

Abstract
This study reports the identification of an endogenous inhibitor of the G protein–gated (KACh) channel and its effect on the KACh channel kinetics. In the presence of acetylcholine in the pipette, KACh channels in inside-out atrial patches were activated by applying GTP to the cytoplasmic side of the membrane. In these patches, addition of physiological concentration of intracellular ATP (4 mM) upregulated KACh channel activity approximately fivefold and induced long-lived openings. However, such ATP-dependent gating is normally not observed in cell-attached patches, indicating that an endogenous substance that inhibits the ATP effect is present in the cell. We searched for such an inhibitor in the cell. ATP-dependent gating of the KACh channel was inhibited by the addition of the cytosolic fraction of rat atrial or brain tissues. The lipid component of the cytosolic fraction was found to contain the inhibitory activity. To identify the lipid inhibitor, we tested the effect of ∼40 different lipid molecules. Among the lipids tested, only unsaturated free fatty acids such as oleic, linoleic, and arachidonic acids (0.2–2 μM) reversibly inhibited the ATP-dependent gating of native KACh channels in atrial cells and hippocampal neurons, and of recombinant KACh channels (GIRK1/4 and GIRK1/2) expressed in oocytes. Unsaturated free fatty acids also inhibited phosphatidylinositol-4,5-bisphosphate (PIP2)-induced changes in KACh channel kinetics but were ineffective against ATP-activated background K1 channels and PIP2-activated KATP channels. These results show that during agonist-induced activation, unsaturated free fatty acids in the cytoplasm help to keep the cardiac and neuronal KACh channels downregulated by antagonizing their ATP-dependent gating. The opposing effects of ATP and free fatty acids represent a novel regulatory mechanism for the G protein–gated K+ channel.
Keywords: acetylcholine; long-chain fatty acid; phosphatidylinositol-4,5-bisphosphate; atria; arachidonic acid
INTRODUCTION
In the heart and brain, binding of neurotransmitters and hormones to their specific GTP binding protein (G protein)–coupled receptors activates an inwardly rectifying K+ channel (KACh channel) via the βγ subunit of Gi/o proteins (Sakmann et al. 1983; Breitwieser and Szabo 1985; Pfaffinger et al. 1985; Andrade et al. 1986; Kurachi et al. 1986; Logothetis et al. 1987). The kinetic properties of KACh channels activated either by an agonist or directly by βγ have been described in many earlier studies. Thus, in cell-attached patches of mammalian atrial cells, KACh channels activated by acetylcholine (ACh) typically show one open state with an open time constant of ∼1 ms and a single-channel conductance of 35–40 pS in symmetrical 140-mM KCl solution (Sakmann et al. 1983; Kurachi et al. 1986; Kim 1991). Longer-lived openings with an open time constant of several milliseconds can be detected (Kim 1991; Nemec et al. 1999), but the frequency of such openings are usually very low in mammalian atrial cell-attached patches. In inside-out patches with ACh in the pipette, KACh channels activated by GTP applied to the cytoplasmic side of the membrane also show predominantly short-lived openings; i.e, an open time constant of ∼1 ms. Interestingly, when 1–4 mM ATP is present together with GTP, the frequency of long-lived openings increases dramatically, leading to a marked increase in channel activity. In the presence of intracellular ATP and GTP, a second open state with a time constant of ∼8 ms can easily be identified from duration histograms (Kim 1991; Hong et al. 1996; Kim and Bang 1999). These results show that KACh channel kinetics observed in cell-attached and inside-out patches with GTP and ATP in the cytoplasmic side are markedly different.
The absence of ATP-induced modification of KACh channels (i.e., long-lived openings and high open probability state) in cell-attached patches has previously led us to hypothesize that an inhibitor of ATP-dependent mechanism exists in the cell (Hong et al. 1996). Such an inhibitor of ATP-dependent mechanism is expected to be critical in regulating the KACh channel activity and thus the cell excitability. In our earlier study, the presence of such an inhibitory substance in the cytoplasm of atrial tissue and subsequently in the cytoplasm of brain tissue was confirmed (Hong et al. 1996). Thus, application of atrial or brain cytosolic fraction to inside-out patches containing ATP-modified KACh channels resulted in complete inhibition of ATP-dependent gating. The putative inhibitor was initially assumed to be a protein, as treatment of the cytosolic fraction with trypsin abolished the inhibitory effect. Our extensive efforts to purify the putative inhibitor protein using conventional protein purification methods were not successful and the identity of the inhibitor has remained unknown. Recently, we noted that the potency of some of the lipids, including phospholipids and free fatty acids, decreased markedly when incubated at 37°C for ∼10 min in air. In our earlier studies to test whether the putative inhibitor was a protein, we had incubated the cytosolic fraction with proteases for ∼30 min at 37°C. This procedure probably inactivated certain lipids as well as proteins. Therefore, we reevaluated the nature of the putative inhibitor by considering the possibility that the inhibitor is a lipid molecule rather than a protein.
This study reports the successful identification of the endogenous inhibitor and its effect on KACh channel kinetics. The inhibitor was determined to be a lipid molecule in the cytoplasm. Tests of various lipid molecules on the KACh channel function showed that only unsaturated free fatty acids such as oleic, linoleic, and arachidonic acids were potent inhibitors of ATP-dependent gating. These findings indicate that downregulation of the KACh current by cytoplasmic unsaturated free fatty acids, achieved by interfering with ATP-mediated changes in KACh channel gating, is an important mechanism for the control of heart rate and synaptic transmission in the brain.
MATERIALS AND METHODS
Cell Preparation
Cultured atrial cells from 1-d-old newborn rats were prepared as described previously (Kim 1991). Animals were used in accordance with the Guide for the Care and Use of Laboratory Animals (No. 85-23; National Institutes of Health). In brief, atrial tissues were cut into small pieces (<1 mm3) with a sharp blade, and incubated in Hank's balanced salt medium containing 0.05% collagenase (Type 2; Worthington Biochemical Corp.) and 0.06% trypsin (from bovine pancreas; Sigma Chemical Co.). After 10 min of agitation, suspended cells were removed and placed in the culture medium consisting of Dulbecco's Modified Eagle's Medium, 10% fetal calf serum, and 0.1% penicillin-streptomycin. Cells were plated on glass coverslips and placed in a 37°C incubator gassed with 5% CO2/95% air for 18–24 h. The CA3 region of the hippocampus from adult rat (Oh et al. 1995) was cut into ∼500-μm-thick slices and placed in a spinner flask containing 10 ml of the same solution with 0.33 mg/ml trypsin and kept at 30°C (Worthington Biochemical Corp.). The enzyme solution was stirred gently and slowly. Slices were placed in 1 ml of solution containing 0.1 mg DNase I (Boehringer Mannheim Biochemicals) and triturated gently with a fire-polished Pasteur pipette. 5–10 μl of the solution containing dissociated cells was placed in the recording chamber and cells were allowed to settle down on glass coverslips. Xenopus oocytes were injected with cRNAs (∼0.5 ng/nl) transcribed from linearized plasmids (pBluescript) containing GIRK1, GIRK2, or GIRK4, as described previously (Kim et al. 1997). Injected oocytes were incubated for ∼2 d at 19°C in Barth's solution. Vitelline membrane was mechanically removed before forming patches.
Electrophysiology
Gigaseals were formed using Sylgard-coated, thin-walled borosilicate pipettes (Kimax) with ∼4 MΩ resistances. Channel currents were recorded with an Axopatch 200 patch-clamp amplifier (Axon Instruments), digitized with a PCM adapter (VR10; Instrutech Corp.), and stored on video tape using a video tape recorder. The recorded signal was filtered at 3 kHz using an eight-pole Bessel filter (−3 dB; Frequency Devices Inc.) and transferred to a Dell computer using the Digidata 1200 interface (Axon Instruments) at a sampling rate of 20 kHz. The filter dead time was ∼100 μs (0.3/cutoff frequency, and therefore events <50 μs will be missed in our analysis. Continuous single-channel currents were then analyzed with the pClamp program without further filtering (Version 6.0.3). Data were analyzed to obtain amplitude histogram and channel activity (nP o). n is the number of channels in the patch, and P o is the probability of a channel being open. nP o was determined from 1–2 min of channel recording. Current tracings shown in figures were filtered at 100 Hz except for expanded tracings, which were filtered at 1 kHz. Data are represented as mean ± SD. Student's t test was used to test for significance between two values at the level of 0.05.
Single Channel Analysis
As every patch contained multiple openings, a maximum likelihood algorithm was used to determine single channel kinetic parameters from idealized patch clamp data (Qin et al. 1996). This well-described method can be used on data containing multiple channel openings and estimates all transition rates. Only patches showing up to four channel opening levels were used for analysis. The best kinetic scheme among six schemes consisting of two or three closed states and two open states was the one shown in Fig. 2 B, as judged by the maximum log likelihood values for each fit (Qin et al. 1996). This model was previously also found to best describe atrial KACh channel kinetics (Nemec et al. 1999). After the rate constants were determined from 3,000–6,000 openings, open time constants were determined by calculating the reciprocal of the transition rate leading away from that state. A scheme with a single open state (Fig. 2 B, dotted line) was used to obtain the mean open time when the two open-time constants were close (within 15%) and maximum log likelihood value for the fit was higher than the value obtained using the scheme with two open states.
Figure 2.

Summary data showing the effect of ATP and cytosolic fraction on channel activity and open time constants. (A) Channel activity (NPo) was determined at various patch conditions. (B) Open time constants were calculated from the rate constants (τo1 = 1/k −4 and τo2 = 1/k −3) obtained by fitting the data to the two kinetic schemes. The kinetic scheme in dotted lines was used to analyze channel kinetics when currents were recorded with GTP or cytosolic fraction. 5 mg/ml protein cytosolic fraction was used for all experiments. Each bar shows the mean ± SD of four values. *Significant difference from the value of the first bar (P < 0.05).
Preparation of Cytosol
Tissues (∼2 g) were homogenized for ∼30 s in ice using a polytron in 5 ml HEPES-buffer containing 50 mM HEPES, 5 mM EDTA, pH 7.4, 0.5 mM dithiothreitol, and 0.1 mM PMSF, and then centrifuged at 3,300 g for 20 min at 4°C (J2-21M, rotor JA-21; Beckman Instruments, Inc.). The supernatant was collected and mixed with same volume of cytoplasmic extract buffer containing 140 mM KCl, 3.0 mM MgCl2, and 30 mM HEPES, pH 7.9. This mixture was centrifuged at 100,000 g for 1 h using an L-60 ultracentrifuge (rotor, 70-TI; Beckman Instruments, Inc.). The supernatant was collected and placed in a dialysis tubing (molecular weight cutoff, 10 kD; Spectrum Medical Industries, Inc.) and dialyzed in 4 liters of solution containing 140 mM KCl, 2 mM MgCl2, 5 mM EGTA, 0.5 mM dithiothreitol, 0.1 mM PMSF, 1 mg/ml leupeptin and 1 mg/ml pepstatin A, and 10 mM HEPES, pH 7.2, for 12 h at 4°C. This cytosolic fraction (20 mg protein/ml) was then stored at −70°C. Protein concentration was determined by Bradford assay. The lipid component of the cytosolic fraction was prepared by vigorous mixing with the same volume of chloroform for ∼1 min. After centrifugation, organic phase was transferred and evaporated completely under nitrogen. Perfusion solution was then added and the mixture sonicated for 5 min before applying to the patch.
Solutions and Materials
The pipette and bath solutions contained 140 mM KCl, 0.5 mM MgCl2, 10 mM HEPES, and 5 mM EGTA, pH 7.2. To change solutions perfusing the cytosolic surface of the inside-out patches, the pipette with the attached membrane was brought to the mouth of the polypropylene tubing through which the desired solution flowed at a rate of ∼1 ml/min. For studies using ATP, amounts of MgCl2 and ATP were determined to produce desired concentrations of free Mg2+ and MgATP using EQCAL software (Biosoft). The free Mg2+ concentration in solutions was always kept constant at 0.5 mM. All experiments were performed at 24–26°C. Free fatty acids were purchased from Sigma Chemical Co., dissolved in chloroform to form a 50-mM stock solution, and stored at −70°C under nitrogen. For free fatty acids supplied in dried powdered form, they were dissolved in either ethanol or DMSO, and subsequently sonicated in the perfusion solution at desired concentrations. ACh, GTP, and ATP were purchased from Boehringer Mannheim Biochemicals. Trypsin (porcine pancreas, Type 2), chymotrypsin, papain, and fatty acid-free albumin were purchased from Sigma Chemical Co. Purified bovine βγ subunit and GTPγS were purchased from Calbiochem Corp. Phosphatidylinositol-4,5-bisphosphate (PIP2), phosphatidylcholine, and phosphatidylethanolamine were purchased from Sigma Chemical Co. Mono- and diacylglycerols were purchased from Biomol and Sigma Chemical Co., and dissolved in chloroform similar to free fatty acids. All other lipids were first dissolved in chloroform and kept at −80°C freezer. The solvent (chloroform) was evaporated and free fatty acids were dissolved by sonication (W-380; Heat Systems-Ultrasonics, Inc.) on ice for ∼10 min in bath recording solution at a desired concentration. The final ethanol or DMSO concentration in the perfusion solution used was <0.1% and had no effect on the KACh channel function.
RESULTS
Presence of an Endogenous Inhibitor of KACh Channel in the Cytoplasm
We first confirmed the marked difference in KACh channel gating kinetics observed in the presence and absence of intracellular ATP using both outside-out and inside-out patches. The potential role of intracellular ATP in agonist-induced KACh current has not been previously studied using outside-out patches. Fig. 1 A shows current recordings from outside-out patches excised from atrial cells. Pipette (intracellular) solution contained GTP (100 μM), or both GTP and ATP (4 mM). These nucleotide concentrations are within the physiological levels found in mammalian heart cells. Extracellular application of 10 μM ACh resulted in activation of KACh channels in both cases as expected, but the channel activity (nP o) was approximately fivefold higher with ATP in the pipette solution than without it. In the presence of GTP alone, only a single open state with an open time constant of 1.0 ± 0.1 ms was sufficient to describe the channel openings. In the presence of GTP and ATP, an additional open state with longer-lived openings was detected (see Fig. 2 B and 3). These results clearly confirm the profound effect of ATP on KACh channel gating that we described in earlier studies. To show that an inhibitor of ATP-dependent gating is present in the cytoplasm of atrial cells, cytosolic fraction (5 mg protein/ml) prepared from rat atrial tissues was included in the pipette along with GTP and ATP. In the presence of the cytosolic fraction, long-lived channel openings were no longer present, indicating that atrial cytosolic fraction contains a substance that inhibits ATP-dependent gating of the KACh channel, and thus reduces the magnitude of KACh channel activation by ACh.
Figure 1.

Inhibition of the ATP-induced changes in KACh channel function by atrial cytosolic fraction (CF). (A) Activation of KACh channels in atrial outside-out patches by 10 μM ACh. Pipette solution contained nucleotides as indicated: 100 μM GTP, 100 μM GTP and 4 mM ATP, or 100 μM GTP, 4 mM ATP, and 5 mg protein/ml CF. (B) With 10 μM ACh in the pipette, KACh channels in inside-out patches were activated by 100 μM GTP. Subsequent effects of 4 mM ATP, atrial CF, and cramming of the patch into an atrial cell cytoplasm are shown. Membrane potential was held at −60 mV and inward currents were recorded.
As reported previously, the mean open time of the KACh channel in cell-attached patches was 1.0 ± 0.1 ms, and very few or no long-lived openings were observed, presumably due to the intact cytosol, which contains the substance that inhibits the ATP-dependent gating (Fig. 1 B). In inside-out patches with ACh in the pipette, GTP applied to the bath solution activated KACh channels, and further addition of ATP (4 mM) produced an approximately fivefold increase in nP o, similar to that observed in outside-out patches. The [ATP]–nP o relationship showed that K 1/2 was 32 ± 8 μM (n = 4) and the Hill coefficient was 1.9 ± 0.2 (n = 3). The effect of ATP did not reverse after its washout for at least 10 min, provided that the patch gigaseals were formed with a mild suction pressure of less than ∼3 mmHg. When the cytosolic fraction (5 mg protein/ml) was added together with GTP and ATP to the bath solution, ATP-induced long-lived openings were no longer observed and the channel activity decreased significantly (Fig. 1 B). In agreement with this result, a rapid inhibition of the ATP effect was observed when the patch containing ATP-modified KACh channels was crammed through the plasma membrane and into the cytosol of a spherically shaped cultured atrial cell (Fig. 1 B). The combined results from outside-out and inside-out patches are summarized in Fig. 2A and Fig. B, which shows changes in channel activity and open time durations, respectively. These results, particularly those obtained using outside-out patches, show that an inhibitor of ATP-dependent gating is present in atrial cytoplasm and that it plays a crucial role in keeping the KACh channel activity downregulated in the presence of an agonist.
As nearly all patches contained multiple openings, we were unable to determine open time durations using the pClamp programs. Therefore, the kinetic scheme shown in Fig. 2 B was used to fit the channel data, from which transition rates between states and open time constants could be extracted (Qin et al. 1996). In the presence of GTP or cytosolic fraction, the channel openings could be better described by the kinetic scheme with only one open state (dotted lines). We were able to obtain a few patches that showed low channel activity and mainly one level of opening, which we used to obtain duration histograms using the pClamp program. As shown in Fig. 3, the open time histograms obtained from channel openings in the presence of GTP and GTP with ATP were best fitted with one and two exponential functions, respectively. For the closed-time histograms, best fits were obtained when three exponential functions were used with or without ATP. These results are consistent with the three closed–two open state model, where the number of open states depends on whether ATP is present or not.
Figure 3.

Open- and closed-time histograms determined from channel recordings from inside-out patches. With ACh present in the pipette, KACh channel was activated by applying GTP 100 μM), or GTP and ATP (4 mM) to the bath solution. Although several channels were present in each patch, the level of activity was low such that mainly one channel was open. Histograms were fitted with one, two, or three exponential functions, and the best fits are shown. In this patch, the open time constants were 1.0 ms with GTP, and 1.2 and 6.7 ms with GTP and ATP. The closed time constants were 1.4, 8.8, and 30.4 ms with GTP and 0.8, 4.9, and 54.4 ms with GTP and ATP. Similar results were obtained in two other patches.
The Endogenous Inhibitor Is a Lipid Molecule
Our previous study suggested that the inhibitor might be a protein as treatment with proteases (with trypsin or α-chymotrypsin for ∼30 min, 37°C) abolished its effect on the ATP-induced changes in KACh channel kinetics. However, we now acknowledge that this suggestion was premature, as incubation of the cytosolic fraction in room air at 37°C for a prolonged period of time (∼30 min) was also found to markedly reduce the modulatory activity of certain lipids on the KACh channel (Kim and Bang 1999). This time, the cytosolic fraction (5 mg protein/ml) was treated with a mixture of proteases containing trypsin, chymotrypsin, and papain (1 mg/ml each) for a shorter period of time to minimize oxidation of lipid molecules (10 min at 37°C). This protease-treated cytosolic fraction still showed an inhibitory activity on the ATP-dependent gating of the KACh channel (Fig. 4 A). Most proteins were degraded by this protease treatment, as judged by SDS-gel electrophoresis, suggesting that the cytosolic inhibitor is not a protein. To test the possibility that lipids such as free fatty acids could be involved in such inhibition, we applied fatty acid–free albumin (50 μg/ml) in the presence of cytosolic fraction. Surprisingly, albumin caused an almost complete reversal of the inhibition of ATP-dependent gating produced by the cytosolic fraction, suggesting that the inhibitor could be a fatty acid or another lipid molecule that binds albumin. To obtain further proof that the inhibitor is a lipid molecule, the lipid component of cytosolic fraction was extracted using chloroform, dried, sonicated in perfusion solution under nitrogen, and directly applied to inside-out patches. No protein was detected in the extracted lipid (<1 μg/ml). As shown in Fig. 4 B, the lipid component extracted from atrial or brain cytosolic fraction was able to fully inhibit the ATP-dependent gating of the KACh channel, and the lipid effect could be removed by adding fatty acid–free albumin (50 μg/ml). The expanded current tracings clearly show the marked changes in the open time duration in the presence and absence of the lipid fraction (see Fig. 5).
Figure 4.
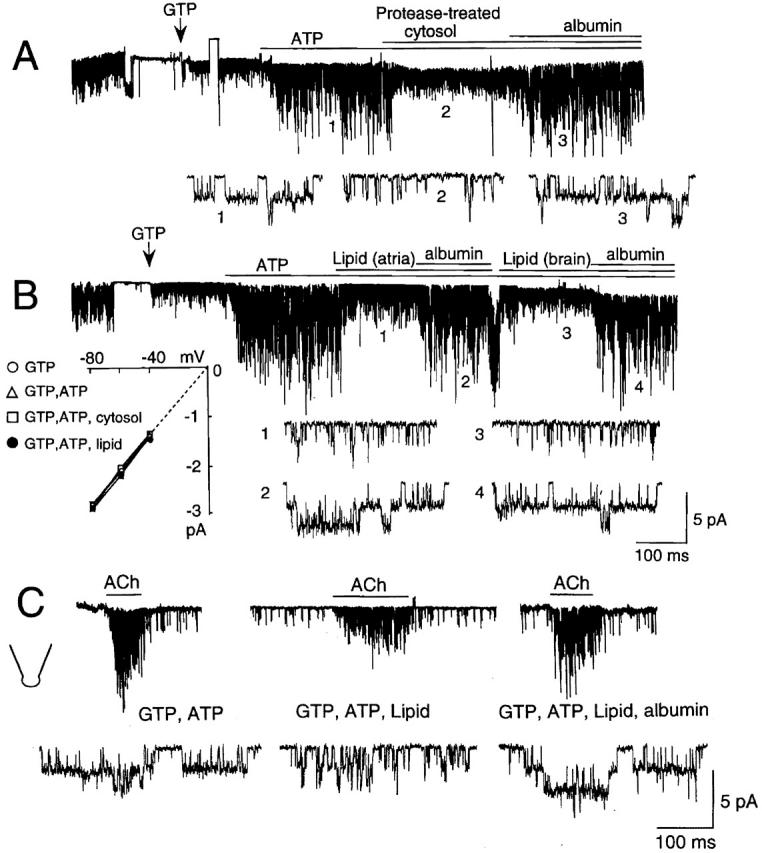
Inhibition of ATP-dependent gating of the KACh channel by protease-treated cytosolic fraction and by the lipid component of the cytosolic fraction (CF). Inside-out patches were formed and membrane potential was held at −60 mV. (A) Inhibition of the ATP effect by protease (mixture of 1 mg/ml trypsin, chymotrypsin, and papain)-treated atrial CF and subsequent reversal of the CF effect by fatty acid–free bovine serum albumin (50 μg/ml). GTP (100 μM) was applied at the arrow and was present throughout the experiment. Expanded current tracings at specific times are shown as indicated by the numbers. (B) Inhibition of the ATP effect by chloroform-extracted (lipid) component of the atrial and brain CF (20 mg protein/ml stock). The concentration of the total lipid has not been determined. Fatty acid–free albumin (50 μg/ml) reversed the inhibition by lipid fraction. Inset shows current–voltage relations (n = 3 each). (C) Activation of KACh channels by ACh in outside-out atrial patches. Pipette solution contained components as indicated. Expanded current tracings show markedly different gating of the KACh channel under different conditions.
Figure 5.

Summary data showing the effect of atrial and brain cytosolic fractions and the chloroform-extracted lipid fraction on KACh channel activity (A) and open time constants (B). CF-P indicates protease-treated cytosolic fraction, and A indicates fatty acid–free albumin. Open time constants were calculated from the rate constants that were determined by fitting the single channel data to the kinetic schemes shown in Fig. 2 B using the QuB analysis. Each bar shows the mean ± SD of four determinations. *Significant difference from the value of the first bar (P < 0.05).
Using an experimental protocol identical to that described in Fig. 1 A, we tested whether the agonist-induced activation of KACh channels is also dependent on the presence of the lipid fraction. In outside-out patches, ACh-elicited KACh channel activity was markedly reduced after addition of the lipid extracted from atrial cytosolic fraction to the pipette, and this was again reversed by 50 μg/ml albumin (Fig. 4 C). Similar results were obtained when the lipid fraction was prepared from brain cytosolic fraction. The combined results obtained from inside-out and outside-out patches are summarized in Fig. 5. For kinetic analysis, we used patches containing two to four channel openings. As described above, the two kinetic schemes were used to obtain open time constants when ATP-induced gating was present. In the presence of cytosolic fraction or its lipid component, ATP-induced long-lived openings were absent and a single open state was sufficient to describe the channel kinetics. The current–voltage relationship of the GTP-activated KACh channel was unaffected by ATP, cytosolic fraction, or the lipid component (Fig. 4 B, inset). These results show that the endogenous factor that downregulates the KACh channel activity by interfering with the ATP-dependent modulation is a lipid substance that binds albumin.
Unsaturated Free Fatty Acids Inhibit ATP-dependent Gating
To identify the lipid molecule that inhibits ATP-dependent gating, we tested many different types of lipid molecules that are normally found in the cell (Table ). A lipid molecule (0.1–50 μM) was applied to the cytoplasmic side of inside-out patches containing ATP-modified KACh channels and concentration-dependent changes in channel activity and open time constants were determined. From these experiments, we found that unsaturated free fatty acids such as oleic, linoleic, arachidonic (AA), eicosapentaenoic, and docosahexaenoic acids were good inhibitors of ATP-dependent gating and thus the channel activity (Fig. 6). In each case, fatty acid-free albumin (50 μg/ml) was able to completely remove the inhibitory effect of free fatty acids. Linoleic acid produced a similar inhibitory effect when KACh channels were activated with purified bovine brain βγ subunit (20 nM; Fig. 6 D), showing that Giα and their downstream signaling molecules were not involved. Fig. 7 A shows concentration-dependent effects of four free fatty acids, a diacylglycerol, and the lipid derived from brain cytosol. The four unsaturated free fatty acids (oleic, linoleic, arachidonic, and docosahexaenoic acids) inhibited KACh channel activity with a K 1/2 of ∼1.5 μM. The Hill coefficients were ∼2 (1.8–2.2), similar to that obtained with the cytosolic lipid, indicating that at least two fatty acid molecules bind the channel or an associated regulatory molecule. The effect of free fatty acids on open time constants calculated from transition rates are shown in Fig. 7 B. Free fatty acids completely inhibited the appearance of the long-open state even in the presence of 4 mM ATP. As expected from the results shown in Fig. 6, when βγ-activated KACh channels were exposed to linoleic or AA (2 μM) first, further addition of 4 mM ATP produced only a small increase in channel activity (1.3 ± 0.1-fold; n = 5; not shown). AA, oleic acid, and linoleic acid (5 μM) added to the pipette solution (extracellular) did not inhibit ATP-dependent gating even after 5 min (n = 4 each). However, when a higher concentration (50 μM) of oleic acid was used, a partial inhibition (65 ± 6%; n = 6) of ATP-dependent gating was present after 5 min, indicating that enough free fatty acids probably crossed the lipid bilayer to reach the intracellular site to inhibit the KACh channel.
Table 1.
Effect of Lipids on ATP-dependent Gating of KACh Channel
| Substance | K 1/2 | |
|---|---|---|
| μM | ||
| Free fatty acids | ||
| Caprylic acid (8:0) | No effect | |
| Palmitic acid (16:0) | No effect | |
| Stearic acid (18:0) | No effect | |
| Palmitoleic acid [16:1 (n-7)] | 10.5 ± 2.4 | |
| Oleic acid [18:1 (n-9)] | 1.5 ± 0.2 | |
| Elaidic acid [t-18:1 (n-9)] | 21.6 ± 4.8 | |
| Linoleic acid [18:2 (n-6)] | 1.2 ± 0.2 | |
| Linoelaidic acid [t,t-18:2 (n-6)] | 3.2 ± 0.6 | |
| α-linolenic acid [18:3 (n-3)] | 6.2 ± 0.8 | |
| Arachidonic acid [20:4 (n-6)] | 1.7 ± 0.2 | |
| Eicosapentaenoic acid [20:5 (n-3)] | 5.6 ± 2.2 | |
| Docosahexaenoic acid [22:6 (n-3)] | 1.4 ± 0.2 | |
| Nervonic acid [24:1 (n-9)] | 12.5 ± 2.1 | |
| Mono- and diacylglycerol | ||
| 1-monooleoylglycerol | 42 ± 6 (nr) | |
| 2-monooleoylglycerol | 48 ± 8 (nr) | |
| 1-monolinoleoylglycerol | 8.4 ± 2.2 (nr) | |
| 1-monopalmitoleoylglycerol | 52 ± 7 (nr) | |
| 1-oleoyl-2-acetylglycerol | 22 ± 3 (nr) | |
| 1,2-dioctanoylglycerol | 2.8 ± 0.4 (nr) | |
| 1,2-dioleoylglycerol | 21 ± 3 (nr) | |
| 1-stearoyl-2-arachidonoyl-sn-glycerol | No effect | |
| 1-stearoyl-2-linoleoyl-sn-glycerol | No effect | |
| Others | ||
| Arachidonic acid methyl ester | 12 ± 3 (nr) | |
| Arachidonyl alcohol | 44 ± 7 (nr) | |
| Phorbol myristate acetate | No effect | |
| Sphingosine (D-form) | No effect | |
| Ceramide (C-2) | No effect | |
| Phosphatidylserine | No effect | |
| Lysophosphatidylethanolamine | No effect | |
| Spermine | No effect | |
| Spermidine | No effect | |
| Glycerophosphate | No effect | |
| Glycerol | No effect | |
| Leukotriene C4 | No effect | |
| Prostaglandin E1 | No effect | |
Figure 6.
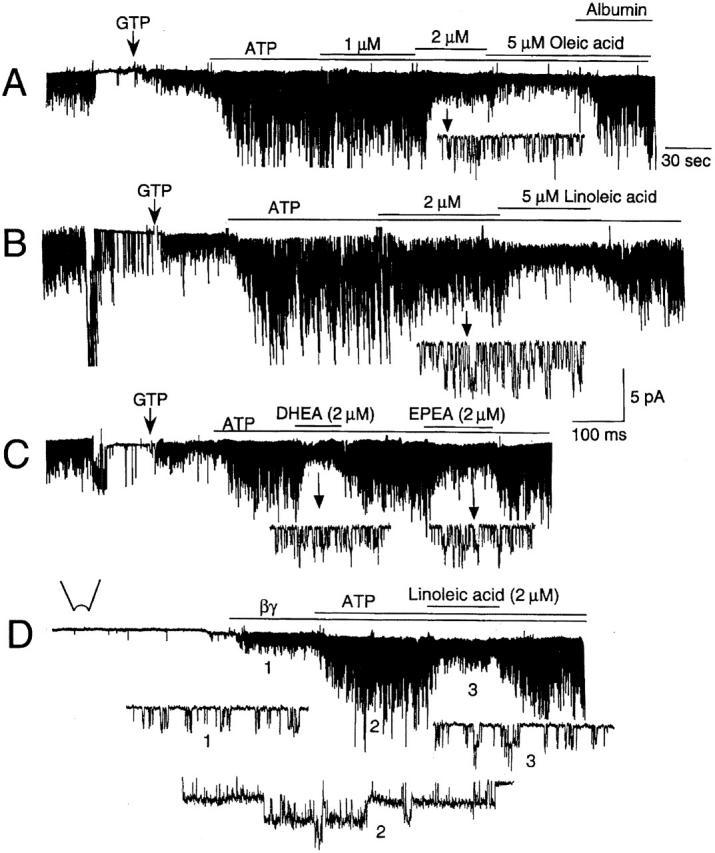
Unsaturated free fatty acids inhibit ATP-dependent gating of KACh channel. Cell-attached patches were first formed with ACh in the pipette and inside-out patches formed subsequently. Membrane potential was held at −60 mV. (A) In the presence of GTP (100 μM) and 4 mM ATP, oleic acid was applied with or without fatty acid–free albumin (100 μg/ml). (B) Similar to A, except that linoleic acid was used. In this patch, opening and fast rundown of several ATP-sensitive K+ channels can be seen. (C) Two different free fatty acids (docosahexaenoic, DHEA, and eicosapentaenoic, EPEA) were used at 2 μM to inhibit the KACh channel activity. (D) In the absence of ACh, βγ (bovine brain, 20 nM) was applied to activate the KACh channels. Linoleic acid (2 μM) reversed ATP-induced modification of the channel kinetics. Expanded current tracings at different times are shown (arrows or numbers).
Figure 7.

Effects of free fatty acids on KACh channel activity and open time constants. Membrane potential was held at −60 mV. (A) To inside-out patches containing ATP-modified KACh channels, a lipid was added at increasing concentrations (0–5 μM). Relative channel activity was determined at each lipid concentration. Data were fitted to the Hill equation nP o = {0.9/[1 + ([FA/K d]N)] + 0.1}, where FA is the lipid species, K d is concentration at which half maximal inhibition occurs, and N is the Hill coefficient (1.8–2.2). Lipid fraction of the cytosol was also tested at different dilutions. Undiluted original cytosolic lipid was prepared by adding 1 ml of bath solution to dried lipids obtained from 1 ml of CF (20 mg protein/ml), and corresponds to the 100 μM free fatty acid on the x scale. (B) Open time constants calculated from transition rates obtained after fitting the data to the two kinetic schemes shown in Fig. 2 B. Each bar is the mean ± SD of four determinations. *Significant difference from the value determined in the absence of ATP.
AA produced the same inhibitory effect in the presence of indomethacin (50 μM; cyclooxygenase inhibitor) or nordihydroguaiaretic acid (10 μM; lipoxygenase inhibitor), indicating that AA itself is the inhibitor (n = 5 each). This is supported by the results that fatty acids that are not substrates for cyclooxygenases or lipoxygenases (i.e., oleic acid) are also potent inhibitors of ATP-dependent gating. Therefore, free fatty acids probably acted directly rather than via their metabolites. Treatment of patches with calphostin-C (protein kinase C inhibitor, 1 μM, 10 min), neomycin (phospholipase C inhibitor, 50 μM, 15 min), or pirenzepine (M1 receptor antagonist, 0.1 μM) also did not affect the inhibition by AA (n = 5 each). These results show that the two enzymes that can be activated by free fatty acids (Meves 1994) and by M1 receptor stimulation are not involved. The inhibitory effects of AA and oleic acid were not affected by a mixture of phosphatase inhibitors (3 mM sodium orthovanadate, 2 mM sodium fluoride, and 0.1 mM zinc sulfate), indicating that phosphatases are also not involved (n = 4 each).
Interestingly, synthetic diacylglycerols and several monoacylglycerols were also effective in inhibiting the ATP-dependent gating, albeit at higher concentrations (Table ). Their inhibitory effect, however, could not be easily removed by washout or fatty acid–free albumin (100 μg/ml). The inhibitory effect of diacylglycerols and monoacylglycerols could be due to long-chain fatty acids present in these molecules. The naturally occurring diacylglycerols (steroyl-arachidonoyl-glycerol and steroyl-linoleoyl-glycerol), however, showed no inhibitory effect even at 100 μM. Methyl ester and alcohol derivatives of arachidonic acid also caused inhibition of KACh channel activity at relatively high concentrations, indicating that the free carboxyl group is not necessary for inhibition of ATP-dependent gating (Table ). Polyamines (Lopatin et al. 1994), which cause inward rectification of K+ channels, also had no effect. Of the lipids listed in Table , only unsaturated free fatty acids showed inhibitory properties nearly indistinguishable from that of cytosolic fraction or its the lipids derived from it. As predicted, cytosolic fraction (5 mg protein/ml) prepared from rat liver, kidney, lung, ventricle, and skeletal muscle were also effective in inhibiting ATP-dependent gating (n = 3 each).
Unsaturated Free Fatty Acids Inhibit PIP2- and Phospholipid-dependent Gating
It has been reported that the ATP-induced change in K+ channel function may occur via generation of PIP2 in the membrane as a result of phosphorylation of PI and PIP by lipid kinases (Hilgemann and Ball 1996; Hilgemann 1997). Although less effective than ATP, intracellularly applied PIP2 was found to increase the number of long-lived openings (τo1, 2–3 ms) and KACh channel activity (Kim and Bang 1999). Therefore, it was of interest to test whether free fatty acids could inhibit PIP2-induced changes in KACh channel kinetics. After activation of KACh channels by applying GTPγS to inside-out patches, 30 μM PIP2 was further added. This resulted in approximately twofold increase in channel activity and the appearance of a second open state with an open time constant of 2.6 ± 0.3 ms (Fig. 8). The effect of PIP2 on the KACh channel was always small compared with that produced by ATP, which produces an approximately fivefold increase in channel activity and τo1 of 7–8 ms. Nevertheless, when AA (2 μM) was applied together with PIP2, AA fully reversed the effect of PIP2 (Fig. 8B and Fig. C). Although not shown, oleic (2 μM) and linoleic (2 μM) acids also inhibited PIP2-dependent gating of the KACh channel (n = 3 each).
Figure 8.

Inhibitory effect of AA on PIP2-induced changes in KACh channel kinetics. After activation of the KACh channels with GTPγS (100 μM), PIP2 (30 μM) was applied to the cytosolic side of the membrane. After ∼1 min, AA was applied along with PIP2, and then only AA was washed off. Expanded current tracings show the mild stimulatory effect of PIP2 on channel activity (B) and open time duration (C). PIP2 induced a second open state with a time constant of 2.6 ms. AA fully reversed these effects of PIP2 (B and C). Each bar is the mean ± SEM of four determinations. *Significant difference from the value determined with GTPγS only.
As phosphatidylcholine (100 μM) and phosphatidylethanolamine (100 μM) have been shown to produce effects similar to PIP2 on KACh channel kinetics (Kim and Bang 1999), we also tested whether AA reverses the changes produced by these two phospholipids using inside-out patches. Our results showed that phosphatidylcholine and phosphatidylethanolamine increased the number of long-lived openings such that τo1 values were 2.5 ± 0.6 and 2.8 ± 0.5 ms, respectively (n = 3). After application of 2 μM AA, only one open state with a time constant of 0.9 ± 0.1 ms was present for both phospholipids. Application of 2 μM linoleic acid resulted in a similar inhibition such that the open time constant was 0.8 ± 0.1 ms (n = 3). Therefore, our results show that unsaturated free fatty acids inhibit the appearance of long-lived openings; i.e., abolish the second open state, regardless of which substance was used to induce it.
Concentration-dependent Effect of Arachidonic Acid on KACh Channel Kinetics
To understand in more detail the effect of a free fatty acid on the KACh channel function, we determined the concentration-dependent effect of AA on transition rates between all connected states in our kinetic scheme. As shown in Fig. 9, KACh channel openings in the cell-attached or in the inside-out state with GTP alone could be described by a linear kinetic scheme with only one open state. The averaged transition rates determined using the QuB analysis from three cells with similar channel activities are shown on the right. Under the experimental conditions of our study, the channel activity in the inside-out patch with 100 μM GTP alone was always lower than that in the cell-attached patch from the same cell. However, the open time constant remained unchanged from that in the cell-attached state (0.9–1.0 ms). Application of 4 mM ATP produced an additional open state with long-lived openings (O1) and also increased the transition rate to the longest closed state (C1). The latter accounts for the many long closings observed in the presence of ATP in inside-out patches, with a mean closed time of 62 ms. At 0.5 μM AA, the primary effect was a decrease in the open time constant (O1; Fig. 10 A), without a change in the transition rate to the longest closed state (C1). At higher AA concentrations, the transition rate to C1 was greatly reduced, in addition to a further decrease in the open time constant (O1). Transition rates between all connected states were affected by AA, indicating that AA acts on both closed and open states. The results show clearly that 4 mM ATP and 2 μM AA together can reestablish the channel kinetics observed in the cell-attached state. A mixture containing 0.5 μM each of oleic acid, linoleic acid, AA, and docosahexaenoic was as effective as 2 μM AA. The effect of AA was voltage-insensitive (Fig. 10 B). Furthermore, the K 1/2 values for inhibition by linoleic acid with pipette solution containing 70, 140, and 210 mM K+ were 1.6 ± 0.4, 1.5 ± 0.3, and 1.7 ± 0.4 μM, respectively (n = 3 each). Therefore, the fatty acid effect is unlikely to involve sites within the pore of the channel.
Figure 9.

Inhibitory effect of arachidonic acid on the KACh channel. A cell-attached patch was formed with ACh in the pipette. After forming an inside-out patch, 100 μM GTP was added to activate the KACh channels. ATP (4 mM) was then added to further increase the channel activity. In the presence of GTP and ATP, 0.5 μM AA was applied and its concentration increased stepwise to 5 μM, and then washed off. Expanded current tracings are shown for different experimental conditions. For each condition, channel data were fitted to one of two kinetic schemes (Fig. 2 B) and transition rates between all connected states were determined. Averaged transition rates determined from three similar patches are shown on the left. The linear scheme was used whenever the open time constants of the two open states were close (i.e., within 15% of each other).
Figure 10.

Effect of AA on open time constants and on the membrane potential–dependent effect on channel activity. (A) Open-time constants were calculated from the transition rates determined from Fig. 9 as described in materials and methods and plotted as a function of [AA]. *Significant difference from the value obtained without AA (P < 0.05). (B) Channel activity was determined at different membrane potentials. The value at −60 mV was taken as 1.0 (GTP + ATP). Each point is the mean ± SEM of four determinations.
Inhibition of KACh Channel Activity by Arachidonic Acid in the Absence of ATP
The inhibition of the ATP-induced increase in mean open time and activity of the KACh channel by unsaturated free fatty acids could be due to a selective block of the ATP-induced effect on the KACh channel. It could also be due to a nonselective effect, affecting the KACh channel regardless of whether the channel is in the ATP-modified state or not. To address this issue, we studied the effect of AA on GTP-activated KACh channels in inside-out patches with ACh in the pipette in the absence of ATP. As described previously, application of 100 μM GTP to the cytoplasmic side of the membrane caused an immediate activation of the KACh channels (Fig. 11 A). AA was then applied to the membrane starting at 0.1 μM and the concentration was increased progressively to 10 μM. Channel activity and mean open times were determined and plotted as a function of AA concentration (Fig. 11C and Fig. D). The concentration at which channel activity decreased by half was 1.6 ± 0.4 μM and the Hill coefficient was 1.7. These values are very close to those obtained from patches in which the KACh channels were studied in the presence of ATP (K 1/2 = 1.7 μM, Hill coefficient = 1.9). AA also caused a significant shortening of the open time duration in the absence of ATP, but only at high concentrations of the free fatty acid. Similar inhibitory effects were observed when purified bovine βγ (100 nM) was used to activate the KACh channel (K 1/2 = 1.5 μM, Hill coefficient = 1.9; mean of two values; data not shown), suggesting that AA may be interfering with βγ-KACh channel interaction to reduce channel activity. The approximately eightfold decrease in the open time constant (τo1) of the ATP-modified KACh channel produced by AA cannot easily be explained by such a mechanism, as 1 μM AA has a negligible effect on the open time constant in the absence of ATP. Changes in [GTP] and, by inference, the amount of βγ available for interaction with the KACh channel, does not affect the open time constant significantly in atrial cells. Therefore, these results suggest that free fatty acids produce two separate effects: a decrease in frequency of opening and inhibition of ATP-induced long-lived openings. Both of these mechanisms are probably involved in AA-induced decrease in channel activity.
Figure 11.

Effect of AA on KACh channel activity and open-time duration in the absence of ATP. (A) KACh channels in an inside-out patch were activated by 100 μM GTP. AA was then added at different concentrations. (B) Channel openings at higher time resolution are shown for each concentration of AA. (C) The channel activity–[AA] relationship is shown. Data were fitted to the Hill equation: nP o = {1/(1 + [AA/K d]N)}, where K d is the concentration at which half maximal inhibition occurs, and N is the Hill coefficient (1.7). K d was 1.6 μM. (D) Open-time constants were determined and plotted as function of [AA]. *Significant difference (P < 0.05). Each point is the mean ± SEM of four determinations.
Effect of Free Fatty Acids on the KACh Channel in Hippocampal Neuron and in Oocytes Expressing GIRK
To determine whether free fatty acids also downregulate KACh channels in neurons and cloned KACh channels expressed in oocytes, we tested their effects on KACh channels in hippocampal pyramidal neurons and in oocytes expressing GIRK1/4 and GIRK1/2. In neurons, 5-hydroxytryptamine (5-HT) was used as agonist to activate the KACh channel as we have done previously (Oh et al. 1995). In the presence of 10 μM 5-HT in the pipette, application of 100 μM GTP to inside-out patches activated the KACh channels with an open time constant of 1.0 ± 0.1 ms (n = 6; Fig. 12 A). Only a single open state was sufficient to describe the channel openings, as assessed from the duration histogram as well as fitting the data to the kinetic schemes with one or two open states. Addition of 4 mM ATP produced appearance of another open state with long-lived openings (Fig. 12 D), similar to that observed in atrial cells. When 2 μM linoleic acid was applied together with GTP and ATP, the long-lived openings were no longer present and channel openings again showed only one open state with a time constant of 1.0 ± 0.1 ms. Oleic acid produced a similar effect on the ATP-modified KACh channel in these neurons (n = 3; Fig. 12 D).
Figure 12.
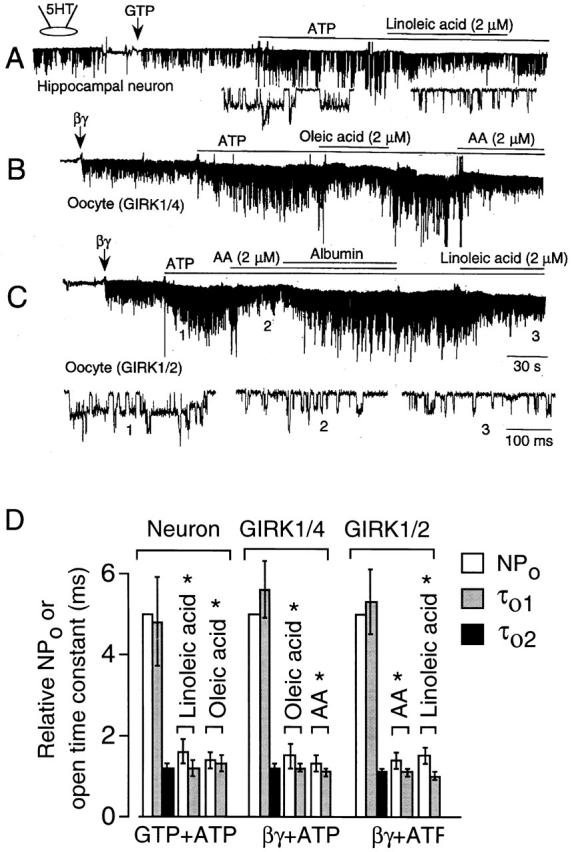
Inhibition of ATP-dependent gating by free fatty acids in neurons, and in oocytes expressing GIRK. Inside-out patches were formed and membrane potential held at −60 mV. (A) 5-HT was used as the agonist. Reversible inhibition by linoleic acid of the ATP-dependent gating of the KACh channels in an adult rat hippocampal neuron is shown with accompanying expanded tracings. (B) The cardiac (GIRK1/4) form of the KACh channel subunits were expressed at low levels in Xenopus oocytes and activated by βγ (50 nM). ATP produced long-lived openings as in atrial cells. Oleic acid, AA, and fatty acid–free albumin (100 μg/ml) were applied as indicated. (C) The neuronal (GIRK1/2) form of the KACh channel subunits were expressed at low levels in Xenopus oocytes and activated by βγ (50 nM). After adding ATP, AA and linoleic acids were applied separately. Albumin was used to reverse the effect of AA. Expanded current tracings show the effect of free fatty acids on the kinetics of channel openings. (D) Summary of changes in relative channel activity and open-time constants determined from above experiments is shown. *Significant difference from the control values obtained in the absence of fatty acid (P < 0.05). Open time constants were calculated from the transition rates. Each bar is the mean ± SD of four determinations.
When expressed in Xenopus oocytes, heteromeric GIRK1/4 that represents the atrial KACh channel also undergoes ATP-dependent gating (Kim et al. 1997). Similar to native KACh channels, oleic and arachidonic acids blocked the ATP-induced changes in GIRK1/4 channel kinetics (Fig. 12 B). Heteromeric GIRK1/2, which represents one form of neuronal KACh channels (Kofuji et al. 1995) was also modified by ATP and free fatty acids in a similar way (Fig. 12 C). Thus, free fatty acids inhibited the appearance of long-lived openings induced by ATP. As in atrial KACh channels, GIRK channels modified by ATP could be described by the same kinetic scheme consisting of three closed and two open states. However, after treatment with a free fatty acid, the channel openings could be fitted better to a scheme with only one open state (Fig. 12 D). Thus, our results show that native KACh and cloned heteromeric GIRK channels expressed in oocytes have nearly identical responses to ATP and free fatty acids.
Free Fatty Acids Do Not Inhibit Background (K1) and ATP-sensitive K+ Channels
Other members of the Kir family of K+ channels including the classical background (IRK, K1) and ATP-sensitive K+ channels (Kir6/SUR and ROMK) have also been shown to be activated by ATP or PIP2 (Takano et al. 1990; Fakler et al. 1994; Baukrowitz et al. 1998; Huang et al. 1998; Shyng and Nichols 1998). Therefore, we asked whether unsaturated free fatty acids also inhibit ATP-induced changes in the function of these Kir channels. To address this question, we studied the effect of ATP and AA using cardiac membrane patches containing either K1 or KATP channels. Fig. 13 A shows an inside-out atrial patch in which KACh channels were first activated with GTP and ACh, and this served as a positive control. Although the K1 channel was not open initially, it was quickly activated by application of 4 mM ATP to the bath solution and remained activated as long as ATP was present. A similar effect of ATP was observed in all seven patches that contained either one or two K1 channels. As expected and shown by expanded current tracings, the marked effects of ATP on the KACh channel activity and open-time duration were also present in this patch. When 2 μM AA was further added, KACh channel activity decreased quickly with associated shortening of the open time constant to 1.0 ± 0.1 ms, but the K1 channel remained fully open and showed no change in its activity (nP o; 0.90 ± 0.04 vs. 0.87 ± 0.06; n = 6). In patches in which ACh and GTP were not used, K1 channels were also activated by intracellular application of ATP, but were not significantly affected by either 2 or 5 μM AA (n = 3 each). Similarly, 2 μM linoleic acid (n = 3), cytosolic fraction (5 mg protein/ml; n = 2), and the lipid fraction (n = 2) failed to significantly influence K1 channel kinetics (data not shown). Thus, these results clearly demonstrate that, at concentrations used here, unsaturated free fatty acids do not affect the ATP-induced activation of the K1 channel, but inhibit that of the KACh channel.
Figure 13.

Effect of AA on ATP- or PIP2-induced changes in K1 and KATP channels in cardiac myocytes. (A) A cell-attached patch was formed with ACh in the pipette. In the inside-out state, GTP was applied first, and then 4 mM ATP added subsequently. ATP activated the K1 channel and augmented KACh channel activity. Further application of 2 μM AA markedly inhibited KACh channel activity but had no effect on the K1 channel. Expanded current tracings show the fully open K1 channel and opening of KACh channels on top of the K1 channel. (B) Formation of the inside-out state of adult rat ventricular cells resulted in activation of several KATP channels that ran down at varying rates. Application of AA during channel rundown resulted in inhibition of KATP channel activity. (C) When PIP2 was applied to prevent rundown, addition of AA failed to inhibit KATP channel activity. The same results were obtained in four other patches. (D) In this patch, KATP channels were allowed to rundown first. ATP was then applied briefly to reactivate the channels. AA also markedly inhibited KATP channel activity in these patches (n = 4).
To test the effect of AA on KATP channels, we used inside-out patches formed from adult rat ventricular cells that usually show many KATP channels in each patch. In Fig. 13 B, six KATP channels were activated upon formation of the inside-out state. In this patch, KATP channel rundown was slow and allowed us to test the effect of AA. AA (5 μM) caused a marked inhibition of the KATP channel activity, and this was irreversible. Although 2 μM AA caused a significant inhibition (62 ± 12% inhibition, n = 3), we used 5 μM AA to obtain a near-maximal inhibition. Linoleic acid (5 μM) also caused a near maximal inhibition. These results show that AA is an effective inhibitor of the KATP channel, as we have described previously (Kim and Duff 1990). As PIP2 causes activation of KATP channels as well as prevents rundown, we tested whether AA inhibits the effect of PIP2 on the KATP channel. As shown in Fig. 13 C, PIP2 applied soon after patch excision prevented rundown of the KATP channels. Interestingly, AA was ineffective in KATP channel inhibition when was PIP2 present, and decreased channel activity only after PIP2 was removed. Therefore, in the presence of PIP2, the effect of 5 μM AA on the KATP channel was clearly different from that on the KACh channel. In Fig. 13 D, we allowed KATP channels to rundown first, and then briefly exposed the membrane to 3 mM ATP. This procedure is well known to reactivate the KATP channels, presumably via formation of PIP2 in the membrane. In this patch, AA also caused a quick inhibition of channel activity, similar to that observed in Fig. 13 B. These results show that 2–5 μM unsaturated free fatty acids affect various inwardly rectifying K+ channels differently, and suggest that ATP- or PIP2-induced changes in Kir channel function probably involve different, nonconserved regions of the channels.
DISCUSSION
Since 1991, when intracellular ATP was first shown to cause marked upregulation of the KACh channel activity in inside-out patches and to increase the number of long-lived openings (Kim 1991), we have suspected that a strong inhibitor of the ATP-induced changes in KACh channel function exists in atrial cells. Our suspicion arose mainly because of the observation that KACh channels in cell-attached patches generally do not show such ATP-dependent gating despite the presence of ATP in the cell. In 1996, we confirmed and reported the existence of such an inhibitor in the cytoplasm of atrial tissue and subsequently in brain tissue. Because treatment of the cytosolic fraction with proteases abolished its activity against the KACh channel, we thought that the endogenous inhibitor was a protein (Hong et al. 1996). We have recently recognized that the activity of the lipids could also be quickly reduced by oxidation when exposed to air at ambient temperatures. Therefore, we reexamined the possibility that the inhibitor is a lipid molecule rather than a protein. We now show evidence that the putative inhibitor is a lipid molecule present in the cytoplasm. Although it was not possible to test every lipophilic molecule that exists in the cell, our results show that certain unsaturated free fatty acids in the cytoplasm are most likely to be the endogenous inhibitor that blocks ATP-dependent gating. This probably explains why we were unable to purify the inhibitor using protein purification methods. An important result of our study is that free fatty acids together with intracellular ATP can reestablish the activity and kinetics of the KACh channels observed in the cell-attached state of intact atrial and neuronal cells.
Are Unsaturated Free Fatty Acids True In Vivo Inhibitors of ATP-dependent Gating?
It would be important to know whether free fatty acids in the cytoplasm are true in vivo inhibitors of the KACh channel. The following points support the role of endogenous free fatty acids in the control of the KACh channel function. (a) Studies using the cytosolic fraction and its lipid component clearly show that an endogenous lipid substance is inhibiting the ATP-dependent gating of the KACh channel. (b) Of the ∼40 different lipophilic compounds tested so far, only unsaturated free fatty acids were found to be potent inhibitors of ATP-dependent gating of the KACh channel with inhibitory properties indistinguishable from that of the cytosolic fraction or its lipid fraction. In earlier studies, intracellular molecules such as cAMP, cGMP, Ca2+, and inositol trisphosphate at physiological concentrations were found to have no significant effect on the KACh channel activity (Hong et al. 1996). However, Ca2+, at concentrations above 30 μM, decreased the open time constant of the KACh channel (Kim 1991). (c) Fatty acid–free albumin completely reversed the inhibitory effect of the cytosolic fraction, the lipid fraction, and unsaturated free fatty acids, and affected the degree of KACh channel activation by ACh in outside-out patches. (d) The biophysical effect of the cytosolic fraction and unsaturated free fatty acids on the ATP dependence of KACh channel gating kinetics were nearly indistinguishable. Taken together, these results provide strong evidence indicating that unsaturated free fatty acids are the in vivo inhibitors of the KACh channel. We do not know exactly which specific free fatty acids are true in vivo inhibitors since free concentrations of different free fatty acids in the cell are not known and difficult to determine (Veerkamp et al. 1991; VanderVusse et al. 1992).
The results of this study suggest that the cytoplasmic concentration of a free fatty acid that inhibits ATP-dependent gating is 1–2 μM. The actual concentration of free fatty acids that inhibits the ATP effect may be much lower, as oxidation of fatty acids probably occurs during preparation and experiment. In support of this, it was found that the effect of AA decreased significantly (K 1/2 = 5.5 ± 1.1 μM, n = 4) after ∼10 min exposure of the AA solution to air and, after 1 hr, K 1/2 was 12.2 ± 3.4 μM (n = 3), although all solutions were kept in ice. We also found that incubation of oleic, linoleic, and linolenic acids at room temperature or at 37°C for 30 min also reduced their inhibitory potency significantly. Different types of fatty acid binding proteins exist at high levels in the cell (∼1 mM), and bind fatty acids with high affinity (K d, ∼1–10 μM; Veerkamp et al. 1991). Therefore, although the concentration of a free fatty acid in the cytoplasm is not known, it is believed to be in the nanomolar range (Veerkamp et al. 1991). Since many unsaturated free fatty acids inhibit the ATP effect, the total free fatty acid concentration could reach near the micromolar range. It is also possible that the free fatty acid concentration near the inner surface of the membrane near the channels is higher than normally assumed, as free fatty acids in the plasma travel through the cell membrane to the cytosol by diffusion partly in the unbound form (Hamilton 1998).
Structural Requirements for Modulation of the KACh Channel by Free Fatty Acids
The inhibitory profile of free fatty acids shows that a long-chain fatty acid with at least one double bond is necessary for blocking ATP-dependent gating. Trans isomers (eladic and linoelaidic acids) were less potent than their cis counterparts, suggesting that some structural elements within the hydrophobic region may be important. Uncharged alcohol and methyl ester forms of AA inhibited ATP-dependent gating irreversibly, suggesting that the carboxyl group of free fatty acids is important for reversibility but is not necessary for inhibition. This supports the idea that the hydrophobic region of free fatty acids may be important for causing inhibition. Mono- and diacylglycerols were also found to be effective inhibitors of ATP-dependent gating, albeit at higher concentrations. The inhibition by mono- and diacylglycerols may be due to the fatty acids in these molecules and their low affinity may be explained by the reduced accessibility due to steric hindrance. Although these synthetic diacylglycerols showed inhibitory effect, naturally occurring diacylglycerols (steroyl-arachidonoyl glycerol and steroyl-linoleoyl-glycerol) were found to have no effect, presumably due to greater steric hindrance. Therefore, these results suggest that all unsaturated free fatty acids may be able to downregulate the KACh channel by inhibiting the ATP-dependent gating. It seems likely, however, that commonly found free fatty acids with high inhibitory potency, such as oleic, linoleic, linolenic, and arachidonic acids, are probably the major players in downregulating the KACh channel activity. The relative lack of effect of free fatty acids from the extracellular side is interesting, considering that fatty acids are thought to cross the membrane rapidly. Therefore, free fatty acids in the plasma are not expected to regulate KACh channel function to any significant degree. Fatty acids in plasma are also mostly bound to albumin (Veerkamp et al. 1991).
Mechanism of Inhibition of KACh Channel Activity by Free Fatty Acids
Our results show that the observed channel activity in cell-attached patches at steady state when ACh is present in the pipette is the sum of the effects produced by GTP (βγ)-induced activation, ATP-induced stimulation, and free fatty acid–induced inhibition. The inhibitory effect of free fatty acid on ATP-dependent gating seems to be the crucial factor that keeps the KACh channel in the short-lived, single open state and maintains low channel activity despite the presence of ATP in the cell. To understand how free fatty acids inhibit ATP-dependent gating, it is first necessary to identify the signaling pathway by which ATP modifies the KACh channel. The evidence from recent studies indicate that PIP2 formed in the membrane via lipid kinases mediates the effect of intracellular ATP on different ion transporters and channels (Hilgemann and Ball 1996; Hilgemann 1997). PIP2 itself does not activate the atrial KACh channel (Kim and Bang 1999), consistent with the result that ATP alone does not activate the KACh channel either. However, PIP2 has been shown to activate cloned inwardly rectifying K+ channels such as IRK1, GIRK2, ROMK1, and Kir6.2/SUR1 and native ATP-sensitive K+ channels (Huang et al. 1998; Sui et al. 1998; Fan and Makielski 1999; Ho and Murrell-Lagnado 1999). Available evidence suggests that the negatively charged PIP2 molecules in the membrane interact with several positively charged residues in the COOH terminus of K+ channels and thereby shifts the conformation of the channel to the “active” state. For the KATP channel, it has been suggested that ATP and PIP2 bind to overlapping sites on the COOH terminus of Kir6.2 and cause PIP2 to influence channel sensitivity to ATP (Baukrowitz et al. 1998; Shyng and Nichols 1998; Fan and Makielski 1999). Thus, the interaction of PIP2 with the K+ channel has been suggested to underlie the ATP-induced changes in the kinetic behavior of the K+ channel.
If PIP2 promotes the active state for all inward-rectifier K+ channels mentioned above by a common mechanism, a blocker of the PIP2 effect would be expected to reduce the active state of these K+ channels. Therefore, we predicted that if unsaturated free fatty acids inhibit ATP- and PIP2-dependent gating of KACh channels, they would also inhibit ATP- or PIP2-induced modification of KATP and K1 channels. Our results in Fig. 13 show, however, that AA (2–5 μM) does not inhibit PIP2-modified KATP channels or ATP-activated background K1 channels. This suggests that the mechanism of ATP- or PIP2-induced effect on the KACh channel may be different from that on K1 and KATP channels. However, recent studies measuring the kinetics of K+ current inhibition by PIP2 antibody show that PIP2–channel interaction is strong for IRK1 and ROMK1, and weak for GIRK (Huang et al. 1998; Zhang et al. 1999). Therefore, the concentrations of free fatty acids used in this study may not be high enough to inhibit PIP2–channel interaction for the K1 and KATP channels. These studies also provided evidence that βγ causes stabilization of the PIP2–-KACh channel interaction that may be directly responsible for channel activation. Thus, the level of PIP2 in the membrane is now believed to be the critical factor that determines the degree of channel activation and open time kinetics when βγ is applied. At a low basal level of PIP2 (no applied ATP), activation by βγ is small and shows only short-lived openings, whereas at high PIP2 levels (applied ATP), channel activity is increased and long-lived openings are induced. Therefore, a likely mechanism by which free fatty acids inhibit the KACh channel activity and shorten the open time duration either in the presence or absence of ATP may involve a reduction of the PIP2–channel interaction.
Interestingly, the KACh channels in bullfrog atrial cells show many long-lived openings (bursts) even in the absence of ATP (Ivanova-Nikolova and Breitwieser 1997). The reasons for the difference in the KACh channel gating in bullfrog and mammalian atrial cells are not evident. However, it is possible that the concentration of unsaturated free fatty acids in bullfrog atrial cells are lower than those in mammalian cells and do not sufficiently inhibit ATP-dependent gating. In this regard, it is interesting to note that the cytoplasmic fraction prepared from oocytes of Xenopus laevis frogs also do not contain sufficient inhibitory activity against ATP-dependent gating of the expressed GIRK1/4 (Kim et al. 1997). Thus, it may be speculated that the control mechanisms of the KACh channel in mammals is different from that of amphibians by virtue of differences in the available free fatty acids in the cytoplasm. In rat atrial cells, the open time duration was reported to depend on the concentration of βγ subunit (Nemec et al. 1999). However, the comparison of the reported effects of ATP and βγ on the open time duration of KACh channels in mammalian atrial cells show that the ATP effect is far greater than that of βγ.
Physiological Significance of the Free Fatty Acid Effect on the KACh Channel
The existence of the dual and opposite effect of intracellular ATP and certain unsaturated free fatty acids on the KACh channel is interesting and curious. Since these molecules provide energy to the cell, it is plausible that the KACh channel activity is influenced by the metabolic state, particularly by the fatty acid metabolism. Small changes in total free fatty acid concentration near 1–2 μM could in principle have a significant effect on KACh channel activity. It is also possible that free fatty acid concentrations are kept at a constant level and do not modulate the KACh channel. Nevertheless, it is clear that without the inhibitory action of free fatty acids, the KACh channels would be in the “high open probability” state due to the ATP-induced increase in channel activity and induction of long-lived openings. Single-channel studies show that in oocytes expressing GIRK1/4, the channels in the cell-attached state are already in the ATP-modified gating mode and show long-lived openings (Kubo et al. 1993; Kim et al. 1997). This is in keeping with the finding that the cytosolic fraction from oocytes has low activity against the ATP-dependent gating of KACh channels in atrial cells (Kim et al. 1997).
In previous studies from our laboratory, it was found that long-lived openings can be observed during the initial several seconds upon formation of cell-attached patches with ACh in the pipette (Kim 1991; Hong et al. 1996). The subsequent fast desensitization of the KACh channel activity in atrial cells was associated with a rapid decrease in the number of long-lived openings, a process that is qualitatively similar to that produced by unsaturated free fatty acids on the ATP-modified KACh channel. The presence of long-lived openings in cell-attached patches after ACh application is difficult to reconcile with the inhibitory action of unsaturated free fatty acids in the cell. Free fatty acids are expected to keep the KACh channels from becoming modified by ATP (or PIP2) such that they open in the low P o mode showing mainly short-lived openings when ACh is applied. At present, we do not have a good explanation for these seemingly contradictory observations. We hope to address this question in the future.
It has been reported recently that endothelin causes inhibition of GIRK when endothelin receptor and GIRK channels are expressed in oocytes (Rogalski et al. 1999). This effect was blocked by inhibitor of PLA2 and mimicked by AA. Therefore, any signaling mechanisms that cause sufficient generation of AA in the cell could reduce KACh channel activity and modulate cell excitability. The potent inhibitory effect of free fatty acid indicates that KACh channel function may also be altered in pathological states such as hypoxia and ischemia, where free fatty acid concentration in the cell is increased (VanderVusse et al. 1992). In the heart, a rise in free fatty acid concentration would be expected to decrease KACh channel activity and therefore increase the heart rate. In the brain, synaptic transmission may be enhanced due to decreased inhibitory input in conditions such as those during ischemia and stroke, where the metabolic state of the cells are altered and free fatty acid concentrations are increased (Bazan et al. 1971). A decrease in free fatty acid concentration in the cell would be expected to augment the response of the KACh channel to intracellular ATP, and thus enhance the vagal effect on the heart and increase synaptic inhibition in the brain. Whether such effects indeed occur in the heart and brain needs further investigation. Our results show that free fatty acids do not work well when applied extracellularly, suggesting that free fatty acids do not cross the atrial cell membrane very easily. However, it is possible that after they crossed the membrane they diffused quickly to the bathing medium.
Arachidonic Acid, Free Fatty Acids and Ion Channel Function
In 1987, AA and its lipoxygenase metabolites were reported to act as second messengers in the modulation of K+ channels in Aplysia neurons (Piomelli et al. 1987). Subsequently, AA was found to activate the KACh channel when applied intracellularly in the absence of GTP and ATP (Kim et al. 1989; Kurachi et al. 1989). Thus, these results are in apparent contradiction to our present study showing that AA and other unsaturated free fatty acids inhibit KACh channel activity. It is now clear that AA or its metabolites are not true activators of the KACh channel. AA-induced slow activation that was observed in the absence of intracellular GTP was probably due to slow destabilization of G proteins and subsequent dissociation of their subunits by high concentrations (10–50 μM) of lipophilic molecules. Consistent with this mechanism, AA was reported to increase the basal GTPγS activation of the KACh current while decreasing the ACh-activated current (Scherer and Breitwieser 1990). Our study shows unambiguously that cytoplasmic AA and unsaturated free fatty acids inhibit KACh channel activity under more normal conditions where GTP and ATP are present.
The role of free fatty acids in ion channel regulation is increasingly recognized as many ion channels are affected by free fatty acids. AA and other long-chain free fatty acids have been shown to affect Na+, Ca2+, and K+ channels in various cell types in different ways (Hallaq et al. 1992; Huang et al. 1992; Fraser et al. 1993; Meves 1994; Devor and Frizzell 1998). More recently, unsaturated free fatty acids have been reported to activate light-sensitive channels (TRP) in Drosophila (Chyb et al. 1999) and mechanosensitive K+ channels in cardiac muscle cells, smooth muscle cells, and neurons (Kim 1992; Kim et al. 1995; Ordway et al. 1995). Polyunsaturated free fatty acids suppress Cl− current (Anderson and Welsh 1990) and voltage-gated K+ (Honore et al. 1994) and Na+ currents (Xiao et al. 1998). In each case, there is evidence to show that free fatty acids act directly and not via their metabolites. There is also increasing evidence indicating that free fatty acids may act directly on channels themselves rather than via changes in the lipid bilayer surrounding the channels (Ordway et al. 1991; Kang and Leaf 1996). For most ion channels, ATP does not appear to be involved in the fatty acid effect. Therefore, the effect of free fatty acids on the KACh channel may involve a different mechanism. Our study on three different inward rectifier K+ channels suggests that free fatty acids exist in the cytoplasm of many types of mammalian cells and that their effects on ion channels may be more specific than previously thought. Therefore, it will be important to understand the cellular mechanisms by which free fatty acids modulate various ion channels and thereby regulate cell function.
Acknowledgments
The authors thank Drs. L.Y. Jan, H.A. Lester, and D.E. Clapham for providing GIRK1, GIRK2, and GIRK4 clones, respectively.
This work was supported by grants from the National Institutes of Health and the American Heart Association.
Footnotes
Dr. Pleumsamran's current address is Department of Physiology, Faculty of Medicine, Chiang Mai University, Chiang Mai 50200, Thailand.
Abbreviations used in this paper: 5-HT, 5-hydroxytryptamine; AA, arachidonic acid; ACh, acetylcholine; KACh channel, G protein–gated channel; PIP2, phosphatidylinositol-4,5-bisphosphate.
References
- Anderson M.P., Welsh M.J. Fatty acids inhibit apical membrane chloride channels in airway epithelia. Proc. Natl. Acad. Sci. USA. 1990;87:7334–7338. doi: 10.1073/pnas.87.18.7334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade R., Malenka R.C., Nicoll R.A. A G protein couples serotonin and GABAB receptors to the same channels in hippocampus. Science. 1986;234:1261–1264. doi: 10.1126/science.2430334. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T., Schulte U., Oliver D., Herlitze S., Krauter T., Tucker S.J., Rippersberg J.P., Fakler B. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 1998;282:1141–1144. doi: 10.1126/science.282.5391.1141. [DOI] [PubMed] [Google Scholar]
- Bazan N.G.J., Bazan H.E.P., Kennedy G.W., Joel C.D. Regional distribution and rate of production of free fatty acids in rat brain. J. Neurochem. 1971;18:1387–1393. doi: 10.1111/j.1471-4159.1971.tb00003.x. [DOI] [PubMed] [Google Scholar]
- Breitwieser G., Szabo G. Uncoupling of cardiac muscarinic and β-adrenergic receptors from ion channels by a guanine nucleotide analogue. Nature. 1985;317:538–540. doi: 10.1038/317538a0. [DOI] [PubMed] [Google Scholar]
- Chyb S., Raghu P., Hardie R.C. Polyunsaturated fatty acids activate the Drosophila light-sensitive channels TRP and TRPL. Nature. 1999;397:255–259. doi: 10.1038/16703. [DOI] [PubMed] [Google Scholar]
- Devor D.C., Frizzell R.A. Modulation of K+ channels by arachidonic acid in T84 cells. I. Inhibition of the Ca(2+)-dependent K+ channel. Am. J. Physiol. 1998;274:C138–C148. doi: 10.1152/ajpcell.1998.274.1.C138. [DOI] [PubMed] [Google Scholar]
- Fakler B., Brandle U., Glowatzki E., Zenner H.-P., Ruppersberg J.P. Kir2.1 inward rectifier K+ channels are regulated independently by protein kinases and ATP hydrolysis. Neuron. 1994;13:1413–1420. doi: 10.1016/0896-6273(94)90426-x. [DOI] [PubMed] [Google Scholar]
- Fan Z., Makielski J.C. Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K(+) Channel. A molecular probe for the mechanism of atp-sensitive inhibition. J. Gen. Physiol. 1999;114:251–270. doi: 10.1085/jgp.114.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser D.D., Hoehn K., Weiss S., MacVicar B.A. Arachidonic acid inhibits sodium currents and synaptic transmission in cultured striatal neurons. Neuron. 1993;11:633–644. doi: 10.1016/0896-6273(93)90075-3. [DOI] [PubMed] [Google Scholar]
- Hallaq H., Smith T.W., Leaf A. Modulation of dihydropyridine-sensitive calcium channels in heart cells by fish oil fatty acids. Proc. Natl. Acad. Sci. USA. 1992;89:1760–1764. doi: 10.1073/pnas.89.5.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J.A. Fatty acid transportdifficult or easy? J. Lipid Res. 1998;39:467–481. [PubMed] [Google Scholar]
- Hilgemann D.W. Cytoplasmic ATP-dependent regulation of ion transporters and channelsmechanisms and messengers. Annu. Rev. Physiol. 1997;59:193–220. doi: 10.1146/annurev.physiol.59.1.193. [DOI] [PubMed] [Google Scholar]
- Hilgemann D.W., Ball R. Regulation of cardiac Na+, Ca2+ exchange and KATP potassium channels by PIP2 . Science. 1996;273:956–959. doi: 10.1126/science.273.5277.956. [DOI] [PubMed] [Google Scholar]
- Ho I.H., Murrell-Lagnado R. Molecular determinants for sodium-dependent activation of G protein-gated K+ channels. J. Biol. Chem. 1999;274:8639–8648. doi: 10.1074/jbc.274.13.8639. [DOI] [PubMed] [Google Scholar]
- Hong S.G., Pleumsamran A., Kim D. Regulation of atrial muscarinic K+ channel activity by a cytosolic protein via G protein–independent pathway. Am. J. Physiol. 1996;270:H526–H537. doi: 10.1152/ajpheart.1996.270.2.H526. [DOI] [PubMed] [Google Scholar]
- Honore E., Barhanin J., Attali B., Lesage F., Lazdunski M. External blockade of the major cardiac delayed-rectifier K+ channel (Kv1.5) by polyunsaturated fatty acids. Proc. Natl. Acad. Sci. USA. 1994;91:1937–1944. doi: 10.1073/pnas.91.5.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.-L., Feng S., Hilgemann D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Huang J.M., Xian H., Bacaner M. Long-chain fatty acids activate calcium channels in ventricular myocytes. Proc. Natl. Acad. Sci. USA. 1992;89:6452–6456. doi: 10.1073/pnas.89.14.6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova-Nikolova T.T., Breitwieser G.E. Effector contributions to Gβγ-mediated signaling as revealed by muscarinic potassium channel gating. J. Gen. Physiol. 1997;109:245–253. doi: 10.1085/jgp.109.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J.X., Leaf A. Evidence that free polyunsaturated fatty acids modify Na+ channels by directly binding to the channel proteins. Proc. Natl. Acad. Sci. USA. 1996;93:3542–3546. doi: 10.1073/pnas.93.8.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. A mechanosensitive K+ channel in heart cellsactivation by arachidonic acid. J. Gen. Physiol. 1992;100:1–20. doi: 10.1085/jgp.100.6.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. Modulation of acetylcholine-activated K+ channel function in rat atrial cells by phosphorylation. J. Physiol. 1991;437:133–155. doi: 10.1113/jphysiol.1991.sp018588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Bang H.W. Modulation of rat atrial G protein–coupled K+ channel function by phospholipids. J. Physiol. 1999;517:59–74. doi: 10.1111/j.1469-7793.1999.0059z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Duff R. Regulation of K+ channels in cardiac myocytes by free fatty acids. Circ. Res. 1990;67:1040–1046. doi: 10.1161/01.res.67.4.1040. [DOI] [PubMed] [Google Scholar]
- Kim D., Lewis D.L., Graziadei L., Neer E.J., Bar-Sagi D., Clapham D.E. G-protein βγ-subunits activate the cardiac muscarinic K channel via phospholipase A2. Nature. 1989;337:557–560. doi: 10.1038/337557a0. [DOI] [PubMed] [Google Scholar]
- Kim D., Sladek C.D., Aguado-Velasco C., Mathiasen J.R. Arachidonic acid activation of a new family of K+ channels in cultured rat neuronal cells. J. Physiol. 1995;484:643–660. doi: 10.1113/jphysiol.1995.sp020693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Watson M., Indyk V. ATP-dependent regulation of a G protein–coupled K+ channel (GIRK1/GIRK4) expressed in oocytes. Am. J. Physiol. 1997;272:H195–H206. doi: 10.1152/ajpheart.1997.272.1.H195. [DOI] [PubMed] [Google Scholar]
- Kofuji P., Davidson N., Lester H.A. Evidence that neuronal G-protein–gated inwardly rectifying K+ channels are activated by Gβγ subunits and function as heteromultimers. Proc. Natl. Acad. Sci. USA. 1995;92:6542–6546. doi: 10.1073/pnas.92.14.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y., Reuveny E., Slesinger P.A., Jan Y.N., Jan L.Y. Primary structure and functional expression of a rat G-protein–coupled muscarinic potassium channel. Nature. 1993;364:802–806. doi: 10.1038/364802a0. [DOI] [PubMed] [Google Scholar]
- Kurachi Y., Ito H., Sugimoto T., Shimizu T., Miki I., Ui M. Arachidonic acid metabolites as intracellular modulators of the G protein–gated cardiac K+ channel. Nature. 1989;337:555–557. doi: 10.1038/337555a0. [DOI] [PubMed] [Google Scholar]
- Kurachi Y., Nakajima T., Sugimoto T. Acetylcholine activation of K channels in cell-free membrane of atrial cells. Am. J. Physiol. 1986;251:H681–H684. doi: 10.1152/ajpheart.1986.251.3.H681. [DOI] [PubMed] [Google Scholar]
- Logothetis D.E., Kurachi Y., Galper J., Neer E.J., Clapham D.E. The βγ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325:321–326. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- Lopatin A.N., Makhina E.N., Nichols C.G. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994;372:366–374. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- Meves H. Modulation of ion channels by arachidonic acid. Prog. Neurobiol. 1994;43:175–186. doi: 10.1016/0301-0082(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Nemec J., Wickman K., Clapham D.E. Gβγ binding increases the open time of IKAChKinetic evidence for multiple Gβγ binding sites. Biophys. J. 1999;76:246–252. doi: 10.1016/S0006-3495(99)77193-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh U., Ho Y.K., Kim D. Modulation of the serotonin-activated K+ channel by G protein subunits and nucleotides in rat hippocampal neurons. J. Membr. Biol. 1995;147:241–253. doi: 10.1007/BF00234522. [DOI] [PubMed] [Google Scholar]
- Ordway R.W., Petrou S., Kirber M.T., Walsh J.V., Jr., Singer J.J. Stretch activation of a toad smooth muscle K+ channel may be mediated by fatty acids. J. Physiol. 1995;484:331–337. doi: 10.1113/jphysiol.1995.sp020668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordway R.W., Singer J.J., Walsh J.V., Jr. Direct regulation of ion channels by fatty acids. Trends Neurosci. 1991;14:96–100. doi: 10.1016/0166-2236(91)90069-7. [DOI] [PubMed] [Google Scholar]
- Pfaffinger P.J., Martin J.M., Hunter D.D., Nathanson N.M., Hille B. GTP-binding proteins couple cardiac muscarinic receptors to a K+ channel. Nature. 1985;317:536–538. doi: 10.1038/317536a0. [DOI] [PubMed] [Google Scholar]
- Piomelli D., Volterra D., Dale N., Siegelbaum S.A., Kandel E.R., Schwartz H., Belardetti F. Lipoxygenase metabolites of arachidonic acid as second messengers for presynaptic inhibition of Aplysia sensory cells. Nature. 1987;328:38–43. doi: 10.1038/328038a0. [DOI] [PubMed] [Google Scholar]
- Qin F., Auerbach A., Sachs F. Estimating single channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys. J. 1996;70:264–280. doi: 10.1016/S0006-3495(96)79568-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski S.L., Cyr C., Chavkin C. Activation of the endothelin receptor inhibits the G protein–coupled inwardly rectifying potassium channel by a phospholipase A2–mediated mechanism. J. Neurochem. 1999;72:1409–1416. doi: 10.1046/j.1471-4159.1999.721409.x. [DOI] [PubMed] [Google Scholar]
- Sakmann B., Noma A., Trautwein W. Acetylcholine activation of single muscarinic K channels in isolated pacemaker cells of the mammalian heart. Nature. 1983;303:250–253. doi: 10.1038/303250a0. [DOI] [PubMed] [Google Scholar]
- Scherer R.W., Breitwieser G.E. Arachidonic acid metabolites alter G protein–mediated signal transduction in heart. Effects on muscarinic K+ channels. J. Gen. Physiol. 1990;96:735–755. doi: 10.1085/jgp.96.4.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S.L., Nichols C.G. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- Sui J.L., Petit-Jacques J., Logothetis D.E. Activation of the atrial KACh channel by the βγ subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proc. Natl. Acad. Sci. USA. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano M., Qin D., Noma A. ATP-dependent decay and recovery of K channels ion guinea-pig cardiac myocytes. Am. J. Physiol. 1990;258:H45–H50. doi: 10.1152/ajpheart.1990.258.1.H45. [DOI] [PubMed] [Google Scholar]
- VanderVusse G.J., Glatz J.F., Stam H.C., Reneman R.S. Fatty acid homeostasis in the normoxic and ischemic heart. Physiol. Rev. 1992;72:881–940. doi: 10.1152/physrev.1992.72.4.881. [DOI] [PubMed] [Google Scholar]
- Veerkamp J.H., Peeters R.A., Maatman R.G.H.J. Structural and functional features of different types of cytoplasmic fatty acid–binding proteins. Biochim. Biophys. Acta. 1991;1081:1–24. doi: 10.1016/0005-2760(91)90244-c. [DOI] [PubMed] [Google Scholar]
- Xiao Y.-F., Wright S.N., Wang G.K., Morgan J.P., Leaf A. Fatty acids suppress voltage-gated Na+ current in HEK293t cells transfected with the α-subunit of the human cardiac Na+ channel. Proc. Natl. Acad. Sci. USA. 1998;95:2680–2685. doi: 10.1073/pnas.95.5.2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., He C., Yan X., Mirshahi T., Logothetis D.E. Activation of inwardly rectifying K+ channels by distinct PtdIns (4,5)P2 interactions. Nat. Cell Biol. 1999;1:183–188. doi: 10.1038/11103. [DOI] [PubMed] [Google Scholar]