

Abstract
In studies of gating currents of rabbit cardiac Ca channels expressed as α1C/β2a or α1C/β2a/α2δ subunit combinations in tsA201 cells, we found that long-lasting depolarization shifted the distribution of mobile charge to very negative potentials. The phenomenon has been termed charge interconversion in native skeletal muscle (Brum, G., and E. Ríos. 1987. J. Physiol. (Camb.). 387:489–517) and cardiac Ca channels (Shirokov, R., R. Levis, N. Shirokova, and E. Ríos. 1992. J. Gen. Physiol. 99:863–895). Charge 1 (voltage of half-maximal transfer, V1/2 ≃ 0 mV) gates noninactivated channels, while charge 2 (V1/2 ≃ −90 mV) is generated in inactivated channels. In α1C/β2a cells, the available charge 1 decreased upon inactivating depolarization with a time constant τ ≃ 8, while the available charge 2 decreased upon recovery from inactivation (at −200 mV) with τ ≃ 0.3 s. These processes therefore are much slower than charge movement, which takes <50 ms. This separation between the time scale of measurable charge movement and that of changes in their availability, which was even wider in the presence of α2δ, implies that charges 1 and 2 originate from separate channel modes. Because clear modal separation characterizes slow (C-type) inactivation of Na and K channels, this observation establishes the nature of voltage-dependent inactivation of L-type Ca channels as slow or C-type. The presence of the α2δ subunit did not change the V1/2 of charge 2, but sped up the reduction of charge 1 upon inactivation at 40 mV (to τ ≃ 2 s), while slowing the reduction of charge 2 upon recovery (τ ≃ 2 s). The observations were well simulated with a model that describes activation as continuous electrodiffusion (Levitt, D. 1989. Biophys. J. 55:489–498) and inactivation as discrete modal change. The effects of α2δ are reproduced assuming that the subunit lowers the free energy of the inactivated mode.
Keywords: cardiac muscle, heterologous expression, continuum model of gating, charge interconversion, charge immobilization
introduction
Voltage-dependent inactivation of Ca channels is an important mechanism of regulation of Ca2+ entry during repetitive stimulation. Its relatively slow kinetics allow for a long-term, direct control of channel availability by transmembrane electric potential. Although inactivation is thought to be a property of the α1 subunit, fine tuning of its voltage sensitivity depends on interactions with auxiliary subunits and with other proteins.
In spite of a resemblance between voltage-dependent inactivation of Ca channels and the so-called slow or C-type inactivation found in the whole superfamily of voltage-gated channels, the structural underpinnings of this process have yet to be identified in Ca channels. Slow inactivation may be characterized by its relatively slow kinetics (τ ≃ 1 s), dependence on extracellular cations, and involvement of the extracellular portions of pore forming S5-S6 linkers (Brum et al., 1988; Hoshi et al., 1990; López-Barneo et al., 1993; Yellen et al., 1994; Balser et al., 1996; Townsend and Horn, 1997). In addition, slow inactivation is believed to result in the appearance of a specific intramembranous charge movement (charge 2; Brum and Ríos, 1987) generated by conformational changes in inactivated channels (Bezanilla et al., 1982; Brum and Ríos, 1987; Shirokov et al., 1992; Olcese et al., 1997). Slow rates of inactivation and recovery are essential for the operational separation of two types of charge. Charge 2 is well defined and separate from charge 1 (the charge that moves in noninactivated or primed channels) because its observable movement proceeds to completion much sooner than inactivation onset or recovery.
In contrast, fast inactivation of Na channels and N-type inactivation of Shaker K channels (ball and chain type; Armstrong and Bezanilla, 1977; Vassilev et al., 1988; Zagotta et al., 1990) recovers rapidly at negative potentials, approximately simultaneously with the inward movement of the intramembranous charge. Because recovery may proceed at rates comparable with that of measurable charge movement, there is no well-defined mode of charge movement that can be ascribed to this type of inactivation. The inward charge movement observed at large negative potentials after an inactivating depolarization occurs in channels as they recover from inactivation (repriming).
Apparently, a ball and chain–type inactivation is not present in Ca channels. It has been shown for several types of Ca channels that the faster component of ionic current decay is driven by ionic current itself. Because neither changes in intracellular calcium (Hadley and Lederer, 1992) nor the ion flow through cardiac channels influence inactivation of gating currents, Shirokov et al. (1993) concluded that Ca2+-dependent inactivation is a separate process, linked to gating currents only indirectly, through channel opening.
We have shown previously that decay of Ba2+ current through L-type Ca channels constituted by α1C and β2a subunits occurs in two phases. The slow phase (τ ≃ 8 s) is associated with voltage-dependent inactivation and is cotemporal with the reduction of available gating charge upon inactivation at positive voltages (Ferreira et al., 1997). We now address in quantitative detail the inactivation of intramembranous charge movement in heterologously expressed α1C channels. Because cardiac Ca channels transiently express at high density in the tsA201 human embryonic kidney cell line, we were able to measure intramembrane charge movements in these channels without using pulse protocols for subtraction of control records. This allowed us to study in detail the effects of conditioning voltage on the movement of voltage sensors, with or without the α2δ subunit, and develop a compact biophysical model that describes voltage-dependent inactivation well. The rate of the slow phase of Ba2+ current decay is three- to fivefold greater in the presence of the α2δ subunit (Ferreira et al., 1997) and is equal to that in native channels. The biophysical model described here reproduces in a parsimonious manner the effects of the α2δ subunit.
methods
Experiments were performed in tsA201 cells grown in DME medium (Sigma Chemical Co., St. Louis, MO) supplemented with 10% FBS (BioWhittaker, Walkersville, MD), 100 U/ml penicillin, and 0.1 mg/ml streptomycin (Sigma Chemical Co.) in 5% CO2. Rabbit α1C, α2δa, and rat β2a cDNAs were subcloned in pCR3, pMT2, and pCMV plasmid vectors, respectively. High purity (A260/A280 ≳ 1.90) large-scale plasmid preparations were obtained using standard protocols (QIAGEN Inc., Chatsworth, CA). Transfections were carried out with 30 μg of each expression plasmid in two combinations (α1C + β2a and α1C + β2a + α2δa) on 100-mm Petri dishes using a calcium phosphate precipitation method (Chien et al., 1995). Electrophysiological recordings were made within 24–48 h after transfection on round nonclustered cells. No sizable ionic or gating currents were observed in tsA201 cells in the absence of transfection. After transfection, the fraction of cells selected and patched that had Ca2+ currents was ∼70%.
Records were obtained by a standard whole-cell patch clamp procedure using an Axopatch 200A amplifier (Axon Instruments, Inc., Foster City, CA) and a 16-bit A/D–D/A converter card (HSDAS 16; Analogic Corp., Peabody, MA) on a PC computer. Patch electrodes were pulled from Corning 7052 glass (Warner Instruments, Hamden, CT) and had resistances of 1.0–1.5 MΩ.
The pipette solution contained (mM): 150 CsOH, 110 glutamate, 20 HCl, 10 HEPES, 5 MgATP, and 10 EGTA, pH 7.6. External recording solutions contained 160 TEA-Cl, 10 Tris, and 10 CaCl2, pH 7.2. To record intramembranous charge movement, the bath solution was replaced with one that included 15 μM of GdCl3. All solutions were adjusted to 300–320 mosmol/Kg. All experiments were carried out at room temperature (∼20°C).
Whole-cell capacitance was 6–15 pF. The time constant of membrane charging typically did not exceed 100 μs. Series resistance, calculated from the capacitive transient, was below 10 MΩ. The single time constant capacitance compensation circuitry of the Axopatch 200A amplifier was routinely used to offset 95–98% of the symmetric capacitive transient. Parameters of the compensation circuitry were set with a 10-mV pulse from the holding potential of −90 mV.
For a 200-mV pulse spanning most of the useful voltage range, the residual linear transient corresponded to a transfer of <10 fC/pF of charge, while maximal intramembranous charge transfer from the expressed channels was ∼100 fC/pF. With this combination of a relatively fast voltage clamp, high level of expression, and effective linear capacitance compensation, we were able to record asymmetric capacitive transients directly, without acquiring control linear transients for further subtraction.
Gating currents were recorded at 1 kHz bandwidth and sampled at 10 kHz. During prolonged pulses, the sampling rate was switched to between 0.05 and 2 kHz. To let the channels recover from inactivation, sets of conditioning and test pulses were separated by 30 s or longer. Charge transfer was calculated as the time integral of the current transient after subtraction of a steady current, which was determined as a 10-ms average, 40 ms after the beginning of the pulse.
Fig. 1 illustrates a typical experiment. The pulse protocol is shown at top. A conditioning pulse of 20 s at 40 mV was applied before each test pulse. Conditioning and test pulses were separated by an interval at −20 mV. Current traces were obtained with different test pulse voltages (V). The charge transferred by the ON transients (•) and the steady current during the test pulse (□) are plotted against voltage in Fig. 1 B. The steady current–voltage relationship was linear in the range from −150 to 50 mV, with steepness corresponding to an input resistance of ∼5 GΩ. The steady current at 0 mV was ∼−3 pA. The charge transfer–voltage relationship was clearly sigmoidal, saturating at extreme voltages. This demonstrates that the contribution of the linear capacitance was small, and validates the evaluation of charge transfer without subtraction of control currents.
Figure 1.

Evaluating quality of analog subtraction of the linear capacitance. (A) Intramembrane charge movement records obtained with analog subtraction of symmetric capacitive transient as described in methods. Pulse protocol is shown at top. Currents were elicited in an α1/β cell by pulses to test voltages (V) between −190 and 90 mV in increments of 20 mV. Every test pulse was preceded by a 20-s long conditioning to 40 mV. Symbols correspond to plots in B. (B) Charge transferred during the ON transient (•) and steady current (□) in traces shown in A, plotted against V.
Data are presented as averages ± SEM. Significance of differences between mean values was evaluated by Student's t test. Voltage distributions and time courses were fitted, respectively, by single Boltzmann and single exponential functions using a nonlinear least-squares routine included in the Sigmaplot software package (SPSS Inc., Chicago, IL).
results
Steady State Distributions of Charge Movement in Primed and Inactivated Channels
To study effects of conditioning depolarization on the voltage dependence of intramembranous charge movement, we used a double pulse protocol illustrated in Fig. 2. First, gating currents were recorded in polarized α1/β cells held at −90 mV (Fig. 2 A). The test pulse was applied from a 50-ms long interpulse at −60 mV. Then the same protocol was applied again but with a 20-s long conditioning pulse to 40 mV preceding each interpulse (Fig. 2 B). Current traces are shown on both panels for a set of test voltages starting at −190 mV in 40-mV increments. There was little charge movement current at voltages more negative than −70 mV in polarized cells, but the conditioning produced a substantial increase of currents at these negative voltages. The charge transferred during the ON transient for the same cell is plotted against membrane potential in Fig. 2 C. When the cell was held at −90 mV, and consequently the channels were available for opening, most of the charge moved during pulses positive to −60 mV (Fig. 2, •). In inactivated channels, by contrast, about half of the charge moved in pulses below −60 mV (Fig. 2, ○). Curves are fits by a shifted Boltzmann function
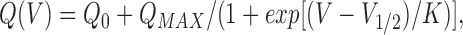 |
1 |
Figure 2.

Conditioning depolarization shifts to negative voltages charge transfer in α1/β cells. (A) Gating current transients in a nonconditioned α1/β cell. Shown are the currents elicited with test pulses to voltages ranging from −190 to 90 mV, in 40-mV intervals, starting from a 50-ms long interpulse at −60 mV. (B) Gating currents in the same cell, conditioned by a 20-s long pulse to 40 mV applied before each test pulse. (C) Charge transfer without (•) and with (○) conditioning, calculated from the records illustrated in A and B and plotted vs. V. Smooth lines are drawn by: Q(V)(nC/μF) = −6 + 50/[1 + exp(V/35 mV)] for primed, and Q(V)(nC/μF) = −26 + 55.5/(1 + exp[(V + 55 mV)/35 mV)] for the inactivated cell.
where Q 0, a negative quantity, is the limit of charge transfer as V tends to −Θ, Q MAX is the difference between the maximal positive transfer and Q 0, V1/2 is the voltage of half-maximal transfer, or transition potential, and K is a steepness constant. The measurable charge movement during the interpulse had ended by the end of the interpulse. Therefore, the changes in charge distribution in inactivated channels were long-lived compared with the time scale of the measurable charge movement.
Addition of the α2δ subunit increased the effect of depolarization on charge transfer. About two thirds of the charge was mobile below −60 mV in inactivated α1/β/α2δ channels (Fig. 3). To compare the effects of conditioning in α1/β and α1/β/α2δ cells, we averaged charge distributions obtained in different cells, normalized as follows. First, Eq. 1 was fitted to individual Q(V) data, and the fitted shift Q 0 was subtracted from Q(V). Data shifted in this way (referred to as “charge distributions”) can be compared without reference to starting voltage. For averaging, the charge distribution was normalized to the individual Q MAX determined in the inactivated cell.1 Averages of normalized distributions are shown in Fig. 4. The curves are Boltzmann fits with parameters listed in Table I. In polarized cells (Fig. 4, •), V1/2 was close to 0 mV, but some 12 mV more negative in α1/β (−8.4 mV) than in α1/β/α2δ cells (3.9 mV). One reason for this small difference is that gating charge in the primed cells is systematically underestimated at voltages positive to 70 mV (and V1/2 is consequently undervalued), due to the presence of nonspecific outward current. With the α2δ subunit present, maximal charge movement increases approximately twofold (Bangalore et al., 1996), which makes the relative error smaller and V1/2 greater.
Figure 3.

Greater negative shift of charge transfer in α1/β/α2δ cells. (A) Gating current transients in a nonconditioned α1/β/ α2δ cell. As in Fig. 2, the records shown are of currents elicited with test pulses to voltages ranging from −190 to 90 mV, in 40-mV intervals, starting from a 50-ms long interpulse at −60 mV. (B) Gating currents in the same cell, conditioned by a 20-s long pulse to 40 mV applied before each test pulse. (C) Charge transfer without (•) and with (○) conditioning, calculated from the records in A and B and plotted vs. V. Smooth lines are drawn by: Q(V)(nC/μF) = −2.5 + 60/ (1 + exp[(V − 7 mV)/25 mV)] for primed, and Q(V)(nC/μF) = −39 + 55/(1 + exp[(V + 85 mV)/32 mV)] for the inactivated cell.
Figure 4.

Effect of conditioning on charge distributions obtained with pulses from −60 mV. (A) Average charge distributions derived from charge transfer functions illustrated in Fig. 2, in nonconditioned (•) and conditioned (○) α1/β cells (n = 8). Smooth lines are fits with Boltzmann equation: Q(V) = Q MAX/ (1 + exp[(V − V1/2)/K)]. Best fit parameters are in Table I. (B) Average charge distributions in α1/ β/α2δ cells (n = 9), derived from charge transfer functions illustrated in Fig. 3.
Table I.
| Composition |
Interpulse voltage (mV) |
n
|
V1/2 (mV) | K (mV) | QMAX | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Conditioned | Reference | Conditioned | Reference | Conditioned | |||||||||||
| α1/β | −60 | 14 | −8.4 ± 2.0 | −65.5 ± 1.7 | 29.6 ± 1.5 | 33.4 ± 1.4 | 0.95 ± 0.02 | 1.00 ± 0.01 | ||||||||
| +40 | 5 | −20.3 ± 3.3 | −89.6 ± 2.9 | 39.2 ± 3.4 | 39.6 ± 2.4 | 0.93 ± 0.05 | 1.02 ± 0.04 | |||||||||
| −150 | 5 | 1.5 ± 4.8 | −17.5 ± 1.7 | 31.6 ± 3.7 | 31.1 ± 1.6 | 1.03 ± 0.06 | 1.00 ± 0.02 | |||||||||
| α1/β/α2δ | −60 | 9 | 3.9 ± 1.2 | −77.3 ± 1.1 | 27.9 ± 0.8 | 32.8 ± 0.9 | 1.02 ± 0.01 | 0.99 ± 0.01 | ||||||||
| +40 | 5 | −21.8 ± 2.4 | −90.4 ± 2.3 | 38.9 ± 2.6 | 30.6 ± 1.9 | 0.96 ± 0.04 | 1.01 ± 0.04 | |||||||||
| −150 | 5 | 2.9 ± 1.5 | −33.9 ± 1.1 | 27.6 ± 1.3 | 28.0 ± 1.0 | 0.93 ± 0.02 | 0.98 ± 0.02 | |||||||||
When charge transfers were measured after the inactivating pulse (Fig. 4, ○), V1/2 was shifted by −57 mV in α1/β cells and by −81 mV in α1/β/α2δ cells. The difference in shifts was statistically significant (P < 0.05). As shown in Table I, inactivation induced only minor changes in steepness and total mobile charge. As was the case for the distribution of charge movement in native cardiomyocytes (Shirokov et al., 1993), the effect of inactivation is best described, with either subunit composition, as a simple shift in the transition potential.
The greater shift in the presence of the α2δ subunit could be the result of a greater extent of inactivation during the conditioning pulse to 40 mV, a slower recovery from inactivation during the interpulse at −60 mV, or both. To test these possibilities, we studied voltage distributions of mobile charge after an inactivating pulse, varying the interpulse voltage.
In the extreme case illustrated in Fig. 5, there was no interpulse. When negative-going test pulses where applied after a 50-ms long step to 40 mV, as shown in Fig. 5 A for an α1/β/α2δ cell, charge transfer was nearly maximal at −100 mV (Fig. 5 C, ▪). After a 20-s long conditioning pulse, charge transfer at −100 mV was only about half-maximal (Fig. 5 C, □). Fig. 5 D plots the corresponding distributions in α1/β/α2δ cells. For comparison, the dashed curves are the best fits in α1/β cells. Addition of the α2δ subunit did not change significantly the transition potentials in noninactivated or inactivated channels. The only significant difference was that charge distribution of inactivated channels was somewhat steeper in the presence of the α2δ subunit. The experiments in Fig. 5 indicate that the difference in distributions after conditioning demonstrated in Fig. 4 was mostly due to a faster recovery of α1/β cells during the interpulse at −60 mV. In the absence of an interpulse allowing recovery, the difference induced by inactivation was about the same with both subunit compositions.
Figure 5.

Effect of conditioning on charge distributions obtained with pulses from 40 mV. (A) Gating currents elicited in an α1/β/α2δ cell. Shown are the currents elicited with test pulses to voltages ranging from −170 to 90 mV in 40-mV intervals, starting from a 50-ms long interpulse at 40 mV. (B) Gating currents in the same cell, elicited by test pulses from a 20-s long conditioning at 40 mV. (C) Average charge distributions in α1/β cells (n = 5), calculated from charge transfers obtained as illustrated in A and B. (▪) Charge distribution after a 50-ms long prepulse to 40 mV. (□) Charge distribution after a 20-s long conditioning. Solid lines are Boltzmann fits through the data. (D) Corresponding average charge distributions in α1/β/α2δ cells (n = 5). Solid lines are Boltzmann fits through the data. Dashed lines copy the fits (shown in C) to average charge distributions without the α2δ subunit. The transition potentials of the distributions obtained after 20-s long conditioning are not significantly different in α1/β/α2δ and α1/β cells (Table I).
When test pulses were applied from an interpulse level of −150 mV, conditioning again led to a negative shift of the charge distribution, now manifested as an increase of gating currents recorded at intermediate voltages, as shown for an α1/β/α2δ cell in Fig. 6, A and B. As shown in Fig. 6 D, the charge distribution in primed cells was affected little by the presence of the α2δ subunit (Fig. 6, •). In conditioned α1/β/α2δ cells, however, the charge distribution was shifted to more negative voltages than in α1/β cells, as a consequence of a slower recovery at −150 mV in the presence of the α2δ subunit. Again, the charge movement currents ended earlier than the interpulse, indicating that even at −150 mV the voltage shift in distribution of mobile charge induced by inactivation recovers much more slowly than the measurable return of the mobile charge.
Figure 6.

Effect of conditioning on charge distributions obtained with pulses from −150 mV. (A) Gating currents elicited in an α1/β/α2δ cell. Shown are the currents elicited with test pulses to voltages ranging from −170 to 90 mV, in 40-mV intervals, starting from a 50-ms long interpulse at −150 mV. (B) Gating currents in the same cell, when the interpulses are preceded by a 20-s long conditioning at 40 mV. (C) Average charge distributions in α1/β cells. (D) Average charge distributions in α1/β/α2δ cells, with the fits to the distributions in α1/β cells represented in dashed trace. The data set is from the same cells as in Fig. 5. Note that the charge distribution in conditioned α1/ β/α2δ cells is considerably more shifted than in α1/β cells (and the fitted transition potentials, listed in Table I, are significantly different).
These experiments demonstrate that in inactivated Ca channels all the charge remains mobile, albeit at voltages more negative than those of activation gating. The concept of modal conversion applicable to slow inactivation of native Na channels (Bezanilla et al., 1982), recombinant Shaker K channels (Olcese et al., 1997), and voltage-dependent inactivation of native Ca channels of skeletal (Brum and Ríos, 1987) and cardiac muscle (Shirokov et al., 1992) is seen to apply to recombinant Ca channels. Gating transitions within the primed mode of the channel produce charge 1 movements, while transitions within the inactivated mode produce sterile charge 2.
The present results suggest that the α2δ subunit has little direct effect on charge 1 and charge 2 movements. Apparently, its main effects are on the kinetics of charge 1–charge 2 interconversion. We confirmed this impression with the experiments described below.
Kinetics of Inactivation of Charge Movement and Effects of the α2δ Subunit
Time-dependent inactivation of Ba2+ current through L-type Ca channels has two kinetic phases. In the absence of the α2δ subunit, prolonged depolarization reduces gating charge mobile above −60 mV with a time course parallel to the slower exponential component of Ba2+ current decay (Ferreira et al., 1997). The time constant of this component in α1/β cells is ∼8 s at 20 mV, while in α1/β/α2δ and in native cells it is ∼2 s. In the experiment illustrated in Fig. 7, we investigated the effect of the α2δ subunit on the onset kinetics of charge reduction. Charge movement was recorded during OFF transients from depolarizations of different duration (protocol at top). In α1/β cells (Fig. 7 A), the OFF gating currents were progressively smaller for increasing test pulse durations up to 20 s. In cells with all three subunits (Fig. 7 B), reduction of the OFF transient saturated after 6 s of depolarization. To average and compare effects in different cells, values of charge moved after the long depolarizations were normalized to the value obtained from the transient after a 45-ms pulse to the same voltage. The averages are plotted in Fig. 7, C and D. Different symbols correspond to different pulse voltages. Reduction of the mobile charge was about three times faster in α1/β/α2δ cells for all voltages. The time constant of the reduction of gating charge at 40 mV in α1/β/α2δ cells was ∼1.7 s, similar to that for the slow phase of Ba2+ current decay in these cells and in native cardiomyocytes.
Figure 7.

The α2δ subunit accelerates inactivation of gating currents. (A) OFF gating current transients in an α1/β cell after different conditioning times at 40 mV. Conditioning times (T) are listed near each plot. (B) OFF gating current transients in an α1/β/α2δ cell after different times at 40 mV. (C) Charge transferred during the OFF transients plotted vs. T. Different symbols correspond to different values V, as listed. Charge transfers are normalized to the value after 0.05 s at the same V and averaged (n = 6 cells). Solid lines are best fits with the equation: Q(T) = [1 − Q(Θ)] exp(−T/τina) + Q(Θ). The best fit values of τina are: 29.0 ± 13.0 s at 0 mV, 10.4 ± 2.6 s at 20 mV, and 7.5 ± 2.3 s at 40 mV. (D) Charge transfers with the same protocol as in C, for α1/β/α2δ cells (n = 7). The best fit values of τina are: 3.2 ± 1.7 s at 0 mV, 2.2 ± 0.6 s at 20 mV, and 1.7 ± 0.3 s at 40 mV.
We studied the effect of α2δ on recovery from inactivation applying a double pulse protocol often used with ionic currents. The experiment is illustrated in Fig. 8. After conditioning, the membrane was kept at the interpulse voltage for a variable time (Tip), and then a test pulse to 50 mV was applied to assess charge movement. Reference test currents (Fig. 8, thick traces) were recorded without conditioning. Gating currents elicited by test pulses from −60 mV took much longer to recover in α1/β/α2δ cells (Fig. 8 B) than in α1/β cells (Fig. 8 A). As shown in Fig. 8, C and D, recovery from inactivation was substantially delayed by the α2δ subunit at every interpulse voltage tested.
Figure 8.

The α2δ subunit slows recovery of gating currents. (A) Gating currents elicited in an α1/β cell by a test pulse to 50 mV from an interpulse of duration Tip = 50 ms at voltage Vip = −60 mV. Thick trace: reference, without conditioning. Thin trace: a 20-s long conditioning at 40 mV preceded the interpulse. Insets: same traces in an expanded time scale. (B) Same protocols as in A, for an α1/β/α2δ cell. (C) Relative charge mobile during ON transients such as those in A, plotted vs. Tip. Different symbols correspond to different Vip. Charge transfers were normalized to the value without conditioning in the same cell, and averaged (n = 7 cells). Solid lines are best fits with: Q(Tip) = 1 − [1 − Q(0)] exp(−Tip/τr). The time constants τr are: 1.15 ± 0.31 s at −20 mV, 0.16 ± 0.04 s at −40 mV, and 0.04 ± 0.02 s at −80 mV. (D) Same as in C, for α1/β/α2δ cells (n = 5). τr is 2.47 ± 0.67 s at −40 mV, 0.61 ± 0.11 s at −60 mV, and 0.17 ± 0.03 s at −80 mV.
In studies of recovery at very negative voltages, it was simpler to record the charge movement of inactivated channels (charge 2), which occurs at potentials negative to −50 mV. For this purpose, a test pulse to −50 mV was applied from an interpulse at more negative voltages (Fig. 9). Because inactivation involves a negative shift in the voltage dependence of charge movement, in the range negative to −50 mV, recovery is associated with a reduction in charge transfer. Given the conservation of total charge (Figs. 4–6), this should be accompanied by an equivalent increase of charge mobile in noninactivated channels (at potentials positive to −50 mV).
Figure 9.

The α2δ subunit slows reduction of charge mobile at negative voltages. (A) Charge movement currents elicited in an α1/β cell by a test pulse to −50 mV from an interpulse of Tip = 50 ms at Vip = −150 mV. (thick trace) Reference. (thin trace) After a 20-s long conditioning at 40 mV. (B) Same protocols as in A, for an α1/β/α2δ cell. (C) Relative increase of charge mobile during ON transients such as those in A, plotted vs. Tip. Different symbols correspond to different Vip. Each increase in charge transfer was normalized to the charge transfer in reference and averaged (n = 4 cells). Solid lines are best fits with: Q(Tip) = Q(0) exp(−Tip/τr). τr is 1.30 ± 0.19 s at −100 mV, 0.52 ± 0.09 s at −150 mV, and 0.32 ± 0.06 s at −200 mV. (D) Same as in C, for α1/β/ α2δ cells (n = 4). τr at −100 mV is 2.4 6 ± 0.63 s, at −150 mV is 1.66 ± 0.53 s, and at −200 mV is 1.87 ± 0.40 s.
Independently of the presence of the α2δ subunit, conditioning caused an approximately twofold increase of charge transfer between −150 and −50 mV (Fig. 9, A and B), if recorded 50 ms after conditioning. Very little of this increase remained after 1 s at −150 mV in α1/β cells, but ∼50% persisted in α1/β/α2δ cells. Fig. 9, C and D, plot the increase of charge mobile between the interpulse voltage and −50 mV, normalized to the charge without conditioning, as a function of interpulse duration at −100 (•), −150 (○), and −200 (▴) mV for α1/β and α1/β/α2δ cells, respectively. Curves are single exponential fits. Without the α2δ subunit, recovery kinetics were strongly voltage dependent in the range explored, and fastest at −200 mV (τ ≃ 300 ms). In contrast, with the α2δ subunit recovery was slow (τ ≃ 2 s) and weakly voltage dependent in this range. The ancillary subunit not only slows the recovery process but also insulates it from the influence of voltage.
discussion
Modal Interconversion of Intramembrane Charge Movement during Inactivation of α1C Channels
The main finding of this study is that voltage-dependent inactivation of heterologously expressed cardiac Ca channels is associated with a large negative shift in the voltage dependence of their charge movement. The intramembranous charge movement in inactivated α1/β channels has a transition potential of ∼−90 mV, which is the same as in native L-type Ca channels (Brum and Ríos, 1987; Shirokov et al., 1992). The charge remains mobile at these voltages until channels recover from inactivation, a first order process with a time constant of ∼300 ms at −200 mV. Because the time scale of charge movement in inactivated α1/β channels (τ ≤ 15 ms) is much faster than recovery from inactivation, the expressed channels exhibit a separate inactivated mode, and the term charge 2 can be applied to the charge mobile at negative voltages in inactivated channels. With α1/β/α2δ channels, the modal separation is even more clear cut.
We showed previously that the onset of inactivation of gating currents in α1/β cells is parallel to a slow phase of Ba2+ current decay (τ ≃ 8 s), and that addition of the α2δ subunit increases the slow rate of ionic current decay, making it similar to that in cardiac cells (τ ≃ 2 s; Ferreira et al., 1997). In agreement with these earlier observations, we now find a time constant of ∼2 s for the reduction of gating currents upon inactivation in cells expressing all three subunits (Fig. 7). Working with native cells at room temperature, we estimated the onset τ of charge interconversion at ≃0.6 s (Shirokov et al., 1993). This estimate may be at fault because of the unavoidable contribution of Na channels to the native gating currents. A discrepancy in the same direction exists for the time course of recovery from inactivation of gating charge (charge 1) in native cells. We reported a time constant of 200 ms for this process (Shirokov et al., 1992), while in the present measurements with expressed channels containing the α2δ subunit τ ≃ 1.5 s (Fig. 9). Again, and for the same reasons, the present measurements must be considered more reliable. Interestingly, in native skeletal muscle, recovery of charge 1 is much slower (τ ≃ 3 s; Brum and Ríos, 1987). The discrepancy in results in native cells could reflect structural differences between the two channels. It could also reflect a better determination, given the vast predominance of dihydropyridine receptors over other sources of intramembranous charge movement in skeletal muscle.
Biophysical Effects of the α2δ Subunit
The present results demonstrate that the α2δ subunit promotes inactivation of gating currents in cardiac L-type Ca channels. We found that the α2δ subunit made the onset of inactivation of intramembrane charge movement three to four times faster, and the recovery from inactivation about five times slower. On the other hand, inactivation of gating currents in L-type Ca channels was much slower than activation gating, even in the presence of the α2δ subunit (τ ≃ 2 s). It is therefore unlikely that the increased rate of inactivation results from primary changes in activation.
In spite of profound effects of the α2δ subunit on inactivation kinetics, the voltage dependence of charge movement in primed and inactivated channels was not significantly changed. In agreement with previous findings (Singer et al., 1991; Welling et al., 1993; Shistik et al., 1995; Bangalore et al., 1996), we found little effect of the α2δ subunit on voltage dependence of activation of ionic currents on α1/β cells (data not shown). In contrast, Felix et al. (1997) reported that the α2δ subunit, added to the α1C in the absence of the β subunit, shifted the activation of ionic currents in tsA201 cells by ∼−10 mV. In Xenopus oocytes, coexpression of α2δ and α1 subunits increased single channel open probability (Shistik et al., 1995), while in HEK 293 cells addition of α2δ to α1 and β subunits speeded up activation and deactivation (Bangalore et al., 1996).
As shown previously (reviewed by Gurnett and Campbell, 1996), the α2δ subunit shifts by ∼−10 mV the steady state inactivation curves of recombinant Ca channels. In light of these observations, our finding that the charge distributions in inactivated channels are unaffected by the α2δ subunit may seem surprising. This and other aspects, however, may be accounted for with biophysical models of voltage-dependent inactivation.
Inactivation of gating currents in Ca channels has been represented by a minimal four state diagram (Scheme I). In it, the “horizontal” transitions are fast and voltage dependent, while (voltage-independent) “vertical” transitions are much slower, which qualifies the pairs of states, C, O, and I*, I, as separate modes that account, respectively, for charge 1 and charge 2. The model is “allosteric”: inactivation and activation occur at separate sites, as the movement of separate gates that influence each other but move individually.
This model already has most of the observed properties, and accommodates the effect of α2δ, which could simply be represented by an equal reduction in the free energy of states I and I* and a decrease in the energy barrier between O and I. It is, however, too oversimplified, failing to account for charge movement between and inactivation from closed states.
Scheme SII represents a generalization of the allosteric model (Marks and Jones, 1992; Kuo and Bean 1994; Olcese et al., 1997), in which activation gating is represented as a multi-step reaction, and inactivation is likely to occur from closed states.
Scheme II.

Scheme SII also features charge 1 and charge 2 modes, an aspect used recently by Olcese et al. (1997) to represent similar observations on gating currents of Shaker K channels. However, it has many parameters that cannot be constrained, especially the number of closed states. For this reason, we generalized these models by incorporating continuum activation gating (Millhauser et al., 1988; Lauger, 1988) in a version of Levitt (1989). This simplified the kinetic scheme, reduced the number of parameters, and gave a more intuitive view of the gating process.
The model equations are presented in detail in the Appendix. Inactivation is described as a voltage-independent reaction, of rate constants kP and kI, between two modes of the channel: P (primed) and I (inactivated). The gating process associated with intramembrane charge movement is represented by a conformational movement along a reaction coordinate x, driven by the electric field (Scheme SIII). Because there are no free energy barriers, the movement is continuous, akin to diffusion. The generalized reaction coordinate projects to one dimension the set of conformations accessible to the channel. A given value of the coordinate characterizes all conformations that take the same amount of energy from the electric field.
Scheme III.

The state of the ensemble of channels is described by two probability density functions, P(x) and I(x). Evolution of these functions is determined by diffusion reaction within the free energy profiles UP(x) and UI(x), which have chemical and electrical additive components. As stated above, at any given transmembrane voltage the electrical energy term will vary linearly with x. An additional assumption, primarily made for simplicity, is that the chemical free energy also depends linearly on x. Therefore, the joint dependence of UP (or UI) on x and V is represented by ruled surfaces (hyperboloids), as in Fig. 10, where the parameters are chosen to simulate data in the presence of α2δ.
Figure 10.

Free energy profiles of the continuum model. Energies UP and UI of the primed and inactivated modes in the model, given by Eq. 3 (Appendix), plotted as a function of transmembrane voltage V and conformational coordinate x. Parameter χ (the value of x at which the planes intersect) is set to 0.55, the value used to simulate the presence of α2δ.
To reproduce the tendency to inactivation at positive and recovery at negative voltages, UP(x) and UI(x) must cross, so that P is favored at the values of x near 0, which are populated at the resting potential, and the opposite occurs at x near 1. Because intermodal transitions are assumed to be intrinsically voltage independent, the transfer of charge as a function of x must be the same in the two modes, or, equivalently, ∂ UP/∂ V = ∂ UI/∂ V. Because voltage cannot directly alter intermodal distribution, the value of x at which P and I are equally probable (intersection of the planes UP(x,V) and UI(x,V) in Fig. 10) will be a constant, χ, independent of V. The continuum model requires four thermodynamic parameters and three kinetic parameters, fewer than even the simple state model of Scheme SI.
Scheme I.

At equilibrium, the distributions satisfy:
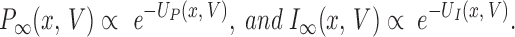 |
2 |
Therefore, in channels held near resting voltages, PΘ(x,V) is large at x close to 0, while IΘ(x,V) is very small. When the voltage is positive, PΘ(x,V) becomes small for all x, while IΘ(x,V) is high at x close to 1. Fig. 11 illustrates the evolution of the system [P(x,t) and I(x,t)] during a pulse from −100 to 0 mV. The initial conditions P(x,0) and I(x,0) were calculated as the equilibrium distributions at −100 mV. The voltage was changed to 0 mV with exponential time course (τm = 0.2 ms). The system evolved with two widely separated time scales, one associated with movements along x and the other with the inactivation transition. The fast process is illustrated in Fig. 11, top. A and B plot P(x,t) and I(x,t) during the first 5 ms of the voltage step. Both P(x,t) and I(x,t) redistribute towards x = 1, reaching a quasi stationary situation, and generating measurable charge movement associated with diffusion along x (Fig. 11 C). After the quasi steady state is reached, probability densities continue to change at a slow rate determined by the inactivation reaction. Fig. 11, bottom, illustrates this slow process. The marked complementary changes in total occupancy of modes P and I (Fig. 11 F) are accompanied by additional diffusion along x. The resulting charge movements are undetectable because of their slow rate, limited by the inactivation reaction. This additional redistribution of charge can be estimated as the difference between steady state charge transfer, calculated from the equilibrium distributions (PΘ and IΘ), and charge transfer determined by numerical integration of the simulated current, as if it were an experimental record.
Figure 11.

Two time domains of inactivation. (A) Initial evolution P(x,t) of the (probability density in the) primed mode, in response to a pulse to 0 mV from a holding potential of −100 mV. (B) Corresponding evolution I(x,t) of the inactivated mode. (C, thin line) Gating currents generated during the pulse. (thick lines) Total modal probabilities, calculated as integrals over x of P and I. Note that the modal probabilities change little during the interval of measurable gating current. (D and E) P(x,t) and I(x,t) in response to the same pulse on a longer time scale (0.05–10 s). (F) Total modal probabilities on the same time scale.
Fig. 12 illustrates the comparison. Gating currents simulated with pulses from −200 mV are shown in Fig. 12 A, left. The areas under ON transients are shown in Fig. 12 B, •. The recordable charge transfer for steps from −200 mV occurs at more positive voltages than the equilibrium distribution of charge Q eq(V) (Fig. 12, curve). The vertical difference between the curve and the symbols corresponds to the additional transfer that occurs slowly as channels inactivate.
Figure 12.


Simulated charge transfers at two holding potentials. (A, left) Gating currents elicited by pulses from –200 mV to different voltages (V). (right) Gating currents from 200 mV. (B) Charge transferred in records in A vs. V. (•) Charge transfer from −200 mV. (○) Charge transfer from 200 mV. (solid line) Equilibrium charge distribution as a function of holding potential. (C) Charge distribution curves, splines through the points in B shifted vertically as described in the text. (thick black line) Distribution at holding potential −200 mV. (thick light grey line) Distribution at holding potential 200 mV. (thick dark grey line) Charge distribution after a 1-s conditioning pulse from −200 to 200 mV, which makes the two modal probabilities approximately equal (0.5). (thin line) Charge distribution generated by Scheme SI after the 1-s conditioning pulse from −200 to 200 mV, with parameters chosen to best reproduce the behavior of the continuum model (see discussion).
Similarly, when pulses are applied from 200 mV, charge mobile in the fast time domain corresponds to redistribution of channels in mode I along the x axis, and the charge transfer (Fig. 12 B, ○) occurs at more negative voltages. Corresponding intramembrane charge movement currents are shown in Fig. 12 A, right.
The steepness of the Boltzmann fits to the simulated charge transfer was ∼25 mV in both primed and fully inactivated channels, corresponding to the transfer of one elementary charge in a single step transition. With the continuum model, the maximal charge transfer had to be set to three elementary charges to simulate such shallow distributions. Interestingly, model-independent estimates of maximal unitary gating charge from gating current noise of Na and K channels provided similar values. 2.4 elementary charges were required assuming that the gating current noise is produced by a number of independent identical particles, which during activation undergo a single irreversible transition (Conti and Stühmer, 1989; Sigg et al., 1994). The continuum model therefore reconciles estimates of elementary charge from microscopic and macroscopic gating current measurements, provided that the macroscopic charge movement is generated by particles with approximately three elementary charges moving independently and with the same half-activation potential. Because 8–12 elementary charges transfer during channel activation, ∼4 such independent particles would be required for channel opening. This would be the case, for example, if the movement of individual S4 segments occurred independently and over the same voltage range, and if all four segments had to move to cause activation.
Charge distributions of squid axon Na, skeletal muscle Ca, and Shaker K channels are less steep after moderate inactivation (Bezanilla et al., 1982; Brum and Ríos, 1987; Olcese et al., 1997). The observation also applied for moderately inactivated cardiac Ca channels in the present work (data not shown). Model simulations of this condition are illustrated in Fig. 12 C. The thick curves are simulated charge distributions in primed and inactivated channels (spline curves through Fig. 12 B, ○ and •). The dark gray curve simulates with the continuum model the measurable charge transfer upon application of negative-going pulses after a 1-s conditioning at 200 mV. In contrast, the four-state model of Scheme SI 2 generated the two-sigmoidal distribution plotted in thin trace, which is close to a linear combination of the fully primed and fully inactivated distributions. Such sharp separation of sigmoidal components was never observed experimentally. Even though the two-modal distributions of charge could be isolated experimentally in the presence of α2δ, the distribution in partially inactivated conditions was not a linear combination of the modal distributions. This is well reproduced by the continuum model.
Simulation of the Effect of the α2δ Subunit
To model the effects of the α2δ subunit on voltage- dependent inactivation, we had only to assume that the subunit stabilizes the inactivated mode I. We specifically assumed that it makes the energy difference between I and P more negative at all values of x. Because of the linearity of UI(x,V), this is equivalent to a decrease in χ, the x value of half inactivation. While inactivation of α1/β channels was simulated with χ = 0.8, α1/ β/α2δ, channels required χ = 0.55 as sole parameter change. The simulated charge distributions of primed and inactivated channels are in Fig. 13. In correspondence with experimental observations (Table I), the transition potentials of simulated charge distributions in fully primed or fully inactivated channels were not affected by the change in χ. (The reason is simple: the voltage distribution of mobile charge within modes is sensitive to the chemical potential gradient, not to an additive constant.) The model also described well the observed difference in the steepness of the charge distributions between primed and conditioned α1/β channels. In conditioned α1/β cells, the steepness of charge distribution measured from an interpulse at 40 mV was ∼40 mV, whereas in nonconditioned cells (−150 mV interpulse) it was ∼30 mV. In conditions simulating α1/β channels (χ = 0.8), the charge distribution of the inactivated channels was also shallower (33 mV) than that of noninactivated channels (25 mV). The difference between the steepness of charge distribution in primed and inactivated α1/β channels is due to the fact that, because of their fast rate of recovery, the charge distribution of inactivated α1/β channels cannot be isolated experimentally.
Figure 13.

Simulation of the effect of α2δ on inactivation of gating currents. (A) Charge distributions, obtained as described for Fig. 12 with pulse patterns illustrated, simulating primed (•) or inactivated α1/β channels (χ = 0.8). The lines represent Boltzmann fits with the following parameters: for primed channels Q MAX = 2.6 e−, V1/2 = 0.9 mV, K = 25.1 mV. For inactivated channels, Q MAX = 2.6 e−, V1/2 = −90.5 mV, K = 32.7 mV. Comparable experimental data are in Fig. 5 A. (B) Same as in A, with χ = 0.55, to simulate α1/β/ α2δ channels. For primed channels, Q MAX = 2.6 e−, V1/2 = 0.8 mV, K = 25.2 mV. For inactivated channels, Q MAX = 2.6 e−, V1/2 = −98.3 mV, K = 25.3 mV. Comparable experimental data are in Fig. 5 B. (C) Onset of reduction of simulated gating currents upon conditioning to three different voltages Vp in α1/β simulations. The fits are given by Q(t) = 1 − [1 − Q(Θ)] exp(−t/τ). Values of fit parameters [Q(Θ) and τ] are: 0.67 and 20.01 s for Vp = 0 mV, 0.38 and 11.12 s for 40 mV, 0.31 and 8.85 s for 80 mV. Comparable experimental data are in Fig. 7 C. (D) Onset of reduction of gating currents in α1/β/α2δ simulations. Values of fit parameters [Q(Θ) and τ] are: 0.27 and 9.78 s for Vp = 0 mV, 0.17 and 3.61 s for 40 mV, 0.16 and 2.68 s for 80 mV. Experimental data are in Fig. 7 D. (E) Recovery from reduction of charge movement in α1/β simulations at three different interpulse voltages Vip. The fits are generated with Q(t) = Q(χ) exp(−t/τ). Corresponding parameters [Q(0) and τ] are: 0.64 and 1.00 s for Vip = −100 mV; 1.39 and 0.341 s for −150 mV; 1.56 and 0.275 s for −200 mV. Experimental data are in Fig. 9 C. (F) Recovery in α1/β/α2δ simulations. Values of fit parameters [Q(Θ) and −] are: 0.75 and 3.96 s for Vip = −100 mV; 1.63 and 1.58 s for −150 mV; 1.83 and 1.20 s for −200 mV. Experimental data are in Fig. 9 D.
The onset kinetics of inactivation in the model were assessed with the pulse protocol used in the experiment of Fig. 7. The corresponding simulated dependencies of amounts of charge movements on the duration of conditioning depolarization are shown in Fig. 13, C and D. Reduction of charge mobile above −50 mV was more rapid for χ = 0.55 (simulating channels with α2δ) than for χ = 0.8, giving rates similar to those obtained experimentally.
Recovery properties of the model were studied with pulse protocols similar to those illustrated in Fig. 9. Fig. 9, E and F, show dependencies of the amount of charge mobile below −50 mV on the interpulse duration. As observed in the experiments with α1/β/α2δ (Fig. 9 D), simulations with χ = 0.55 (Fig. 13 F) exhibit a slow recovery rate and are weakly voltage dependent at voltages below −150 mV. In agreement with the observations in α1/β cells (Fig. 9 C), with χ = 0.8 recovery is three to five times faster (Fig. 13 E), approaching the speed of recordable charge movement. In all, the continuum model reproduces well the effects of α2δ, under the hypothesis that the subunit changes a single parameter of energy distribution.
Modal Separation
With χ = 0.55, the model behaves similarly to the channels in the presence of α2δ, evolving with two well- defined time scales: a fast one associated with measurable charge movement and a slow one determined by inactivation. This separation of time scales is a requisite for a well-defined charge 2, which can be ascribed unequivocally to mode I.
When χ is close to 1 (simulating the absence of the α2δ subunit) the I ↔ P conversion becomes very fast at negative voltages, and the separation between time scales becomes less clear cut. At −200 mV, the most negative potential that is consistently accessible, recovery in simulations of channels lacking α2δ has a τ = 0.28 s (Fig. 13 E), in good agreement with the experiment (0.32 s, Fig. 9 C), and well beyond the time needed to complete the measurable movement of charge (∼50 ms). The fastest possible recovery is achieved from x = 0 (a condition that requires forbiddingly large negative potentials) and proceeds with a time constant of ∼0.16 s. This is close to but greater than the time of charge movement, so that modal separation still prevails in simulations of channels without α2δ, even at experimentally inaccessible negative potentials.
If the rate constant of the I ↔ P reaction (see Appendix, Eq. 11) was just an order of magnitude greater than the value determined for channels without the α2δ subunit, modal separation would break down. This would be reflected in the appearance of a substantial slow component in the charge movement during recovery at intermediate negative voltages, cotemporal with I → P transitions. Such a component, not observed experimentally in Ca channels, is a distinctive feature of the gating current in Na channels, the “remobilization” component observed when channels are reprimed at −130 mV (Armstrong and Bezanilla, 1977). For the continuum model to simulate such fast inactivation, it is necessary that the inactivation processes further stabilize the mobile charged moieties in the trans (open) position. In that case, some values of x close to 0 will not be populated in the inactivated mode at intermediate negative voltages.
Likewise, Kuo and Bean (1994) were able to simulate onset and recovery of fast inactivation in Na channels with the model illustrated by Scheme SII. Therefore, both modal interconversion, documented in the present work, and the so-called charge immobilization phenomena that accompany fast inactivation can be reproduced with general allosteric models of the types represented by Schemes II and III.
The preceding considerations are relevant to whether voltage-dependent inactivation in L-type Ca channels is slow (C-type) or fast (N-type). Inactivation appears to be slow because it exhibits clear modal separation.
On the other hand, there is an important distinction between voltage-dependent inactivation of Ca channels and slow inactivation of Na and K channels. Whereas in these channels recovery from slow inactivation takes many seconds, in L-type Ca channels recovery would be fast, were it not for the stabilizing effect of the α2δ subunit on the voltage-inactivated states. Structure–function studies are required for establishing the mechanism of this stabilization.
Acknowledgments
We thank Drs. Duanpin Chen (Rush University) for discussions and help with the continuum model, and Werner Melzer (University of Ulm, Ulm, Germany) for many comments on the manuscript.
This work was supported by a Scientist Development Grant from the American Heart Association (to R. Shirokov) and by National Institutes of Health grant AR-43113 (to E. Ríos).
appendix
The continuum model of inactivation, represented in Scheme SIII, comprises the following set of equations. The energies of modes P and I, in dimensionless expressions are:
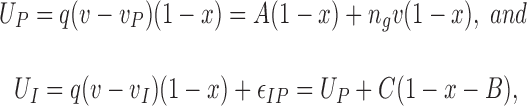 |
3 |
where q = Q/e is the total charge transfer (in number of electrons), v is dimensionless voltage, equal to Ve/ k BT, v P and v I are the transition potentials of the corresponding modes, εIP is the energy difference between U I and U P at x = 1, and A, B, C, and n g are constants (Levitt, 1989).
To emphasize that at x = χ ≡ 1 − B the energy difference U IP changes its sign, Eq. 3 can be rewritten as
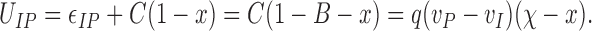 |
4 |
The steady state probability densities are defined as follows:
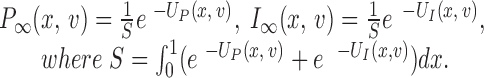 |
5 |
The steady state charge distribution Q eq(V) is calculated numerically from
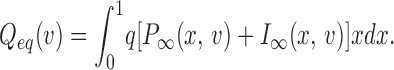 |
6 |
Transitions between the two modes are described by the following set of differential equations:
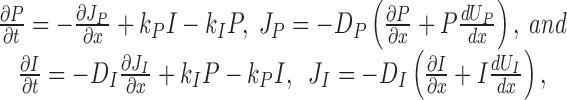 |
7 |
where D P and D I are generalized diffusion coefficients. The fluxes satisfy reflective boundary conditions:
 |
8 |
The rate constants of the interconversion are defined by the energy profiles. For a symmetrical barrier, the rates are
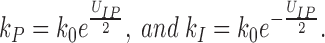 |
9 |
Eqs. 7 and 8 were solved using a fully implicit finite differencing scheme with a band diagonal system of linear equations. The band diagonal system was solved by the bandec and banbks routines of Press et al. (1992) (The computer program for simulation [DOS and X-Win versions] can be obtained by e-mail request to rshiroko@rush.edu). The x grade was 50. The time steps were automatically adjustable depending on accuracy of solution. The accuracy of the solution was determined as deviation of the total probability from 1 and it was set at 3%. For simulations we used: q = 3, v P = 0, v I = −4 (equivalent to −100 mV at room temperature), D P = 100 s−1, D I = 50 s−1, k 0 = 0.05 s−1, and χ = 0.8 for α1/β channels or χ = 0.55 for α1/β/α2δ channels.
The transfer of mobile charge (from an all-in starting distribution) is calculated by
 |
10 |
consistent with the definition of the potentials (Eq. 3). The charge movement current is i g(t) = dQ/dt.
From Eqs. 4 and 9, the time constant of the I ↔ P transition (τIP) is limited by
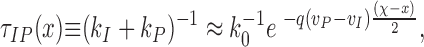 |
11 |
or ln(τIP k 0) ≈ −6 (χ − x) for the parameters used. Because the difference between v P and v I is substantial, the time constant τIP is small compared with k 0, when χ is close to 1 and at x is close to 0.
Footnotes
Dr. Ferreira's permanent address is Depto. Biofisica, Facultad de Medicina, Montevideo, Uruguay CP 11800. Dr. Shirokov's permanent address is A.A. Bogomoletz Institute of Physiology, Kiev, Ukraine 252024.
For example, charge transfer (nC/pF, V in mV) in the cell illustrated in Fig. 3 is fitted by Q(V) = −2.5 + 59.6/{1 + exp[(V − 6.8)/25.0]} in the nonconditioned case, and Q(V) = −38.9 + 55.0/{1 + exp[(V + 85.0)/31.8]} after conditioning. The normalized distributions in the same cell are, respectively: Q′(V) = 1.08/{1 + exp[(V − 6.8)/25.0]}, and Q′(V) = 1.00/{1 + exp[(V + 85.0)/31.8]}.
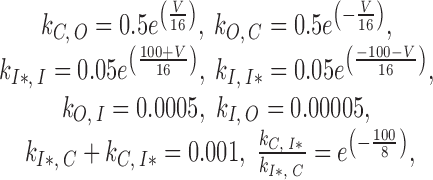 |
references
- Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangalore R, Mehrke G, Gingrich K, Hofmann F, Kass RS. Influence of L-type Ca channel α2δ subunit on ionic and gating current in transiently transfected HEK 293 cells. Am J Physiol. 1996;270:H1521–H1528. doi: 10.1152/ajpheart.1996.270.5.H1521. [DOI] [PubMed] [Google Scholar]
- Balser JR, Nuss HB, Romashko D, Marban E, Tomaselli GT. Functional consequences of lidocaine binding to slow-inactivated sodium channels. J Gen Physiol. 1996;107:643–658. doi: 10.1085/jgp.107.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F, Taylor RE, Fernandez J. Distribution and kinetics of membrane dielectric polarization. I. Long-term inactivation of gating currents. J Gen Physiol. 1982;79:21–40. doi: 10.1085/jgp.79.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum G, Ríos E. Intramembrane charge movement in frog skeletal fibers. Properties of charge 2. J Physiol (Camb) 1987;387:489–517. doi: 10.1113/jphysiol.1987.sp016586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum G, Fitts R, Pizarro G, Ríos E. Voltage sensors of the frog skeletal muscle membrane require calcium to function in excitation–contraction coupling. J Physiol (Camb) 1988;398:475–505. doi: 10.1113/jphysiol.1988.sp017053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien A, Zhao X, Shirokov R, Puri T, Chang CF, Sun D, Ríos E, Hosey M. Roles of membrane-localized β subunit in the formation and targeting of functional L-type Ca2+channels. J Biol Chem. 1995;270:30036–30044. doi: 10.1074/jbc.270.50.30036. [DOI] [PubMed] [Google Scholar]
- Conti F, Stühmer W. Quantal charge redistributions accompanying the structural transitions of sodium channels. Eur Biophys J. 1989;17:53–59. doi: 10.1007/BF00257102. [DOI] [PubMed] [Google Scholar]
- Felix R, Gurnett C, De Waard M, Campbell K. Dissection of functional domaines of the voltage-dependent Ca2+ channel α2δ subunit. J Neurosci. 1997;17:6884–6891. doi: 10.1523/JNEUROSCI.17-18-06884.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira G, Yi J, Ríos E, Shirokov R. Ion-dependent inactivation of barium current through L-type Ca channels. J Gen Physiol. 1997;109:449–461. doi: 10.1085/jgp.109.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurnett C, Campbell K. Transmembrane auxiliary subunits of voltage-dependent ion channels. J Biol Chem. 1996;271:27975–27978. doi: 10.1074/jbc.271.45.27975. [DOI] [PubMed] [Google Scholar]
- Hadley RW, Lederer WJ. Ca2+ and voltage inactivate Ca2+channels in guinea-pig ventricular myocytes through independent mechanisms. J Physiol (Camb) 1991;444:257–268. doi: 10.1113/jphysiol.1991.sp018876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shakerpotassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Kuo C-C, Bean BP. Na+channels must deactivate to recover from inactivation. Neuron. 1994;12:819–829. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- Lauger P. Internal motions in proteins and gating kinetics of ionic channels. Biophys J. 1988;53:877–884. doi: 10.1016/S0006-3495(88)83168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt D. Continuum model of voltage-dependent gating. Macroscopic conductance, gating current, and single-channel behavior. Biophys J. 1989;55:489–498. doi: 10.1016/S0006-3495(89)82842-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shakerpotassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Marks T, Jones SW. Calcium currents in the A7r5 smooth muscle-derived cell line. An allosteric model for Ca channel activation and dihydropyridine agonist action. J Gen Physiol. 1992;99:367–390. doi: 10.1085/jgp.99.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millhauser GL, Salpeter EE, Oswald RE. Diffusion models of ion-channel gating and the origin of power low distributions from single-channel recording. Proc Natl Acad Sci USA. 1988;85:1503–1507. doi: 10.1073/pnas.85.5.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcese R, Latorre R, Toro L, Bezanilla F, Stefani E. Correlation between charge movement and ionic current during slow inactivation in Shaker K+channels. J Gen Physiol. 1997;110:579–589. doi: 10.1085/jgp.110.5.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press, W.H., B.P. Flannery, S.A. Teukolsky, and W.T. Vetterling. 1988–1992. Numerical recipes in C: the art of scientific computing. Cambridge University Press. 50–54.
- Shistik E, Ivanina T, Puri T, Hosey M, Dascal N. Ca2+ current enhancement by α2/δ and β subunits in Xenopus oocytes: contribution of changes in channel gating and α1protein level. J Physiol (Camb) 1995;489:55–62. doi: 10.1113/jphysiol.1995.sp021029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigg D, Stefani E, Bezanilla F. Gating current noise produced by elementary transitions in Shakerpotassium channels. Science. 1994;264:578–582. doi: 10.1126/science.8160016. [DOI] [PubMed] [Google Scholar]
- Singer D, Biel M, Lotan I, Flockerzi V, Hofmann F, Dascal N. The roles of the subunits in the function of the Ca channel. Science. 1991;253:1553–1557. doi: 10.1126/science.1716787. [DOI] [PubMed] [Google Scholar]
- Shirokov R, Levis R, Shirokova N, Ríos E. Two classes of gating current from L-type Ca channels in guinea pig ventricular myocytes. J Gen Physiol. 1992;99:863–895. doi: 10.1085/jgp.99.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokov R, Levis R, Shirokova N, Ríos E. Ca2+-dependent inactivation of cardiac L-type Ca channels does not affect their voltage sensor. J Gen Physiol. 1993;102:1005–1030. doi: 10.1085/jgp.102.6.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend C, Horn R. Effect of alkali metal cations on slow inactivation of cardiac Na+channels. J Gen Physiol. 1997;110:23–33. doi: 10.1085/jgp.110.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- Welling A, Bosse E, Cavalié A, Bottlender R, Ludwig A, Nastainczyk W, Flockerzi V, Hofmann F. Stable coexpression of Ca channel α1, β and α2/δ subunits in a somatic cell line. J Physiol (Camb) 1993;471:749–765. doi: 10.1113/jphysiol.1993.sp019926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G, Sodickson D, Chen T-Y, Yurman ME. An engineered cysteine in the external mouth of a K channel allows inactivation to be modulated by metal binding. Biophys J. 1994;66:1068–1075. doi: 10.1016/S0006-3495(94)80888-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shakerpotassium channels by peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]