

Abstract
N-type voltage-gated calcium channel activity in rat superior cervical ganglion neurons is modulated by a variety of pathways. Activation of heterotrimeric G-proteins reduces whole-cell current amplitude, whereas phosphorylation by protein kinase C leads to an increase in current amplitude. It has been proposed that these two distinct pathways converge on the channel's pore-forming α1B subunit, such that the actions of one pathway can preclude those of the other. In this study, we have characterized further the actions of PKC on whole-cell barium currents in neonatal rat superior cervical ganglion neurons. We first examined whether the effects of G-protein–mediated inhibition and phosphorylation by PKC are mutually exclusive. G-proteins were activated by including 0.4 mM GTP or 0.1 mM GTP-γ-S in the pipette, and PKC was activated by bath application of 500 nM phorbol 12-myristate 13-acetate (PMA). We found that activated PKC was unable to reverse GTP-γ-S–induced inhibition unless prepulses were applied, indicating that reversal of inhibition by phosphorylation appears to occur only after dissociation of the G-protein from the channel. Once inhibition was relieved, activation of PKC was sufficient to prevent reinhibition of current by G-proteins, indicating that under phosphorylating conditions, channels are resistant to G-protein–mediated modulation. We then examined what effect, if any, phosphorylation by PKC has on N-type barium currents beyond antagonizing G-protein–mediated inhibition. We found that, although G-protein activation significantly affected peak current amplitude, fast inactivation, holding-potential–dependent inactivation, and voltage-dependent activation, when G-protein activation was minimized by dialysis of the cytoplasm with 0.1 mM GDP-β-S, these parameters were not affected by bath application of PMA. These results indicate that, under our recording conditions, phosphorylation by PKC has no effect on whole-cell N-type currents, other than preventing inhibition by G-proteins.
Keywords: G-protein, inactivation, L-type calcium channel, phorbol ester, phosphorylation
INTRODUCTION
N-type voltage-gated calcium channel activity in rat superior cervical ganglion (SCG) neurons is modulated by a variety of mechanisms. G-protein–coupled, membrane-delimited pathways have been shown to decrease whole-cell barium current amplitude by selective inhibition of N-type current (Plummer et al. 1991; for review, see Hille et al. 1995). There are several features of membrane-delimited inhibition. This inhibition is mediated by heterotrimeric G-proteins, either activated via G-protein–coupled receptors (Wanke et al. 1987; Plummer et al. 1991), or stimulated directly by GTP (Plummer et al. 1989; Ikeda 1991). G-protein–inhibited whole-cell currents characteristically exhibit slowed or “reluctant” voltage-dependent activation (Bean 1989). Finally, G-protein–mediated inhibition can be blocked by dialysis of the cell with GDP-β-S, or relieved temporarily by application of a strong positive voltage step (Bean 1989; Ikeda 1991).
The rate of reinhibition of currents facilitated by prepulse application is related to the concentration of activated G-proteins. This was demonstrated with increasing concentrations of GTP-γ-S (Lopez and Brown 1991), neurotransmitter (Erlich and Elmslie 1995), or free intracellular Gβγ (Zamponi and Snutch 1998). Each of these studies showed that higher levels of G-protein activity resulted in faster rates of reinhibition following facilitation, thereby suggesting that prepulses lead to dissociation of the G-protein from the channel.
Additional modulation of N-type calcium channel activity exists via phosphorylation by protein kinase C. Activation of PKC by phorbol esters leads to an enhancement of whole-cell current amplitude in sympathetic neurons, as well as an attenuation of subsequent transmitter-induced, membrane-delimited inhibition (Swartz 1993; Swartz et al. 1993; Zhu and Ikeda 1994). Additionally, PKC activation reverses tonic G-protein–mediated inhibition in adult SCG neurons (Swartz 1993; Zhu and Ikeda 1994), suggesting that one effect of PKC phosphorylation on whole-cell currents is a relief of G-protein–mediated inhibition. The cytoplasmic linker between the first and second domains of the α1B subunit of the channel has been proposed as one possible site for convergence of these pathways (Zamponi et al. 1997; Hamid et al. 1999).
Previous data (Swartz 1993; Swartz et al. 1993; Zhu and Ikeda 1994) have implicated PKC activity in blocking or reversing G-protein-mediated inhibition. Moreover, it has been suggested that G-protein binding to the channel will block the effects of PKC activation (Swartz 1993). Although these findings suggest mutual exclusivity between these pathways, conclusive evidence to support this hypothesis has not been documented previously. Our results demonstrate that phosphorylation by PKC is sufficient to block G-protein–mediated inhibition. In addition, we provide evidence that G-protein binding is sufficient to prevent PKC-induced enhancement of whole-cell current amplitude. Finally, we examined the effect of activating PKC on whole-cell currents in the absence of G-protein–mediated inhibition. Our results show that, when GDP-β-S is included in the pipette solution, phosphorylation by PKC is without effect on whole-cell current amplitude, voltage-dependent activation, fast inactivation, or holding potential–dependent inactivation. Taken together, these data support a model of mutual exclusivity and suggest that the primary role of phosphorylation by PKC, in this cell type, is to prevent the channel from exhibiting reluctant gating.
METHODS
Cell Preparation and Culture
SCG were dissected from 1–4 d old Sprague-Dawley rats (Charles River Laboratories) and dissociated by trituration (Hawrot and Patterson 1979). SCG neurons were used for these experiments because ∼80–90% of the whole-cell barium current is N-type (Plummer et al. 1989). Moreover, this cell type has been shown previously to be sensitive to modulation by G-proteins and phosphorylation by protein kinase C (Swartz 1993; Zhu and Ikeda 1994). Cells were maintained in 5% CO2 at 37°C. The culture medium consisted of DMEM supplemented with 7.5% fetal bovine serum, 7.5% calf serum, 100 IU/ml penicillin, 0.1 mg/ml streptomycin, 4 mM l-glutamine (all from Sigma Chemical Co.), and 0.2 μg/ml nerve growth factor (Bioproducts for Science). After dissociation, cells were plated on poly-l-lysine–coated glass coverslips and incubated for at least 4 h before recording. Spherical neurons lacking visible processes were selected for recording, and used within 16 h of preparation.
Electrophysiology
Barium currents were recorded using the whole-cell configuration of an Axopatch 200B patch-clamp amplifier (Axon Instruments). Except where noted, voltage steps were applied every 4 s from a holding potential of −90 mV. When used, prepulses preceded the test pulse by 5 ms. Currents were recorded at 20–24°C, passed through a four-pole low-pass Bessel filter at 1 kHz, and then digitized at 5 kHz with a 1401 plus interface (Cambridge Electronic Design), except the activation data, which were filtered at 5 kHz and digitized at 20 kHz. Data were collected using the Patch software suite, version 6.3 (Cambridge Electronic Design), and stored on a personal computer for off-line analysis. Capacitive currents were corrected online, and leak currents were subtracted using a scaled-up hyperpolarizing pulse. Pipettes were pulled (PB-7 puller; Narishige) from borosilicate capillary tubes (2-000-210; Drummond Scientific) and heat-polished just before use (MF-9 microforge; Narishige), leading to pipette tip resistances ranging from 2 to 2.5 MΩ. For most recordings, pipette tips were coated with Sylgard (Dow Corning) to minimize capacitance. Drugs were applied via gravity-driven bath perfusion, with an estimated time to complete bath exchange of 5–10 s.
The control bath solution consisted of (mM): 125 NMDG-aspartate, 10 HEPES, 20 barium-acetate, 0.0005 tetrodotoxin, pH 7.5 (296 mOsm). The pipette solution contained (mM): 122 cesium-aspartate, 10 HEPES, 10 EGTA, 5 MgCl2, 4 ATP (disodium salt), 0.4 GTP (sodium salt), pH 7.5 (293 mOsm); where indicated, GTP was substituted with 0.1 mM of either GTP-γ-S or GDP-β-S (lithium salts).
Transmitters were excluded from the bath, and G-proteins were directly activated by including GTP or GTP-γ-S in the pipette solution (Ikeda 1991). This allowed us to avoid receptor desensitization (Huganir and Greengard 1990), and achieve a reproducible steady state level of G-protein activation. To minimize modulation of channel activity by a pertussis-toxin–insensitive, calcium-dependent diffusible second-messenger pathway, 10 mM EGTA was included in the pipette solution (Hille et al. 1995).
Pharmacology
Phorbol 12-myristate 13-acetate (PMA) and 4-α-phorbol 12-myristate 13-acetate (4-α-PMA) were obtained from Research Biochemicals, Inc. GTP-γ-S and GDP-β-S were obtained from either Research Biochemicals, Inc. or Sigma Chemical Co. The PKC inhibitor bisindolylmaleimide I (BLM) was obtained from Calbiochem Corp., and ω-conotoxin GVIA (CTX) was from Bachem. All other chemicals and reagents were obtained from Sigma Chemical Co. Stock solutions of tetrodotoxin and CTX were prepared in double-distilled water; stock solutions of PMA, 4-α-PMA, and BLM were prepared in DMSO. Currents obtained in control bath solution containing the maximal final concentration of DMSO (0.124%) were indistinguishable from solutions lacking DMSO (not shown).
Data Analysis
Analysis software included Patch 6.3, Microsoft Excel 97, and Origin 5.0 (Microcal Software, Inc.). Current amplitude was measured isochronically for all recordings. Data are presented as mean ± SEM. Statistical significance was determined using a Student's two-tailed, paired t test or a two-way t test for two means; data were considered significantly different if P < 0.05. Sample size is given in parentheses within the figures, unless provided elsewhere. Fraction remaining was measured as the ratio of current amplitude at the end of the test pulse to the amplitude at the onset of the test pulse (see Fig. 3 A); this method of measuring fast inactivation has been described previously as residual fraction of peak current (de Leon et al. 1995). The Boltzmann fits presented in Fig. 8, and the data shown in Table , were calculated using the equation:
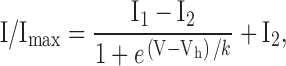 |
where I/Imax is normalized current, V is voltage in millivolts, Vh is the voltage at half-maximal current, k is the slope of activation in millivolts per e-fold change in current, and I1 and I2 are the minimum and maximum values of I, respectively.
Figure 3.
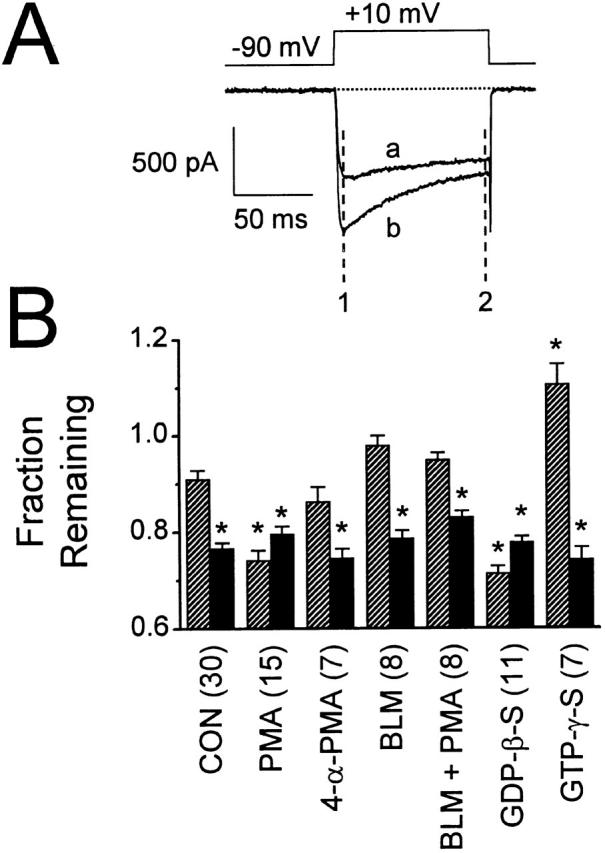
Prepulses and PMA both increase voltage-dependent fast inactivation. (A) Fast inactivation was measured as the fraction of the initial inward current (1) that remains at the end of a 100-ms test pulse (2). (a) Control current elicited by the voltage command shown; (b) current elicited after bath application of 500 nM PMA. (B) Summary of fraction remaining, as defined in A, for unfacilitated (no prepulse, hatched columns) and facilitated (prepulse to +80 mV, solid columns) whole-cell currents. In control solution, application of a prepulse reduced the fraction remaining from 0.91 ± 0.02 to 0.77 ± 0.01 (n = 30). Bath application of 500 nM PMA (n = 15) led to a reduction in unfacilitated fraction remaining (0.74 ± 0.02), whereas facilitated currents in PMA were not significantly different than facilitated control currents (0.80 ± 0.01). 500 nM 4-α-PMA, 100 nM BLM, and PMA after BLM were all without effect. As with PMA, dialysis with GDP-β-S (n = 11) reduced the unfacilitated fraction remaining (0.71 ± 0.01), but was without effect on facilitated currents (0.78 ± 0.01). In contrast, dialysis with GTP-γ-S (n = 7) greatly increased the unfacilitated fraction remaining (1.11 ± 0.04), but, as with all other treatments, was without effect on facilitated currents (0.74 ± 0.02). *P < 0.05 compared with unfacilitated control.
Figure 8.
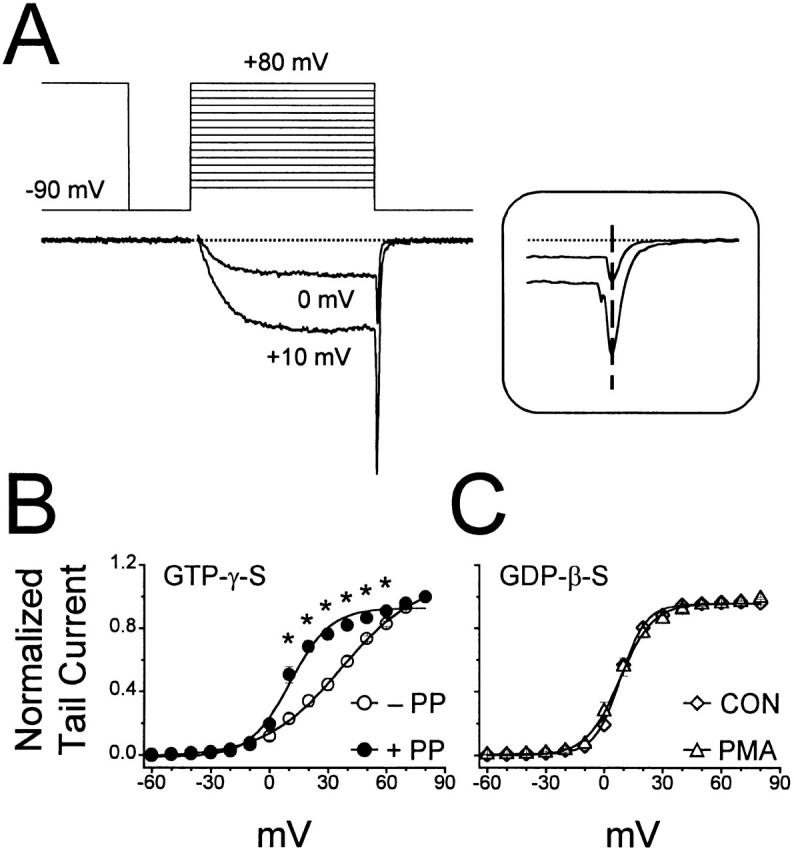
Activation of PKC does not alter the voltage dependence of activation of whole-cell barium currents. (A) Activation plots were generated using a voltage command consisting of 15-ms pulses to incremental test potentials, either with or without a 100-ms prepulse to +80 mV. Shown are two currents elicited, at the voltage indicated, with GDP-β-S in the pipette solution. Fast tail current amplitudes (A, inset) were measured at the dashed line, normalized, and then plotted against test potential (B). Boltzmann fits (see methods) were applied to the data (solid lines), and all curves were fit with a Chi-square value of <0.002. (B) When 0.1 mM GTP-γ-S was included in the pipette solution, the threshold for activation was approximately −20 mV (−PP, ○). Applying prepulses (+PP, •) was without effect on the threshold of activation, but the voltage eliciting half-maximal activation was shifted negative. *P < 0.01 versus no prepulse. (C) When the pipette solution contained 0.1 mM GDP-β-S, unfacilitated control (⋄) and PMA (500 nM, ▵) activation plots were virtually indistinguishable. See Table for values and sample sizes.
Table 1.
Boltzmann Analysis of Activation Curves
| Bath solution | Pipette solution | Prepulse | n | Vh* | k ‡ | |
|---|---|---|---|---|---|---|
| Control | ○§ | GTP-γ-S | − | 6 | 36.9 | 19.8 |
| Control | • | GTP-γ-S | + | 6 | 10.6 | 10.4 |
| BLM | GTP-γ-S | − | 6 | 41.9 | 19.0 | |
| BLM | GTP-γ-S | + | 6 | 19.0 | 14.0 | |
| Control | ⋄ | GDP-β-S | − | 21 | 8.3 | 6.6 |
| Control | GDP-β-S | + | 11 | 7.2 | 5.6 | |
| BLM | GDP-β-S | − | 9 | 5.1 | 6.7 | |
| BLM | GDP-β-S | + | 6 | −1.3 | 3.9 | |
| PMA | ▵ | GDP-β-S | − | 13 | 8.0 | 8.6 |
| PMA | GDP-β-S | + | 5 | 1.8 | 7.0 | |
| 4-α-PMA | GDP-β-S | − | 7 | 2.9 | 4.9 | |
| 4-α-PMA | GDP-β-S | + | 7 | 1.2 | 4.1 |
*Voltage eliciting half-maximal activation (mV). ‡Slope constant (mV/e-fold change). §Symbols correspond to Fig. 8
RESULTS
Neonatal Rat SCG Neurons Exhibit Tonic G-Protein–mediated Inhibition
To examine the effects of PKC on whole-cell currents, it was necessary to first confirm that, under our recording conditions, tonic inhibition of whole-cell currents by G-proteins could be observed, and that activating PKC could block this inhibition. Tonic inhibition was observed in neonatal rat SCG neurons in the absence of G-protein–coupled receptor activation, when GTP was included in the pipette solution. Stepping from −90 to +10 mV (Fig. 1 A) elicited an inward current whose amplitude was greatly facilitated if this step was preceded by a pulse to +80 mV (Fig. 1 B, left, and Fig. 2). 4 s later, current amplitude had returned to baseline, consistent with time-dependent recovery of tonic G-protein–mediated inhibition (Lopez and Brown 1991; Zamponi and Snutch 1998).
Figure 1.
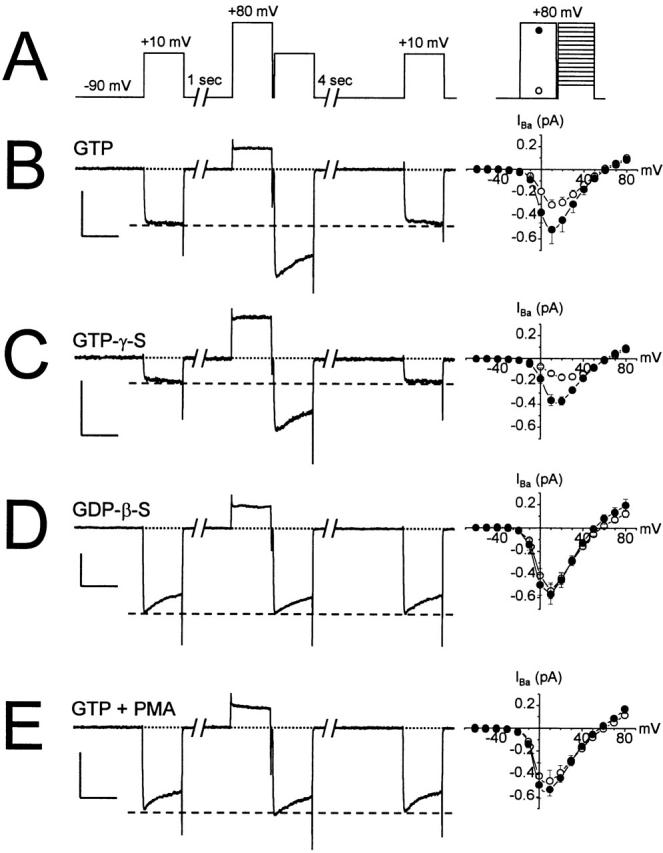
Modulation of whole-cell barium currents by G-proteins and PMA. (A) The voltage protocol used to elicit the currents shown in B–E. (Left) Whole-cell sweeps; (right) mean current–voltage plots elicited with (•, n = 7–20) or without (○, n = 6–12) prepulses to +80 mV. In this and subsequent figures, the delay between the prepulse and test pulse is 5 ms, and error bars not visible are contained within the symbols. (B) Whole-cell currents were elicited in control bath solution with GTP in the pipette. (C) GTP-γ-S was substituted for GTP in the pipette solution. (D) GDP-β-S was substituted for GTP in the pipette. (E) The same cell shown in B, 2 min after bath application of 500 nM PMA. For all sweeps, the horizontal calibration bars are 100 ms, and the vertical calibration bars are 500 pA.
Figure 2.

Summary of facilitation, expressed as the ratio of current amplitude after a prepulse to current amplitude before a prepulse. Control facilitation was 1.71 ± 0.06 (n = 34). Application of 500 nM PMA reduced facilitation to 1.01 ± 0.02 (n = 19). In contrast, 500 nM 4-α-PMA, 100 nM BLM, and PMA after BLM were all without significant effect. Substituting 0.1 mM GDP-β-S for GTP in the pipette solution decreased facilitation to 0.91 ± 0.02 (n = 11), whereas substituting 0.1 mM GTP-γ-S for GTP increased facilitation to 2.76 ± 0.14 (n = 9). After a 10-min incubation with 1 μM CTX, control facilitation was lost (0.92 ± 0.03, n = 12), and subsequent application of PMA was without further effect (0.91 ± 0.04, n = 4). *P < 0.001 compared with control.
The increase in current amplitude following a prepulse was voltage dependent, as shown by the current–voltage analysis (Fig. 1 B, right). Maximal facilitation was observed from 0 to +10 mV, and dropped to undetectable levels as the test pulse became more positive, consistent with voltage-dependent relief of G-protein–mediated inhibition (Bean 1989; Ikeda 1991). Because both maximal inward current and maximal facilitation were observed at a test potential of +10 mV, this voltage was used for subsequent experiments.
Tonic inhibition of whole-cell currents displayed additional characteristics that have been described previously. In addition to decreased current amplitude, G-protein–inhibited whole-cell currents also exhibited slowed activation kinetics and increased facilitation; these effects were more pronounced when GTP-γ-S was substituted for GTP in the pipette solution (Fig. 1 C and 2). In contrast, G-protein–mediated inhibition was minimized by dialysis of the cell with GDP-β-S (Fig. 1 D). This was reflected by a loss of prepulse facilitation (Fig. 2).
In addition to slowing voltage-dependent activation, modulation by G-proteins has also been shown to decrease voltage-dependent fast inactivation (Netzer et al. 1994). To confirm that the currents recorded under our conditions also displayed a decrease in fast inactivation as a result of modulation by G-proteins, we quantified inactivation by measuring the fraction of initial inward current remaining at the end of the test pulse (Fig. 3; see also Fig. 1). As expected, currents elicited in control bath solution, with GTP in the pipette, showed little inactivation, and application of a prepulse greatly increased fast inactivation. Dialyzing the cell with GDP-β-S had essentially the same effect on fast inactivation as prepulses. In contrast, dialysis with GTP-γ-S greatly decreased inactivation, although application of a prepulse was sufficient to increase fast inactivation to the same level as with GDP-β-S.
Taken together, these results verify that, under the conditions used in this study, inhibition of whole-cell currents by G-proteins is readily observable. Moreover, the effects of this inhibition on current amplitude, facilitation, and kinetics can be completely reversed either by applying prepulses or by including GDP-β-S in the pipette solution.
PMA Enhances Whole-Cell N-Type Currents by Preventing Tonic G-Protein–mediated Inhibition
PKC activation has been shown previously to enhance whole-cell currents and lead to a reduction in G-protein–mediated inhibition in SCG neurons (Swartz 1993; Zhu and Ikeda 1994), thereby minimizing prepulse facilitation. Activating PKC had similar effects under our recording conditions. When the phorbol ester PMA (500 nM) was added to the bath of the cell shown in Fig. 1 B, current amplitude significantly increased (Fig. 1 E and 4). Application of a prepulse after PMA treatment had no effect on amplitude (Fig. 1 E and 2), consistent with previous findings that PMA reduces G-protein–mediated inhibition and prepulse facilitation. PMA increased current amplitude over a range of test potentials (Fig. 1 E, right), consistent with a loss of voltage-dependent inhibition. In addition to modulating both current amplitude and facilitation, PMA also affected fast inactivation, significantly decreasing the fraction remaining to a level not significantly different than that observed after dialysis with GDP-β-S (Fig. 1 and Fig. 3).
If PKC activation is sufficient to account for the increased current amplitude, decreased facilitation, and altered kinetics observed in cells that show tonic inhibition, then we would predict that these three parameters should change along a similar time course. This was addressed by measuring each of these parameters in a single recording (Fig. 5). Indeed, as expected, after application of PMA to the bath, the changes observed in facilitation and fraction remaining closely paralleled the change in unfacilitated current amplitude.
Figure 5.

The effects of PMA on current amplitude, facilitation, and fast inactivation follow a parallel time course in a single whole-cell recording. (A and C) Open symbols represent currents elicited without a prepulse; closed symbols represent currents elicited after a prepulse to +80 mV. 500 nM PMA was applied to the bath where indicated by the solid bar. (A) Time course of peak inward current. (B) Time course of facilitation, as calculated in Fig. 2. (C) Fraction remaining was calculated as in Fig. 3 and plotted against time.
To confirm that the effects observed after PMA application were due to activation of PKC, we conducted two control experiments (Fig. 2 Fig. 3 Fig. 4). First, the inactive PMA analogue 4-α-PMA (Van Duuren et al. 1979) was applied to the bath to determine whether any of PMA's effects on currents were due to nonspecific actions of phorbol esters. 4-α-PMA was without effect on facilitation, current amplitude, or fast inactivation. Second, to determine whether PMA's effects were due to selective activation of PKC, the PKC-specific inhibitor BLM (Toullec et al. 1991) was applied to the bath before application of PMA. BLM effectively blocked PMA's ability to alter facilitation, current amplitude, and inactivation, indicating that PMA's effects on whole-cell currents are mediated selectively through PKC activation.
Figure 4.

Summary of fold change in peak inward current amplitude for unfacilitated (no prepulse, hatched columns) and facilitated (prepulse to +80 mV, solid columns) whole-cell currents elicited at +10 mV. 500 nM PMA (n = 18) increased unfacilitated currents by 1.31 ± 0.05-fold, but was without effect on facilitated currents (0.92 ± 0.04-fold). 500 nM 4-α-PMA was without effect, and 100 mM BLM blocked PMA's effects. Application of PMA after dialysis with GDP-β-S (n = 8) was without effect (1.03 ± 0.09-fold for unfacilitated, and 0.94 ± 0.06-fold for facilitated). After a 10-min preincubation in 1 μM CTX (n = 8), PMA caused slight but significant increases in both unfacilitated and facilitated currents (1.14 ± 0.02-fold and 1.16 ± 0.02-fold, respectively). *P < 0.005 compared with control.
Because neonatal rat SCG neurons contain additional types of calcium currents, the changes in whole-cell currents observed after PMA application could be due to actions on currents other than N-type. Therefore, to examine the effects of PMA on non–N-type currents, cells were incubated in a solution containing the N-type calcium channel blocker CTX for 10 min before patch-clamp recording (Fig. 2 and Fig. 4). CTX was excluded from the recording solutions to allow washout of any reversible block of non–N-type calcium channels (McCleskey et al. 1987; Plummer et al. 1989). Consistent with neonatal SCG neurons expressing mostly N-type calcium current, CTX blocked 80–85% of the whole-cell current (not shown). As previously demonstrated in adult rat SCG neurons (Ikeda 1991), facilitation of whole-cell currents was lost after treatment with CTX (Fig. 2), consistent with G-protein–mediated inhibition being restricted to N-type current. Even after N-type channels were blocked, however, bath application of 500 nM PMA caused a slight but significant increase in the amplitude of currents elicited both with and without a prepulse (Fig. 4). There was no significant difference between the fold change of unfacilitated and facilitated currents, suggesting that PMA's effect on non–N-type current is independent of voltage. These data are consistent with previous studies of rat CA3 hippocampal neurons (Swartz 1993), frog sympathetic neurons (Yang and Tsien 1993), and adult rat SCG neurons (Zhu and Ikeda 1994), in which phorbol esters increased current amplitudes after application of CTX, suggesting that non–N-type calcium channel activity might also be modulated by protein kinase C. This increase was ∼15% of currents already inhibited at least 80% by CTX treatment. Therefore, the contribution of non–N-type calcium current in response to PMA in untreated cells can be considered negligible and is not considered further.
G-Protein–mediated Inhibition Blocks PKC's Effect on Whole-Cell Currents
We observed that PMA only affected whole-cell currents that demonstrated G-protein–mediated inhibition, causing a relief of that inhibition (Fig. 1 and Fig. 4). Moreover, PMA appeared to act faster when prepulses were applied during the recording. This is consistent with the previous observation that relatively long prepulses were required to demonstrate PMA's effect on G-protein–mediated inhibition (Swartz 1993). Because prepulses are thought to cause the dissociation of the G-protein from the channel (Lopez and Brown 1991; Zamponi and Snutch 1998), we examined whether phosphorylation by PKC occurs only when the channel is not associated with a G-protein. To address this, we measured the effect of prepulses on PMA's ability to enhance maximally inhibited currents; i.e., when GTP-γ-S was included in the pipette solution.
After membrane breakthrough, current amplitude rapidly decreased (Fig. 6 A). This decrease was due to influx of GTP-γ-S and subsequent activation of G-proteins, as application of a prepulse was sufficient to restore current amplitude. Currents were then elicited without prepulses, to minimize dissociation of G-protein–channel interactions. In the absence of prepulses, PMA was essentially without effect on whole-cell current amplitude (Fig. 6 C). Even after 8 min of stimulation with PMA, considerable G-protein–mediated inhibition remained, as prepulses could still facilitate current amplitude (Fig. 6 A).
Figure 6.
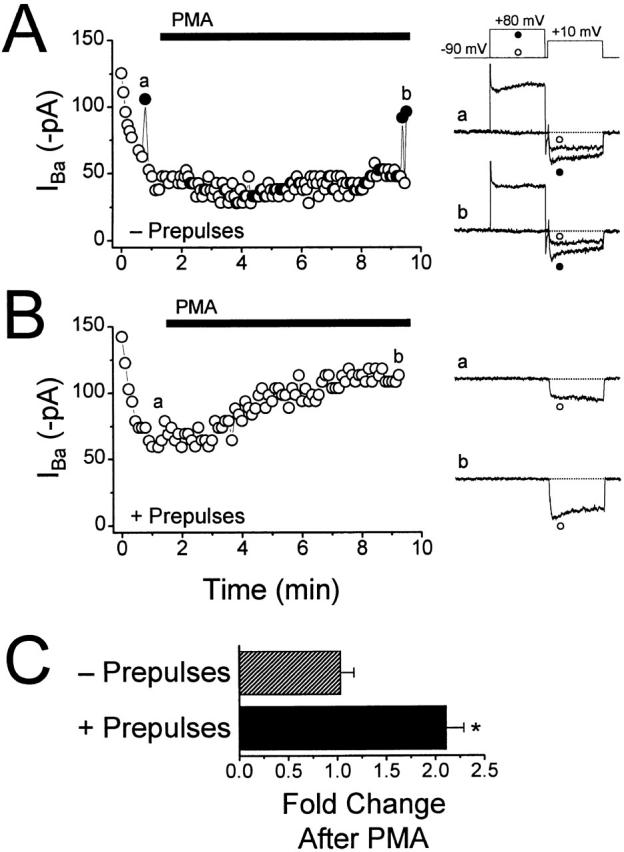
G-protein–mediated inhibition blocks PMA's effects on whole-cell currents. (A–B) Currents were elicited by stepping to +10 mV, and peak inward current was plotted against time (left). GTP-γ-S was included in the pipette solution, and 500 nM PMA was applied to the bath as indicated by the solid bars. At the right are individual sweeps from the times indicated. (A) Currents were elicited without applying prepulses, except where indicated by •, which were evoked following a prepulse to +80 mV. (B) In a second experiment, currents were elicited using the voltage protocol shown in A; prepulses were applied every other sweep, and currents evoked without a prepulse are plotted. (C) Summary of the fold change in current amplitude after application of PMA. In the absence of prepulses, PMA caused a 1.04 ± 0.13-fold change in current amplitude (n = 4). In contrast, when alternating test pulses were preceded by prepulses, applying PMA increased current amplitude 2.12 ± 0.17-fold (n = 4). *P < 0.005 compared with those without prepulses.
In separate experiments, currents were recorded in which every other test pulse to +10 mV was preceded by a pulse to +80 mV (Fig. 6 B). Under these conditions, application of PMA was sufficient to enhance whole-cell current amplitude. Because GTP-γ-S increases the level of available Gβγ subunits in the cytoplasm, we would expect PMA to have a slower effect when recording with GTP-γ-S than with GTP. We therefore measured the time constants of PMA's effect when prepulses were applied under both conditions. The time constant observed was 3.85 ± 0.69 min (n = 13) with GTP, and 5.18 ± 0.68 min (n = 3) with GTP-γ-S. In addition, because GTP-γ-S leads to a greater inhibition of current than GTP (Fig. 1 and Fig. 2), we would expect PMA to cause a greater enhancement of whole-cell currents after dialysis of GTP-γ-S. As predicted, when prepulses were applied throughout the recording, PMA increased current amplitude approximately twofold (Fig. 6 C), compared with 1.3-fold with GTP (Fig. 4). These results indicate that phosphorylation occurred only after G-proteins were displaced from the channel. Together with the finding that phosphorylation by PKC prevents G-protein–mediated inhibition, these findings support mutual exclusivity between G-protein binding and phosphorylation.
GDP-β-S Precludes PKC-mediated Enhancement of Current Amplitude
The above data indicate that PKC activation prevents G-protein–mediated inhibition. However, it is unclear whether PKC affects whole-cell currents in additional ways. Therefore, we next examined whether PKC modulates whole-cell currents in the absence of G-protein activity. When GDP-β-S was included in the pipette, we observed an increase in current amplitude after membrane breakthrough (not shown), consistent with previously published results (Netzer et al. 1994). This increase is believed to be the result of a loss of tonic G-protein–mediated inhibition, a hypothesis supported by the complete loss of observable facilitation (Fig. 1 D and 2). Once this effect reached steady state (typically within 1 min of membrane breakthrough), we studied the effect of activating PKC on whole-cell currents. Unlike the significant increase observed when GTP or GTP-γ-S was included in the pipette, PMA had no significant effect on current amplitude when GDP-β-S was used (Fig. 4).
Holding Potential–induced Inactivation of Whole-Cell Currents Is Not Affected by PKC Activation
Although it did not affect current amplitude, PKC activation might affect other properties of the current. Thus, we next examined whether phosphorylation has an effect on holding potential–dependent inactivation. Data were collected from 100-ms test pulses to +10 mV, preceded by 2.2-s prepulses to varying potentials. For these experiments, no attempt was made to isolate fast inactivation from steady state inactivation; hence, all inactivation measured with this protocol was defined as holding potential–induced inactivation (Fox et al. 1987; Jones and Marks 1989; Patil et al. 1998). Normalized inactivation curves generated with this protocol are shown in Fig. 7 A. When GDP-β-S was included in the pipette, normalized current amplitude decreased as holding potential became less negative, reaching a minimum of ∼0.3 at around +20 mV. A slight but statistically significant recovery from inactivation was observed as the holding potential became more positive; this is consistent with previous reports of “U”-shaped inactivation curves recorded under similar conditions (Patil et al. 1998). Bath application of PMA was without effect on holding potential–induced inactivation, suggesting that phosphorylation, in the absence of G-protein activation, has no effect on inactivation. In contrast, including GTP in the pipette led to a lower degree of holding potential–induced inactivation, suggesting a possible role of G-protein modulation in protecting the channels from inactivation.
Figure 7.
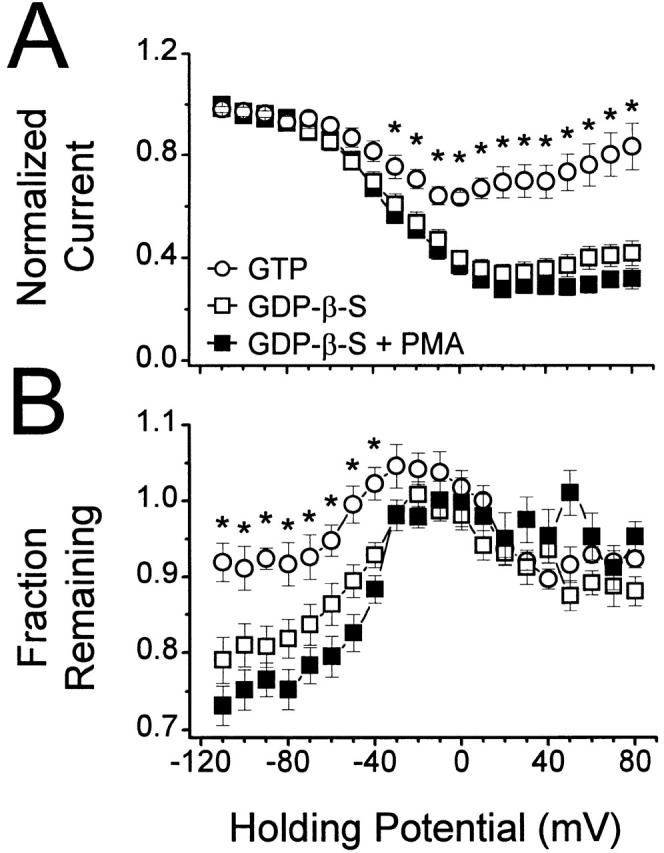
Activation of PKC does not affect holding potential–dependent inactivation. Currents were elicited by first applying a 2.2-s prepulse of varying voltage, followed 5-ms later by a 100-ms step to +10 mV. ○ were obtained with GTP in the pipette (n = 6), □ were obtained with 0.1 mM GDP-β-S substituted for GTP (n = 7), and ▪ were obtained with 0.1 mM GDP-β-S in the pipette and 500 nM PMA in the bath (n = 6). The symbol key pertains to both A and B. (A) Normalized holding potential–induced inactivation plots were generated by measuring the peak inward current during test pulses to +10 mV from each holding potential, and then dividing by the maximum current obtained. No statistically significant difference exists between the two sets of GDP-β-S data. (B) The fraction of current remaining was calculated as in Fig. 3. No statistically significant difference exists between the two sets of GDP-β-S data. *P < 0.05 versus GDP-β-S and versus GDP-β-S + PMA.
Changes in holding potential can also affect fast inactivation (Patil et al. 1998). Therefore, analyzing the same recordings presented in Fig. 7 A for fraction remaining provided an additional means of examining the effects of phosphorylation on inactivation in the absence of modulation by G-proteins. Fig. 7 B shows that, at negative holding potentials, cells containing GTP showed little fast inactivation, whereas cells containing GDP-β-S showed significantly more, consistent with the data presented in Fig. 3. At less negative holding potentials, fast inactivation decreased, reaching a fraction remaining of ∼1. Applying PMA to cells dialyzed with GDP-β-S was without significant effect on fraction remaining, demonstrating that phosphorylation by PKC, in itself, does not affect inactivation of channels.
Stimulation of PKC Does Not Affect Voltage-dependent Activation of Whole-Cell Currents
Our data suggest that G-protein–mediated inhibition shifts the voltage dependence of current activation to more positive voltages (Fig. 1). Moreover, previous data (Zhu and Ikeda 1994) demonstrated a voltage-dependent increase in tail current amplitude after application of PMA. Based on these findings, we hypothesized that this increase was due to a block of G-protein–mediated inhibition. To test this, voltage-dependent activation was examined under various recording conditions (Fig. 8 and Table ). First, activation was measured when endogenous G-proteins were maximally stimulated by dialysis with GTP-γ-S (Fig. 8 B). Consistent with relief of G-protein–mediated inhibition, prepulses significantly facilitated voltage-dependent activation without shifting the threshold of activation. Cells dialyzed with GDP-β-S (Fig. 8 C) displayed voltage-dependent activation that was similar to cells recorded with prepulses after GTP-γ-S dialysis (Table ). This is consistent with a loss of G-protein–mediated inhibition. Subsequent application of PMA had no effect on voltage-dependent activation, supporting the hypothesis that Zhu and Ikeda 1994 reported increase in tail current amplitude was due to the loss of G-protein–mediated inhibition. More importantly, these results indicate that activation of PKC, in the absence of G-protein–mediated inhibition, does not affect activation of whole-cell currents.
DISCUSSION
In this study, we sought to determine what effect, if any, activation of PKC by PMA has on whole-cell barium currents in neonatal rat SCG neurons, in the absence of G-protein modulation. Before proceeding with this analysis, however, we established that, under our recording conditions, whole-cell currents can undergo voltage-dependent G-protein–mediated inhibition, and that this inhibition is restricted to N-type currents (Plummer et al. 1991). Moreover, dialysis with GDP-β-S was sufficient to minimize this inhibition. We also confirmed that, in cells dialyzed with GTP, activation of PKC reduced prepulse facilitation, enhanced current amplitude, and increased fast inactivation (Swartz 1993; Swartz et al. 1993). In addition, PMA reversed tonic inhibition, as demonstrated by comparing the effects of PMA with the effects of either applying prepulses or dialyzing the cell with GDP-β-S. Finally, as demonstrated previously (Zhu and Ikeda 1994), we found that activation of PKC enhanced non–N-type currents present in this cell type.
Having confirmed the presence of tonic G-protein–mediated inhibition, and its modulation by PKC, we next examined whether these two mechanisms can preclude one another. By examining the effect of PMA on current amplitude in the absence of prepulses, we demonstrated that G-protein–mediated inhibition is sufficient to block the effects of PKC activity. Moreover, our findings are consistent with the hypothesis that activation of PKC is sufficient to block G-protein–mediated inhibition. These results support a model of mutual exclusivity between phosphorylation and G-protein–mediated inhibition, consistent with previously published results (Swartz 1993; Zhu and Ikeda 1994; Zamponi et al. 1997; Hamid et al. 1999).
Lastly, we examined whether PKC activation in the absence of G-protein–mediated inhibition causes additional modulation of whole-cell currents. When inhibition was first minimized by including GDP-β-S in the pipette solution, bath application of PMA was without significant effect on current amplitude, fast and holding potential–dependent inactivation, or voltage-dependent activation, suggesting that PKC's only role in modulating N-type currents is to block G-protein–mediated inhibition. These results are somewhat surprising since multiple putative PKC consensus sites are present on the pore-forming α1B subunit (Dubel et al. 1992). However, these findings are consistent with some recombinant studies that indicate that phosphorylation of select sites in the I–II linker of the α1B subunit can account for the loss of inhibition by G-proteins (Zamponi et al. 1997; Hamid et al. 1999).
N-type calcium channels inhibited by G-proteins have been termed “reluctant” by Bean 1989, reflecting the channel's diminished response to changes in membrane potential (Wanke et al. 1987; Bean 1989; Ikeda 1991; Patil et al. 1996). Because G-protein binding to the channel is sufficient to block PKC's effects, “reluctant” can be further defined as “reluctant and P-resistant,” indicating that the channel is not only reluctant to open, but also resistant to phosphorylation by PKC (Fig. 9). This G-protein–bound form of the channel may be important in mediating readily reversible modulation of neurotransmitter release (Koh and Hille 1997).
Figure 9.

Phosphorylation by PKC and modulation by G-proteins are mutually exclusive. Unmodulated N-type calcium channels are willing and available for either G-protein modulation or phosphorylation by PKC (Willing/Available). G-protein binding leads to a reluctant channel that is resistant to phosphorylation (Reluctant/P-Resistant). Alternatively, phosphorylation by PKC leads to a willing channel that is resistant to G-protein interactions (Willing/G-Resistant). Note that shifting from P-reluctant to G-reluctant (and vice versa) requires a transition through the willing and available form of the channel.
In contrast, channels not inhibited by G-proteins have been called “willing,” indicating a more rapid response to changes in membrane potential (Bean 1989). Channel activity can shift from reluctant to willing with the application of a strong positive voltage pulse (Bean 1989; Ikeda 1991), or by a train of action potentials (Williams et al. 1997), leading to facilitated whole-cell currents. Our results indicate the existence of at least two distinct willing forms of the channel, with phosphorylation by protein kinase C serving as the molecular switch between them (Fig. 9). Channels not modulated by either G-proteins or phosphorylation constitute one willing form, which we call “willing and available,” as these channels are willing to open, and are available for modulation by either G-protein activation or phosphorylation by PKC. Subsequent phosphorylation by PKC drives the channel into a second willing form, called “willing and G-resistant,” meaning willing to open, but resistant to G-protein modulation. In contrast to the P-resistant form, this G-resistant form would allow for long-term facilitation of calcium currents in synaptic membranes.
In conclusion, we have confirmed that N-type calcium channel activity in neonatal rat SCG neurons undergoes voltage-dependent G-protein–mediated inhibition. In addition, stimulation of PKC enhances whole-cell barium currents by blocking this inhibition. Moreover, when G-proteins are activated with GTP-γ-S, enhancement by PMA only occurs after prepulses, indicating that G-proteins must dissociate from the channel to observe the effects of phosphorylation by PKC. Finally, we have demonstrated that, under our recording conditions, there appears to be no functional effect of phosphorylation by PKC on N-type calcium channel activity beyond causing a long-term block of G-protein–mediated inhibition. Because of the existence of many PKC consensus sites on the N-type calcium channel (Dubel et al. 1992), future studies using other recording conditions might reveal additional effects of phosphorylation.
Acknowledgments
We thank Drs. Alex Dopico and Liwang Liu for critiquing various versions of this manuscript.
This publication was made possible by support from the National Institutes of Health (grant NS34195) and its contents are solely the responsibility of the authors and do not necessarily reflect the official view of these granting agencies. A.R. Rittenhouse is the recipient of an Established Investigator Award from the American Heart Association (grant 9940225).
Footnotes
Portions of this work were previously published in abstract form (Barrett, C.F., and A.R. Rittenhouse. 1998. J. Gen. Physiol. 112:18a).
Abbreviations used in this paper: BLM, bisindolylmaleimide I; CTX, ω-conotoxin GVIA; SCG, superior cervical ganglion.
References
- Bean B.P. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- de Leon M., Wang Y., Jones L., Perez-Reyes E., Wei X., Soong T.W., Snutch T.P., Yue D.T. Essential Ca(2+)-binding motif for Ca(2+)-sensitive inactivation of L-type Ca2+ channels. Science. 1995;270:1502–1506. doi: 10.1126/science.270.5241.1502. [DOI] [PubMed] [Google Scholar]
- Dubel S., Starr T., Hell J., Ahlijanian M., Enyeart J., Catterall W., Snutch T. Molecular cloning of the alpha-1 subunit of an omega-conotoxin-sensitive calcium channel. Proc. Natl. Acad. Sci. USA. 1992;89:5058–5062. doi: 10.1073/pnas.89.11.5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich I., Elmslie K.S. Neurotransmitters acting via different G proteins inhibit N-type calcium current by an identical mechanism in rat sympathetic neurons. J. Neurophysiol. 1995;74:2251–2257. doi: 10.1152/jn.1995.74.6.2251. [DOI] [PubMed] [Google Scholar]
- Fox A.P., Nowycky M.C., Tsien R.W. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. J. Physiol. 1987;394:149–172. doi: 10.1113/jphysiol.1987.sp016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamid J., Nelson D., Spaetgens R., Dubel S.J., Snutch T.P., Zamponi G.W. Identification of an integration center for cross-talk between protein kinase C and G protein modulation of N-type calcium channels. J. Biol. Chem. 1999;274:6195–6202. doi: 10.1074/jbc.274.10.6195. [DOI] [PubMed] [Google Scholar]
- Hawrot E., Patterson P.H. Long-tern culture of dissociated sympathetic neurons. Methods Enzymol. 1979;58:574–584. doi: 10.1016/s0076-6879(79)58174-9. [DOI] [PubMed] [Google Scholar]
- Hille B., Beech D.J., Bernheim L., Mathie A., Shapiro M.S., Wollmuth L.P. Multiple G-protein–coupled pathways inhibit N-type Ca channels of neurons. Life Sci. 1995;56:989–992. doi: 10.1016/0024-3205(95)00038-8. [DOI] [PubMed] [Google Scholar]
- Huganir R.L., Greengard P. Regulation of neurotransmitter receptor desensitization by protein phosphorylation. Neuron. 1990;5:555–567. doi: 10.1016/0896-6273(90)90211-w. [DOI] [PubMed] [Google Scholar]
- Ikeda S.R. Double-pulse calcium channel current facilitation in adult rat sympathetic neurones. J. Physiol. 1991;439:181–214. doi: 10.1113/jphysiol.1991.sp018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S.W., Marks T.N. Calcium currents in bullfrog sympathetic neurons. II. Inactivation. J. Gen. Physiol. 1989;94:169–182. doi: 10.1085/jgp.94.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh D.S., Hille B. Modulation by neurotransmitters of catecholamine secretion from sympathetic ganglion neurons detected by amperometry. Proc. Natl. Acad. Sci. USA. 1997;94:1506–1511. doi: 10.1073/pnas.94.4.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez H.S., Brown A.M. Correlation between G protein activation and reblocking kinetics of Ca2+ channel currents in rat sensory neurons. Neuron. 1991;7:1061–1068. doi: 10.1016/0896-6273(91)90350-9. [DOI] [PubMed] [Google Scholar]
- McCleskey E.W., Fox A.P., Feldman D.H., Cruz L.J., Olivera B.M., Tsien R.W., Yoshikami D. Omega-conotoxindirect and persistent blockade of specific types of calcium channels in neurons but not muscle. Proc. Natl. Acad. Sci. USA. 1987;84:4327–4331. doi: 10.1073/pnas.84.12.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzer R., Pflimlin P., Trube G. Tonic inhibition of neuronal calcium channels by G proteins removed during whole-cell patch-clamp experiments. Pflügers Arch. 1994;426:206–213. doi: 10.1007/BF00374773. [DOI] [PubMed] [Google Scholar]
- Patil P.G., Brody D.L., Yue D.T. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–1038. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Patil P.G., de Leon M., Reed R.R., Dubel S., Snutch T.P., Yue D.T. Elementary events underlying voltage-dependent G-protein inhibition of N-type calcium channels. Biophys. J. 1996;71:2509–2521. doi: 10.1016/S0006-3495(96)79444-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer M.R., Logothetis D.E., Hess P. Elementary properties and pharmacological sensitivities of calcium channels in mammalian peripheral neurons. Neuron. 1989;2:1453–1463. doi: 10.1016/0896-6273(89)90191-8. [DOI] [PubMed] [Google Scholar]
- Plummer M.R., Rittenhouse A., Kanevsky M., Hess P. Neurotransmitter modulation of calcium channels in rat sympathetic neurons. J. Neurosci. 1991;11:2339–2348. doi: 10.1523/JNEUROSCI.11-08-02339.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz K.J. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neuronsdisruption of G protein–mediated inhibition. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- Swartz K.J., Merritt A., Bean B.P., Lovinger D.M. Protein kinase C modulates glutamate receptor inhibition of Ca2+ channels and synaptic transmission. Nature. 1993;361:165–168. doi: 10.1038/361165a0. [DOI] [PubMed] [Google Scholar]
- Toullec D., Pianetti P., Coste H., Bellevergue P., Grand-Perret T., Ajakane M., Baudet V., Boissin P., Boursier E., Loriolle F. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Van Duuren B.L., Tseng S.S., Segal A., Smith A.C., Melchionne S., Seidman I. Effects of structural changes on the tumor-promoting activity of phorbol myristate acetate on mouse skin. Cancer Res. 1979;39:2644–2646. [PubMed] [Google Scholar]
- Wanke E., Ferroni A., Malgaroli A., Ambrosini A., Pozzan T., Meldolesi J. Activation of a muscarinic receptor selectively inhibits a rapidly inactivated Ca2+ current in rat sympathetic neurons. Proc. Natl. Acad. Sci. USA. 1987;84:4313–4317. doi: 10.1073/pnas.84.12.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S., Serafin M., Muhlethaler M., Bernheim L. Facilitation of N-type calcium current is dependent on the frequency of action potential-like depolarizations in dissociated cholinergic basal forebrain neurons of the guinea pig. J. Neurosci. 1997;17:1625–1632. doi: 10.1523/JNEUROSCI.17-05-01625.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Tsien R.W. Enhancement of N- and L-type calcium channel currents by protein kinase C in frog sympathetic neurons. Neuron. 1993;10:127–136. doi: 10.1016/0896-6273(93)90305-b. [DOI] [PubMed] [Google Scholar]
- Zamponi G.W., Bourinet E., Nelson D., Nargeot J., Snutch T.P. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- Zamponi G.W., Snutch T.P. Decay of prepulse facilitation of N type calcium channels during G protein inhibition is consistent with binding of a single Gbeta subunit. Proc. Natl. Acad. Sci. USA. 1998;95:4035–4039. doi: 10.1073/pnas.95.7.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Ikeda S.R. Modulation of Ca2+-channel currents by protein kinase C in adult rat sympathetic neurons. J. Neurophysiol. 1994;72:1549–1560. doi: 10.1152/jn.1994.72.4.1549. [DOI] [PubMed] [Google Scholar]