

Abstract
The D4/S4-5 interhelical region plays a role in sodium channel fast inactivation. Examination of S4-5 primary structure in all domains suggests a possible amphipathic helical conformation in which a conserved group of small hydrophobic residues occupies one contiguous surface with a more variable complement of nonpolar and polar residues on the opposite face. We evaluated this potential structure by replacing each residue in D4/S4-5 of the rat SkM1 skeletal muscle sodium channel with substitutions having different side chain properties. Of the 63 mutations analyzed, 44 produced functional channels. P1473 was intolerant of substitutions. Nonpolar substitutions in the conserved hydrophobic region were functionally similar to wild type, while charged mutations in this region before P1473 were nonfunctional. Charged mutations at F1466, M1469, M1470, and A1474, located on the opposite surface of the predicted helix, produced functional channels with pronounced slowing of inactivation, shifted voltage dependence of steady-state inactivation, and increased rate of recovery from inactivation. The substituted-cysteine-accessibility method was used to probe accessibility at each position. Residues L1465, F1466, A1467, M1469, M1470, L1472, A1474, and F1476C were easily accessible for modification by sulfhydryl reagents; L1464, L1468, S1471, and L1475 were not accessible within the time frame of our measurements. Molecular dynamics simulations of residues A1458 to N1477 were then used to explore energetically favorable local structures. Based on mutagenesis, substituted-cysteine-accessibility method, and modeling results, we suggest a secondary structure for the D4/S4-5 region in which the peptide chain is α-helical proximal to P1473, bends at this residue, and may continue beyond this point as a random coil. In this configuration, the entire resultant loop is amphipathic; four residues on one surface could form part of the binding site for the inactivation particle.
Keywords: sodium channel inactivation, mutagenesis, cysteine accessibility, structural modeling
introduction
Voltage-gated sodium channels play a major role in the propagation of action potentials in excitable tissues (Hodgkin and Huxley, 1952). Although sodium channels may contain an α and one or two smaller β subunits, the major functional elements of the channel are contained in the large (∼260 kD) alpha subunit. Sodium channel alpha subunits share structural features with a superfamily of voltage-gated channels that includes Ca++ channels and K+ channels. The sodium channel's alpha subunit has four large internal repeat domains (D1–D4), each containing six putative transmembrane helices (S1–S6) (Noda et al., 1984). The S4 segments in each domain act as voltage sensors, moving outward upon depolarization (Greenblatt et al., 1985; Stuhmer et al., 1989; Yang and Horn, 1995; Mannuzzu et al., 1996; Yang et al., 1996). The intracellular segment between D3 and D4 (ID3-4)1 serves as part of the fast inactivation gate (Vassilev et al., 1988; Patton et al., 1992; West et al., 1992; Eaholtz et al., 1994), which occludes the open pore of the channel from the cytoplasmic side after activation (Bezanilla and Armstrong, 1977; Armstrong and Bezanilla, 1977).
In the sodium channel, three hydrophobic residues ID3-4 have been shown to be critical for fast inactivation (West et al., 1992); charged residues in this region appear to play a less consistent role (Moorman et al., 1990; Patton et al., 1992). According to current models, inactivation requires that this ID3-4 region, or the structurally homologous amino terminal inactivation ball in potassium channels (Hoshi et al., 1990), binds to a receptor region on the cytoplasmic surface of the channel. For potassium channels, evidence suggests that the S4-5 interhelical region of each subunit forms part of this receptor surface, since mutations in this region that alter local charge also interfere with inactivation (Isacoff et al., 1991) or change the affinity of the channel for the inactivation ball peptide (Holmgren et al., 1996). Some mutations in the S4-5 linker of potassium channels also alter ion permeability, implying that this segment is located near the cytoplasmic end of the ion permeation pathway (Slesinger et al., 1993). Residues in the sodium channel D4/S4-5 linker have recently been shown to play an important role in fast inactivation in this channel as well (Tang et al., 1996; Mitrovich et al., 1996; Smith and Goldin, 1997). Several mutations that produce human paramyotonia congenita are located in the D3/S4-5 region (A1156T, I1160V); these mutations also affect the rate of channel inactivation and recovery from inactivation (Yang et al., 1994; Ji et al., 1995).
The amino acid sequence in the S4-5 region of sodium channels, although variable between domains, is highly conserved among isoforms within each domain. Furthermore, helical wheel analysis comparing all domains suggests common structural features that are not apparent from sequence alignment. These include a potential amphipathic helical conformation containing on one surface a region of conserved small nonpolar residues and on the opposite surface a group of more variable nonpolar and polar residues. Such an amphipathic helix could be located at the cytoplasmic end of the channel pore, with the nonpolar surface interacting with adjacent hydrophobic helices and the opposite surface forming part of the inactivation gate binding site.
In an effort to test this hypothetical structure and to probe the role of D4/S4-5 in channel inactivation, we have carried out a mutagenic analysis of D4/S4-5 in the SkM1 sodium channel by introducing multiple substitutions at each residue between I1461 and N1477. For cysteine substitutions at each position, accessibility to aqueous sulfhydryl-modifying reagents was also assessed. Potential secondary structure of the D4/S4-5 peptide was then studied by molecular modeling using dynamic simulations techniques. Taking into consideration the results of our mutagenesis, substituted-cysteine-accessibility method (SCAM) analysis, and modeling, we propose that this region forms an amphipathic α-helix at the inner mouth of the ion pore, and that a cluster of accessible residues on the variable surface of the helix forms part of the binding site for ID3-4 during channel inactivation.
materials and methods
Site-directed Mutagenesis
The site-specific mutations in the rat SkM1 channel were created using the Altered Sites II (Promega Corp., Madison, WI) protocol with minor modifications. The full-length SkM1 channel was originally cloned into the pRC/CMV vector (Invitrogen Corp., San Diego, CA) and divided into three cassettes. The COOH-terminal cassette encompassing D4/S4-5 (pSr1CT), which was cloned into pAlter-1 between the EcoRI and BamHI sites in its polylinker, encodes sodium channel sequences between the SstII site at 4355 and 5822 at the 3′ terminal EcoRI site. The single-stranded DNA generated from the pSr1CT vector is the noncoding strand and the primers used to generate the mutations are coding. Primers differing at one position from the original sequence extended 10–12 bp on either side of the mutation site, those differing in two positions extended 15–19 bp on either side of the site, and those differing in three positions extended 20–25 bp on either side of the site. Primers were phosphorylated before use.
Mutagenesis was carried out using single-stranded DNA as the template and 5 pmol of mutagenic primer. After mutagenesis, four to six colonies were selected, minipreps (Promega Corp.) were carried out, and DNA was sequenced by the dideoxy method using Sequenase v2.0 (USB Biologicals, Cleveland, OH) to at least 80 bp on either side of the mutation. Two or more independent clones for each mutation were inserted into the full-length sodium channel in the pCI vector (Promega Corp.), sequenced, and used for electrophysiological analysis.
Transient Expression in Cultured Cells
tsA-201 cells in culture were transiently transfected with 20–40 μg SkM1 sodium channel α-subunit DNA and 5–10 μg of DNA encoding the CD8 receptor using the standard calcium-phosphate method. Cotransfection of CD8 allowed us to perform visual localization of transfected cells (Margolskee et al., 1993; Jurman et al., 1994). Transfected cells were maintained overnight in 5% CO2 in DMEM (GIBCO BRL, Gaithersburg, MD) with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, followed by replacement with fresh medium and additional incubation for 8 h at 5% CO2 before splitting. 24–36 h after transfection, the cells were split by brief trypsinization and repeated trituration through a fine-bore pipette tip and replated for electrophysiological analysis.
Electrophysiology
Macroscopic currents were recorded from tsA-201 cells in the whole-cell configuration. Cells were equilibrated to room temperature (18–20°C) for 5–10 min, and then washed twice with extracellular solution containing (mM): 150 NaCl, 2 KCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES (Sigma Chemical Co., St. Louis, MO), pH 7.4. Dynabeads (DYNAL A.S., Oslo, Norway) precoated with antibody to CD8 were added and cells with more than five bound dynabeads were selected for patching. With wild-type (WT) cDNA, this method gives close to 100% yield of current-expressing cells. Recordings were performed 2–4 d after transfection using an Axopatch-1B amplifier (Axon Instruments, Foster City, CA). Data were filtered at 5 kHz and acquired with pCLAMP software (Axon Instruments). Electrodes were filled with (mM) 35 NaCl, 105 CsF, 10 EGTA, 10 HEPES, pH 7.4. Patch electrode resistance after 80% compensation was 0.5–1.5 mΩ. All recording protocols were performed at least 10 min after obtaining the whole-cell configuration to insure stabilization of currents. Cells expressing peak inward currents between 1 and 20 nA were selected for further analysis. Mutants that produced currents <1 nA were considered inactive since currents below this level could not be reliably distinguished from variable endogenous currents that can be up to 1 nA in control tsA-201 cells. For subsequent data analysis, we used PClamp6 (Axon Instruments), Origin (Microcal Software, Inc., Northampton, MA), and SigmaPlot (Jandel Scientific, Corte Madera, CA) software. Data analysis was carried out as previously described (Ji et al., 1996).
Modification by Sulfhydryl Reagents
Sulfhydryl reagents, sodium (2-sulfonatoethyl)methanethiosulfonate (MTSES) or [2-(trimethylammonium)ethyl]methanethiosulfonate (MTSET) (Toronto Research Chemicals, Inc., Toronto, Alberta, Canada), were added to the patch pipet at a concentration of 2 mM and allowed to diffuse from the pipet into the cell under whole-cell configuration. A 20-ms control pulse from a holding potential of −120 to −10 mV was applied every 30 s to monitor inactivation kinetics.
Structural Modeling
Structural modeling of the S4-5 peptide was carried out using the programs “InsightII” (BIOSYM/Molecular Simulation, San Diego, CA) and “Grasp” (Nicholls et al., 1991) implemented on a Silicon Graphics work station. For the structures shown in Fig. 8, molecular dynamics simulations (including energy minimization) were carried out by the “Discover” molecular mechanics module of InsightII, using the consistent valence force field (CVFF), in water, at 300°K temperature, with a time step of 1.0 fs, at constant density, and under the assumption that the preferred starting structure for D4/S4-5 is an α-helix up to P1473, with no initial constraints on the region distal to this proline. Typically, simulations were followed over ∼100,000 iterations, corresponding to a total time interval of 0.1 ns. The Grasp software package was used for graphical visualization of the resulting structures.
Figure 8.
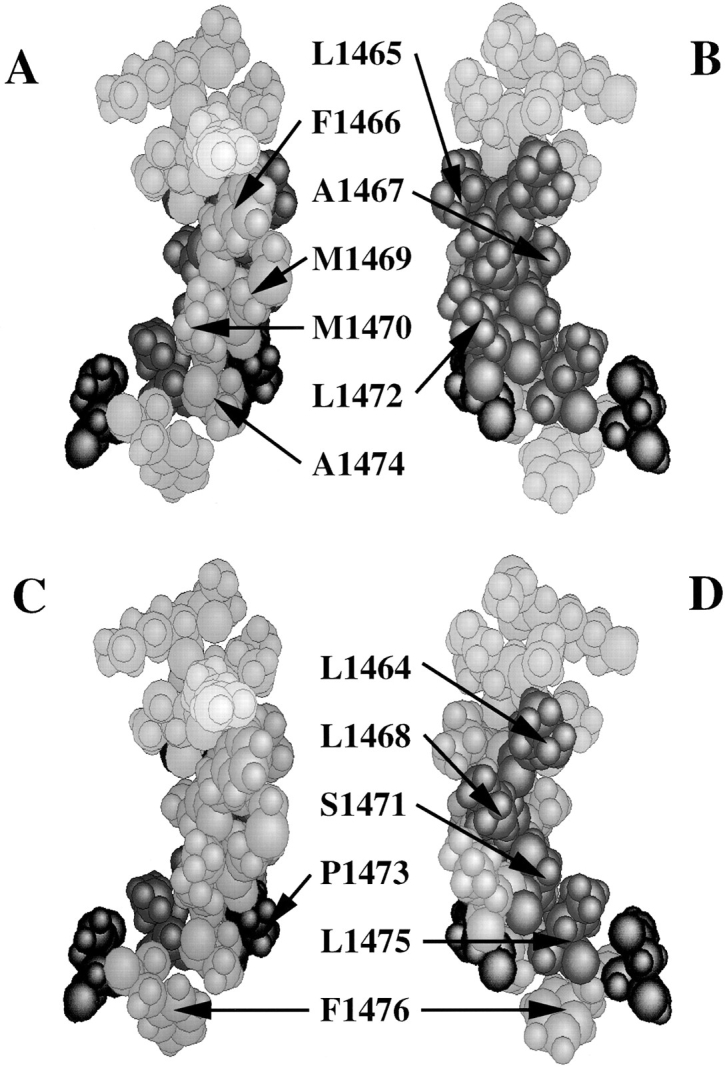
Molecular dynamics simulations for the D4/S4-5 segment, with initial conditions of α-helix before P1473 and random coil beyond that point, reached equilibrium after ∼50,000 iterations (0.05-ns simulation time), with a structure that subsequently remained stable between 0.05 and 0.1 ns (0.1 ns total simulation time). In this structure, residues I1461 to L1472 remain in an α-helix; the structure bends at P1473 and residues distal to it are in an extended chain. Atomic models of the structure after 0.1 ns of simulation are shown. Residues where charged substitutions prolonged channel inactivation are shown in A (green). These residues include F1466, M1469, M1470, and A1474, where charged mutations most strongly destabilized the inactivated state; this cluster of residues is postulated to form part of the inactivation particle binding site. The distribution of residues that are easily accessible for MTSES or MTSET modification (C, green) parallels that of mutations affecting inactivation. Residues where charged substitution produced nonfunctional channels, shown in B (red), are concentrated on the opposite surface of the structure; this surface also contains the residues that are inaccessible to cysteine modification (D, red) and corresponds to the conserved nonpolar surface identified in the initial helical wheel projection (see Fig. 1).
results
Although many of the proposed interhelical regions in each sodium channel repeat domain are poorly conserved, the S4-5 interhelical loop in any single domain shows a high degree of amino acid sequence homology among various sodium channel isoforms. One potential secondary structure for all or part of this region might be an α-helix, and helical wheel analysis of the central 18 amino acid residues in this region does suggest additional common structural features among domains that are not apparent in the primary structure (Fig. 1). These features include a conserved sector of small hydrophobic residues that occupies one contiguous surface of a potential helix in all four domains; the opposite face of this helix is less well conserved and contains various charged and polar residues in different repeat domains, suggesting an amphipathic structure. In our discussion, this face is referred to as the variable surface of the proposed helix.
Figure 1.

Helical wheel projection of the S4-5 region in domains 1–4 of the SkM1 sodium channel, aligned with respect to a common serine residue at the location arbitrarily designated 12 on the helical wheel. Residues marked with an asterisk are conserved in that location for the various rat brain and muscle sodium channels. This analysis suggests the presence of a conserved region of small nonpolar residues (gray) opposite a more variable nonpolar and polar surface, consistent with an amphipathic helical structure. The mutations introduced for this study at each location in D4/S4-5 are indicated.
We introduced mutations that differed in charge, size, polarity, or aromaticity into positions of the primary sequence in D4/S4-5 between I1461 and N1477. Mutations were evaluated for kinetic properties after transient expression in tsA-201 cells. A total of 63 mutations was created at 17 sites (Fig. 1). Of these 63 mutations, 19 were nonfunctional (Table I). The kinetic properties of the remaining 44 mutants were analyzed using whole-cell patch clamp techniques (Tables II and III).
Table I.
Mutations in D4/S4-5 Producing Nonfunctional Channels
| L1464 E (14) | L1472 D (14); K (16) | |
| L1465 E (10) | P1473 A (7); C (14); E (7); G (7) | |
| A1467 D (7) | A1474 L (21) | |
| L1468 E (65); K (50) | L1475 A (33); K (94) | |
| S1471 D (41); F (37); K (33); M (52) | N1477 C (8) |
Number of analyzed cells is shown in parentheses. For each mutant, at least two independent clones were analyzed in at least two independent transfections.
Table II.
Inactivation Kinetics of D4/S4-5 Mutants
| Position-mutation | Tau inactivation | Steady state inactivation | Recovery from inactivation (−110 mV) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| K d | Slope | |||||||||
| n | ms | mV | mV | ms | ||||||
| WT | (17) | 0.56 ± 0.04 | −81.7 ± 2.1 | 5.6 ± 0.2 | 3.6 ± 0.1 | |||||
| R1462-E | (10) | 0.59 ± 0.06 | −86.0 ± 2.5 | 10.0 ± 0.7 | 1.9 ± 0.2** | |||||
| R1462-K | (14) | 0.53 ± 0.05 | −81.9 ± 1.8 | 6.2 ± 0.3 | 2.7 ± 0.2** | |||||
| R1462-L | (6) | 0.61 ± 0.05 | −82.5 ± 2.8 | 8.6 ± 0.2 | 2.8 ± 0.1 | |||||
| T1463-E | (4) | 0.31 ± 0.05* | −93.4 ± 2.9** | 7.1 ± 0.6 | 3.0 ± 0.4 | |||||
| F1466-E | (7) | 1.8 ± 0.2** | −66.8 ± 1.5** | 7.6 ± 0.9 | 2.0 ± 0.2** | |||||
| F1466-W | (4) | 0.56 ± 0.04 | −84.4 ± 2.4 | 5.4 ± 0.3 | 4.5 ± 0.2 | |||||
| L1468-A | (9) | 0.43 ± 0.02* | −77.8 ± 2.5 | 5.6 ± 0.1 | 2.7 ± 0.3 | |||||
| L1468-F | (10) | 0.64 ± 0.05 | −88.8 ± 2.4* | 7.8 ± 0.4 | 3.6 ± 0.4 | |||||
| L1468-M | (7) | 0.66 ± 0.05 | −80.0 ± 2.5 | 6.7 ± 0.3 | 3.2 ± 0.2 | |||||
| L1468-S | (9) | 0.66 ± 0.04 | −74.5 ± 1.5 | 7.2 ± 0.4 | 1.7 ± 0.1** | |||||
| M1469-E | (4) | 2.3 ± 0.2** | −51.2 ± 2.8** | 11.7 ± 2.8 | 1.4 ± 0.2** | |||||
| M1469-K | (5) | 2.6 ± 0.5** | −64.9 ± 7.5** | 10.2 ± 1.4 | 1.5 ± 0.5** | |||||
| M1469-L | (4) | 0.46 ± 0.06 | −78.9 ± 2.7 | 4.7 ± 0.2 | 4.2 ± 0.4 | |||||
| M1470-E | (9) | 2.3 ± 0.2** | −66.0 ± 3.0** | 7.4 ± 0.9 | 1.8 ± 0.1** | |||||
| M1470-F | (4) | 0.41 ± 0.03 | −86.4 ± 3.4 | 7.0 ± 0.3 | 2.9 ± 0.7 | |||||
| M1470-K | (7) | 1.7 ± 0.1** | −67.4 ± 5.3** | 5.2 ± 0.1 | 1.4 ± 0.1** | |||||
| M1470-L | (10) | 0.47 ± 0.05 | −92.8 ± 2.9** | 6.4 ± 0.4 | 8.1 ± 0.8** | |||||
| S1471-A | (5) | 0.43 ± 0.06 | −88.5 ± 1.2 | 5.4 ± 0.4 | 4.5 ± 0.4 | |||||
| S1471-T | (8) | 0.66 ± 0.05 | −80.2 ± 3.5 | 6.6 ± 0.5 | 4.1 ± 0.4 | |||||
| L1472-A | (9) | 0.64 ± 0.04 | −72.4 ± 1.3* | 4.5 ± 0.3 | 3.3 ± 0.3 | |||||
| L1472-F | (6) | 0.69 ± 0.01 | −71.2 ± 1.7* | 5.7 ± 0.3 | 3.0 ± 0.3 | |||||
| L1472-T | (8) | 0.58 ± 0.02 | −79.9 ± 1.2 | 6.1 ± 0.3 | 4.3 ± 0.4 | |||||
| A1474-D | (10) | 2.9 ± 0.3** | −60.2 ± 2.8** | 8.0 ± 1.0 | 2.5 ± 0.2* | |||||
| A1474-K | (8) | 0.65 ± 0.07 | −70.2 ± 2.2** | 6.1 ± 0.7 | 1.4 ± 0.1** | |||||
| L1475-E | (9) | 0.73 ± 0.07* | −75.5 ± 4.4 | 7.5 ± 0.1 | 2.5 ± 0.3* | |||||
| L1475-F | (4) | 0.38 ± 0.06 | −78.8 ± 3.4 | 7.2 ± 0.5 | 1.9 ± 0.2* | |||||
| F1476-E | (9) | 0.59 ± 0.06 | −106.7 ± 2.3** | 7.2 ± 0.7 | 7.5 ± 1.8** | |||||
| F1476-K | (6) | 0.94 ± 0.12** | −92.2 ± 1.8** | 8.8 ± 1.9 | 18.1 ± 4.2** | |||||
| F1476-W | (5) | 0.41 ± 0.06 | −93.3 ± 4.5* | 7.2 ± 1.2 | 5.2 ± 0.8 | |||||
Results show mean ± SEM. Number of analyzed cells is shown in parenthesis. Inactivation time constants were obtained by single exponential fitting of current traces obtained from voltage pulses to −10 mV from a holding potential of −120 mV. Results of Boltzmann fit of steady state inactivation curves are shown. Mutants that showed a significant difference compare with WT in a t test at the level of 0.05 are marked with one asterisk and at the level of 0.01 with two asterisks.
Table III.
Inactivation Properties of Cysteine Mutants
| Position-mutation | Tau inactivation (−10 mV) | Steady state inactivation | Recovery from inactivation (−110 mV) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Kd | Slope | |||||||||
| n | ms | mV | mV | ms | ||||||
| WT | (17) | 0.56 ± 0.04 | −81.7 ± 2.1 | 5.6 ± 0.2 | 3.8 ± 0.3 | |||||
| I1461-C | (7) | 0.44 ± 0.01 | −90.6 ± 2.7* | 6.1 ± 0.3 | 10.5 ± 2.9** | |||||
| R1462-C | (9) | 0.43 ± 0.03* | −82.6 ± 2.3 | 7.2 ± 0.2 | 2.1 ± 0.1** | |||||
| T1463-C | (5) | 0.52 ± 0.02 | −91.3 ± 2.4* | 6.5 ± 0.5 | 4.4 ± 0.2 | |||||
| L1464-C | (6) | 0.95 ± 0.12* | −84.2 ± 2.7 | 9.2 ± 0.8 | 7.2 ± 1.3** | |||||
| L1465-C | (6) | 0.76 ± 0.04* | −71.2 ± 3.3** | 6.7 ± 0.5 | 1.8 ± 0.1** | |||||
| F1466-C | (4) | 0.78 ± 0.16 | −68.1 ± 3.6** | 7.4 ± 0.3 | 2.0 ± 0.2** | |||||
| A1467-C | (6) | 0.67 ± 0.1 | −90.4 ± 3.0* | 6.4 ± 0.3 | 6.3 ± 1.0** | |||||
| L1468-C | (5) | 0.64 ± 0.09 | −91.1 ± 2.1 | 8.1 ± 0.3 | 4.1 ± 0.4 | |||||
| M1469-C | (6) | 0.90 ± 0.05** | −75.6 ± 2.0 | 5.7 ± 0.4 | 3.5 ± 0.5 | |||||
| M1470-C | (7) | 0.52 ± 0.5 | −87.4 ± 2.3 | 6.2 ± 0.4 | 6.1 ± 0.4** | |||||
| S1471-C | (5) | 0.50 ± 0.1 | −91.0 ± 2.9* | 5.7 ± 0.3 | 8.0 ± 1.7** | |||||
| L1472-C | (7) | 0.52 ± 0.06 | −85.1 ± 3.2 | 5.0 ± 0.7 | 3.9 ± 0.8 | |||||
| P1473-C | (14) | NR | ||||||||
| A1474-C | (5) | 0.60 ± 0.03 | −79.3 ± 1.8 | 5.3 ± 0.2 | 3.4 ± 0.4 | |||||
| L1475-C | (5) | 0.37 ± 0.02* | −76.8 ± 10.5 | 7.5 ± 1.0 | 1.8 ± 0.4* | |||||
| F1476-C | (7) | 0.47 ± 0.03 | −80.6 ± 1.8 | 7.0 ± 0.4 | 3.0 ± 0.2 | |||||
| N1477-C | (8) | NR | ||||||||
Results show mean ± SEM. Number of analyzed cells are shown in parenthesis. Inactivation time constants (Tau) were obtained by single exponential fitting of current traces obtained from voltage pulses to −10 mV (inward current) and to +60 mV (outward current) from a holding potential of −120 mV. Results of Boltzmann fit of steady state inactivation curves are shown. Mutants that showed a significant difference compare to WT in a t test at the level of 0.05 are marked with one asterisk and at the level of 0.01 with two asterisks.
Useful information can be gleaned from the distribution of the 19 inactive mutants. None of the mutations introduced at P1473 produced functional channels, and this proline may mark a critical transition in the protein secondary structure. All large nonpolar and charged substitutions at S1471 were inactive; only small conservative substitutions were tolerated, suggesting a size-sensitive aspect to the region occupied by this side chain. Mutations that introduced a charge into the conserved hydrophobic sector of the proposed helix before P1473 (shaded area in Fig. 1) eliminated normal channel function (Table I; L1464E, L1465E, A1467D, L1468E/K, S1471D/K, and L1472D/K), while small uncharged substitutions in this region produced channels with kinetic characteristics similar to WT channels (Table II). Most charged substitutions in the opposite surface of the proposed helix were functional, although often with markedly abnormal kinetics. Small polar or nonpolar mutations in residues on this variable face had effects that differed considerably with location and specific amino acid substitution.
Among all mutations in D4/S4-5 that produced functional channels with abnormal kinetics (Table II), the most marked changes were observed in the kinetics of channel inactivation (Fig. 2). However, small effects on channel activation were also seen for some substitutions. For example, mutation F1466E slightly altered the current-voltage (I/V) relationship, shifting the peak of inward current to more depolarized potentials, while mutation M1470E shifted the peak to more hyperpolarized potentials (Fig. 3 A). Similar shifts were observed for the conductance-voltage (G/V) relationships (Fig. 3 B). The remaining mutations showed I/V and G/V relationships comparable to WT. Although we did not make corrections for liquid junction potentials, all of the tested mutations exhibited reversal potentials close to the theoretically predicted one, implying little change in ionic selectivity.
Figure 2.

Normalized responses of WT and selected mutant sodium channels induced by depolarization to −10 mV from a holding potential of −120 mV. Charged mutations in the variable region, exemplified by M1470E and A1474D, caused a pronounced slowing of inactivation with little change in activation, while substitution of the small hydrophobic leucine at M1470 accelerated the rate of inactivation.
Figure 3.

(A) Current-voltage relationship (I/V) of WT and selected mutants. Reversal potentials for WT and all mutants were ∼2,863 mV, in good agreement with the calculated reversal potential, suggesting that these mutations have little effect on cation selectivity. Some mutations produced small shifts in the peak of the I/V curve; the most prominent are shown here. The F1466E mutation shifted the curve toward more depolarized potentials, while M1470E produced a small hyperpolarizing shift. (B) Conductance–voltage relationship of WT and selected mutants. The largest depolarizing shifts were observed for A1474K, F1466E, and T1463E. M1470E introduces a small hyperpolarizing shift. In all cases, however, the slope of the Boltzmann fit is not significantly different from WT. The midpoint and the slope of Boltzmann fit were −35.5 ± 0.4 and 6.7 ± 0.4 for WT, −23.4 ± 0.5 and 10.0 ± 0.5 for 63E, −27.4 ± 0.3 and 8.5 ± 0.4 for 66E, and −44.2 ± 0.3 and 5.3 ± 0.2 for 70E.
Effects of D4/S4-5 Mutations on Channel Inactivation
The most dramatic effects of mutations in D4/S4-5 were on the kinetics of inactivation (Table II). We observed the largest effects for substitutions at F1466, M1469, M1470, and A1474, while smaller effects were seen with mutations at T1463, L1475, and F1476. In all cases, even when inactivation was markedly slowed, the decay of the sodium current was well fitted with a single exponent. Time constants derived from these exponential fits are plotted in Fig. 4 A for several representative mutations. Charged mutations at positions F1466, M1469, M1470, and A1474 shared several kinetic features. These mutations produced a striking increase in the inactivation time constant (τh), a depolarizing shift in the midpoint of the steady state inactivation (h∞•/V) curve (Fig. 4 B), and an acceleration in recovery from inactivation (τrec) (Fig. 5). Effects of charged mutations at M1469 and M1470 did not reflect the sign of the charge that was introduced, since prolongation of inactivation, acceleration of recovery from inactivation, and depolarizing shift of the h∞•/V curve were observed with both positive and negative substitutions at these positions. Both negative A1474D and positive A1474K substitutions produced a large depolarizing shift in the h∞•/V curve, although A1474D had a much greater effect on τh and a smaller effect on τrec. Charged substitutions at M1470 strongly slowed inactivation, increased τrec, and shifted h∞•/V in the depolarizing direction, while introduction of the nonpolar residues L or F produced changes in the opposite direction; h∞•/V was shifted in the hyperpolarizing direction while inactivation was accelerated slightly and, in the case of M1470L, recovery was slowed.
Figure 4.

The most prominent effects of mutations in D4/S4-5 were observed on the kinetics of inactivation. (A) Voltage dependence of the inactivation time constant. Time constants (τh) were obtained by single exponential fitting to the falling phase of the sodium current. The largest effects were observed for substitutions at F1466, M1469, M1470, and A1474. Both positively and negatively charged mutations at M1470 dramatically slowed inactivation at all voltages, while the small nonpolar M1470L mutation accelerated τh (see Table II). (B) Voltage dependence of steady state inactivation (h∞•/V) for WT and selected mutants. Inactivation was induced by a 500-ms prepulse to the plotted voltage. The data were fitted by the Boltzmann equation. In general, charged mutations that slowed inactivation and accelerated recovery produced shifts in the h∞•/V curve in the depolarizing direction (M1469E/K, M1470E/K, and A1474D/K), while those that accelerated inactivation and slowed recovery shifted the h∞•/V curve in the hyperpolarizing direction (R1462E, T1463E, and F1476E/K).
Figure 5.

Recovery from inactivation for selected mutations. (A) Time course of recovery from inactivation at −110 mV. Recovery was measured using a double pulse protocol: the first pulse was used to induce inactivation and, after various delays, a second pulse was applied to assess the extent to which channels had recovered from inactivation. Mutations such as F1466E, M1470E, and A1474D that slowed inactivation and shifted the voltage dependence of inactivation in the depolarizing direction also accelerated the rate of recovery from inactivation. Introduction of the nonpolar leucine at M1470, which had an action opposite to M1470E on inactivation rate and voltage dependence, also produced the converse effect on recovery, slowing the rate at all potentials. Mutations at other locations, such as F1476K (shown), cause marked slowing of recovery from inactivation. (B) Voltage dependence of the recovery time constant (τrec) for WT and selected mutants. For all mutations studied, voltage dependence of τrec was retained.
The effects of substitutions at F1476 did not follow this pattern. Mutations at this residue produced the largest hyperpolarizing shifts in h∞•/V that we observed (−106.7 ± 2.3 mV for F1476E; −81.7 ± 2.1 mV for WT), in contrast to the depolarizing shift in h∞•/V seen with charged mutations at F1466, M1469, M1470, and A1474. Charged substitutions at F1476 had little effect on τh, but lysine substitution caused a fivefold slowing in τrec. Substitution of the aromatic side chain in tryptophan for phenylalanine actually accelerated inactivation, with τh at +60 mV 44% of WT and recovery kinetics comparable with WT. However, steady state inactivation for the tryptophan was also shifted 11.6 mV in the hyperpolarizing direction.
In general, charged mutations that slowed inactivation and accelerated recovery from inactivation produced shifts in the h∞•/V curve in the depolarizing direction (F1466E, M1469E/K, M1470E/K, and A1474D/K), while those that slowed recovery shifted the h∞•/V curve in the hyperpolarizing direction (R1462E, T1463E, M1470L, S1471A/C, and F1476E/K). The slopes of the h∞•/V curves were relatively insensitive to mutations in the D4/S4-5 region. Exceptions were R1462E and M1469E/K, where approximately twofold increases in slope were observed.
The effects of mutations in the hydrophobic surface distal to P1473 were not consistent with the predictions derived from a simple helical structure. For example, mutations at position L1475 differed in effect from those at adjacent locations in a predicted helix such as L1464, L1465, L1468, S1471, and L1472. Unlike mutations at these five locations, charged substitutions at L1475 were functional, with slight slowing in inactivation, while bulky noncharged substitutions accelerated inactivation. Mutations at the previous (A1474) and subsequent (F1476) positions exhibited both charge and size selectivity.
When referenced to the helical wheel projection for the S4-5 region, charged mutations that resulted in nonfunctional channels are segregated to one half of the helix (Fig. 6). Locations at which charged mutations produced functional channels with significant effects (P < 0.01) on the kinetics of inactivation are found on the other half of the helix. One location that does not fit easily into this analysis is L1475; lysine substitution at this site creates a nonfunctional channel, yet replacement with glutamic acid results in only slightly prolonged inactivation kinetics and slightly accelerated recovery from inactivation. This residue is distal to proline 1473, which may mark a transition in helical structure within this region.
Figure 6.

Locations where charged mutations of either sign produced nonfunctional channels occupy one half of the helical projection (•), including the conserved nonpolar face identified in Fig. 1 (shaded gray). Nonpolar substitutions at these sites were functional; most showed little effect on inactivation kinetics. Residues where charged substitutions produced significant changes (P < 0.01) in the rate of channel inactivation are located on the opposite side of the helix (○). All mutations of P1473 (X) produced nonfunctional channels. Charged mutations at L1475 (position 16), distal to this proline, differed from those in the shaded region that precedes the proline; lysine substitution at L1475 produced a nonfunctional channel while glutamic acid replacement resulted in normal expression with only minor changes in kinetics (◑).
Accessibility of Cysteine Mutations
Cysteine mutations were created at each position between I1461 and N1477. The effects of these cysteine substitutions on channel kinetics are shown in Table III. Cysteine substitution was tolerated at all positions except for P1473 and N1477. After documenting the effect of each mutant on channel kinetics, we examined the result of exposure to the sulfhydryl reagents MTSES and MTSET.
Since the S4-5 region is located on the cytoplasmic surface of the channel in current models of tertiary structure, modifying reagents were introduced into the internal solution of the patch pipette and allowed to diffuse into tsA-201 cells expressing each mutant after the whole-cell recording configuration was achieved. Because of this, the rate-limiting step in channel modification for the most easily accessible residues was equilibration of the cytoplasm with the modifying reagent rather than reaction with the cysteine residue. For cysteine mutants that showed significant change in kinetics with sulfhydryl reagents, the first detectable alteration occurred within 5 min of achieving whole-cell configuration, and typically continued to develop during the 20–40-min observation interval (Fig. 7 B). Residues that we considered to be inaccessible showed no change in kinetics over exposure periods as long as 40 min (Fig. 7 A).
Figure 7.

Accessibility of cysteine mutations was determined by evaluating modifications induced in inactivation kinetics after exposure of the cytoplasmic surface of the channel to MTSET or MTSES. (A) Cysteine mutations at some locations showed no response to these sulfhydryl reagents, although charged mutations at the same location produced significant alterations in kinetics; L1468C is shown as an example. These residues were considered inaccessible to the reagents under the conditions of our experiments. Currents were elicited by pulses from −120 to −10 mV applied at 1-min intervals after attaining the whole-cell configuration. Currents were filtered at 5 kHz and normalized. (B) Cysteine mutations at other locations, such as M1470C, showed rapid and progressive changes in kinetics after exposure to sulfhydryl reagents, indicative of modification. These residues were considered accessible in our analysis. A third group of cysteine mutants showed no effect with exposure to MTSES or MTSET, but also exhibited little change in kinetics when the same position was directly substituted with a charged residue. These locations were considered uninformative. (C) Summary of the effects on τh of cysteine mutants exposed to MTSES and MTSET. For each mutant, the ratio of τh in the cysteine mutant in the absence of modifying reagents to the observed τh after exposure to these reagents at a holding potential of −120 mV is plotted. Residues considered inaccessible are shown in black; those that were accessible are cross-hatched. White bars are uninformative. *Significant differences (P < 0.01) from the behavior of wild-type channels after similar exposure to these reagents.
In general, where MTSES and MTSET produced alterations in kinetics, the changes were similar to those observed with the corresponding charged mutations at the same location (compare Table III and Fig. 7 C). When we analyzed the effects of exposure to MTSES and MTSET on τh in those of cysteine mutants and compared them with the effects of charged substitutions in the mutagenesis, we found three qualitative groups of modification effects. Cysteine mutants at some positions showed a rapid change in τh during the first 20 min after initiation of exposure to intracellular sulfhydryl-modifying reagents at a holding potential of −120 mV (Fig. 7 B). When changes in inactivation kinetics were analyzed relative to the unmodified cysteine mutant, they were significant at the level of P < 0.01 when compared with changes in WT channels exposed to the same reagent. We considered these residues to be easily accessible to the modifying reagents (F1466C, A1467C, M1469C, and M1470C for both MTSES and MTSET reagents; L1465C, L1472C, A1474C, and F1476C only for MTSES). Other cysteine mutants showed no change in kinetics after exposure to the sulfhydryl reagents for periods up to 40 min, although dramatic effects were seen with direct substitution of charged residues at the same location (Fig. 7 A); we assume that these positions are inaccessible to the polar modifying reagents within the time frame of our assay (L1464C, L1468C, S1471C, and L1475C for both MTSES and MTSET; L1465C and L1472C for MTSET). A third group of cysteine mutants did not show significant effects on τh after MTSES/MTSET exposure, but also exhibited little change in inactivation kinetics when charged residues were substituted at the same location. Since we cannot assume that successful modification by MTSES or MTSET would alter inactivation kinetics, we interpreted these mutants as not informative (I1461C, R1462C, and T1463C). For residues before P1473, locations judged to be inaccessible by these criteria were clustered in the conserved hydrophobic region of the helical wheel projection, while residues that were easily modified by MTSES/MTSET lie mostly in the opposite surface of the predicted structure, consistent with an amphipathic helical conformation. We recognize that this analysis is qualitative. It is possible that some residues that show no change in kinetics in our study might eventually do so under different experimental conditions, suggesting a very slow reaction rate rather than absolute inaccessibility.
Structural Modeling
To develop a better working model of potential conformations in the D4/S4-5 region than that provided by helical wheel projection, molecular dynamics simulations were carried out on residues 1458–1477 using the InsightII structural modeling program (see materials and methods). This method has been used extensively to evaluate the dynamic motion and stability of potential structures in proteins and peptides (Brooks, 1995; Friesner and Gunn, 1996), and has been shown between local structure predicted by this method and that determined using physical methods in potential transmembrane helices of sodium channels (Doak et al., 1996). For our purposes, molecular dynamics simulations were used only to evaluate the feasibility of a proposed structure for the D4/S4-5 region.
Based on the results of our mutational and SCAM analyses, an initial condition of α-helical structure before P1473 was chosen with no subsequent structural constraints. After equilibration during the first 50,000 iterations (0.05 ns), convergence in total energy was obtained. During the subsequent 0.05 ns of simulation, a stable structure was reached in which residues 1458– 1473 maintained an amphipathic α-helical conformation with a contiguous nonpolar surface formed by residues I1461, L1464, L1465, L1468, S1471, and L1472 (Fig. 8). In this hypothetical structure, the side chains of residues R1462, T1463, F1466, M1469, and M1470 lie in proximity on the opposite side of the helix. The structure bends at P1473 and continues as an extended chain beyond that point. When modeled as an extended chain, the region beyond P1473 also has an amphipathic nature, with a nonpolar face that includes L1475 in the same plane as the nonpolar face of the preceding helix. Residues where mutations produced the largest effects on inactivation (Fig. 8 A, green) have side-chains that are clustered on one surface of the model, while the nonfunctional charged mutations are preferentially located on the opposite nonpolar surface (Fig. 8 B, red). Locations considered inaccessible by our SCAM analysis are found in this nonpolar face (Fig. 8 D, red), while easily accessible residues line the opposite side, both proximal and distal to the termination of the helix at P1473 (Fig. 8 C, green).
discussion
In this study, our goal was to probe the local secondary structure of the D4/S4-5 region and its general role in channel inactivation by analyzing multiple mutations throughout this region, rather than to investigate kinetic mechanisms through a detailed evaluation of several mutations at a few sites. Our mutational analysis was initially driven by the following observations: (a) the S4-5 region in each repeat domain exhibits a higher degree of sequence conservation among isoforms than most other interhelical regions; (b) the sequence in this region displays conserved topography when analyzed for potential helical organization; (c) mutations in this interhelical region of potassium and sodium channels affect inactivation kinetics (Isacoff et al., 1991; Holmgren et al., 1996; Tang et al., 1996; Mitrovich et al., 1996); and (d) natural mutations in this region of the human SkM1 channel are associated with diseases of excitation in muscle (Yang et al., 1994).
When the S4-5 region from all four sodium channel repeat domains is examined in helical wheel projection, a potential surface containing conserved small nonpolar residues is apparent. On the opposite side of the helix is a region of less-conserved residues with side chain properties that are more variable between domains (referred to below as the variable surface). This organization is consistent with an amphipathic helix that might lie at the interface between a hydrophobic membrane environment and the cytoplasm. For residues 1461–1473 in D4/S4-5, both our mutation and cysteine modification data support such a conformation. In general, charged substitutions that we made in the conserved nonpolar face were not functional, while those on the variable surface typically did function, but with kinetic effects that were position and substitution dependent. Proline 1473 appears to mark a critical transition in structure—none of our mutations at this location produced functional channels and the effects of mutations beyond this proline do not conform to a simple helical pattern.
These results in the SkM1 S4-5 can be compared with observations on the S4-5 region in the Shaker K+ channel. When the K+ channel S4-5 sequence is aligned in helical projection with SkM1 channel by indexing to the conserved serine near the center of each loop (Shaker = S392; SkM1 = S1471), it is apparent that the Shaker channel shares a similar potential amphipathic helical structure (Fig. 9). On the basis of measured reactivity rates for cysteine mutants, Holmgren et al. (1996) concluded that their results with the Shaker S4-5 before M393 were consistent with an α-helical structure, a possibility also raised by Isacoff et al. (1991). Isacoff et al. (1991) reported that the E395N mutation in Shaker eliminated fast inactivation; in helical projection, E395 is homologous with A1474 in the SkM1 channel, one of four positions where alterations in charge markedly slowed fast inactivation. Holmgren et al. (1996) identified another Shaker S4-5 residue, A391, where modifications that introduced charge greatly reduced channel affinity for the inactivation ball peptide; charged mutations or modifications of either sign at the homologous F1466 in SkM1 also produce prominent slowing in fast inactivation, consistent with a reduction in affinity for the inactivation particle. The concordance of observations on K+ and Na+ channel mutations when analyzed with respect to an amphipathic helical conformation suggests that this local structure may be conserved throughout this class of voltage-gated ion channels.
Figure 9.

Comparison of helical wheel projections of the SkM1 D4/S4-5 and the Shaker potassium channel S4-5, aligned with respect to a conserved serine (Shaker S392; SkM1 S1471) near the middle of each. The K+ channel S4-5 exhibits a nonpolar face similar to that found in Na+ channel S4-5 regions. Two residues in the Shaker S4-5 where modification produced large changes in inactivation (shaded ovals) have been identified by Isacoff et al. (1991) (E357) and Holmgren et al. (1996) (A395). These locations are structurally homologous to SkM1, M1469, and A1474, where comparable large effects on inactivation were found with modification. The structural features discussed here for the SkM1 channel may be shared by other members of the voltage-gated ion channel family.
A Potential Structure for D4/S4-5
Molecular dynamics simulations of residues 1458–1477 in the D4/S4-5 region confirms that the residues proximal to P1473 can form a stable α-helix. Residues distal to this proline may prefer an extended chain configuration, although the small number of residues in this distal segment makes interpretation more difficult. While an extended chain here is a stable low energy configuration, a helical structure COOH terminal to this region might stabilize a local helical structure in this region as well. In the structure shown, residues 1461–1472 form an extension of the S4 helix toward the cytoplasm. Proline 1473 may provide the bend that interrupts the helix and turns the loop back toward S5. This conformation is consistent with helical content in the eel D4/S4-S4-5 and D4/S4-5 peptides estimated by circular dichroism (CD) (Brullemans et al., 1994; Helluin et al., 1996). This CD analysis also indicated a helix-coil transition within the eel D4/S4-5, elicited with changing solvent polarity, that the authors associate with the homologous proline in that peptide.
When viewed in this way (Fig. 8), one surface of the proposed structure is occupied by small nonpolar residues where charged mutations produced nonfunctional channels. In contrast, residues where mutations caused the largest effects on the kinetics of inactivation have their side chains oriented on the opposite surface. This hypothetical structure is also supported by our cysteine accessibility data; those cysteines located on the nonpolar surface are not easily accessible for modification, while most on the opposite surface are readily modified in the resting state. In the context of the complete channel, the variable surface of the D4/S4-5 helix and extended chain could face the vestibule of the pore inner mouth, while the opposite nonpolar surface may interact with hydrophobic surfaces of other domain helices.
In the structure model shown in Fig. 8, the side-chains of F1466, M1469, M1470, and A1474 residues occupy contiguous locations on the variable surface of the structure. Their proximity, as well as the similarity of kinetic effects for charged mutants, suggests that the four might contribute to a common site of interaction with the ID3-4 inactivation domain. Introduction of a charge at this site could destabilize the bound, inactivated conformation, slow channel inactivation, and accelerate recovery from inactivation. Since modifications of either charge at these four locations shift h∞•/V in the same direction, local charge in this region is not likely to contribute directly to the binding. Substitution of the aromatic tryptophan for phenylalanine (F1466W) produces a channel with inactivation kinetics very close to wild type while cysteine substitution markedly affects inactivation, suggesting that it is the aromatic nature, rather than the size, of the side chain that is critical for the interaction of the inactivation particle with the binding site (Dougherty, 1996). This is consistent with the results of mutagenesis in ID3-4, where neutralization of charged residues have relatively little impact on channel inactivation (Moorman et al., 1990; Patton et al., 1992) and the largest effects are seen with modification of three adjacent nonpolar residues (West et al., 1992).
We suggest that the nonpolar surface of S4-5 in each domain or subunit slides along its contacts with the nonpolar surface(s) of adjacent domain helices in response to conformational changes associated with activation and S4 outward movement (Yang and Horn, 1995; Yang et al., 1996). The opposite, variable surfaces of S4-5 in each domain or subunit then contributes to the formation of a binding site for the inactivation gate.
Mutations in D4/S4-5 Predominantly Affect Channel Inactivation
Residues F1466, M1469, M1470, and A1474 appear to be particularly important for channel inactivation in SkM1. Charged mutations at these locations, while functional, markedly slowed inactivation, increased τrec, and shifted the midpoint of the h∞•/V relationship in the depolarizing direction. Conversely, leucine substitution at either M1469 or M1470 accelerated inactivation; M1470L also caused a hyperpolarizing shift in steady state inactivation. Tang et al. (1996) recently showed that the double mutants MM/QQ and MM/AA in the human heart sodium channel, at sites that correspond to M1469 and M1470 in SkM1, produced the same kinetic effect as our charged substitutions at these two methionines. Our data indicate that these methionines are independently involved in the inactivation process; charged substitutions at either location produced a fourfold increase in τh at −10 mV and a depolarizing shift in h∞•/V. Mitrovich et al. (1996) modified four locations in D4/S4-5 of the human muscle sodium channel (hSkM1) and reported that the F1473S mutation (homologous to F1466 in SkM1) caused a slowing in τh and depolarizing shift in h∞•/V comparable to the effects at F1466 shown here. Mutations at the conserved serine (S1478A/C) produced identical effects to those we observed for the homologous S1471A/C mutations; the effects of charged or large nonpolar substitutions at this site in hSkM1 were not reported but were uniformly lethal in SkM1. The only discrepancy between the two studies concerns the L1482A (L1475A in SkM1) mutation; while this mutation had little effect in hSkM1, we found no channel function in multiple independent clones of this substitution in the rat SkM1 background.
Smith and Goldin (1997) carried out a glutamine screen of the S4-5 regions in D2 and D3 of the rat brain sodium channel. While most glutamine substitutions had little effect on inactivation, A1329N in D3/S4-5 did alter inactivation kinetics. Charged mutations at this residue that slowed fast inactivation could be partially compensated by reciprocal mutations in F1489 of the ID3-4 region. Mutations at A1329 in the rat brain channel D3/S4-5 that slowed inactivation reduced rather than accelerated the rate of recovery from inactivation. This combination of effects is comparable to the results we observed with the L1464C mutation in SkM1 D4/S4-5, which is located at the homologous position in our helical projection. However, L1464C appears to be inaccessible for modification by sulfhydryl reagents in SkM1 D4/S4-5, suggesting that it is not likely to be available for binding to the inactivation particle.
Summary
We have analyzed each of the residues between I1461 and N1477 in D4/S4-5 through multiple substitutions and SCAM. Our data support a direct role for this region in forming the binding surface for the inactivation particle, and identify four residues that play a particularly important part in this process. Analysis of our results suggests a structure for the region that includes an amphipathic helix continuing from D4/S4 to P1473, with a critical transition at this proline. Translocation of the nonpolar surface of the resultant S4-5 loop along complementary surfaces of adjacent domain helices in response to voltage-driven S4 movement may lead to the organization of the opposite variable surface, facing the pore inner vestibule, into a receptor for the inactivation particle. Many of these structural features may be shared with voltage-gated potassium channels.
Acknowledgments
We are grateful to Drs. Richard Horn and Mark Rich for their helpful comments, to Dr. Kim Sharp for assistance with molecular modeling, and to Ms. Martha Sholl and Ms. Huan Ying Zhou for technical assistance.
This work was supported in part by National Institutes of Health grants NS-18013, NS-08075, and P30CA16520-20.
Abbreviations used in this paper
- ID3-4
intracellular segment between D3 and D4
- MTSES
sodium (2-sulfonatoethyl)methanethiosulfonate
- MTSET
[2-(trimethylammonium)ethyl]methanethiosulfonate
- SCAM
substituted-cysteine-accessibility method
- WT
wild type
Note Added in Proof
Since this manuscript was submitted for publication, two papers have appeared that describe analysis of structure in the D4/S4-5 regions of the human skeletal muscle sodium channel (Lerche et al., 1997. J. Physiol. 505:345–352) and the rat brain IIa sodium channel (McPhee et al., 1998. J. Biol. Chem. 273:1121–1129). Using different experimental approaches, both groups report data consistent with an α-helical structure in this region.
references
- Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CL., III Methodological advances in molecular dynamics simulations of biological systems. Curr Opin Struct Biol. 1995;5:211–215. doi: 10.1016/0959-440x(95)80078-6. [DOI] [PubMed] [Google Scholar]
- Brullemans M, Helluin O, Dugast J-Y, Molle G, Duclohier H. Implication of segment S45 in the permeation pathway of voltage-dependent sodium channels. Eur Biophys J. 1994;23:39–49. doi: 10.1007/BF00192204. [DOI] [PubMed] [Google Scholar]
- Doak DG, Mulvey D, Kawaguchi K, Villalain J, Campbell ID. Structural studies of synthetic peptides dissected from the voltage-gated sodium channel. J Mol Biol. 1996;258:672–687. doi: 10.1006/jmbi.1996.0278. [DOI] [PubMed] [Google Scholar]
- Dougherty DA. Cation-π interactions in chemistry and biology: a new view of benzene, phe, tyr, and trp. Science. 1996;271:163–167. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- Eaholtz G, Scheuer T, Catterall W. Restoration of inactivation and block of open sodium channels by an inactivation gate peptide. Neuron. 1994;12:1041–1048. doi: 10.1016/0896-6273(94)90312-3. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Gunn JR. Computational studies of protein folding. Annu Rev Biophys Biomol Struct. 1996;25:315–342. doi: 10.1146/annurev.bb.25.060196.001531. [DOI] [PubMed] [Google Scholar]
- Greenblatt RE, Blatt Y, Montal M. The structure of the voltage-sensitive sodium channel. Inferences derived from computer-aided analysis of the Electrophorus electricuschannel primary structure. FEBS Lett. 1985;193:125–134. doi: 10.1016/0014-5793(85)80136-8. [DOI] [PubMed] [Google Scholar]
- Helluin O, Breed J, Duclohier H. Polarity-dependent conformational switching of a peptide mimicking the S4–S45 linker of the voltage-sensitive sodium channel. Biochim Biophys Acta. 1996;1279:1–4. doi: 10.1016/0005-2736(95)00270-7. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. The components of membrane conductance in the giant axon of Loligo. J Physiol (Camb) 1952;116:473–496. doi: 10.1113/jphysiol.1952.sp004718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren M, Jurman ME, Yellen G. N-type inactivation and the S4–S5 region of the Shaker K+channel. J Gen Physiol. 1996;108:195–206. doi: 10.1085/jgp.108.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanism of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Isacoff EY, Jan YN, Jan LY. Putative receptor for the cytoplasmic inactivation gate in the Shaker K+channel. Nature. 1991;353:86–90. doi: 10.1038/353086a0. [DOI] [PubMed] [Google Scholar]
- Ji, S., A.L. George, R. Horn, and R.L. Barchi. 1995. Sodium channel mutations and disorders of excitation in human skeletal muscle. In Ion Channels and Genetic Diseases. The Rockefeller University Press, New York. 61–76. [PubMed]
- Ji S, George AL, Horn R, Barchi RL. Paramyotonia congenita mutations reveal different roles for segments S3 and S4 of domain D4 in hSkM1 sodium channel gating. J Gen Physiol. 1996;107:183–194. doi: 10.1085/jgp.107.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurman ME, Boland LM, Liu Y, Yellen G. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 1994;17:876–881. [PubMed] [Google Scholar]
- Margolskee RF, McHendry-Rinde B, Horn R. Panning transfected cells for electrophysiological studies. Biotechniques. 1993;15:906–911. [PubMed] [Google Scholar]
- Mannuzzu LM, Moronne MM, Isacoff EY. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- Mitrovich N, Lerche H, Heine R, Fleischhaue R, Pika-Hartlaub U, Hartlaub U, George AL, Jr, Lehmann-Horn F. Role in fast inactivation of conserved amino acids in the IV/S4–S5 loop of the human muscle Na+channel. Neurosci Lett. 1996;214:9–12. doi: 10.1016/0304-3940(96)12866-4. [DOI] [PubMed] [Google Scholar]
- Moorman JR, Kirsch GE, Brown AM, Joho RH. Changes in sodium channel gating produced by point mutations in a cytoplasmic linker. Science. 1990;250:688–691. doi: 10.1126/science.2173138. [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N, et al. Primary structure of Electrophorus electricussodium channel deduced from cDNA sequence. Nature. 1984;312:121–127. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- Patton DE, West JW, Catterall WA, Goldin AL. Amino acid residues required for fast Na+-channel inactivation: charge neutralizations and deletions in the III–IV linker. Proc Natl Acad Sci USA. 1992;89:10905–10909. doi: 10.1073/pnas.89.22.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slesinger PA, Jan YN, Jan LY. The S4–S5 loop contributes to the ion-selective pore of potassium channels. Neuron. 1993;11:739–749. doi: 10.1016/0896-6273(93)90083-4. [DOI] [PubMed] [Google Scholar]
- Smith MR, Goldin AL. Interaction between the sodium channel inactivation linker and domain III S4–S5. Biophys J. 1997;73:1885–1895. doi: 10.1016/S0006-3495(97)78219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channnel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Tang L, Kallen RG, Horn R. Role of an S4–S5 linker in sodium channel inactivation probed by mutagenesis and a peptide blocker. J Gen Physiol. 1996;108:89–104. doi: 10.1085/jgp.108.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Ji S, Zhou M, Ptacek LJ, Barchi RL, Horn R, George AL. Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proc Natl Acad Sci USA. 1994;91:12785–12789. doi: 10.1073/pnas.91.26.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Horn R. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 1995;15:213–218. doi: 10.1016/0896-6273(95)90078-0. [DOI] [PubMed] [Google Scholar]
- Yang N, George AL, Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]