

Abstract
Gating of the cystic fibrosis transmembrane conductance regulator (CFTR) involves a coordinated action of ATP on two nucleotide binding domains (NBD1 and NBD2). Previous studies using nonhydrolyzable ATP analogues and NBD mutant CFTR have suggested that nucleotide hydrolysis at NBD1 is required for opening of the channel, while hydrolysis of nucleotides at NBD2 controls channel closing. We studied ATP-dependent gating of CFTR in excised inside-out patches from stably transfected NIH3T3 cells. Single channel kinetics of CFTR gating at different [ATP] were analyzed. The closed time constant (τc) decreased with increasing [ATP] to a minimum value of ∼0.43 s at [ATP] >1.00 mM. The open time constant (τo) increased with increasing [ATP] with a minimal τo of ∼260 ms. Kinetic analysis of K1250A-CFTR, a mutant that abolishes ATP hydrolysis at NBD2, reveals the presence of two open states. A short open state with a time constant of ∼250 ms is dominant at low ATP concentrations (10 μM) and a much longer open state with a time constant of ∼3 min is present at millimolar ATP. These data suggest that nucleotide binding and hydrolysis at NBD1 is coupled to channel opening and that the channel can close without nucleotide interaction with NBD2. A quantitative cyclic gating scheme with microscopic irreversibility was constructed based on the kinetic parameters derived from single-channel analysis. The estimated values of the kinetic parameters suggest that NBD1 and NBD2 are neither functionally nor biochemically equivalent.
Keywords: ATP-binding cassette transporter, cystic fibrosis, patch-clamp, gating mechanism, single-channel kinetics
introduction
The cystic fibrosis transmembrane conductance regulator (CFTR)1 (Riordan et al., 1989) is a member of the ATP-binding cassette (Riordan et al., 1989) or traffic ATPase (Ames and Lecar, 1992) superfamily. Proteins in this superfamily harvest the energy from the hydrolysis of ATP to transport a variety of substrates across the cell membrane (Kuchler and Thorner, 1992). Like other members in this family, CFTR is predicted to contain two membrane spanning domains, each composed of six putative membrane-spanning segments and two nucleotide binding domains (NBD1 and NBD2). However, unlike other members, CFTR has a regulatory domain containing multiple consensus sites for phosphorylation via PKA; functionally, CFTR itself is a plasma membrane chloride channel (e.g., Anderson et al., 1991a; Bear et al., 1992).
It is thought that regulation of CFTR channel activity is through PKA-dependent phosphorylation of the regulatory (R) domain. Although a prerequisite for activation, PKA phosphorylation of the R domain in itself is not sufficient for opening of the channel. Once the R domain is phosphorylated, nucleotide interaction with the NBDs is coupled to the opening and closing (i.e., gating) of the channel. The NBDs contain a highly conserved region known as the Walker A and Walker B motifs. It is believed that this region forms a close association with the phosphates of bound nucleoside triphosphates (Saraste et al., 1990). From the sequence of the NBDs, it is predicted that they not only bind, but hydrolyze nucleotides. Biochemical experiments using purified CFTR have demonstrated that the protein functions as an ATPase (Bear et al., 1992). Molecular modeling by sequence comparison and functional studies of CFTR have suggested that the function of NBDs (especially NBD2) in CFTR parallels that of a G protein (Hwang et al., 1994; Carson and Welsh, 1995; Manavalan et al., 1995; Senior and Gadsby, 1997). Furthermore, recent studies have shown that the purified NBDs can hydrolyze ATP and GTP (Ko and Pedersen, 1995; Randak et al., 1997). The functional implication of these biochemical results is that CFTR may use the free energy of ATP hydrolysis to drive conformational changes associated with gating transitions.
Previous work on CFTR gating by Hwang et al. (1994) and Gunderson and Kopito (1995) has established that nucleotide binding and hydrolysis at the NBDs is important in channel gating. However, each group proposed a different role for the two NBDs in gating of the channel. Hwang et al. (1994) proposed that hydrolysis at NBD1 is coupled to channel opening, while subsequent binding at NBD2 stabilizes the open conformation; hydrolysis of ATP at NBD2 triggers channel closing. In contrast, Gunderson and Kopito (1995) proposed that nucleotide binding and hydrolysis at NBD1 is only a prerequisite for channel opening; subsequent nucleotide binding at NBD2 is actually responsible for channel opening and hydrolysis at this second ATP binding site is responsible for channel closure. Both models are supported by the observations that the nonhydrolyzable ATP analogues, such as AMP-PNP and ATPγS, fail to open CFTR channels, but dramatically increase the open time in the presence of ATP (Anderson et al., 1991b; Gunderson and Kopito, 1994; Hwang et al., 1994). The fact that CFTR mutants with impaired nucleotide hydrolysis at NBD2 present a prolonged channel open time (Carson et al., 1995; Gunderson and Kopito, 1995; Wilkinson et al., 1996) also supports these models.
We studied gating of CFTR in excised inside-out patches from NIH3T3 cells stably transfected with wild-type (wt) or K1250A-CFTR. In wt-CFTR, both the mean open and closed times depend on [ATP]. This concentration dependence of the mean open time is more prominent in K1250A-CFTR, a mutant CFTR of which the conserved lysine residue in the Walker A motif of NBD2 is converted to alanine. Our kinetic data are consistent with a cyclic gating scheme that violates microscopic reversibility due to an input of energy from ATP hydrolysis.
materials and methods
Cell Culture and Electrophysiology
Both wt (Berger et al., 1991) and K1250A (lysine to alanine mutation) CFTR channels were stably expressed in NIH3T3 cells (NIH3T3-CFTR and NIH3T3-K1250A, respectively). NIH3T3-K1250A cells stably expressing K1250A-CFTR were established using the retroviral vector pLJ (a generous gift from Dr. Mitchell Drumm, Case Western Reserve University, Cleveland, OH). Both cell lines were grown at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM glutamine and 10% calf serum. For patch-clamp recording, cells were passaged and grown on small, sterile glass chips in 35-mm tissue culture dishes. Before recording, glass chips were transferred to a continuously perfused chamber located on the stage of an inverted microscope (Olympus Corp.).
Patch-clamp pipette electrodes were made using a two-stage vertical puller (Narishige). The pipette tips were fire-polished with a homemade microforge to ∼1 μM external diameter, resulting in a pipette resistance of 3–6 MΩ in the bath solution. CFTR channel currents were recorded at room temperature with an EPC-9 patch-clamp amplifier (HEKA Electronik), filtered at 100 Hz with a built-in three-pole Bessel filter (Frequency Devices Inc.), and stored on videotape. Unless otherwise mentioned, data were subsequently played back at a refiltered frequency of 25 Hz with an eight-pole Bessel filter and captured onto a hard disk at a sampling rate of 50 Hz. Pipette potential was held at 50 mV in reference to the bath. Downward deflections represent channel openings.
The pipette solution contained (mM): 140 N-methyl-d-glucamine chloride (NMDG-Cl), 2 MgCl2, 5 CaCl2, and 10 HEPES, pH 7.4 with NMDG. Cells were perfused with (mM): 150 NaCl, 2 MgCl2, 1 EGTA, 5 glucose, and 5 HEPES, pH 7.4 with 1 N NaOH. In excised inside-out patch mode, the superfusion solution contained (mM): 150 NMDG-Cl, 10 EGTA, 10 HEPES, 8 Tris, and 2 MgCl2, pH 7.4 with NMDG. Immediately after excision, patch pipettes were moved to a small homemade chamber where a complete solution change could be accomplished within 3–5 s.
MgATP, salts, and buffers were purchased from Sigma Chemical Co. The catalytic subunit of PKA was purchased from Promega Corp. AMP-PNP was purchased from Calbiochem Corp. All nucleotides were dissolved in the NMDG-Cl solution used for excised studies at a stock concentration of 250 mM. When millimolar ATP was used in the superfusion solution, noticeable acidification occurred, so the pH of superfusion solutions were readjusted to 7.4 with NMDG after addition of ATP.
Kinetic Analysis
Macroscopic kinetic analysis.
The mean current amplitude of the macroscopic CFTR channel current was estimated by averaging the current over a 20–60-s stretch with Igor software (Wavemetrics). Curve fits of the time courses for deactivation by washout of AMP-PNP and ATP from patches containing wt-CFTR or the time courses for deactivation by washout of ATP from patches containing K1250A-CFTR were obtained by using the Igor software.
Data filtering and sampling.
Our goal is to understand gating transitions of CFTR related to ATP hydrolysis, which likely occurs on a time scale in the range of hundreds of milliseconds to seconds (Dousmanis, 1996; Bear et al., 1997). However, short-lived transitions of tens of milliseconds or less are commonly observed in CFTR (Haws et al., 1992; Fischer and Machen, 1994; Winter et al., 1994; Mathews et al., 1998). These “flickers” are more prevalent in cell-attached patches (Haws et al., 1992), are voltage- dependent (Fischer and Machen, 1994), and may be due to blockade of the channel by anions on the cytoplasmic side (Haws et al., 1992; Linsdell and Hanrahan, 1996; Ishihara and Welsh, 1997). To eliminate these flickers from our analysis, we filtered our signals at a relatively low cutoff frequency (−3 dB) of 25 Hz using an eight-pole Bessel filter. In addition, open events are defined as intervals separated by closings of 80 ms or greater. We found that using a cutoff time of <80 ms (e.g., Carson et al., 1995; Li et al., 1996) results in estimates of gating transitions that are too fast to be consistent with ATP hydrolysis-dependent gating (see discussion). Our 25 Hz filter setting results in a 10–90% rise time of 12 ms; therefore, ATP-dependent gating transitions are likely to be unaffected by the filter, whereas flickers with durations of 10 ms or less are greatly attenuated.
Single-channel kinetic analysis.
Single-channel P o was calculated by dividing the summed open time with the total time (∼4 min). For dwell time analysis of ATP-gated CFTR, we used the previously described method (Baukrowitz et al., 1994; Wang et al., 1998). The probability density function (pdf) of the closed time distribution was generated from pooled closed events. This pdf consists of a sum of two exponential components. From the theoretical model (see Fig. 8, Scheme 3) and all the kinetic parameters (see Table I), the areas and time constants of these two components were found by using a routine written by Dr. Gillis using MATLAB software (The MathWorks) following standard “Q-matrix” techniques (Colquhoun and Hawkes, 1995a).
Figure 8.
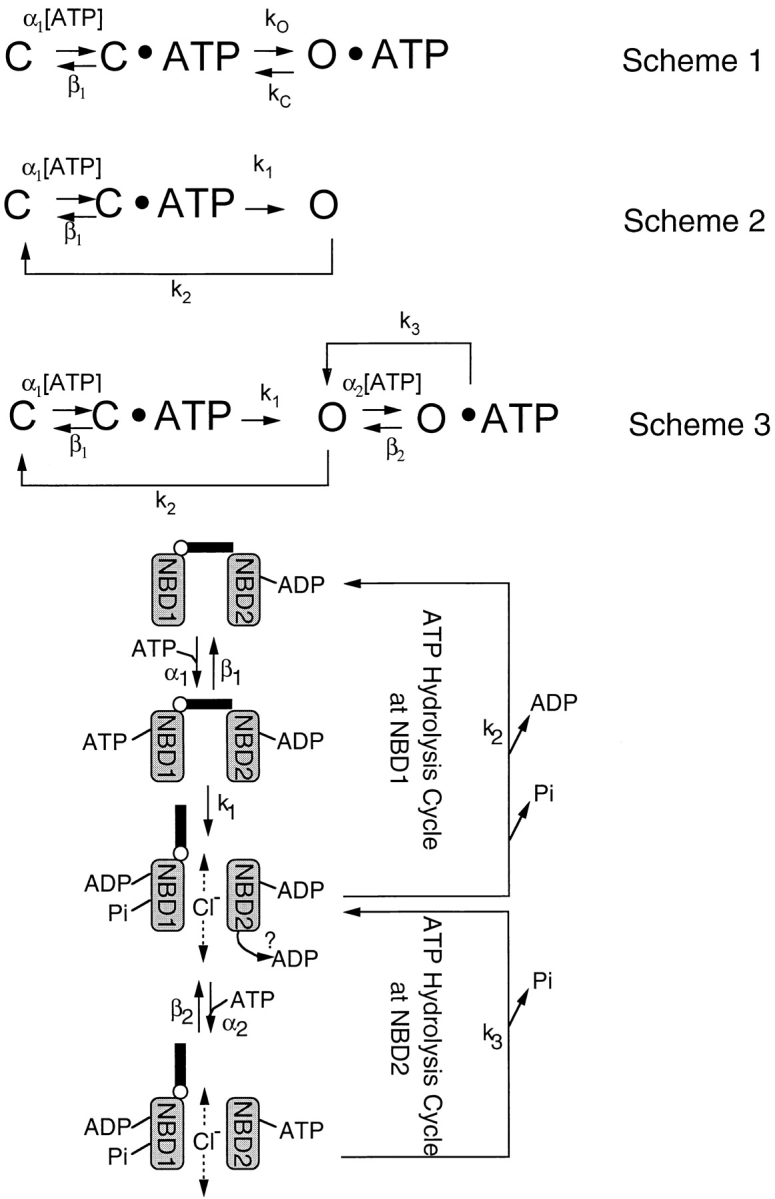
CFTR gating schemes. Scheme 1 is a linear model based on traditional ligand-gated channel gating schemes. Scheme 2 is a cyclic model for CFTR channel gating, but does not take into account the stabilization of the open state via ATP action at NBD2. Scheme 3 is a cyclic model for CFTR gating with two coordinating ATP binding sites. The cartoon depicts a hypothetical graphic presentation of the kinetic Scheme 3 (see text for details).
Table I.
Summary of Estimated Values for the Kinetic Parameters of CFTR Gating
| Parameters | Estimated values | Sources of value | ||
|---|---|---|---|---|
| k 1 | 1.92 s−1 | 1/ τc [ATP→ ∞] from Fig.4 A | ||
| k 2 | 3.85 s−1 | 1/τo [ATP → 0] Fig. 4 B | ||
| α1 | 75600 −1 s−1 * | Fit of Eq. 1 to Fig. 4 A | ||
| β1 | 6.5 s−1 * | Fit of Eq. 1 to Fig. 4 A | ||
| K d1 | 86 μM* | β1/α1 | ||
| β2 | 0.03 s−1 | Decay time constant upon washout of AMP-PNP, Fig. 7 | ||
| α2 | 109 M −1 s−1‡ | P o of wt-CFTR with AMP-PNP/ATP mixtures, Fig. 7 | ||
| K d2 | 275 μM‡ | β2/α2 | ||
| k 3 | 1.78 s−1‡ | Fit of Eq. 2 to data of Fig. 2 C |
Absolute values of α1 and β1 are less certain than the ratio (K d1).
The certainty of these values is low because of the apparently limited impact of NBD2 on P o in our experiments, however, α1 >> α2 and K d2 > K d1.
Curve fitting of the τc vs. ATP and single-channel P o vs. ATP relationships was performed using the Igor software and macros written to quantitatively describe these relationships (see discussion).
results
ATP Dose–Response Relationship of Phosphorylated CFTR
CFTR gating was studied in excised inside-out patches from NIH3T3 cells stably transfected with CFTR cDNA. In agreement with previous reports (Anderson et al., 1991a; Berger et al., 1991), the addition of ATP alone to the excised inside-out patches from NIH3T3-CFTR cells resulted in little channel activity, suggesting negligible basal phosphorylation of CFTR before patch excision. Application of the catalytic subunit of PKA (50 U/ml) with ATP (2.75 mM) activated a macroscopic current of ∼2.2 pA in this patch (Fig. 1 A). Although this macroscopic current is the result of the activation of multiple channels, it is not so large as to be unable to resolve some of the single-channel steps. Since the single-channel amplitude is ∼0.4 pA and the P o under this condition is ∼0.4 (see below), it is estimated that 14 CFTR channels are present in this patch. Upon removal of both PKA and ATP, CFTR channel current rapidly decays in a monotonic manner. Readdition of ATP in the absence of PKA elicited a current magnitude approximately the same as that with ATP plus PKA, suggesting little dephosphorylation over that time span. Although we occasionally observed “rundown” of CFTR channels over long recording, presumably due to dephosphorylation by membrane-associated phosphatases, this is seldom a major problem in our attempt to quantify CFTR gating since patches showing significant rundown are discarded. Note an absence of any channel opening events for ∼15 s when ATP was completely removed. With the presence of 14 channels in the patch, this result suggests a negligible opening rate (<0.004 s−1) in the absence of ATP.
Figure 1.
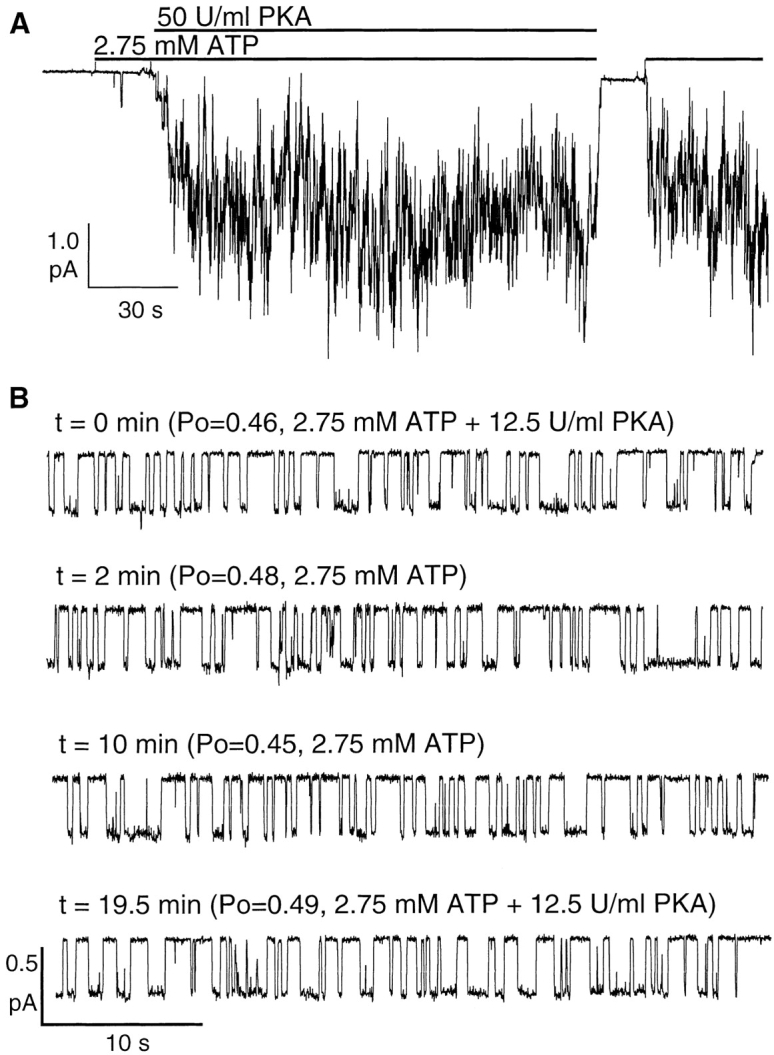
ATP is required to open PKA-phosphorylated CFTR in excised in side-out patches. (A) A continuous current trace showing activation of CFTR channels with PKA and ATP, and lack of channel openings in the absence of ATP. (B) A semicontinuous recording of a single CFTR channel in the presence of PKA plus ATP or ATP alone. Each stretch of single-channel recording is 45 s.
It is known that the degree of CFTR phosphorylation can affect ATP-dependent gating (Chang et al., 1993; Rich et al., 1993; Hwang et al., 1994; Mathews et al., 1998; Wang et al., 1998), and that the presence of membrane-associated protein phosphatases can hamper accurate assessment of CFTR gating (e.g., Carson and Welsh, 1993; Hwang et al., 1994). We are fortunate that significant dephosphorylation of CFTR in the membrane patches does not occur in most of our experiments. Thus, when a steady state CFTR channel activity is reached upon addition of PKA and ATP into excised inside-out patches, CFTR channels should be highly phosphorylated and remain phosphorylated even when PKA is removed from the bath. This provides an opportunity to examine the ATP-dependent gating of strongly phosphorylated CFTR. Fig. 1 B shows a semicontinuous recording of a single CFTR in the presence or absence of PKA. The P o of the channel in the presence of PKA and ATP (2.75 mM) was 0.46. 2 or 10 min after removal of PKA, the P o remained unchanged (0.48 and 0.45, respectively). A second application of PKA at 19.5 min after PKA was removed did not alter the P o significantly. Thus, the CFTR channel behavior we observed likely reflects the gating pattern of strongly phosphorylated CFTR.
To study ATP-dependent gating, we monitored the activity of phosphorylated CFTR channels in the presence of different concentrations of ATP. CFTR chloride channels in excised inside-out patches were first phosphorylated with PKA and ATP. To obtain an accurate dose–response relationship, we bracketed the experiments at tested concentrations of ATP with the responses of 2.75 mM ATP. Any minor rundown (<20%) was compensated for by comparing the channel activity in the presence of the tested concentration to the average activity in the presence of 2.75 mM ATP immediately before and after each tested concentration. The results of an experiment were disregarded when any rundown >20% was detected during a bracketed period. To ensure that we are observing the activity of fully phosphorylated CFTR throughout our experiments, we periodically compared the channel activity in the presence of 2.75 mM ATP to the activity in the presence of 2.75 mM ATP plus PKA (see Fig. 1 B). We assumed little or no dephosphorylation of the protein if no further increase in channel activity was detected upon the readdition of PKA.
Fig. 2 A shows a representative trace with multiple phosphorylated CFTR channels exposed to two different concentrations of ATP. The mean current amplitude in the presence of 0.5 mM ATP is ∼80% that in the presence of 2.75 mM ATP. Fig. 2 B demonstrates the dose response of ATP-dependent gating by normalizing the macroscopic current at different concentrations of ATP to the current level at 2.75 mM. Since no alteration in single-channel amplitude is detected at different ATP concentrations (data not shown), it is concluded that the changes in the macroscopic current amplitude is due to alterations in the P o of individual channels. To demonstrate the effects of the ATP concentration on the single-channel P o, similar experiments were performed in patches containing a single CFTR channel. In these experiments, analysis was performed on recordings of strongly phosphorylated channels at least 4 min in length for each ATP concentration tested. Similar to the macroscopic current amplitude, ATP increased the single-channel P o in a concentration-dependent manner (Fig. 2 C). The plot of single-channel P o vs. ATP concentration is very similar to the macroscopic dose–response relationship (Fig. 2 D), suggesting that gating of CFTR is independent of the number of channels in the patches and that the macroscopic dose response is exclusively due to changes in gating at different ATP concentrations. To understand how changes in the ATP concentration affect CFTR gating, single-channel kinetics were examined.
Figure 2.

The ATP concentration dependence of CFTR channel activity. (A) A continuous current trace showing the effects of different ATP concentrations on CFTR channel activity. (B) The dose–response relationship between [ATP] and macroscopic CFTR channel current. For each patch (containing a different number of CFTR), current was normalized to the average of the current achieved with 2.75 mM ATP immediately before and after the tested concentration. (C) The dose–response relationship between [ATP] and the single-channel P o. The smooth curve represents the fit of the data points to Eq. 2 (see discussion for details). (D) Overlaid plots of B and C. All data points are mean ± SEM of four or more values obtained from different patches at each concentration of ATP.
ATP-dependent Gating of CFTR
Dwell time analysis of CFTR channel activity in patches containing a single CFTR channel was performed. Fig. 3 A shows representative single-channel traces at four different [ATP]. It should be noted that openings that last for hundreds of milliseconds are interrupted by closings that vary with [ATP]. Even at 10 μM ATP, when there are only 10 openings in the 64-s trace shown, the openings are well dispersed and do not appear to occur in clusters. In addition, as the concentration of ATP is increased, some longer openings, although rare, can be seen.
Figure 3.

Effects of [ATP] on CFTR gating. (A) Single-channel traces of PKA-phosphorylated CFTR at different concentrations of ATP as marked. (B) Cumulative dwell time analysis of the traces shown in A.
Fig. 3 B shows cumulative dwell time histograms from the data shown in Fig. 3 A. The closed time constant (τc) and open time constant (τo) were estimated from single exponential fit of the cumulative closed and open time histograms at each [ATP]. The relationship between τc and [ATP] follows a saturating function with no significant change in τc at ATP concentrations higher than 1.0 mM (Fig. 4 A). This result suggests that at low micromolar concentrations of ATP, channels are not ATP-bound all the time; therefore, binding of ATP imposes a rate-limiting step in channel opening. However, once the binding of ATP to CFTR is saturated at ∼1 mM, the rate-limiting step in the opening of CFTR channels is some concentration-independent, post-binding event(s). To examine the closed time distribution in detail, we pooled all closed events in four patches in the presence of 100 or 500 μM ATP and generated the pdf of the closed time distribution (Fig. 5, A and B). The closed time pdf resembles a single exponential distribution with the important exception that the maximal number of events is not at the minimal binned interval. The paucity of short-lived closings is particularly apparent at 100 μM ATP and is indicative of the presence of irreversible steps in ATP-dependent gating of CFTR (Colquhoun and Hawkes, 1995b; see discussion).
Figure 4.

ATP concentration dependence of the mean open time and the mean closed time. Both open (τo) and closed (τc) time constants were obtained from at least five patches in the presence of different concentrations of ATP. (A) ATP concentration dependent of the closed time constant. The smooth curve represents fit of the data points to the approximate relationship from Eq. 1 (see text for details). The squares represent exact solutions using Q matrix techniques to Fig. 8, Scheme 3, with the parameters listed in Table I. (B) ATP concentration dependence of the open time constant.
Figure 5.

Probability density function of the closed time distributions at 0.1 (A) or 0.5 (B) mM ATP. The smooth curves represent the predicted pdf using kinetic parameters derived for Fig. 8, Scheme 3 (see text for details). Insets show the expanded graphs that contain the first 20 bins.
Fig. 4 B demonstrates that τo increases with increasing [ATP] (Fig. 4 B). This ATP-dependent change in τo is inconsistent with the model proposed by Gunderson and Kopito (1995). In the model of Gunderson and Kopito (1995), the lifetime of the open state is determined by the hydrolysis rate of bound ATP at NBD2; therefore, τo is not predicted to change with [ATP]. In contrast, a prolongation of τo with increasing [ATP] is consistent with the model of Hwang et al. (1994) because it includes two open states with ATP binding at NBD2 leading to occupancy of the long-lived open state (see discussion). Since this latter model predicts a prolongation of open times with increasing [ATP] due to ATP binding to NBD2, we next examined [ATP]-dependent gating of K1250A-CFTR, a mutant in which the conserved lysine in the Walker A motif of NBD2 is converted to alanine. This mutant CFTR has been shown to assume prolonged openings at 1 mM ATP presumably due to diminished hydrolysis of ATP at NBD2 (Carson et al., 1995; Gunderson and Kopito, 1995).
Gating of K1250A-CFTR channels was examined in the presence of either 10 μM or 2.75 mM ATP. Fig. 6 A shows that a K1250A-CFTR channel, preactivated with PKA and ATP (not shown), was “locked” in an open state with 2.75 mM ATP and the channel closed ∼2 min after ATP washout. In the same patch, very brief openings were seen when 10 μM ATP was applied. These short openings are the first evidence that the channel can close in the absence of ATP hydrolysis by NBD2 (see below). The single channel amplitude, obtained from the all-point histograms (not shown), of K1250A-CFTR channels opened with millimolar ATP is about the same as that for brief openings in the presence of 10 μM ATP (Fig. 6 B). Note the presence of short flickering closures even in the absence of ATP (Fig. 6 A), indicating that these closings are not coupled to an ATP hydrolysis cycle. Therefore, to quantify ATP-dependent gating events, these flickering closings should be excluded (see materials and methods).
Figure 6.

ATP concentration dependence of the channel open time for K1250A-CFTR. (A) A continuous current trace of K1250A-CFTR in the presence or absence of ATP. The channel had been activated with PKA and ATP before the start of the trace. This mutant channel remains open for 2 min after removal of ATP. However, the same channel shows brief openings at 10 μM ATP. (B) Single channel amplitudes of K1250A-CFTR (obtained from all point histograms of 30 s recordings) at 10 μM or 2.75 mM ATP. (C) The open time histogram of K1250A-CFTR at 10 μM ATP. Pooled open times from four patches were analyzed. This distribution was fitted with a single exponential function. (D) Slow closing of PKA-phosphorylated K1250A-CFTR. The expanded trace shows stepwise closing of individual channels after ATP is removed. (E) Relaxation of the ensemble current. Macroscopic relaxation of the K1250A-CFTR channel current was constructed from multiple washouts of ATP from the same patch. A single exponential fit of the current relaxation yields a time constant of 181.28 ± 0.25 s for deactivation in the experiment shown.
To quantify the brief openings of K1250A-CFTR in the presence of 10 μM ATP, dwell time analysis of the cumulative open time from three different patches was performed. The cumulative histogram of pooled open times in the presence of 10 μM ATP could be fitted with a single-exponential function yielding a τo of 0.25 ± 0.01 s (Fig. 6 C), which is close to the mean open time of wt-CFTR in the presence of 10 μM ATP. Since the “locked open” time of K1250A-CFTR is apparently very long (Fig. 6 A), it will be very difficult to collect enough events for dwell time analysis. Even if we obtain patches containing a single K1250A-CFTR channel, the flickering closures in locked open state may interfere with the analysis. Although excluding closing intervals <80 ms is appropriate for eliminating flickers in analysis of wt-CFTR, this exclusion is not sufficient to eliminate flickers from analysis of K1250A-CFTR because the ratio of flickers to “true” closings (gating) of CFTR is much higher. We therefore estimate the time constant for the locked open state from the macroscopic current decay upon washout of 2.75 mM ATP. At this concentration, most of K1250A-CFTR channels are locked open, as can be judged from the small magnitude of macroscopic current fluctuations (Fig. 6 D). The mean open lifetime of ATP-locked channels should be the same as the time constant for the current decay upon washout of ATP since the opening rate in the absence of ATP is negligible. To obtain a better estimation of the decay time constant, we repeated the ATP washout several times in the same patch and fitted the decay phase of ensemble macroscopic current with a single exponential function (Fig. 6 E). From three different patches, the exponential decay of the macroscopic current upon washout of ATP yields a time constant of 162.6 ± 31.8 s (our solution change has a dead time of ∼3 s; see materials and methods). These results suggest that, at low micromolar [ATP], K1250A-CFTR can assume brief openings with a time constant close to that of wt-CFTR at equivalent [ATP], but this mutant CFTR can be locked open for minutes at millimolar ATP. Assuming ATP is not hydrolyzed by the NBD2 of K1250A-CFTR, the slow closing rate reflects a slow dissociation of ATP from CFTR (presumably from NBD2). If the brief openings of K1250A-CFTR at 10 μM ATP represent open channel conformations without NBD2 being occupied, this observation suggests that CFTR can close even when ATP acts exclusively on NBD1 (see Carson et al., 1995; also see discussion).
To examine the rate of nucleotide unbinding from NBD2 of wt-CFTR, we used the nonhydrolyzable ATP analogue, AMP-PNP, which “locks open” CFTR presumably because it cannot be hydrolyzed by NBD2 to allow channel closure. To estimate the off rate of AMP-PNP from NBD2, we examined the time course of channel closure after washing out ATP/AMP-PNP mixtures that produce the locked open state. In Fig. 7, 0.5 mM ATP was applied to elicit macroscopic PKA-phosphorylated CFTR channel current with a mean current amplitude of 1.16 pA in this patch. Addition of 0.5 mM AMP-PNP in the continued presence of ATP caused a 25% increase of steady state current. Once a new level of activity was obtained, both ATP and AMP-PNP were washed out, resulting in a biphasic decay of channel activity (compare Figs. 1 A and 2 A). At this one-to-one ratio of ATP and AMP-PNP, most of the channels are not locked open, as evidenced by the magnitude of current fluctuations at the steady state. In fact, from the slow closing steps after washout of nucleotides, it is estimated that only 1 or 2 channels had been locked open by AMP-PNP. In the same patch, however, 2.75 mM AMP-PNP caused a threefold increase in the macroscopic current elicited with 0.5 mM ATP. In spite of an approximately sixfold higher concentration of AMP-PNP, a significant number of channels are not locked open because large current fluctuations can be seen. Compared with the current decay phase upon removal of 0.5 mM AMP-PNP, however, more slow channel closing steps (representing closings from the locked open state) were observed. Although our solution change does not allow us to assess the fast phase of current decay, the slow phase of the current decay, which lasts for tens of seconds, can be quantified. To generate a macroscopic current decay, we again used ensemble CFTR current from the same patch. The slow phase of the current decay upon removal of 0.5 or 2.75 mM AMP-PNP was fitted with a single exponential function yielding a similar time constant of ∼30 s. This concentration-independent decay rate is consistent with the idea that the rate-limiting step for the current decay is the dissociation of AMP-PNP from NBD2, a concentration-independent process. These results also support the hypothesis that ATP binding to NBD2 stabilizes the open channel conformation. This stabilizing effect is greatly magnified when hydrolysis at NBD2 is eliminated, either through chemical modification of the binding molecule (AMP-PNP) or molecular alteration of the CFTR protein itself (i.e., K1250A mutation).
Figure 7.

Slow closing of AMP-PNP locked open wt-CFTR. A continuous current trace showing the effects of AMP-PNP on ATP-opened wt-CFTR. Ensemble currents were constructed from the slow component of the decay from multiple washouts in the same patch. The ensemble current decay could be fitted with a single exponential function yielding a time constant for deactivation of 32.33 ± 0.43 and 29.36 ± 0.07 s for the washout of 0.5 mM and 2.75 mM AMP-PNP, respectively. Multiple ensemble deactivations from different patches yielded an average time constant for the slow deactivation phase of 34.9 ± 13.7 s (n = 6) for 2.75 mM AMP-PNP and 33.2 ± 14.4 s (n = 6) for 0.5 mM AMP-PNP.
discussion
Kinetically Distinct States Differentiate Gating Model
Several linear equilibrium models have been proposed to explain ATP-dependent gating of CFTR (Gunderson and Kopito, 1994; Venglarik et al., 1994; Winter et al., 1994). These models, all similar to models used to explain the kinetics of ligand-gated channels, do not consider the fact that CFTR is an ATPase and the likelihood that ATP hydrolysis provides the free energy that drives gating transitions. Since the free energy generated from hydrolysis of a single ATP molecule is >10 kT, it seems reasonable to model some of the ATP hydrolysis-driven conformational changes as essentially irreversible. Before we focus our discussion on asymmetrical, cyclic schemes that have been proposed to describe CFTR gating, we will first examine the feasibility of a simple equilibrium kinetic scheme by using our single-channel data.
Assuming that ATP binding, but not hydrolysis, at NBD1 opens the channel, a simple scheme for a typical ligand-gated channel can be derived (Fig. 8, Scheme 1; e.g., Gunderson and Kopito, 1994; Venglarik et al., 1994; Winter et al., 1994). According to this scheme, the transitions between C·ATP and O·ATP reflect the short flickers that we exclude from our analysis by omitting closings <80 ms. Thus, the only transitions we measure are between states C and C·ATP, and Fig. 8, Scheme 1, predicts that our measured mean closed time is inversely proportional to [ATP] and therefore should approach 0 for high [ATP]. This is inconsistent with our observation of a minimum closed time of ∼400 ms at high [ATP] (Fig. 4 A). In addition, Scheme 1 obeys microscopic reversibility and thus predicts that the pdf of the closed time distribution has two decay time constants. Our results with a rising phase in the pdf of the closed time distribution (i.e., a short time constant with a negative amplitude, Fig. 5, A and B) is inconsistent with Scheme 1 being an adequate model to explain CFTR channel gating.
Three cyclic gating models for CFTR gating have been proposed whereby ATP hydrolysis at NBD1 is a prerequisite for channel opening (Hwang et al. 1994; Carson et al., 1995; Gunderson and Kopito 1995). These models differ in their proposed roles for the two NBDs' participation in gating of the channel. Hwang et al. (1994) and Baukrowitz et al. (1994) proposed that for every ATP molecule hydrolyzed at NBD1, one opening event occurs (a strict coupling); subsequent binding at NBD2 stabilizes the open conformation; ATP hydrolysis at NBD2 leads to channel closure. Their model also suggests that channels can close even without a functional interaction between ATP and NBD2, although data supporting this notion are lacking. In contrast, Gunderson and Kopito (1995) propose that nucleotide binding and hydrolysis at NBD1 is only a prerequisite for channel opening; subsequent nucleotide binding at NBD2 is actually responsible for channel opening, and hydrolysis at NBD2 causes channel closure. The model proposed by Carson et al. (1995) is very similar to that by Hwang et al. (1994) except that hydrolysis at NBD2 is obligatory for channel closing, a feature shared by the model proposed by Gunderson and Kopito (1995). All three models are supported by the observations that the nonhydrolyzable ATP analogues, such as AMP-PNP and ATPγS, fail to open CFTR channels, but dramatically increase the open time in the presence of ATP (Hwang et al., 1994; Carson et al., 1995; Gunderson and Kopito, 1995). The fact that mutations of the CFTR protein that impair nucleotide hydrolysis at NBD1 or NBD2 result in prolongation of channel closed or open times (Fig. 6; Gunderson and Kopito, 1995; Carson et al., 1995; compare Wilkinson et al., 1996) also supports these models.
A critical observation in our current work for differentiating these models is the ATP concentration dependence of the mean open time (Fig. 4 B). The model proposed by Gunderson and Kopito (1995) predicts that the mean open time is independent of [ATP] because the termination of an opening event is controlled by hydrolysis of the bound ATP at NBD2, which is an [ATP]-independent parameter. However, the model proposed by Hwang et al. (1994) predicts a second prolonged open state when ATP binds to NBD2. Thus, two open time constants reflecting the life times of these two open states are expected. We are unable to clearly separate two exponential components in the open dwell time histograms for wt-CFTR (e.g., Fig. 3 B). Nevertheless, we observe a shift of the mean open time as [ATP] is increased. Our failure to detect a double exponential distribution of open times does not necessarily eliminate the possibility of two open states. If the difference in the mean lifetime of two open states is small and/or the transition between two open states is slow, then technically it will be difficult to collect sufficient events to resolve these two time constants. Furthermore, our exclusion of “flickers” with the 80 ms minimal duration criterion may also distort the resolution of the two components. We clearly resolve two open times with K1250A-CFTR and demonstrate a dramatic [ATP] dependence of the channel open time for this mutant CFTR. One interesting observation is that at 10 μM ATP, the mean open time for K1250A-CFTR is ∼250 ms, a value very close to that for wt-CFTR at the equivalent [ATP]. Thus, although this NBD2 mutant CFTR is able to open for minutes at high concentrations of ATP, it can assume very brief opening (∼250 ms) at low micromolar [ATP]. This observation is expected if the on rate of ATP binding to NBD1 is much larger than that for NBD2, and therefore at a low concentration of ATP, ATP interacts with NBD1 exclusively. Based on this interpretation, the brief opening with K1250A-CFTR is coupled to hydrolysis of one ATP molecule at NBD1 and subsequently the channel can close without ATP hydrolysis at NBD2. This conclusion is in conflict with the model for the obligatory role of ATP hydrolysis at NBD2 in channel closings (Gunderson and Kopito, 1995; Carson et al., 1995). This same observation that the open time of K1250A-CFTR depends on [ATP] is also inconsistent with the proposal that ATP binding at NBD2 opens the channel (Gunderson and Kopito, 1995). According to this latter model, every opening of K1250A-CFTR should last for minutes.
Construction of a Cyclic Gating Scheme
From the above discussion, we conclude that the model proposed by Hwang et al. (1994) provides a reasonable background to construct a more quantitative scheme with the kinetic parameters obtained from the present work. First, the spontaneous opening of phosphorylated CFTR (C in Fig. 8, Scheme 2) in the absence of ATP is negligible (e.g., Fig. 1 A), suggesting that only ATP-bound channels (C·ATP in Scheme 2) can advance to the open state. Since the input of energy is involved in opening the channel, we propose that the transition from the ATP-bound closed state to the open state (O in Fig. 8, Scheme 2) is a relatively irreversible process. Since NBD1 alone allows channel opening as well as closing (see above), we propose that the state O sojourns in C directly. This will be another irreversible step since the C to O transition (i.e., opening CFTR in the absence of ATP) is negligible. Fig. 8, Scheme 2, summarizes the coupling of ATP hydrolysis at NBD1 to channel gating. At [ATP] ≥ 1 mM, the ATP binding step is saturated. A maximal opening rate of 1.92 s−1, the reciprocal of the averaged closed time at millimolar [ATP] (Fig. 4 A), should be equal to k 1, the intrinsic opening rate for an ATP-bound channel. Although the open time constants at different [ATP] are the weighed average of the lifetimes for the two open states, O and O·ATP (see below), the mean open time at a very low [ATP] should represent a closing of the channel through ATP action at NBD1 only. We estimate this short open time constant by extrapolating the first five data points in Fig. 4 B to 0 mM ATP. The reciprocal of this short open time constant (0.26 s) should be equal to k 2 (3.85 s−1), the intrinsic closing rate of the open channel by NBD1. The fact that this short open time constant is very close to the mean open time of K1250A-CFTR at 10 μM ATP further supports this assignment.
Assuming a steady state condition for Fig. 8, Scheme 2, we obtained Eq. 1 (Zeltwanger, 1998) to describe the relationship between [ATP] and τc (Fig. 4 A). Fitting Eq. 1 to the data of Fig. 4 A yields values of 75,600 s−1 M−1 and 6.5 s−1 for α1 and β1, respectively. It should be noted that, although changing α1 or β1 individually alters the best fit drastically, when the K d1 (β1/α1) is kept constant (86 μM), the best fit does not change significantly with parallel changes in α1 and β1 over a 20-fold range (data not shown). Thus, the absolute values for α1 and β1 are less certain than the value of 86 μM for K d1. However, α1 is not likely smaller than 1,000 M−1 s−1 since the fitted curve starts to deviate from the actual data points at low [ATP] (not shown). Even though Eq. 1 is based on an assumption of steady state (which, strictly speaking, cannot be true for a single molecule), the values for α1 and β1 are likely appropriate for two reasons. First, the mean closed times at different [ATP] found from the exact solution of the Q matrix (Colquhoun and Hawkes, 1995a; Fig. 4 A, □), were very close to the closed times calculated from Eq. 1 (Fig. 4 A, solid line). Second, these values for k 1, k 2, α1, and β1 also reasonably fit the pdf of the closed time distributions (including the rising phase and the position of the peak) at 0.1 and 0.5 mM ATP (Fig. 5, smooth curves):
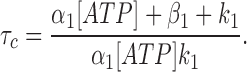 |
1 |
To expand Fig. 8, Scheme 2, to incorporate the function of NBD2 (Fig. 8, Scheme 3), we assume that ATP can only bind to the open conformation of CFTR (Hwang et al., 1994). We also assume that ATP hydrolysis at NBD2, instead of closing the channel directly, enables the channel to close through the state O (Fig. 8, Scheme 3, and Hwang et al., 1994). This assumption, although somewhat arbitrary, simplifies the biochemical interpretation of the gating transitions (see below). As discussed above, the rate-limiting step for channel closing from the AMP-PNP locked open state is likely the dissociation rate of AMP-PNP. Since the structure of AMP-PNP is very similar to that of ATP, we assume that the off rate for ATP (β2 in Fig. 8, Scheme 3) at NBD2 for wt-CFTR is the same as that for AMP-PNP. Our estimation of the off rate for AMP-PNP from the macroscopic current decay upon removal of nucleotides is 0.03 s−1. A small β2 value suggests that channel closings from O·ATP almost always sojourn in the state O via hydrolysis and dissociation of the hydrolytic products from NBD2.
Since the off rate of AMP-PNP at NBD2 is very slow, AMP-PNP locked open state can be considered an absorbing state. A reasonably fast on rate of AMP-PNP should lead to most channels locked in that state. However, even at 2.75 mM AMP-PNP, not all channels were locked open (Fig. 7), suggesting a very slow on rate of AMP-PNP (and presumably also ATP) to NBD2. The steady state P o for CFTR in the presence of 0.5 mM ATP is ∼0.28. A 3.18-fold increase of the macroscopic current by addition of 2.75 mM AMP-PNP indicates a P o of 0.89 in the presence of 0.5 mM ATP and 2.75 mM AMP-PNP (Fig. 7). This P o is determined by the fraction of channels in the open state and the fraction of channels in the locked open state. Since the opening rate and closing rate of CFTR at 0.5 mM ATP, and the closing rate of AMP-PNP locked open channels are known (Figs. 4 and 7), the association rate constant for AMP-PNP is estimated to be 109 s−1 M−1. Assuming ATP has similar on and off rates as AMP-PNP to NBD2, an ATP dissociation constant, K d2 (β2/α2), of 275 μM is obtained for NBD2. Finally, we used the equation (Eq. 2) that describes the relationship between the steady state P o and [ATP] to obtain the k 3 value (see Zeltwanger, 1998 for detailed derivation). Fitting Eq. 2 with the data using all the kinetic parameters derived above yields a k 3 value of 1.78 s−1 (Fig. 2 C, smooth line). This value has some degree of uncertainty because P o is not very sensitive to NBD2 function in our excised patch experiments. Table I summarizes all the estimated values for the kinetic parameters and their experimental origins. It seems surprising that even at 10 mM ATP, the on rate of ATP to NBD2 is ∼1 s−1, suggesting a minimal contribution of NBD2 to P o of CFTR in our experiments using excised inside-out patches. This puzzling lack of role of NBD2 may be related to some of the discrepancies among different reports discussed below.
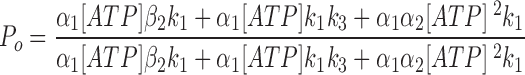 |
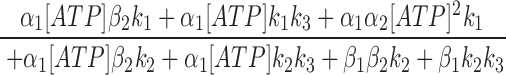 |
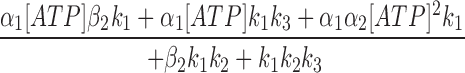 |
2 |
Comparison of Kinetic Values with Previous Studies
A discrepancy exists between our maximal P o measured in excised inside-out patches (∼0.5, Fig. 1 B) and previous reports with higher P o values. When CFTR gating was examined in excised giant patches from cardiac myocytes, a P o ≥ 0.8 can be achieved in the presence of PKA and 0.5 mM ATP (e.g., Fig. 3 B in Hwang et al., 1994). This is not likely due to species or tissue differences because a similar high P o was observed with human wt-CFTR incorporated into lipid bilayers (Gunderson and Kopito, 1994). In fact, the P o of wt-CFTR in cell- attached patches from Calu-3 cells, a human airway epithelial cell line, can be >0.9 in the presence of cAMP stimulation (Haws et al., 1994). This latter result is especially enigmatic. In cell-attached patches, in addition to the ATP-dependent gating events, phosphorylation/dephosphorylation events need to be considered in determining P o (Hwang et al., 1997). Since only phosphorylated CFTR channels can be opened by ATP, a P o of ∼0.5 in inside-out patches (Fig. 1 B) for the strongly phosphorylated CFTR should impose a maximal P o of ∼0.5 for CFTR activity measured in cell-attached patches where at least one more closed state (dephosphorylated closed state) should be considered in calculating P o. Furthermore, the channel open times in cell-attached patches can be easily resolved into two components (Hwang et al., 1997), and the longer time constant is greater than the mean open time at 10 mM ATP in excised patches from the current study. One interesting possibility is that a cytosolic factor, which affects CFTR gating, is missing in our excised patches but remains in other systems (Hwang et al., 1994; Gunderson and Kopito, 1994).
Numerous reports (Li et al., 1993; Gunderson and Kopito, 1994; Carson et al., 1994, 1995; Winter et al., 1994; Li et al., 1996; Dousmanis, 1996; Sheppard et al., 1997; Mathews et al., 1998) on CFTR gating have generated many different numerical values for the kinetic parameters. This difference likely reflects the difference in methods used for kinetic analysis. Importantly, different cutoffs are used to define CFTR gating events, resulting in a wide range of the time constants (2 ms–3 s). Interestingly, the steady state P o values reported for wt-CFTR in those reports were not very different (0.28– 0.65). Very few studies have assigned those time constants to specific kinetic events, perhaps because there often is an inconsistency between kinetic parameters, determined by dwell time analysis, and the independently measured steady state P o. For example, Li et al. (1996), using 60 ms as a cutoff, reported a P o of 0.48 at 1 mM ATP for purified wt-CFTR incorporated into lipid bilayers, whereas the kinetic analysis shows a mean burst duration (τb) and a mean interburst duration (τib) of 600 and 125 ms, respectively, which would predict a P o of 600/(600 + 125), or 0.83. An earlier report from the same group showed a P o of 0.28 with a τb of 0.36 s and τib of 3.35 s (Li et al., 1993). Clearly, these values of the kinetic parameters cannot easily be reconciled with the P o value. Carson et al. (1995), using 20 ms as a cutoff, reported a mean burst time of ∼1 s for K1250A-CFTR. This number is evidently an underestimation of the true ATP-coupled open time for K1250A-CFTR as a continuous burst of opening that lasts for minutes is observed even when ATP is removed (Fig. 6 A). On the other hand, we believe that the method we used for macroscopic as well as microscopic kinetic analysis provides a reasonable approximation of the [ATP]-dependent changes in P o (Fig. 2).
Biochemical Implications
Other than the amino acid sequences in the conserved Walker A and B motifs, NBD1 and NBD2 share only ∼30% sequence homology (Riordan et al., 1989). Our results, as well as others, suggest that, functionally, they play very different roles in CFTR gating. These distinct functional roles for CFTRs NBD1 and NBD2 also distinguish CFTR from p-glycoprotein, another member of the ATP-binding cassette (ABC) transporter family. The two NBDs in p-glycoprotein share 66% sequence homology and there is no evidence so far to support that NBDs in p-glycoprotein are functionally distinct. Biochemical studies suggest that two NBDs of p-glycoprotein may alternate in catalyzing ATP hydrolysis reactions (reviewed by Senior and Gadsby, 1997). This is not very surprising for members of the ABC family other than CFTR since structural (perhaps functional) symmetry is recently proposed from the x-ray crystallographic structure of His permease (Hung et al., 1998). Thus, although CFTR shares topological similarities with other proteins in this family, numerous differences may have emerged during evolution.
Our data also suggest differences in ATP binding affinity between NBD1 and NBD2. For NBD1, ATP has a much faster on as well as off rate. However, the association rate constant for NBD2 is slower than diffusion by several orders of magnitude, suggesting the rate constant, α2, obtained from our analysis does not truly represent the association rate constant for a simple bimolecular reaction. One possible explanation for this apparent slow on rate of ATP to NBD2 can be inferred from the analogy with G proteins. If the function of NBD2 indeed parallels that of a G protein, ADP, the hydrolytic product of ATP hydrolysis, may be entrapped after hydrolysis. In analogy to the G protein function, occupancy of this ADP molecule in NBD2 may prevent ATP binding. If this analogy is applied to our model (Fig. 8, Scheme 3), then the state O contains an ADP molecule in NBD2. A similar proposal was recently postulated by Senior and Gadsby (1997). Thus, the apparent slow on rate for ATP binding to NBD2 is caused by the slow dissociation of ADP, and this slow off rate sets an upper limit for ATP association with NBD2.
As discussed above, our kinetic data fail to explain some of the previous observations that the P o of CFTR can reach 0.8–0.9 (e.g., Hwang et al., 1994; Gunderson and Kopito, 1994; Haws et al., 1994). We do not know the mechanism that can account for these discrepancies. Perhaps the hypothetical factor that we lose upon patch excision accelerates the ATP/ADP exchange rate. Thus, much of the role of NBD2 in determining P o is lost in excised inside-out membrane patches. Again, in analogy to G proteins, for example, GDP binds very tightly to the G protein Ras, but the GTP/ GDP exchange rate is greatly increased by the presence of the Sos protein (Chardin et al., 1993).
Two irreversible steps in gating transitions are proposed to be coupled to ATP hydrolysis at NBD1 (Fig. 8, Scheme 2). A paucity of short closings in the pdf of the closed time (Fig. 5, A and B) is predicted with a scheme such as Scheme 2 that violates microscopic reversibility. Baukrowitz et al. (1994) proposed that CFTR channels open after ATP is hydrolyzed. According to this proposition, we speculate that the splitting of the γ-phosphate from ATP yields one irreversible step with the rate constant, k 1, and the release of the hydrolytic products, phosphate or ADP or both, can generate a second irreversible step with the rate constant, k 2 (Fig. 8). Then the intrinsic hydrolysis rate for NBD1 is k 1 k 2/(k 1 + k 2) = 1.28 s−1. This value is comparable to the reported hydrolysis rate of ∼1 s−1 per CFTR molecule (Li et al., 1996). Our model also suggests that the ATP hydrolysis reactions in NBD1 and NBD2 are coupled. Hydrolysis of ATP at NBD2 requires occurrence of an earlier ATP hydrolysis reaction at NBD1; while part of the ATP hydrolysis cycle at NBD1 (the O → C transition) can be disrupted by ATP occupancy at NBD2 (Fig. 8). One prediction from this model is that abolition of the ATP hydrolysis rate at NBD2 will affect the overall ATP hydrolysis by CFTR. Assuming the opening rate of K1250A-CFTR is the same as that of wt-CFTR, our kinetic data suggest a maximal ATP hydrolysis rate of ∼0.005 s−1, which is 1/200 of that for wt-CFTR. Recent preliminary data that K1250A-CFTR has a drastically reduced rate of ATP hydrolysis support this coupled ATP turnover hypothesis (Ramjeesingh et al., 1998). One should note, however, that our model, although sufficient to explain numerous biophysical and biochemical results, simply provides a testable framework (Fig. 8) for future exploration to advance our understanding of the biochemical basis of CFTR gating via ATP hydrolysis.
Acknowledgments
This work was supported by the National Institutes of Health (HL-53445) and the Cystic Fibrosis Foundation. F. Wang is a recipient of the Postdoctoral Fellowship from the American Heart Association, Missouri Affiliate.
Abbreviations used in this paper
- CFTR
cystic fibrosis transmembrane conductance regulator
- NMDG
N-methyl-d-glucamine
probability density function
- wt
wild type
references
- Ames GF, Lecar F. ATP-dependent bacterial transporters and cystic fibrosis: analogy between channels and transporters. FASEB J. 1992;6:2660–2666. doi: 10.1096/fasebj.6.9.1377140. [DOI] [PubMed] [Google Scholar]
- Anderson MP, Berger HA, Rich DP, Gregory RJ, Smith AE, Welsh MJ. Nucleoside triphosphates are required to open the CFTR channel. Cell. 1991a;67:775–784. doi: 10.1016/0092-8674(91)90072-7. [DOI] [PubMed] [Google Scholar]
- Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mulligan RC, Smith AE, Welsh MJ. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991b;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Hwang T-C, Nairn AC, Gadsby DC. Coupling of CFTR Cl−channel gating to an ATP hydrolysis cycle. Neuron. 1994;12:473–482. doi: 10.1016/0896-6273(94)90206-2. [DOI] [PubMed] [Google Scholar]
- Bear CE, Li C, Kartner N, Bridges RJ, Jensen TJ. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR) Cell. 1992;68:809–818. doi: 10.1016/0092-8674(92)90155-6. [DOI] [PubMed] [Google Scholar]
- Bear CE, Li C, Galley K, Wang Y, Garami E, Ramjeesingh M. Coupling of ATP hydrolysis with channel gating by purified, reconstituted CFTR. J Bioenerg Biomembrs. 1997;29:465–473. doi: 10.1023/a:1022435007193. [DOI] [PubMed] [Google Scholar]
- Berger HA, Anderson MP, Gregory RJ, Thompson S, Howard PW, Maurer RA, Mulligan R, Smith AE, Welsh MJ. Identification and regulation of the cystic fibrosis transmembrane conductance regulator-generated chloride channel. J Clin Invest. 1991;88:1422–1431. doi: 10.1172/JCI115450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MR, Welsh MJ. 5′-Adenylylimidodiphosphate does not activate CFTR chloride channels in cell-free patches of membrane. Am J Physiol. 1993;265:L27–L32. doi: 10.1152/ajplung.1993.265.1.L27. [DOI] [PubMed] [Google Scholar]
- Carson MR, Travis SM, Winter MC, Sheppard DN, Welsh MJ. Phosphate stimulates CFTR Cl−channels. Biophys J. 1994;67:1867–1875. doi: 10.1016/S0006-3495(94)80668-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MR, Travis SM, Welsh MJ. The two nucleotide-binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. J Biol Chem. 1995;270:1711–1717. doi: 10.1074/jbc.270.4.1711. [DOI] [PubMed] [Google Scholar]
- Carson MR, Welsh MJ. Similarity between CFTR and GTP-binding proteins. Biophys J. 1995;69:2443–2448. doi: 10.1016/S0006-3495(95)80113-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang XB, Tabcharani JA, Hou YX, Jensen TJ, Kartner N, Alon N, Hanrahan JW, Riordan JR. Protein kinase A (PKA) still activates CFTR chloride channel after mutagenesis of all 10 PKA consensus phosphorylation sites. J Biol Chem. 1993;25:11304–11311. [PubMed] [Google Scholar]
- Chardin P, Camonis JH, Gale NW, Van Aelst L, Schlessinger J, Wigler MH, Bar-Sagi D. Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science. 1993;260:1338–1343. doi: 10.1126/science.8493579. [DOI] [PubMed] [Google Scholar]
- Colquhoun, D., and A.G. Hawkes. 1995a. A Q-matrix cookbook. In Single-Channel Recording. 2nd ed. B. Sakmann and E. Neher, editors. Plenum Publishing Corp., New York. 589–633.
- Colquhoun, D., and A.G. Hawkes. 1995b. The principles of the stochastic interpretation of ion-channel mechanisms. In Single-Channel Recording. 2nd ed. B. Sakmann and E. Neher, editors. Plenum Publishing Corp., New York. 397–482.
- Dousmanis, A.G. 1996. The CFTR (cystic fibrosis transmembrane conductance regulator) channel: anion permeation and regulation by adenylyl cyclase and ATP hydrolysis. Ph.D. Dissertation, Rockefeller University, New York.
- Fischer H, Machen TE. CFTR displays voltage dependence and two gating modes during stimulation. J Gen Physiol. 1994;104:541–566. doi: 10.1085/jgp.104.3.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson KL, Kopito RR. Effects of pyrophosphate and nucleotide analogs suggest a role for ATP binding and hydrolysis in cystic fibrosis transmembrane regulator channel gating. J Biol Chem. 1994;269:19349–19353. [PubMed] [Google Scholar]
- Gunderson KL, Kopito RR. Conformational states of CFTR associated with channel gating: the role of ATP binding and hydrolysis. Cell. 1995;82:231–239. doi: 10.1016/0092-8674(95)90310-0. [DOI] [PubMed] [Google Scholar]
- Haws C, Krouse ME, Xia Y, Gruenert DC, Wine JJ. CFTR channels in immortalized human airway cells. Am J Physiol. 1992;263:L692–L707. doi: 10.1152/ajplung.1992.263.6.L692. [DOI] [PubMed] [Google Scholar]
- Haws C, Finkbeiner WE, Widdicombe JH, Wine JJ. CFTR in Calu-3 human airway cells: channel properties and role in cAMP-activated Cl−conductance. Am J Physiol. 1994;266:L502–L512. doi: 10.1152/ajplung.1994.266.5.L502. [DOI] [PubMed] [Google Scholar]
- Hung L-W, Wang IX, Nikaido K, Liu P-Q, Ames GF-L, Kim S-H. Crystal structure of the ATP-binding subunit of an ABC transporter. Nature. 1998;396:703–707. doi: 10.1038/25393. [DOI] [PubMed] [Google Scholar]
- Hwang T-C, Nagel G, Narin AC, Gadsby DC. Regulation of gating of CFTR Cl channels by phosphorylation and ATP hydrolysis. Proc Natl Acad Sci USA. 1994;91:4698–4702. doi: 10.1073/pnas.91.11.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang T-C, Wang F, Yang IC, Reenstra WW. Genistein potentiates wild-type and delta F508-CFTR channel activity. Am J Physiol. 1997;273:C988–C998. doi: 10.1152/ajpcell.1997.273.3.C988. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Welsh MJ. Block by MOPS reveals a conformation change in the CFTR pore produced by ATP hydrolysis. Am J Physiol. 1997;273:C1278–C1289. doi: 10.1152/ajpcell.1997.273.4.C1278. [DOI] [PubMed] [Google Scholar]
- Ko YH, Pedersen PL. The first nucleotide binding fold of the cystic fibrosis transmembrane conductance regulator can function as an active ATPase. J Biol Chem. 1995;270:22093–22096. doi: 10.1074/jbc.270.38.22093. [DOI] [PubMed] [Google Scholar]
- Kuchler K, Thorner J. Secretion of peptides and proteins lacking hydrophobic signal sequences: the role of adenosine triphosphate–driven membrane translocators. Endocr Rev. 1992;13:499–514. doi: 10.1210/edrv-13-3-499. [DOI] [PubMed] [Google Scholar]
- Li C, Ramjeesingh M, Reyes E, Jensen T, Chang X, Rommens JM, Bear CE. The cystic fibrosis mutation (delta F508) does not influence the chloride channel activity of CFTR. Nat Genet. 1993;3:311–316. doi: 10.1038/ng0493-311. [DOI] [PubMed] [Google Scholar]
- Li C, Ramjeesingh M, Wang W, Garami E, Hewryk M, Lee D, Rommens JM, Galley K, Bear CE. ATPase activity of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1996;271:28463–28468. doi: 10.1074/jbc.271.45.28463. [DOI] [PubMed] [Google Scholar]
- Linsdell P, Hanrahan JW. Flickery block of single CFTR chloride channels by intracellular anions and osmolytes. Am J Physiol. 1996;271:C628–C634. doi: 10.1152/ajpcell.1996.271.2.C628. [DOI] [PubMed] [Google Scholar]
- Manavalan P, Dearborn DG, McPherson JM, Smith AE. Sequence homologies between nucleotide binding regions of CFTR and G-proteins suggest structural and functional similarities. FEBS Lett. 1995;366:87–91. doi: 10.1016/0014-5793(95)00463-j. [DOI] [PubMed] [Google Scholar]
- Mathews CJ, Tabcharani JA, Chang XB, Jensen TJ, Riordan JR, Hanrahan JW. Dibasic protein kinase A sites regulate bursting rate and nucleotide sensitivity of the cystic fibrosis transmembrane conductance regulator chloride channel. J Physiol (Camb) 1998;508:365–377. doi: 10.1111/j.1469-7793.1998.365bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramjeesingh M, Li C, Garemi E, Galley KA, Huan LJ, Wang Y, Bear CE. A novel model for coupling of CFTR ATPase activity with channel function. Pediatr Pulmonol. 1998;S17:201. . (Abstr.) [Google Scholar]
- Randak C, Neth P, Auerswald EA, Eckerskorn C, Assfalg-Machleidt I, Machleidt W. A recombinant polypeptide model of the second nucleotide-binding fold of the cystic fibrosis transmembrane regulator functions as an active ATPase, GTPase and adenylate kinase. FEBS Lett. 1997;410:180–186. doi: 10.1016/s0014-5793(97)00574-7. [DOI] [PubMed] [Google Scholar]
- Rich DP, Berger HA, Cheng SH, Travis SM, Saxena M, Smith AE, Welsh MJ. Regulation of the cystic fibrosis transmembrane conductance regulator Cl−channel by negative charge in the R domain. J Biol Chem. 1993;268:20259–20267. [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem BS, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plasvsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Saraste M, Sibbald PR, Wittinghofer A. The P-loop-a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci. 1990;15:430–434. doi: 10.1016/0968-0004(90)90281-f. [DOI] [PubMed] [Google Scholar]
- Senior AE, Gadsby DC. ATP hydrolysis cycles and mechanism in p-glycoprotein and CFTR. Semin Cancer Biol. 1997;8:143–150. doi: 10.1006/scbi.1997.0065. [DOI] [PubMed] [Google Scholar]
- Sheppard DN, Robinson KA. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl−channels expressed in a murine cell line. J Physiol (Lond) 1997;503:333–346. doi: 10.1111/j.1469-7793.1997.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venglarik CJ, Schultz BD, Frizzell RA, Bridges RJ. ATP alters current fluctuation of cystic fibrosis transmembrane conductance regulator: evidence for a three-state activation mechanism. J Gen Physiol. 1994;104:123–146. doi: 10.1085/jgp.104.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zeltwanger S, Yang IC, Narin AC, Hwang T-C. Actions of genistein on cystic fibrosis transmembrane conductance regulator channel gating. Evidence for two binding sites with opposite effects. J Gen Physiol. 1998;111:477–490. doi: 10.1085/jgp.111.3.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DJ, Mansoura MK, Watson PY, Smit LS, Collins FS, Dawson DC. CFTR: the nucleotide binding folds regulate the accessibility and stability of the activated state. J Gen Physiol. 1996;107:103–119. doi: 10.1085/jgp.107.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter MC, Sheppard DN, Carson MR, Welsh MJ. Effect of ATP concentration on CFTR Cl−channels: a kinetic analysis of channel regulation. Biophys J. 1994;66:1398–1403. doi: 10.1016/S0006-3495(94)80930-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeltwanger, S. 1998. Gating of cystic fibrosis transmembrane conductance regulator (CFTR) chloride channels by nucleoside triphosphates. Ph.D. Dissertation, University of Missouri, Columbia, MO.