

Abstract
Inward rectifying K channels are essential for maintaining resting membrane potential and regulating excitability in many cell types. Previous studies have attributed the rectification properties of strong inward rectifiers such as Kir2.1 to voltage-dependent binding of intracellular polyamines or Mg to the pore (direct open channel block), thereby preventing outward passage of K ions. We have studied interactions between polyamines and the polyamine toxins philanthotoxin and argiotoxin on inward rectification in Kir2.1. We present evidence that high affinity polyamine block is not consistent with direct open channel block, but instead involves polyamines binding to another region of the channel (intrinsic gate) to form a blocking complex that occludes the pore. This interaction defines a novel mechanism of ion channel closure.
Keywords: spider venom toxin, polyamine, inward rectification, K channels
introduction
Strong inward rectifier K channels such as Kir2.1 (IRK1) play a key role in setting the resting membrane potential and regulating excitability in heart, neurons, and many other cell types (Doupnik et al., 1995). The strong inward rectification property has been previously attributed to intracellular Mg and polyamines directly plugging the channel pore at membrane potentials positive to the K equilibrium potential, causing voltage-dependent open channel block (Matsuda et al., 1987; Vandenberg, 1987; Ficker et al., 1994; Lopatin et al., 1994; Stanfield et al., 1994; Taglialatela et al., 1994; Fakler et al., 1995). Detailed analysis of block by spermine, however, requires a fairly complex model to account for all the voltage dependence and kinetic features (Lopatin et al., 1995). Moreover, open channel block as the sole mechanism of inward rectification has been questioned in a recent study in which Kir2.1 channels were reconstituted into lipid bilayers (Aleksandrov et al., 1996). In the absence of Mg or polyamines, the channels still exhibited rectification. Mg or polyamines enhanced inward rectification, but the mechanism of block was more complex than could be modeled by simple open channel block.
We have previously described an intrinsic pH-sensitive gating mechanism of inward rectification in Kir2.1 in the absence of Mg or polyamines (Shieh et al., 1996). This was manifested as a persistent voltage- dependent inactivation of outward currents in excised patches after prolonged washout in polyamine- and Mg-free solution, which was accelerated by raising intracellular pH to 9.0. The intrinsic gating mechanism is kinetically much slower than polyamine or Mg block and is most obvious only with large depolarizations (to more than +40 mV), explaining why it was only subtly apparent in some previous studies and assumed to represent residual polyamine block (Ficker et al., 1994; Lopatin et al., 1994).
Spider venom polyamine toxins, philanthotoxin and argiotoxin, have been shown to block N-methyl-d-aspartate receptors extracellularly (Brackley et al., 1993; Williams, 1993; Donevan and Rogawski, 1996). Since polyamines cause inward rectification in Kir channels, we investigated the effects of these toxins on the strong inward rectifier channel Kir2.1 (IRK1) to obtain further mechanistic insight into polyamine-induced inward rectification. The toxins are structurally similar to spermine at one end and have a bulky hydrophobic aromatic group at the other (see Fig. 1 A). By studying competitive interactions between polyamines and these polyamine toxins, we present evidence that polyamine block is more complex than direct open channel block. Our results suggest that direct open channel block accounts for low affinity block by spermine and spermidine, but high affinity block by these polyamines involves their binding to another region of the channel (intrinsic gate) to form a blocking complex that occludes the pore.
Figure 1.

Polyamines and polyamine toxins. (A) Chemical structures of spermine, spermidine, and the polyamine toxins philanthotoxin and argiotoxin. (B) Dose–response curves of block of wild-type Kir2.1, D172N, and E224G by spermine (left) and philanthoxin (•, ○, and ▵) or argiotoxin (▾, right). Each data point is the mean ± SEM for n = 4–6 patches. The giant excised inside-out patches were all superfused for >5 min with Mg- and polyamine-free bath solution before spermine or phi-lanthotoxin exposure. Currents were measured at the end of a 200-ms voltage clamp pulse to +40 mV, from a holding potential of −40 mV. Superimposed curves are least-squares fits to a Hill equation, with the numbers indicating K 0.5 values. The Hill coefficients ranged from 0.62 to 0.90 for spermine and from 0.55 to 0.70 for philanthoxin.
methods
Molecular Biology
In vitro T7-transcribed cRNA (Ambion Inc.) was injected (50– 5,000 ng) into stage V–VI Xenopus oocytes isolated by partial ovariectomy under tricaine anaesthesia. PCR mutagenesis was carried out by the overlap extension technique (Ho et al., 1989).
Electrophysiology
Patch clamp studies were performed at room temperature on giant excised inside-out patches, as described previously (Shieh et al., 1996). The patch electrode contained (mM): 100 KOH, 1.8 CaCl2, and 5 HEPES, pH 7.4 with 100 MES. The standard bath solution contained (mM): 100–104 KCl + KOH, 5 EDTA, and 5 HEPES, adjusted to pH 7.2 with MES. Polyamines and polyamine toxins were added directly to the bath solution. Rapid bath solution changes (<1 s) were accomplished using a rapid solution exchange device (Shieh et al., 1996). All current traces shown in the figures were leak corrected by subtracting currents recorded with 30 mM tetraethylammonium in the bath solution, which blocked Kir2.1 currents completely (Shieh et al., 1996). Curve-fitting and model simulations were performed using Sigmaplot version 2.0 (Jandel Scientific) and SCoP version 3.5 (Simulation Resources, Inc.).
results
Polyamine Toxins Block Kir2.1 Channels
Kir2.1 channels were heterologously expressed in Xenopus oocytes. When applied to the cytoplasmic surface of giant inside-out patches, both philanthotoxin and argiotoxin blocked outward currents through Kir2.1 channels in a voltage-dependent manner. At +40 mV, the concentrations of philanthotoxin and argiotoxin producing half-maximal block (K 0.5) were 47 nM (n = 4 patches), and 37 nM (n = 4), respectively (Fig. 1 C). Unlike N-methyl-d-aspartate receptors, spermine blocked Kir2.1 channels with higher affinity (K 0.5 of 5.9 nM at +40 mV) than either toxin (Fig. 1 B). Since earlier mutagenesis studies have defined two key negatively charged amino acids, D172 in the M2 region and E224 in the COOH terminus, as regulators of polyamine- and Mg-induced inward rectification in Kir2.1 (Wible et al., 1994; Taglialatela et al., 1995; Yang et al., 1995), we also tested the effects of philanthotoxin on the mutants D172N and E224G. Both mutants were considerably less sensitive to philanthotoxin block than wild-type Kir2.1, with K 0.5 values of 1,220 nM for D172N and 7,090 nM for E224G (Fig. 1 C). These findings are generally consistent with the long-pore plugging model of polyamine block (Lopatin et al., 1995), in which polyamines such as spermine are proposed to insert lengthwise into the channel pore. The polyamine end of the toxin could also insert lengthwise, but would be unable to insert crosswise because of the large hydrophobic end.
Interactions between Polyamines and Polyamine Toxins
We next investigated interactions between spermine and philanthotoxin block. A fixed concentration of philanthotoxin (30 nM) was chosen that partially blocked steady state outward current at +40 mV, by 44% (Fig. 2 A). In the presence of 30 nM philanthotoxin, the concentration of spermine required to block the remaining current by 50% (K 0.5) increased from 5.9 to 78 nM (n = 5 patches; Fig. 2 B). The same effect was seen with the trivalent polyamine spermidine (Fig. 2 E), whose K 0.5 increased from 31 nM in the absence of philanthoxin to 342 nM (n = 5 patches) when 30 nM philanthotoxin was present. Finally, argiotoxin (10 nM) also interfered with spermine block of outward Kir2.1 currents to a comparable extent as philanthotoxin, increasing the K 0.5 for spermine from 5.1 to 53 nM (n = 5 patches) at +40 mV (Fig. 2 F).
Figure 2.

Competitive inhibition of polyamine block by polyamine toxins. (A) Representative traces from the same giant excised inside-out patch showing block of wild-type Kir2.1 channel by various concentrations of spermine (S) in the absence (left) or presence (right) of 30 nM philanthotoxin (T). Recording conditions and voltage-clamp protocol as in Fig. 1. (B–D) Dose–response of outward currents at +40 mV (after 200 ms) to spermine in Kir2.1 wild type (B), D172N (C), and E224G (D), in the absence (○) or presence (•) of philanthotoxin. The gray crosses show the normalized data for philanthoxin + spermine, to better illustrate the shift in K 0.5. The superimposed curves are least-squares fits to the Hill equation. Hill coefficients ranged from 0.7 to 0.9. (E) Dose–response curves for wild-type Kir2.1 currents to spermidine (Spd) in the absence (○) and presence (•, gray crosses, normalized) of 30 nM philanthoxin. Hill coefficients ranged from 0.42 to 0.51. (F) Dose–response curve of wild-type Kir2.1 currents at +40 mV to spermine in the absence (○) or presence (•, gray crosses, normalized) of 10 nM argiotoxin (AT). Hill coefficients ranged from 0.77 to 0.99. Data points in B–F represent the mean ± SEM for n = 4–6 patches.
The ability of polyamine toxins to desensitize Kir2.1 channels to block by spermine or spermidine to this degree is difficult to reconcile with a direct open channel block mechanism in which toxin and polyamine molecules compete independently for binding site(s) in the pore. In the long-pore plugging model (Lopatin et al., 1995; Fig. 3 A), it is hypothesized that two spermine molecules directly occupy the pore, producing either shallow (B1) or deeply bound (B2 and B3) states. We incorporated two additional blocked states to represent direct block by a single toxin molecule occupying the pore (the B4 state), and a toxin molecule capping a deeply bound spermine molecule (the B5 state). 10 rate constants (five equilibrium constants) connect the various states. At a concentration of toxin producing 44% block, this model does not produce a large shift in the K 0.5 for spermine (Fig. 3 B). A small increase in the K 0.5 for spermine (1.8-fold) can be produced if the B5 state is disabled (Fig. 3 C). As the affinity of the toxin for the B5 state is increased, however, the K 0.5 for spermine progressively shifts to lower rather than higher values. The same predictions were obtained with simpler models of direct open channel block, with totals of two to four blocked states. These results indicate that block of Kir2.1 channels by polyamines and polyamine toxins is more complex than a direct open channel block mechanism (Lopatin et al., 1995).
Figure 3.

Long pore plugging model of inward rectification. (A) Reaction scheme for the “long pore” model of direct open channel block by spermine (S) proposed by Lopatin et al. (1995) (shaded area), modified to include direct competitive block by philanthotoxin (T). Cartoons illustrate states in which the channel pore is open (O), blocked by spermine (B1–B3), philanthotoxin (B4), or both spermine and philanthotoxin (B5). K 1–5 represent equilibrium constants (k reverse/k forward) for the various transitions. See text for details. (B–C) Fits of the direct block model in A to the experimental data for the spermine–philanthotoxin interaction in wild-type Kir2.1 shown in Fig. 3 B, where ○ are spermine alone, • are spermine + philanthotoxin, and gray crosses are normalized spermine + philanthotoxin. Model parameters in B: K 1 = 7.75 × 10−5 M, K 2 = 7.75 × 10−5 M, K 3 = 7.75 × 10−5 M, K 4 = 47 × 10−9 M, K 5 = 47 × 10−9 M. In C, the B5 state was disabled by increasing K 5 to 103 M. In neither case could the model reproduce quantitatively the increase in K 0.5 for spermine in the presence of philanthotoxin.
Can the Intrinsic Gating Mechanism Account for the Interaction between Polyamines and Polyamine Toxins?
An intrinsic gating mechanism has previously been proposed to explain inward rectification of K channels (Oliva et al., 1990). Recently we (Shieh et al., 1996) and others (Aleksandrov et al., 1996) have characterized an intrinsic gating mechanism in the cloned Kir2.1 channels in which persistent inactivation of outward current occurs in the absence of polyamines or Mg. Fig. 4 shows the time course of inactivation of outward Kir2.1 currents in giant inside-out patches excised into a polyamine- and Mg-free solution. Over the first 5 min, there was a time-dependent decrease in the rate of inactivation of outward current (measured at +60 mV), previously attributed to the washout of endogenous polyamines (Ficker et al., 1994; Lopatin et al., 1994; Fakler et al., 1995; Yang et al., 1995), but thereafter no further change occurred. During the polyamine- and Mg-free washout period, if Kir2.1 currents were first inactivated by depolarizing the membrane to +60 mV, and then rapidly unblocked by hyperpolarizing to −30 mV, a subsequent depolarization to +60 mV still produced inactivation of outward current. This seems inconsistent with a blocking molecule lodged in the pore of the channel, since it should diffuse away once released from its blocking site during hyperpolarization, and not be available to reblock the channel upon the next depolarization (unless a compartmentalized pool of poorly diffusible polyamines is postulated). An alternative explanation is that polyamines bind to and enhance the effectiveness of an intrinsic gate. The polyamine molecule could remain tethered to the intrinsic gate when released from the pore by hyperpolarization, and thereby be available to reocclude the pore during the subsequent depolarization.
Figure 4.
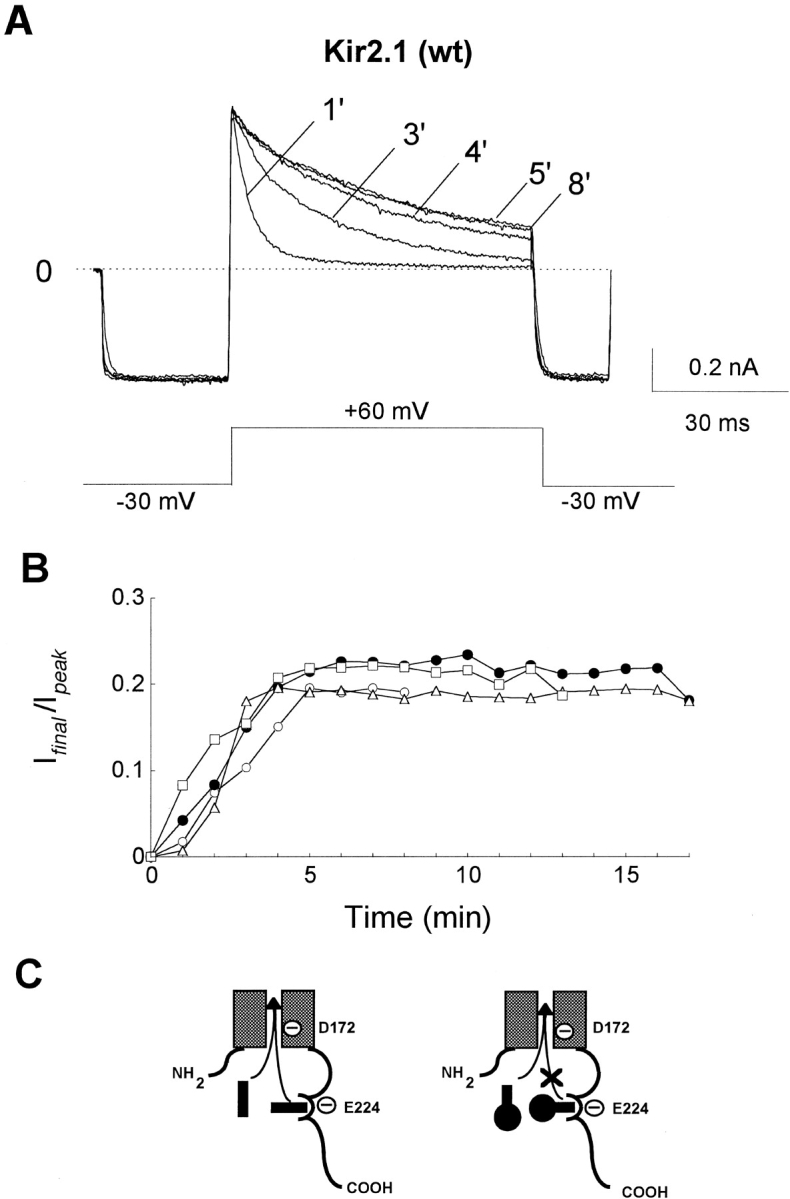
Time course of inactivation of outward currents in wild-type Kir2.1 channels. (A) Macroscopic currents were recorded from a giant inside-out patch excised from a Xenopus oocytes expressing wild-type Kir2.1 channels, while superfusing with Mg and polyamine-free bath solution. At the times indicated after patch excision, outward wild-type Kir2.1 currents elicited by voltage clamp pulses to +60 mV from a holding potential of −30 mV showed progressively slower inactivation, reaching a steady state after 5 min. Despite identical amplitudes of inward current at −30 mV (indicating complete unblock), endogenous polyamines still remained available to reblock the current upon depolarization to +30 mV, albeit at a slower rate (e.g., 1- vs. 3-min traces). (B) Rate of inactivation of outward current in four patches. Fraction of noninactivating current (I final/I peak) at the end of a 60-ms voltage clamp to +40 mV is plotted for each patch. The rate of inactivation reached steady state by 5 min of washout. (C) Hypothetical schema of channel block by polyamines (left) or polyamine toxins (right) using an intrinsic gate mechanism. Unlike spermine, it is assumed that the hydrophobic head of the toxin molecule is too large to block the pore when its polyamine end is tethered to the intrinsic gate. Conjectured locations of two negatively charged residues D172 and E224 are shown at the pore docking site and at the polyamine-binding site on the intrinsic gate, respectively.
To determine whether this type of mechanism could account for the philanthotoxin-spermine interaction, we examined the model schematically illustrated in Figs. 4 C and 5 A. We hypothesize that, due to its linear structure with four symmetrically spaced positive charges (Fig. 1 A), spermine binds at one end to the intrinsic gate, and the other end to a docking site in the ion-conducting pathway (Fig. 4 C). This mechanism is assumed to account for the high affinity voltage-dependent block by spermine, whereas direct interaction of free, untethered spermine with the pore docking site produces lower affinity block. Philanthotoxin, however, can only block Kir2.1 channels when its spermine-like end lodges in the docking site in the pore (Fig. 4 C). If the spermine-like end of philanthotoxin binds to the intrinsic gate, the hydrophobic end is too bulky to reach and interact with the docking site in the pore. This hypothetical schema predicts an interaction between polyamine toxins and spermine: if, in the presence of a polyamine toxin, the putative site in the intrinsic gate to which spermine binds is occupied by the spermine-like end of the polyamine toxin molecule, then spermine will no longer have access to its high affinity blocking mechanism. Thus, spermine's ability to block Kir2.1 channels with high affinity should be reduced.
The simplest model of interaction between a polyamine toxin (T), spermine (S), and an intrinsic gate is shown in Fig. 5 A. Three open states reflect either nothing (O1), spermine (O2), or a toxin molecule (O3) bound to the intrinsic gate site, with the pore docking site empty. There are seven possible blocked states: the B1 state represents high affinity block by the spermine-intrinsic gate complex occluding the pore. The B2, B3, and B4 states represent an untethered spermine molecule bound to the docking site in the pore (direct low affinity block) with either nothing, spermine, or a toxin molecule, respectively, bound to the intrinsic gate. The B5, B6, and B7 states represent the analogous conditions for a toxin molecule directly blocking the pore. As in the long-pore plugging model (Fig. 3 A), 10 rate constants connect the various states, represented in the diagram by their corresponding equilibrium constants K 1–K 5. Based on the difference in affinities for spermine block between wild-type Kir2.1 and the E224G mutation (Fig. 1 B), we chose K 0.5 for high affinity block by the spermine-intrinsic gate complex to be ∼1,000× higher than direct block by untethered spermine molecules. By altering the affinity of the toxin for the intrinsic gate (i.e., adjusting the K 3 equilibrium constant), the experimentally observed decreased affinity of spermine block in the presence of toxin is readily reproduced by the model, and yields an acceptable fit to the data (Fig. 5 B). The increase in K 0.5 in the presence of toxin is explained by the O3 state being favored as the affinity of the toxin for the intrinsic gate is increased, shifting the equilibrium away from spermine's high affinity blocking pathway (O1 → O2 → B1). In contrast, a direct open channel block mechanism, in which both toxin and spermine compete independently for a binding site in the pore, provides no means for the toxin to shift the equilibrium towards an open state, and so cannot account for the observed shift in the K 0.5 for spermine in the presence of philanthotoxin. Although we have not tested for toxin–toxin interactions, we would predict minimal interaction between different polyamine toxins, since neither would have access to the high affinity blocking route coupled to the intrinsic gate.
Figure 5.

Intrinsic gate model of inward rectification. (A) The simplest reaction scheme showing spermine (S) and philanthotoxin (T) competition for an intrinsic gate and a pore-docking site in the Kir2.1 channel. Cartoons illustrate the blocked states (B1–B7) and the O3 state in which the toxin is bound to the intrinsic gate, preventing spermine's access to its high affinity blocking pathway (O1 → O2 → B1). K 1–5 represent equilibrium constants (k reverse/k forward) for the various transitions. Shaded area represents states involved in spermine block in the absence of toxin, see Fig. 3 A for comparison. See text for further details. (B–D) Fits of the intrinsic gate model in A to the experimental data for the spermine–philanthotoxin interaction shown in Fig. 3, B–D, where ○ are spermine alone, • are spermine + philanthotoxin, and gray crosses are normalized spermine + philanthotoxin. Model parameters for wild-type Kir2.1 (B): K 1 = 7.75 × 10−5 M, K 2 = 7.75 × 10−5 M, K 3 = 5.8 × 10−9 M, K 4 = 6 × 10−9 M, K 5 = 47 × 10− 9M; for D172N (C): K 1 = 7.75 × 10−5 M, K 2 = 115 × 10−5 M, K 3 = 132 × 10−9 M, K 4 = 89 × 10−6 M, K 5 = 1,220 × 10−9 M; for E224G (D): K 1 = 103 M, K 2 = 103 M, K 3 = 103 M, K 4 = 6,030 × 10−9 M, K 5 = 7,090 × 10−9 M.
The Polyamine-Polyamine Toxin Interaction in Kir2.1 Channels with Mutated Rectification Sites
The philanthotoxin–spermine competition experiments strongly support the role of an intrinsic gate in high affinity polyamine block of Kir2.1 channels. Since D172 and E224 have both been shown to be important in regulating the sensitivity of Kir2.1 to block by polyamines and Mg (Taglialatela et al., 1995; Yang et al., 1995; Abrahms et al., 1996), we also examined the philanthotoxin–spermine interaction in the D172N and E224G mutants. For D172N, the K 0.5 for spermine at +40 mV increased from 5.9 to 90 nM (n = 4), and for philanthotoxin, from 47 to 1,220 nM (n = 6), compared with wild-type Kir2.1 (Fig. 1, B and C). In the presence of 700 nM philanthotoxin, which blocked outward current at +40 mV by 40%, the K 0.5 for spermine increased from 90 to 1,050 nM (Fig. 2 C). A possible interpretation of these findings is that D172 regulates binding of polyamines and toxin to the pore docking site (Fig. 4 C). By collectively destabilizing the binding to this site of the spermine-intrinsic gate complex, untethered spermine and philanthotoxin, D172N should exhibit lower affinity block by both spermine and philanthotoxin. Assuming that the highest affinity block still occurs via the spermine-intrinsic gate complex, however, the ability of philanthotoxin to decrease the affinity of spermine block should still be preserved. All of these findings were obtained in the D172N mutant. By appropriately adjusting the equilibrium constants to reflect the lower affinities at the pore docking site, these effects were readily simulated in the intrinsic gating model (Fig. 5 C). In contrast, the direct block model (Fig. 3 A) again failed to reproduce the philanthotoxin–spermine interaction (not shown).
For E224G, the K 0.5s for block by spermine and philanthotoxin were also both increased, from 5.9 to 6,030 nM (n = 4) and from 47 to 7,090 nM (n = 5), respectively (Fig. 1, B and C). However, in the presence of 3,000 nM philanthotoxin, which blocked outward current at +40 mV by 31%, the increase in the K 0.5 of spermine block, as seen in both wild-type Kir2.1 and the D172N mutant, was eliminated (6,030 nM in the absence of philanthotoxin versus 4,800 nM in its presence, n = 5). A possible interpretation of these findings is that E224 primarily regulates polyamine binding to the intrinsic gate. If the E224G mutation impairs binding of spermine and philanthotoxin to the intrinsic gate, then block can occur only through direct interaction of untethered spermine or philanthotoxin with the pore docking site (near D172). The state diagram for the E224G mutant then reduces to a direct open channel block model containing only one open state (O1 in Fig. 5 A) and two blocked states (B2 and B5); i.e., a simplified version of Fig. 3 A. In this case, the spermine–philanthotoxin interaction should be markedly reduced. Exclusive assignment of E224's role to the polyamine binding site on the intrinsic gate, however, does not account for the increase in the K 0.5 for philanthotoxin block in the E224G mutant. Thus, E224 must also contribute to the stability of philanthotoxin and, perhaps, spermine binding to the pore region. This is consistent with previous observations showing that the E224G mutation affects single channel properties through the Kir2.1 pore (Yang et al., 1995).
discussion
These findings support the intrinsic gate model of spermine block hypothesized in Figs. 4 C and 5 A, in which the D172 residue regulates the docking site in the pore, and E224 regulates the polyamine binding site on the intrinsic gate in addition to influencing binding of polyamine toxins (and probably also polyamines) in the pore. The evidence can be summarized as follows: (a) an intrinsic gating mechanism of inward rectification in Kir2.1 has been previously identified (Aleksandrov et al., 1996; Shieh et al., 1996), which persists indefinitely after washout of endogenous internal polyamines or Mg (Fig. 4 B). (b) Polyamines remain available to reblock Kir2.1 channels after they are released from their blocking site in the pore by hyperpolarization (Fig. 4 A), suggesting that they remain tethered to another region of the channel from which they dissociate slowly. (c) Polyamine toxins, such as philanthotoxin and argiotoxin, interfere with the ability of spermine or spermidine to block Kir2.1 channels (Fig. 2). This effect cannot be explained by a direct open channel block mechanism (Fig. 3 A), but is readily accounted for by an intrinsic gate model (Fig. 5 A). In the latter, high affinity block is due to a spermine-intrinsic gate complex, and low affinity block by direct open channel block of untethered spermine molecules. (d) The D172N mutation reduces the affinity of the channel for block by both spermine and philanthotoxin, but does not eliminate the spermine–philanthotoxin interaction, suggesting its major effect is destabilization of the pore docking site for all three blocking entities (the spermine-intrinsic gate complex, untethered spermine, and philanthotoxin). (e) The E224 mutation eliminates the spermine–philanthoxin interaction, consistent with destabilization of spermine's binding to the intrinsic gate. The assignment of D172 to the pore docking site, and E224 primarily to the intrinsic gate binding site may explain why two such widely separated amino acids play key roles in rectification.
Several limitations of this study should be recognized. We cannot absolutely exclude the possibility of a compartmentalized pool of polyamines remaining after extensive washing of excised patches. However, such a pool would have to have multiple time constants with fast and very slow (>>15 min) components (see Fig. 4 B), and cannot account for the philanthotoxin–spermine interaction by a pure open channel block mechanism (see Fig. 3). In the model proposed in Fig. 5 A, the analysis was simplified by omitting the blocked state corresponding to the intrinsic gate unoccupied by a spermine molecule, even though outward current was significantly attenuated at the end of the voltage clamp pulse to +40 mV in the absence of polyamines or Mg (Fig. 2 A). Long voltage clamp pulses (200 ms) were therefore used to allow this process to reach quasi-steady state before measuring the effects of polyamines and polyamine toxins on the remaining current. Our analysis was restricted to equilibrium conditions at +40 mV, and we have not yet evaluated whether the intrinsic gate model can account for the complex kinetics and voltage dependencies characterized extensively by Lopatin et al. (1995). The substantial block by the intrinsic gating mechanism itself (in the absence of polyamines) complicates this analysis. Whether Mg or other polyamines, such as putrescine, bind to or use this putative intrinsic gate mechanism has also not yet been determined. Although we think it likely based on earlier analysis of Kir2.1 mutants (Lee et al., 1997), we have not proven that the intrinsic gate that weakly blocks outward current in the absence of polyamines is the same structure to which polyamines bind to cause high affinity block. The molecular configuration of the putative intrinsic gate region and even number of intrinsic gates per channel (given the homotetrameric structure of Kir2.1) remains purely speculative; it is solely for clarity and convenience that the intrinsic gate has been drawn as a single tethered gating particle in Figs. 4 C and 5 A. In fact, the dual effect of the E224G mutation on both the polyamine binding site on the intrinsic gate and its docking site in the pore is difficult to reconcile physically with such a simple model. At first glance, Fig. 5 A appears complicated. However, the polyamine block mechanism (shaded area) has the same number of rate constants and only one additional state compared with the direct block model Fig. 3 A. Incorporating the toxin–spermine interaction adds four additional states compared with the direct block model, but retains the same number of rate constants. Finally, whether the intrinsic gate mechanism also applies to other members of the strong inward rectifier K+ channel family is currently unknown.
In summary, these findings support the idea that a region of the Kir2.1 channel acts as an intrinsic gate that binds a polyamine molecule such as spermine or spermidine to induce strong inward rectification. Multiple positive charges on the polyamine molecule are likely to facilitate the voltage-dependent interaction of the intrinsic gate–polyamine complex with a pore docking site. Lower affinity block is produced by direct open channel block by untethered polyamines. The intrinsic gate mechanism retains the important feature of the strong inward rectifiers, relief of inward rectification by elevated K, hypothesized to result from destabilization of the polyamine-intrinsic gate docking site in the pore by external K ions. In view of recent structural information about pore dimensions of inward rectifier K channels (Doyle et al., 1998), the intrinsic gate is an appealing modification to the direct block long pore plugging mechanism (Lopatin et al., 1995), since it requires only one spermine molecule, either tethered or untethered, to block the pore. With the distance from the internal surface to the selectivity filter estimated at 3 nm (Doyle et al., 1998), a single spermine molecule (2 nm in length; Lopatin et al., 1995) occupying this section of the pore is reasonable, but more problematic for a direct block mechanism requiring two spermine molecules stacked lengthwise on top of one another (4 nm long) to fit into this region.
Acknowledgments
We thank Drs. K. Philipson, F. Bezanilla, and C.G. Nichols for critically reading the manuscript and Dr. Yujuan Lu for expert technical support.
Supported by National Institutes of Health grants RO1 HL60025, RO1 HL36729, and P50 HL52319, by an American Heart Association, Greater Los Angeles Affiliate Grant-in-Aid (to S.A. John) and Research Fellowship (to J.-K. Lee), and by the Laubisch Fund and Kawata Endowment.
Footnotes
Jong-Kook Lee and Scott A. John contributed equally to this work and should be considered co-first authors.
Dr. Lee's current address is Department of Circulation, Research Institute of Environmental Medicine, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8601 Japan.
references
- Abrahms CJ, Davies NW, Shelton PA, Stanfield PR. The role of a single aspartate residue in ionic selectivity and block of a murine inward rectifier K+channel Kir2.1. J Physiol (Camb) 1996;493:643–649. doi: 10.1113/jphysiol.1996.sp021411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrov A, Velimirovic B, Clapham DE. Inward rectification of the IRK1 K+channel reconstituted in lipid bilayers. Biophys J. 1996;70:2680–2687. doi: 10.1016/S0006-3495(96)79837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackley PTH, Bell DR, Choi S-K, Nakanishi K, Usherwood PNR. Selective antagonism of native and cloned kainite and NMDA receptors by polyamine-containing toxins. J Pharmacol Exp Ther. 1993;266:1573–1580. [PubMed] [Google Scholar]
- Donevan SD, Rogawski MA. Multiple actions of arylalkylamine arthropod toxins on the N-methyl-d-aspartate receptor. Neuroscience. 1996;70:361–375. doi: 10.1016/0306-4522(95)00342-8. [DOI] [PubMed] [Google Scholar]
- Doupnik CA, Davidson N, Lester HA. The inward rectifier potassium channel family. Curr Opin Neurobiol. 1995;5:268–277. doi: 10.1016/0959-4388(95)80038-7. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Fakler B, Brandle U, Glowatzki E, Weidemann S, Zenner HP, Ruppersberg JP. Strong voltage-dependent inward rectification of inward rectifier K channels is caused by intracellular spermine. Cell. 1995;80:149–154. doi: 10.1016/0092-8674(95)90459-x. [DOI] [PubMed] [Google Scholar]
- Ficker E, Taglialatela M, Wible BA, Henley CM, Brown AM. Spermine and spermidine as gating molecules for inward rectifier K+channels. Science. 1994;266:1068–1072. doi: 10.1126/science.7973666. [DOI] [PubMed] [Google Scholar]
- Ho S, Hont HD, Pullen JK, Pease LR. Site directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Lee JK, John SA, Lu Y, Weiss JN. Polyamines cause inward rectification in Kir2.1 (IRK1) channels by forming a tethered blocking particle with an intrinsic gate. Circulation. 1997;96:I-426. [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994;372:366–369. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. The mechanism of inward rectification of potassium channels: “long-pore plugging” by cytoplasmic polyamines. JGen Physiol. 1995;106:923–955. doi: 10.1085/jgp.106.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda H, Saigusa A, Irisawa H. Ohmic conductance through the inwardly rectifying K channel and blocking by Mg2+ . Nature. 1987;325:156–159. doi: 10.1038/325156a0. [DOI] [PubMed] [Google Scholar]
- Oliva C, Cohen IS, Pennefather P. The mechanism of rectification of iK1 in canine Purkinje myocytes. J Gen Physiol. 1990;96:299–318. doi: 10.1085/jgp.96.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh RC, John SA, Lee J-K, Weiss JN. Inward rectification of IRK1 expressed in Xenopusoocytes: effects of intracellular pH reveal an intrinsic gating mechanism. J Physiol (Camb) 1996;494:363–376. doi: 10.1113/jphysiol.1996.sp021498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield PR, Davies NW, Shelton PA, Khan IA, Brammar WJ, Standen NB, Conley EC. The intrinsic gating of inward rectifier K+channels expressed from the murine IRK1 gene depends on voltage, K and Mg. J Physiol (Camb) 1994;475:1–7. doi: 10.1113/jphysiol.1994.sp020044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela M, Ficker E, Wible BA, Brown AM. C-terminus determinants for Mg2+ and polyamine block of the inward rectifier K+channel IRK1. EMBO (Eur Mol Biol Organ) J. 1995;14:5532–5541. doi: 10.1002/j.1460-2075.1995.tb00240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela M, Wible BA, Caporaso R, Brown AM. Specification of pore properties by the carboxyl terminus of inwardly rectifying K+channels. Science. 1994;264:844–847. doi: 10.1126/science.8171340. [DOI] [PubMed] [Google Scholar]
- Vandenberg CA. Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc Natl Acad Sci USA. 1987;84:2560–2564. doi: 10.1073/pnas.84.8.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wible BA, Taglialatela M, Ficker E, Brown AM. Gating of inwardly rectifying K+channels localized to a single negatively charged residue. Nature. 1994;371:246–249. doi: 10.1038/371246a0. [DOI] [PubMed] [Google Scholar]
- Williams K. Effects of Agelenopsis aperta toxins on the N-methyl- d-aspartate receptor: polyamine-like and high affinity antagonist actions. J Pharmacol Exp Ther. 1993;266:231–236. [PubMed] [Google Scholar]
- Yang J, Jan YN, Jan LY. Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+channel. Neuron. 1995;14:1047–1054. doi: 10.1016/0896-6273(95)90343-7. [DOI] [PubMed] [Google Scholar]