagent Bz-MTS: analysis of IFM-ICM mutation. Biochem Biophys Res Commun. 1997;233:606–610. doi: 10.1006/bbrc.1997.6510. [DOI] [PubMed] [Google Scholar]
- Chahine M, George AL, Jr, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Chen L-Q, Santarelli V, Horn R, Kallen RG. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J Gen Physiol. 1996;108:549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depp MR, Goldin AL. Probing S4–S5 regions of the rat brain sodium channel for the fast inactivation particle receptor site. Biophys J. 1996;70:A317. [Google Scholar]
- Durell SR, Guy HR. Atomic scale structure and functional models of voltage-gated potassium channels. Biophys J. 1992;62:238–247. doi: 10.1016/S0006-3495(92)81809-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton DC, Brodwick MS, Oxford GS, Rudy B. Arginine-specific reagents remove sodium channel inactivation. Nature. 1978;271:473–476. doi: 10.1038/271473a0. [DOI] [PubMed] [Google Scholar]
- Featherstone DE, Richmond JE, Ruben PC. Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys J. 1996;71:3098–3109. doi: 10.1016/S0006-3495(96)79504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleig A, Fitch JM, Goldin AL, Rayner MD, Starkus JG, Ruben PC. Point mutations in IIS4 alter activation and inactivation of rat brain IIA Na channels in Xenopus oocyte macropatches. Pflügers Arch. 1994;427:406–413. doi: 10.1007/BF00374254. [DOI] [PubMed] [Google Scholar]
- Fleischhauer R, George AL, Lerche H, Mitrovic N, Lehmann-Horn R. Electrophysiological characterization of a novel myotonia causative mutation in the human muscle sodium channel. Biophys J. 1996;70:A318. [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hartmann HA, Tiedeman AA, Chen SF, Brown AM, Kirsch GE. Effects of III–IV linker mutations on human heart Na+channel inactivation gating. Circ Res. 1994;75:114–122. doi: 10.1161/01.res.75.1.114. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. The components of membrane conductance in the giant axon of Loligo. J Physiol (Camb) 1952;116:473–496. doi: 10.1113/jphysiol.1952.sp004718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren M, Jurman ME, Yellen G. N-type inactivation and the S4–S5 region of the Shaker K+channel. J Gen Physiol. 1996;108:195–206. doi: 10.1085/jgp.108.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Isacoff EY, Jan YN, Jan LY. Putative receptor for the cytoplasmic inactivation gate in the Shaker K+channel. Nature. 1991;353:86–90. doi: 10.1038/353086a0. [DOI] [PubMed] [Google Scholar]
- Kallen RG, Cohen SA, Barchi RL. Structure, function and expression of voltage-dependent sodium channels. Mol Neurobiol. 1993;7:383–428. doi: 10.1007/BF02769184. [DOI] [PubMed] [Google Scholar]
- Kellenberger S, West JW, Scheuer T, Catterall WA. Molecular analysis of the putative inactivation particle in the inactivation gate of the brain-type IIA Na+channel. J Gen Physiol. 1997;109:589–605. doi: 10.1085/jgp.109.5.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keynes RD. A new look at the mechanism of activation and inactivation of voltage-gated ion channels. Proc R Soc Lond B Biol Sci. 1992;249:107–112. doi: 10.1098/rspb.1992.0091. [DOI] [PubMed] [Google Scholar]
- Keynes RD. The kinetics of voltage-gated ion channels. Q Rev Biophys. 1994;27:339–434. doi: 10.1017/s0033583500003097. [DOI] [PubMed] [Google Scholar]
- Kontis KJ, Rounaghi A, Goldin AL. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J Gen Physiol. 1997;110:391–401. doi: 10.1085/jgp.110.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson HP, Baker OS, Dhillon DS, Isacoff EY. Transmembrane movement of the Shaker K+channel S4. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- Lerche H, Mitrovic N, Dubowitz V, Lehmann-Horn F. Paramyotonia congenita: the R1448P Na+channel mutation in adult human skeletal muscle. Ann Neurol. 1996;39:599–608. doi: 10.1002/ana.410390509. [DOI] [PubMed] [Google Scholar]
- Lerche H, Peter W, Fleischhauer R, Pika-Hartlaub U, Malina T, Mitrovic N, Lehmann-Horn F. Role in fast inactivation of the IV/S4-S5 loop of the human muscle Na+channel probed by cysteine mutagenesis. J Physiol (Camb) 1997;505:345–352. doi: 10.1111/j.1469-7793.1997.345bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman ER, Hess P, Weaver F, Koren G. Voltage-sensing residues in the S4 region of a mammalian K+channel. Nature. 1991;353:752–756. doi: 10.1038/353752a0. [DOI] [PubMed] [Google Scholar]
- Logothetis DE, Kammen BF, Lindpaintner K, Bisbas D, Nadal-Ginard B. Gating charge differences between two voltage-gated K+channels are due to the specific charge content of their respective S4 regions. Neuron. 1993;10:1121–1129. doi: 10.1016/0896-6273(93)90060-5. [DOI] [PubMed] [Google Scholar]
- Lopez GA, Jan YN, Jan LY. Hydrophobic substitution mutations in the S4-sequence alter voltage-dependent gating in Shaker K+channels. Neuron. 1991;7:327–336. doi: 10.1016/0896-6273(91)90271-z. [DOI] [PubMed] [Google Scholar]
- Margolskee RF, McHendry-Rinde B, Horn R. Panning transfected cells for electrophysiological studies. Biotechniques. 1993;15:906–911. [PubMed] [Google Scholar]
- Marten I, Hoshi T. Voltage-dependent gating characteristics of the K+channel KAT1 depend on the N and C termini. Proc Natl Acad Sci USA. 1997;94:3448–3453. doi: 10.1073/pnas.94.7.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur R, Zheng J, Yan Y, Sigworth FJ. Role of the S3–S4 linker in Shaker potassium channel activation. J Gen Physiol. 1997;109:191–199. doi: 10.1085/jgp.109.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack K, Tanouye MA, Iverson LE, Lin JW, Ramaswami M, McCormack T, Campanelli JT, Mathew MK, Rudy B. A role for hydrophobic residues in the voltage-dependent gating of Shaker K+channels. Proc Natl Acad Sci USA. 1991;88:2931–2935. doi: 10.1073/pnas.88.7.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A critical role for transmembrane segment IVS6 of the sodium channel alpha subunit in fast inactivation. J Biol Chem. 1995;270:12025–12034. doi: 10.1074/jbc.270.20.12025. [DOI] [PubMed] [Google Scholar]
- McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A role for intracellular loop IVS4–S5 of the Na+channel alpha-subunit in fast inactivation. Biophys J. 1996;70:A318. [Google Scholar]
- Moorman JR, Kirsch GE, Brown AM, Joho RH. Changes in sodium channel gating produced by point mutations in a cytoplasmic linker. Science. 1990;250:688–691. doi: 10.1126/science.2173138. [DOI] [PubMed] [Google Scholar]
- O'Leary ME, Chen L-Q, Kallen RG, Horn R. A molecular link between activation and inactivation of sodium channels. J Gen Physiol. 1995;106:641–658. doi: 10.1085/jgp.106.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxford GS, Wu CH, Narahashi T. Removal of sodium channel inactivation in squid giant axons by n-bromoacetamide. J Gen Physiol. 1978;71:227–247. doi: 10.1085/jgp.71.3.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papazian DM, Shao XM, Seoh SA, Mock AF, Huang Y, Wainstock DH. Electrostatic interactions of S4 voltage sensor in Shaker K+channel. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- Patlak J. Molecular kinetics of voltage-dependent Na+channels. Physiol Rev. 1991;71:1047–1080. doi: 10.1152/physrev.1991.71.4.1047. [DOI] [PubMed] [Google Scholar]
- Perozo E, Santacruz-Toloza L, Stefani E, Bezanilla F, Papazian DM. S4 mutations alter gating currents of Shaker K channels. Biophys J. 1994;66:345–354. doi: 10.1016/s0006-3495(94)80783-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas E, Armstrong C. Sodium conductance activation without inactivation in pronase-perfused axons. Nat New Biol. 1971;229:177–178. doi: 10.1038/newbio229177a0. [DOI] [PubMed] [Google Scholar]
- Schonherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol (Camb) 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ. Voltage gating of ion channels. Q Rev Biophys. 1994;27:1–40. doi: 10.1017/s0033583500002894. [DOI] [PubMed] [Google Scholar]
- Smith MR, Goldin AL. Interaction between the sodium channel inactivation linker and domain III S4-S5. Biophys J. 1997;73:1885–1895. doi: 10.1016/S0006-3495(97)78219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Tang L, Kallen RG, Horn R. Role of an S4–S5 linker in sodium channel inactivation probed by mutagenesis and a peptide blocker. J Gen Physiol. 1996;108:89–104. doi: 10.1085/jgp.108.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlau H, Heinemann SH, Stuhmer W, Pongs O, Ludwig J. Amino terminal-dependent gating of the potassium channel rat eagis compensated by a mutation in the S4 segment. J Physiol (Camb) 1997;502:537–543. doi: 10.1111/j.1469-7793.1997.537bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDongen AM, Frech GC, Drewe JA, Joho RH, Brown AM. Alteration and restoration of K+channel function by deletions at the N- and C-termini. Neuron. 1990;5:433–443. doi: 10.1016/0896-6273(90)90082-q. [DOI] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, George AL, Jr, Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- Yang N, Horn R. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 1995;15:213–218. doi: 10.1016/0896-6273(95)90078-0. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Aldrich RW. Voltage-dependent gating of Shaker A-type potassium channels in Drosophila muscle. J Gen Physiol. 1990;95:29–60. doi: 10.1085/jgp.95.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]