

Abstract
To investigate possible effects of adrenergic stimulation on G protein–activated inwardly rectifying K+ channels (GIRK), acetylcholine (ACh)-evoked K+ current, IKACh, was recorded from adult rat atrial cardiomyocytes using the whole cell patch clamp method and a fast perfusion system. The rise time of IKACh was 0.4 ± 0.1 s. When isoproterenol (Iso) was applied simultaneously with ACh, an additional slow component (11.4 ± 3.0 s) appeared, and the amplitude of the elicited IKACh was increased by 22.9 ± 5.4%. Both the slow component of activation and the current increase caused by Iso were abolished by preincubation in 50 μM H89 {N-[2-((p -bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide, a potent inhibitor of PKA}. This heterologous facilitation of GIRK current by β-adrenergic stimulation was further studied in Xenopus laevis oocytes coexpressing β2-adrenergic receptors, m2 -receptors, and GIRK1/GIRK4 subunits. Both Iso and ACh elicited GIRK currents in these oocytes. Furthermore, Iso facilitated ACh currents in a way, similar to atrial cells. Cytosolic injection of 30–60 pmol cAMP, but not of Rp-cAMPS (a cAMP analogue that is inhibitory to PKA) mimicked the β2-adrenergic effect. The possibility that the potentiation of GIRK currents was a result of the phosphorylation of the β-adrenergic receptor (β2AR) by PKA was excluded by using a mutant β2AR in which the residues for PKA-mediated modulation were mutated. Overexpression of the α subunit of G proteins (Gαs) led to an increase in basal as well as agonist-induced GIRK1/GIRK4 currents (inhibited by H89). At higher levels of expressed Gαs, GIRK currents were inhibited, presumably due to sequestration of the β/γ subunit dimer of G protein. GIRK1/GIRK5, GIRK1/GIRK2, and homomeric GIRK2 channels were also regulated by cAMP injections. Mutant GIRK1/GIRK4 channels in which the 40 COOH-terminal amino acids (which contain a strong PKA phosphorylation consensus site) were deleted were also modulated by cAMP injections. Hence, the structural determinant responsible is not located within this region. We conclude that, both in atrial myocytes and in Xenopus oocytes, β-adrenergic stimulation potentiates the ACh-evoked GIRK channels via a pathway that involves PKA-catalyzed phosphorylation downstream from β2AR.
Keywords: G protein–activated inwardly rectifying K+ channels , protein kinase A, heterologous facilitation, cardiomyocytes, Xenopus
INTRODUCTION
Adaptation of cardiac output by the sympathetic nerve acts through potentiation of voltage-dependent Ca2+ and Na+ channel function resulting from the activation of the stimulatory α subunit of G proteins (Gαs), adenylyl cyclase, and subsequent phosphorylation of these channels by cAMP-dependent protein kinase (PKA; Osterrieder et al. 1982; Cachelin et al. 1983; Frohnwieser et al. 1997). Acetylcholine (ACh), released from the vagus nerve, on the other hand, negatively regulates heart rate through inhibitory, pertussis toxin sensitive, G-proteins. Gαi inhibits adenylyl cyclase and subsequently PKA, while direct activation of G-protein–activated inwardly rectifying K+ channels (GIRK) is achieved by simultaneous release of the β/γ subunit dimer of G protein (Gβγ) (for review, see Dascal 1997). Little is known about any possible connection of the sympathetic pathway to membrane hyperpolarizing effectors such as GIRKs. Several reports do describe regulation of GIRKs and/or IKACh by (a) transphosphorylation of GDP via nucleosidediphosphate kinase, (b) directly by intracellular nucleotides, and/or (c) by protein phosphorylation/dephosphorylation (reviewed by Dascal 1997). Kim 1990 found that β-adrenergic stimulation and exposure of inside-out patches derived from rat atrial cells to the catalytic subunit of PKA enhanced IKACh, but in other preparations PKA effects on IKACh were absent ( Trautwein et al. 1982; Nargeot et al. 1983; Wang and Lipsius 1995). We recently found that GTPγS-activated Gαs attenuates G βγ-induced activation of GIRK channels expressed in Xenopus oocytes in a membrane-delimited manner ( Schreibmayer et al. 1996). This raises the possibility that such inhibition may occur in atrial myocytes when sympathetically stimulated. The current study investigates the effects of β-adrenergic stimulation on GIRK channels in native rat atrial cells and in the Xenopus laevis oocyte expression system. We find that, in atrial myocytes as well as in Xenopus oocytes, the main effect of β-adrenergic stimulation is an enhancement of the ACh-induced GIRK current via PKA-catalyzed phosphorylation. The target for this phosphorylation is still unclear, but it is downstream from the β-adrenergic receptor.
MATERIALS AND METHODS
Electrophysiology
Atrial cells were enzymatically disaggregated from hearts of adult Sprague-Dawley rats as described (Dascal et al. 1993) and stored in the incubator for 1–3 d at 37°C in M199 under 95% O 2/5% CO2 until electrophysiological recordings were performed. Cells were then pipetted into a plastic petri dish with a diameter of 35 mm, mounted to a Peltier element thermostated object holder of an inverted microscope (IM35; Carl Zeiss, Inc.). Patch-clamp current recordings were performed with an Axopatch 1D amplifier (Axon Instruments) using fire-polished pipettes with an open tip resistance of 2–4 MΩ, pulled from standard hematocrit capillaries (564, L/M-3P-A puller; List Electronik). Whole cell current recordings were obtained by rupturing the membrane patch under the tip of the glass pipette by suction. Pipette solutions for perforated patch recordings were made by mixing 100 μl of a nystatin stock solution (50 mg/ml nystatin dissolved in DMSO in the original solution) with 10 ml pipette solution, sonicated and filtered through 200-μm ultrafilters before use. The amount of nystatin needed to get electrical perforation within 1–2 min after gigaseal formation varied within a range of 1–5× and depended on the atrial cell preparation. Current recordings were obtained by keeping the cell's membrane potential constant at −80 mV and superfusing with HP medium (see below) containing 10−5 M acetylcholine and/or 10−6 M isoproterenol. H89 {N -[2-((p -Bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide} was introduced into the cytosol by adding 5 × 10−5 M to the pipette solution. In addition to a regular, gravitation driven slow perfusion with HP, we also used a fast perfusion system (ALA Scientific) that allowed the fast exchange of medium surrounding the myocyte within 1 s. Current traces were Bessel low-pass filtered at 20 Hz and digitized at 50 Hz using the Labmaster-TL-1 interface (Axon Instruments) connected to a standard Pentium PC using Axotape software (Axon Instruments). The traces were analyzed using Fetchan 6.0 (Axon Instruments). For the evaluation of onset kinetics of IKACh, only those cells were selected whose washout kinetics of acetylcholine indicated efficient fast superfusion (i.e., the time course of washout was exponential). The rising phase of I KACh was fitted to standard one- or two-exponential equations using Sigmaplot (Jandel Scientific). Xenopus laevis oocytes were prepared as described (Dascal and Lotan 1992), and the following amounts of cRNA were injected (ng/oocyte): 0.1–1.5 muscarinic m2-receptor (m2R), 0.05–0.75 wild-type β-adrenergic receptor (β2ARwt), 0.05–0.75 β2ARPF, 0.0025–0.2 GIRK1 wt, 0.0025–0.0075 GIRK1ΔC40, 0.0125–0.2 GIRK4wt, 0.0125–0.0375 GIRK4 ΔC40, 0.075–0.75 GIRK2wt, 0.02–2 Gαs, 1.5 Gβ2, 1.5 Gγ1, 10 CFTR. To knock out the endogenously existing GIRK5 subunit, 80 ng/oocyte of the phosphothioated antisense oligonucleotide KHA2 was injected together with the cRNAs (Hedin et al. 1996). Oocytes were kept in NDE at 19–21°C in an incubator for 3–10 d before electrophysiological recordings. Oocytes were placed in a recording chamber that allowed superfusion with extracellular medium by gravity flow at 19–21°C and currents were recorded with the two-electrode voltage clamp method using agarose cushion electrodes ( Schreibmayer et al. 1994) and a Geneclamp 500 amplifier (Axon Instruments). The membrane potential was kept constant at −80 mV and the resulting current was measured first in regular ND96 solution, and then in a high-K+ extracellular medium (HK) with and without various neurotransmitters. Cytosolic injection of reagents was performed as described (Frohnwieser et al. 1997). Current traces were low pass filtered at 10 Hz and digitized at 50 Hz using a TL-1-125 interface (Axon Instruments) connected to a 486 compatible PC (Steiner Computers) using the Axotape software (Axon Instruments). Evaluation of current recordings was performed using the Fetchan 6.0. Preincubation of oocytes in H89 was performed by placing the oocytes for 2–3 h in NDE containing 5 × 10−5 M H89 before experimentation (see Perets et al. 1996). Oocytes were Pertussis toxin (PTX) treated by injecting 100 or 200 ng/oocyte of the A-protomer of the toxin (Alomone Laboratories) 3–16 h before the experiment ( Sharon et al. 1997).
Molecular Biology
Plasmid vectors were constructed, grown in bacteria, isolated, and linearized using standard procedures (Sambrook et al. 1989). cRNA was synthesized as described (Dascal and Lotan 1992). The following plasmid vectors were used as described previously: m2 R, β2AR (Fidler-Lim et al. 1995); Gαs (Blumenstein et al. 1999); GIRK1 wt, GIRK2wt, GIRK4wt ( Silverman et al. 1996); CFTR ( Uezono et al. 1993); Gβ2 and Gγ1 (Jing et al. 1999). The cDNA of β2AR PF was obtained from Y. Daaka (Daaka et al. 1997) and subcloned into the pGEMHE vector (Liman et al. 1992). To create GIRK1ΔC40 and GIRK4ΔC40, we performed a PCR procedure with forward and reverse primers containing the desired parts of the coding sequence of each cDNA preceded or followed, respectively, by restriction enzyme recognition sequences. The PCR products were digested with the restriction enzymes and ligated into the EcoRI and HindIII sites (GIRK1ΔC40) and Sma1 and XbaI sites (GIRK4ΔC40) of pGEMHE.
Data Normalization and Statistics
To normalize our data (to eliminate scatter introduced by batch-to-batch variation of protein expression in oocyte preparations, different ratios of basal to acetylcholine induced currents for different GIRK isoform combinations or mutants), all currents were expressed as a percentage of the basal HK-induced current of the GIRK1/GIRK4 heterooligomeric channel of a given experimental day and batch of oocytes. Different groups were compared using the unpaired Student's t test (Sigmaplot 2.0; Jandel Scientific). In some instances, the calculated average value of current increase in percent was tested for a significant difference from zero by assuming a normal distribution ( Murray and Spiegel 1961).
Solutions
The composition of the solutions used was as follows (mM): HP: 136 KCl, 4 NaCl, 2 MgCl2, 10 HEPES, buffered with NaOH to pH 7.4; pipette solution: 120 K+/aspartate, 20 NaCl, 2 MgCl2 , 11 EGTA, 1 CaCl2, 2 ATP, 0.1 GTP, 10 HEPES, buffered with KOH to pH 7.4; ND96: 96 NaCl, 2 KCl, 1 MgCl2, 1 CaCl 2, 5 HEPES, buffered with NaOH to pH 7.4; NDE: same as ND96, but contained 2.5 mM pyruvate, 0.1% antibiotics (G-1397, 1,000× stock; Sigma Chemical Co.) and 1.8 CaCl2 ; HK: 96 KCl, 2 NaCl, 1 MgCl2, 1 CaCl2, 5 HEPES, buffered with KOH to pH 7.4.
Chemicals
All chemicals used were reagent grade throughout. Reagents for molecular biology were purchased from Boehringer-Mannheim or MBI Fermentas.
RESULTS
IKACh of freshly dissociated rat atrial cells was elicited by superfusion of the cells with 10−5 M acetylcholine in HP solution. The intracellular solution contained 2 mM ATP to allow the production of cAMP. β2AR coactivation by coapplication of 10−6 M isoproterenol enhanced IKACh by 20–25% (Fig. 1A and Fig. B). Furthermore, the onset kinetics of IKACh were changed: when only acetylcholine was present, the activation kinetics followed a monoexponential function with a time constant in the range of 0.4 s; an additional, slower time constant in the range of 10 s appeared upon isoproterenol coapplication (Fig. 1B and Fig. C; Table ). When isoproterenol was applied after the onset of I KACh, an additional increase of IKACh could also be observed, which was comparable in size to the β-adrenergic effect upon coapplication. This additional increase of IKACh was also observed when the whole-cell recording was performed using the perforated patch technique with nystatin, leaving the macromolecular composition of the cytosol intact (Fig. 1 B). Application of 10−6 M isoproterenol alone did not result in any detectable IKACh or increase in inward current (n = 5, data not shown). When H89, a specific inhibitor of PKA, was included in the patch pipette, the β-adrenergic effects, both on magnitude as well as on kinetics of IKACh, disappeared (Fig. 1).
Figure 1.

Effect of β-adrenergic stimulation on IKACh of rat atrial cells. (A) Original whole cell current recordings at a membrane holding potential of −80 mV. IKACh was induced by superfusion with ACh (10 μmol/liter) or ACh plus Iso (1 μmol/liter) in the absence (top) and in the presence (bottom) of 50 μmol/liter H89. (B) Statistics of the effect of Iso on IKACh amplitude, shown as percent increase compared with IKACh induced by ACh alone. Calculated average values ± SEM are shown; the number of individual cells is given in parenthesis. (* and **) Mean value deviates significantly (P < 0.05 and 0.01) from the H89 treated group. (Left to right) Effect of Iso coapplied with ACh (control, H89 treatment); additional IKACh induced by Iso application during ACh (control, IKACh recorded with nystatin perforated patch, H89 treatment). (C) Effect of Iso coapplication on kinetics of I KACh activation. (Left to right) Regular IKACh induced by ACh superfusion; IKACh induced by ACh coapplied with Iso; same as previous, but in the presence of 50 μmol/liter H89. (Dotted line) Monoexponential fit through the rising phase of I KACh. (Broken line) Biexponential fit, comprising a fast and an additional slow time constant.
Table 1.
Effect of Isoproterenol on rise time of IKACh
| Ach | Iso | H89 | τfast | τslow | n | Afast |
|---|---|---|---|---|---|---|
| + | − | − | 443 ± 85 ms | nd | 4 | / |
| + | + | − | 649 ± 139 ms | 11.4 ± 3.0 s | 4 | 0.78 ± 0.07 |
| + | + | + | 354 ± 4 ms | nd | 3 | / |
| + | − | + | 351 ± 36 ms | nd | 4 | / |
Concentrations used (μmol/liter): 10 ACh, 1 Iso, 50 H89. nd, slow component not detectable.
To investigate the details of the signal transduction pathway leading to the β-adrenergic effect on IKACh, we used a heterologous expression system, the Xenopus laevis oocyte. To assess the ability of heterologously expressed β2AR to activate PKA in Xenopus oocytes, we coexpressed β2AR together with the CFTR, a Cl− channel that is a known target for PKA phosphorylation (Anderson et al. 1991; Cheng et al. 1991). Even without coexpression of exogenous Gαs, isoproterenol produced a marked increase in inward current, which was equal in magnitude to CFTR inward currents produced by cAMP injections (Fig. 2A and Fig. B). Furthermore, isoproterenol-induced CFTR currents were prevented and/or blocked by cytosolic injections of Rp-cAMPS ( Fig. 2 A; summarized in C), demonstrating that the isoproterenol response was entirely due to PKA stimulation and that Rp-cAMPS injections are indeed effective in blocking endogenous PKA in Xenopus laevis oocytes.
Figure 2.
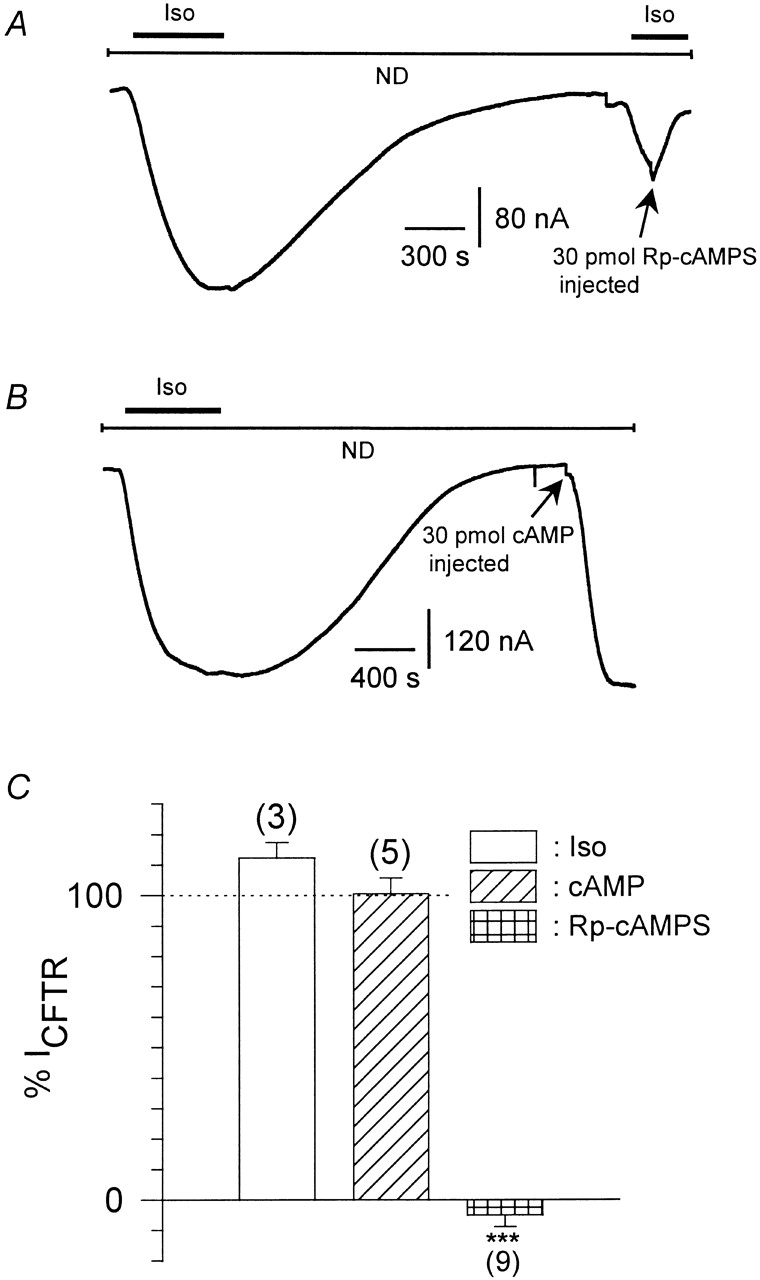
Stimulation of PKA by β2AR in Xenopus oocytes. (A) Original current trace from an oocyte expressing CFTR Cl− channels and β2AR recorded at a holding potential of −80 mV in ND96 solution. Upon isoproterenol superfusion (1 μmol/liter), CFTR channels were stimulated by PKA phosphorylation, what could be reversed by washing out of the agonist. During the second application of Iso, 30 pmol Rp-cAMPS were injected into the cytosol, abolishing and reversing the effect of agonist. (B) Same as in A, but instead of the second Iso pulse, 30 pmol cAMP was injected, stimulating CFTR channels to the same extend as the Iso pulse. (C) Statistics of stimulation of CFTR by agonist and/or cAMP and analogues normalized to a first, control pulse of Iso (taken as 100%). Calculated average values ± SEM are shown; the number of individual oocytes is given in parenthesis. (***) Mean value deviates significantly (P < 0.001) level from the group injected with stimulatory cAMP analogues. (Iso) After washing out of the first Iso pulse, 5 nl H2O were injected into the oocyte, and then the magnitude of CFTR currents elicited by a second pulse of Iso was measured. (cAMP) After the first pulse of Iso superfusion, 30–60 pmol cAMP (n = 3) or Sp-cAMPS (n = 2) were injected into the cytosol and the resulting CFTR currents were measured. (Rp-cAMPS) Same as cAMP, but Rp-cAMPS was injected instead of stimulatory cyclic nucleotides.
Modulation of GIRK channels by β-adrenergic receptors was studied in Xenopus oocytes heterologously expressing muscarinic m2 -receptor, β-adrenergic receptors, and two subunits of G-protein–activated, inwardly rectifying K+ channels (GIRK1 and GIRK4, respectively). Heteromeric GIRK1/GIRK4 channels are believed to constitute the atrial GIRK (see Dascal 1997), although homomeric GIRK4 channels may also contribute to total IKACh (Corey et al. 1998). Currents were recorded at −80 mV, first in a high-Na+, low-K+ physiological medium (ND96), which was then exchanged to a medium with high external K+, low-Na+ concentration (HK; leading to a basal inward current, termed IHK). When isoproterenol was applied together with acetylcholine, a marked increase of IACh was observed compared with the application of ACh alone (Fig. 3 A). Application of isoproterenol (Iso) when ACh was already present in the bathing solution led to a marked additional increase of IACh. In contrast to native rat atrial cells, application of isoproterenol alone during HK led to a small but clearly detectable increase in inward current, termed IIso ( Fig. 3 B). These effects of Iso were absent in oocytes that expressed GIRK and m2R, but were not injected with cRNA encoding β2ARs. It has been reported that G-protein βγ-subunits released from β-adrenergic receptors can directly activate GIRK channels in Xenopus oocytes and atrial cells, provided that exogenous Gβ and/or Gαs are overexpressed (Fidler-Lim et al. 1995; Bender et al. 1998; Sorota et al. 1999). To exclude direct Gβγ effects and to distinguish them from effects put forth by the cytosolic second messenger branch, 3′5′-cAMP was directly injected into the oocytes during electrophysiological recording. An enhancement of inward current resulted both in the absence and the presence of acetylcholine (Fig. 3C and Fig. E). The current voltage relation of cAMP-induced currents clearly revealed inward rectification (Fig. 3 F). Furthermore, cAMP effects on IHK were absent when cRNA encoding GIRK channels was not injected into the oocytes. The enhancement of the basal activity of GIRK channels by cAMP injection was not altered by β2AR coexpression [59.6 ± 7.5% (n = 55, see Fig. 3 D) vs. 59.7 ± 7.2% (n = 9, data not shown)]. Injection of Rp-cAMPS, a cAMP analogue and inhibitor of PKA, did not enhance the basal inward currents. This fact indicates that nonspecific effects of cyclic nucleotides, unrelated to PKA activation, were absent. To better understand the mechanisms of modulation of GIRK via β2AR and Gαs in Xenopus oocytes and to substantiate the finding that PKA might stimulate basal and agonist-induced GIRK currents in the oocytes, we overexpressed Gα s at different concentrations in addition to GIRK1, GIRK4, and m2R. At low doses of injected cRNA encoding Gαs, basal and ACh-evoked GIRK currents were enhanced, while at higher doses a suppression of both current components was observed. Interestingly, the increase in both basal and agonist-induced currents was inhibited by preincubation of the oocytes in 5 × 10−5 mol/liter H89, while the suppression at higher expression levels of Gαs was H89 insensitive (Fig. 4). These results suggest that the basal activity of the overexpressed Gαs is sufficient to activate adenylyl cyclase, and thus PKA, and to cause an increase in GIRK activity. At higher doses, the free Gαs most likely sequesters Gβγ, thus causing a decrease in GIRK activity.
Figure 3.
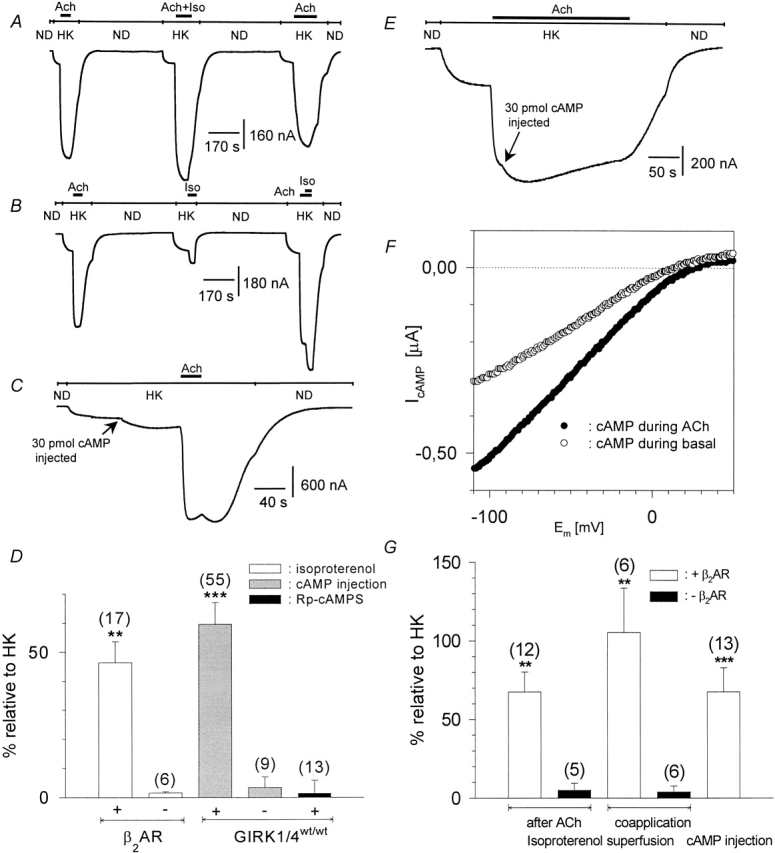
β2-adrenergic facilitation of heterologously coexpressed GIRK currents and cAMP effects in Xenopus laevis oocytes. (A) Original current trace recorded at a membrane holding potential of −80 mV. ND and HK denote changes of the superfusion medium from ND96 to HK, bars denote superfusion with agonists. (B) Same as in A, but Iso was applied alone or during ACh. (C) Same as in A, but 30 pmol cAMP was injected during HK before ACh superfusion. (D) Statistics of the effects of isoproterenol superfusion and/or cytosolic injection of cAMP and Rp-cAMPS on basal current amplitude, expressed as percentage of basal I HK. Calculated average values ± SEM are shown; the number of individual cells is given in parenthesis. (** and ***) Mean value deviates significantly (P < 0.01 and 0.001) from the corresponding control group (i.e., oocytes not expressing heterologous β2AR in the case of Iso effects) or cAMP injection into native oocytes in the case of cAMP effects. (Left to right) Iso-induced current on basal HK (with or without coexpressed β2AR), 30–60 pmol cAMP injection during basal HK, cAMP injection into native oocytes, injection of 30–60 pmol Rp-cAMPS during basal HK. (E) Same as in C, but cAMP was injected during ACh. (F) Current–voltage relation of the cAMP-induced currents as measured by a triangular voltage pulse (f = 1 Hz). (○) Current induced by cytosolic cAMP injection on basal current, obtained by subtraction of the basal current before cAMP injection from basal current after cAMP injection. (•) Current induced by cAMP injections on ACh-induced currents. (G) Statistics of the effects of isoproterenol superfusion and/or cytosolic injection of cAMP on ACh-induced current amplitude, expressed as a percentage of basal IHK. Calculated average values ± SEM are shown; the number of individual cells is given in parenthesis. (** and ***) Mean value deviates significantly ( P < 0.01 and 0.001) from the corresponding control group (i.e., oocytes not expressing heterologous β2AR in the case of Iso effects) or cAMP injection into native oocytes in the case of cAMP effects. (Left to right) Iso effect during ACh (with or without β 2AR), Iso effect upon coapplication with ACh (with or without β2AR), cAMP injection during ACh.
Figure 4.
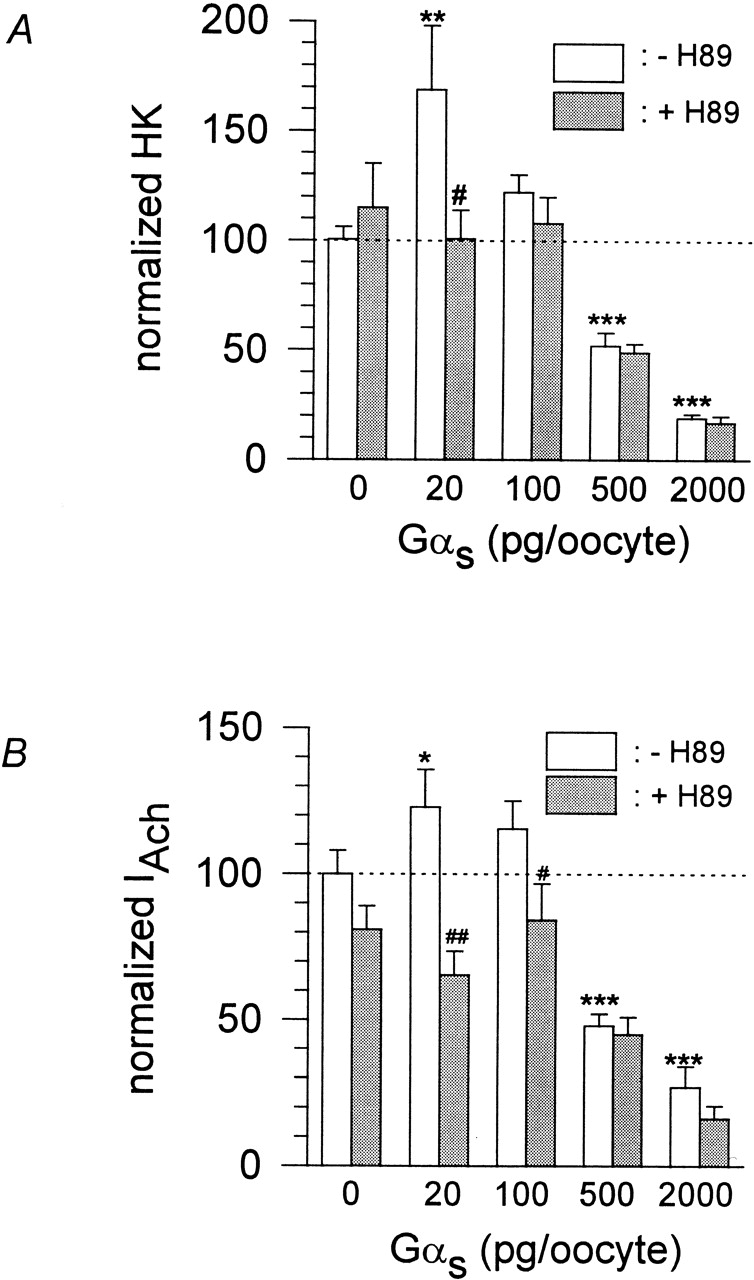
Effect of heterologous Gαs overexpression on basal and agonist-induced GIRK currents. (A) Statistics of effect of Gαs overexpression on basal current, normalized to basal IHK current of control (no Gαs overexpressed). Calculated average values ± SEM from two different batches of oocytes are shown (three to five oocytes per batch). Individual experiments were performed either in the absence (white bars) or after preincubation (gray bars) of oocytes in 50 μmol/liter H89. (# and ##) Mean value deviates significantly (P < 0.05 and 0.01) from the corresponding basal current (same amount of Gαs cRNA injected), but without H89 preincubation. (*, **, and ***) Mean value deviates significantly (P < 0.05, 0.01, and 0.001) from the basal current without Gα s overexpression. (B) Same as in A, but agonist-induced GIRK currents are normalized to IACh of control (no Gαs overexpressed).
In the intact atrium, stimulation of GIRK occurs via PTX-sensitive G proteins of the Gi/Go family (reviewed by Dascal 1997). Recently, it has been discovered that β 2-adrenergic receptors switch their G-protein selectivity from Gαs to Gαi/Gαo when β 2AR is phosphorylated by PKA (Daaka et al. 1997). This raised the possibility that the β-adrenergic stimulation of GIRK could result from a direct activation by PTX-sensitive G protein via the PKA phosphorylated β2AR. Therefore, oocytes expressing β2ARwt, m2R, and GIRK1/GIRK4 channels were pretreated with PTX to block the Gi/Go family proteins. While basal inward currents were reduced by the PTX treatment (indicating some contribution of basal-active inhibitory G proteins to the resting inward current), the current induced by isoproterenol was reduced slightly (Fig. 5). Furthermore, instead of β2ARwt, we expressed a mutant β2 -adrenergic receptor in which the PKA-dependent selectivity switch was disrupted (β2ARPF; Daaka et al. 1997). Interestingly, β2 -adrenergic receptor stimulation persisted (Fig. 5A and Fig. B). PTX treatment of oocytes expressing the mutant receptor resulted in a decrease of basal, agonist-independent current similar to the one observed with wild-type receptors, but the isoproterenol-evoked current was not affected.
Figure 5.
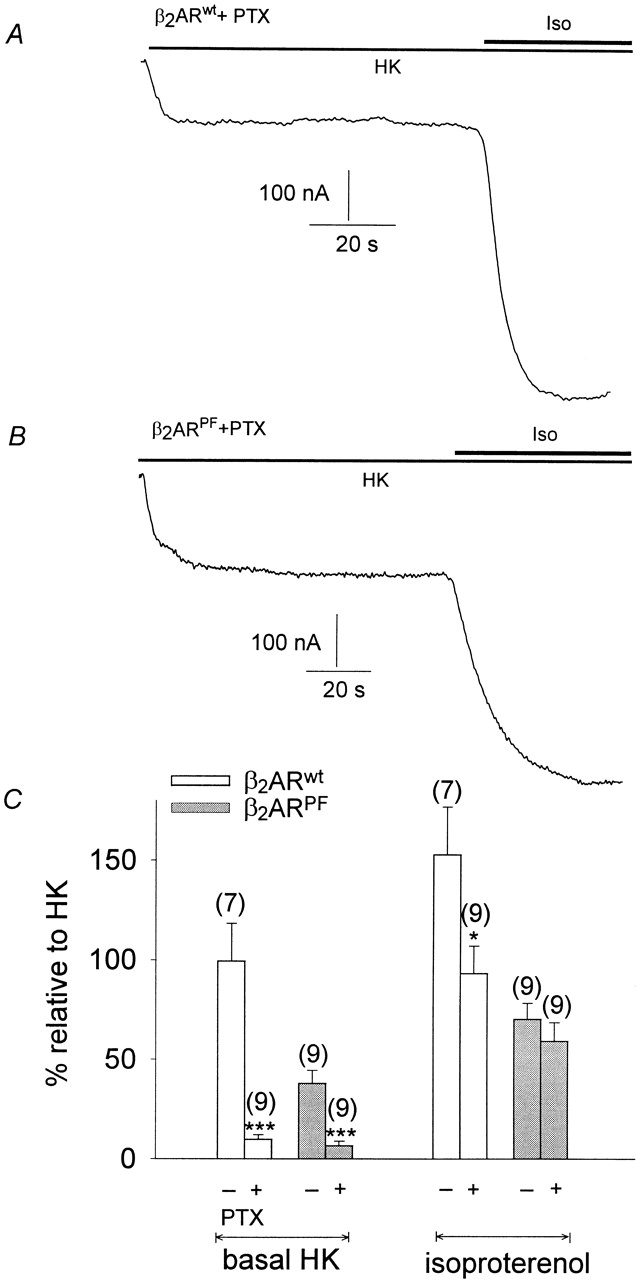
β2-adrenergic facilitation of GIRK currents is not produced by coupling to inhibitory G-proteins. (A) Original current traces recorded at −80 mV. GIRK currents were induced by superfusion of the oocytes with 1 μmol/liter isoproterenol. (Top) Current trace from an oocyte expressing β2ARwt and treated with PTX. (Bottom) Same as in the top, but the oocyte expressed β2AR PF instead of β2ARwt. (B) Statistics of the effect of PTX treatment and/or β2AR mutation on GIRK facilitation, expressed as a percentage of basal IHK of the control (no PTX treatment, β2ARwt expressed). (Empty bars) Oocytes expressing β2ARwt; (hatched bars) oocytes expressing β2ARPF. (+ and −) Oocytes incubated or not treated with PTX. Calculated average values ± SEM from three different batches of oocytes (three oocytes/batch) are shown. (* and ***) Mean value deviates significantly ( P < 0.05 and 0.001) from the corresponding control group; i.e., oocytes without PTX treatment.
Phosphorylation of seven-helix receptors is an important mechanism for the regulation of receptor activity (Pitcher et al. 1998). To discriminate between PKA effects on the receptor(s) and on the channel, GIRK1/GIRK4 channels were expressed without m2R and β2 AR. cAMP injections were performed to test for PKA effects on basal GIRK currents. As evident from Fig. 6, basally active GIRK channels respond to cAMP injections with the usual increase in activity (39.0 ± 7.5% of basal HK; n = 12; mean value differs statistically significant from zero at the P < 0.001 level).
Figure 6.

cAMP effects in the absence of heterologously coexpressed seven-helix receptors. Original current trace recorded at −80 mV derived from an oocyte expressing exclusively GIRK1/GIRK4 proteins. During HK superfusion, 50 pmol cAMP was injected.
To investigate whether other isomeric forms of GIRK channels are also modulated by PKA, we expressed different subunit compositions: GIRK1/GIRK5, GIRK1/GIRK2, and the homooligomeric GIRK2 channels. For combinations not containing GIRK5, the endogenous GIRK5 subunit was eliminated by coinjection of a subunit-specific antisense oligonucleotide (Hedin et al. 1996). When only cRNA encoding the GIRK1 isoform was injected with the oligonucleotide, the resulting IACh (flowing via the GIRK1/GIRK5 channels) was greatly reduced when compared with oocytes injected with the same amount of GIRK1 cRNA, but without the oligonucleotide (Fig. 7 D). Coinjection of the oligonucleotide with GIRK1/GIRK4 cRNAs also reduced IACh to some extent, indicating that in the previous experiments GIRK1/GIRK5 heterooligomeric channels still contributed to the acetylcholine response. As can be seen from Fig. 7, heterooligomeric GIRK1/GIRK2, GIRK1/GIRK4, and GIRK1/GIRK5, as well as homooligomeric GIRK2 channels respond to cytosolic cAMP injections, both under basal and under agonist-stimulated conditions. The cAMP effect on the homooligomeric GIRK2 channels was even significantly larger than on GIRK1/GIRK4 channels. The carboxy-terminal part of the GIRK subunit is involved in Gβγ binding (Huang et al. 1997), is important for gating ( Luchian et al. 1997), and contains a PKA phosphorylation consensus site (Dascal 1997). Hence, truncated forms of GIRK1 and GIRK4 were generated in which the last 40 amino acids were deleted. These constructs, however, still contained the Gβγ binding regions. Cytosolic cAMP injections enhanced currents of mutant channels comprised of both GIRK1ΔC40/GIRK4ΔC40 . The cAMP-induced currents had the following magnitude, when normalized to the basal current of the oocytes expressing GIRK1wt /GIRK4wt: cAMP injections on basal currents: GIRK1 ΔC40/GIRK4ΔC40, 33.3 ± 8.7% (n = 9), compared with 66.1 ± 8.0% (n = 40) for GIRK1 wt/GIRK4wt; cAMP injections during ACh superfusion: GIRK1ΔC40/GIRK4ΔC40, 67.2 ± 28.0% (n = 6) compared with 100.1 ± 22.9% (n = 13) for GIRK1wt/GIRK4wt. All cAMP effects deviate significantly from zero at the P < 0.001 level. Hence, the distal COOH terminus (last 40 aminoacids) of the GIRK channel is not the structural determinant that mediated the PKA effect.
Figure 7.

cAMP effects on GIRK isoforms. (A) Original current trace recorded at −80 mV from an oocyte expressing m2R and GIRK1. GIRK currents are therefore produced exclusively from the GIRK1/GIRK5 heterooligomeric assembly. cAMP was injected during basal HK current. (B) Current trace recorded from an oocyte expressing GIRK1/GIRK2 channels (endogenous GIRK5 was knocked out with the KHA2 antisense oligonucleotide). cAMP was injected during ACh superfusion. (C) Same as in B, but the oocyte expressed homooligomeric GIRK2 channels. (D) Effect of antisense oligonucleotide (KHA2) coinjected with either GIRK1 or GIRK1/GIRK4 cRNA on IACh currents. Calculated average values ± SEM are shown; the number of individual cells is given in parenthesis. (* and ***) Mean value deviates significantly (P < 0.05 and 0.001) from the corresponding control group (KHA2 not coinjected). (E) Statistics of cAMP effects on various GIRK subunit combinations on basal IHK currents as well as on agonist-induced currents (IACh). Calculated average values ± standard error of the effect normalized to basal IHK produced by the GIRK1/GIRK4 combination of a given experimental day are shown; the number of individual oocytes are in parenthesis. (** and ***) Mean value of the experimental group deviates significantly (P < 0.01 and 0.001) from zero.
DISCUSSION
Our results clearly show that IKACh in native atrial myocytes derived from adult rats is enhanced by β-adrenergic stimulation. This result strongly supports the finding of Kim 1990 that application of isoprenaline to newborn rat atrial cells increased I KACh channel activity in cell-attached membrane patches. In Xenopus laevis oocytes, heterologously expressed β2 -adrenergic receptors are able to activate GIRKs ( Fidler-Lim et al. 1995; and this study). In view of the results of the current study, only part of this β-adrenergic activation in the heterologous expression system can be attributed to a direct Gβγ effect. Furthermore, in native atrial cells, direct G-protein activation by β2ARs has been shown to occur only after heterologous overexpression of Gαs ( Sorota et al. 1999), of Gβ1 subunits ( Bender et al. 1998), or after antisense oligonucleotide knockout of GRK2 (Wellner-Kienitz et al. 1999). Kim 1990 suggested that β-adrenergic activation of I KACh is produced by Gαs-induced PKA phosphorylation, but in other preparations, PKA effects on IKACh were absent (Trautwein et al. 1982; Nargeot et al. 1983; Wang and Lipsius 1995). In our preparation, the slow onset kinetics as well as inhibition by H-89 indicate that the isoproterenol-induced increase of IKACh can be attributed to phosphorylation by PKA after activation of β2 -adrenergic receptors. Interestingly, isoproterenol was ineffective in the absence of acetylcholine in native atrial cells, suggesting that both Gβγ dimers and PKA phosphorylation are required for GIRK activation.
To study the molecular mechanism of PKA-induced GIRK stimulation in more detail, we employed the Xenopus laevis oocyte expression system. First, the CFTR Cl− channels were expressed as reporters of PKA activity, since PKA activation of these channels is well documented (Anderson et al. 1991; Cheng et al. 1991). Cytosolic injections of cAMP or Sp-cAMPS effectively activated CFTR currents to the same extent as heterologously coexpressed β2 AR (Fig. 2). This finding clearly shows that the coexpressed β2AR, via the oocyte's endogenous Gαs, is able to activate PKA to the same extent as direct cytosolic injections of the second messenger. Furthermore, cytosolic injections of Rp-cAMPS (a selective inhibitor of PKA) completely abolished β-adrenergic stimulation of CFTR, demonstrating that PKA activity is completely blocked under these conditions. Further evidence for PKA regulation of GIRK was derived using this system: intracellular injection of cAMP, but not of Rp-cAMPS, stimulated GIRK currents to the same extend as heterologously coexpressed β2AR (Fig. 3). This finding is substantiated by the fact that modest overexpression of Gαs leads to an increase of basal (agonist independent) as well as acetylcholine-activated GIRK current that can be inhibited by H89, a selective blocker of PKA (Fig. 4). At higher levels of overexpression, however, GIRK activity decreases, most likely by sequestering endogenously available G βγ subunits (see Fidler-Lim et al. 1995). So we conclude that PKA phosphorylation facilitates GIRK channel opening either by direct phosphorylation of the channel protein or of an auxiliary cofactor that interacts with the channel. Desensitization of β2 AR has been shown to occur via two distinct protein kinases: β-adrenergic receptor kinase phosphorylates the β2AR, thereby triggering binding of β-arrestin to the receptor, which, in turn, terminates further catalytic activation of heterotrimeric G-proteins (for review, see Pitcher et al. 1998). On the other hand, PKA phosphorylation of the β2AR changes the specificity of the receptor from the stimulatory to the inhibitory (PTX-sensitive) heterotrimeric G-protein (Daaka et al. 1997). Since GIRK channels are activated predominantly via inhibitory, PTX-sensitive G-proteins, PKA might promote β-adrenergic stimulation of GIRK channels by the latter mechanism. In our experiments, PTX treatment reduced basal GIRK currents (both when wild-type and mutant β2 AR's were expressed), showing that inhibitory PTX-sensitive G-proteins contribute to basal activity in the Xenopus laevis oocyte system. Activation of GIRK currents via both β2AR wt and β2ARPF, however, took place with or without PTX treatment. This fact clearly indicates that activation of GIRK via β2AR does not occur primarily via Gαi /Gαo. On the other hand, PTX treatment significantly reduced isoproterenol-induced GIRK currents in the case of β2 ARwt (but not of β2ARPF), indicating that coupling of β2ARwt to Gα i/Gαo also contributes to some extent. Hence, I Iso in Xenopus laevis oocytes is comprised of direct G-protein effects (both PTX sensitive and insensitive) in addition to the part contributed by PKA. Chen and Yu 1994 found that 8-bromoadenosine 3′,5′-cyclic monophosphate incubation, as well as cytosolic injection of the catalytic subunit of PKA, reduced desensitization of GIRK channels that were expressed in Xenopus oocytes. This effect was attributed to PKA effects on the coexpressed μ-opioid receptor. Since cAMP injections into oocytes expressing GIRK1/GIRK4 channels without any coexpressed seven-helix receptors were also effective (Fig. 6), we conclude that PKA did not act via the seven-helix receptors in our case.
A strong PKA phosphorylation consensus site has been reported to exist in the very end of the carboxy terminus of the GIRK1 subunit ( Dascal 1997). Hence, truncated forms of both GIRK1 and GIRK4 were generated, which were still functional but were missing the last 40 amino acids containing this site. Our results clearly demonstrate that mutant channels react to cAMP injections essentially in the same manner as the wild type. These data indicate that PKA might exert its effect either via another, hitherto unidentified, site located on one of the channels subunits, or via an unknown cytosolic or membrane delimited cofactor that serves as the direct target for PKA.
In native atrial cells, a heterooligomeric protein comprising the GIRK1 and GIRK4 subunits produces most of IKACh. From our study, it becomes evident that GIRK1/GIRK4 heterooligomeric channels are subject to PKA stimulation in native atrial cells as well as in the heterologous expression system. In addition, we were able to demonstrate that other GIRK subunit combinations are subject to PKA-induced facilitation: GIRK1/GIRK5, GIRK1/GIRK2, and even homooligomeric GIRK2 channels were found to be regulated by PKA in the same manner. This finding indicates that we are dealing with a phenomenon of broad functional significance for GIRK channel physiology. Stimulatory and inhibitory G-proteins are thought to produce opposing effects on cardiac excitability under normal physiological conditions. In the present study, however, this is not the case. The physiological role of such heterologous facilitation could be understood as follows: in the heart, β-adrenergic stimulation is known to act via voltage-dependent Ca2+ and Na+ channels, thereby promoting depolarization, excitability, and contraction ( Osterrieder et al. 1982; Cachelin et al. 1983; Frohnwieser et al. 1997). In the absence of a simultaneous facilitation of membrane-hyperpolarizing inwardly rectifying K+ currents, the membrane-depolarizing effect might lead not only to an increase in heart rate and force of contraction, but also to abnormal patterns of conduction of excitation and subsequent arrhythmias. A concomitant enhancement of IKACh would stabilize the heartbeat under these conditions. The exact physiological role of this newly discovered mechanism, however, remains to be elucidated in further studies.
Acknowledgments
Ms. C. Lorenz (Graz, Austria) provided excellent technical support. The authors thank K. Groschner (Graz, Austria), A. Kovoor (Pasadena, CA), and L. Weigl (Vienna, Austria) for critically reading the manuscript. We thank Y. Daaka and R. Lefkowitz for the cDNA of β2ARPF .
This study was supported by the Austrian Science Foundation (SFB708, P11560-MED, P13724-GEN), the Austrian National Bank (OENB7716), the National Institutes of Health (GM56260), and the Israel Basic Research Fund.
Note added in proof: During the review process of this paper, Medina et al. reported at the 1999 APS conference, “Biology of Potassium Channels: From Molecules to Disease,” that the GIRK1 subunit itself is phosphorylated in vitro by PKA (Medina, I., G. Krapivinsky, P. Kovoor, L. Krapivinsky, and D.E. Clapham. 1999. Channel phosphorylation is required for IKACh activation by G βγ. Physiologist. 42:A-17).
Footnotes
Dr. Uezono's present address is Department of Pharmacology, Miyazaki Medical College, School of Medicine, Kiyotake 889-1692, Miyazaki, Japan.
Abbreviations used in this paper: ACh, acetylcholine; β 2AR: β2-adrenergic receptor; Gα , α subunit of G proteins; Gβγ, β/γ subunit dimer of G protein; GIRK, G protein–activated inwardly rectifying K+ channel; HK, high-K+ extracellular medium; Iso, isoproterenol; m2R, muscarinergic acetylcholine receptor subtype 2; PKA, cAMP- activated protein kinase; PTX, pertussis toxin.
References
- Anderson M.P. , Berger H.A. , Rich D.P , Gregory R.J , Smith A.E. , Welsh M.J. Nucleoside triphosphates are required to open the CFTR chloride channel. Cell. 1991 ;67:775–784 . doi: 10.1016/0092-8674(91)90072-7. [DOI] [PubMed] [Google Scholar]
- Bender K. , Wellner-Kienitz M.-C. , Meyer T. , Pott L. Activation of muscarinic K+ current by β-adrenergic receptors in cultured atrial myocytes transfected with β1 subunit of heterotrimeric G proteins. FEBS Lett. 1998 ;439:115–120 . doi: 10.1016/s0014-5793(98)01350-7. [DOI] [PubMed] [Google Scholar]
- Blumenstein Y. , Ivanina T. , Shistik E. , Bossi E. , Peres A. , Dascal N. Regulation of Ca2+ channels by coexpression of G αs in Xenopus oocytes. FEBS Lett. 1999 ;444:78–84 . doi: 10.1016/s0014-5793(99)00035-6. [DOI] [PubMed] [Google Scholar]
- Cachelin A.B. , de Peyer J.E. , Kokubun S. , Reuter H. Ca2+ channel modulation by 8-bromocyclic AMP in cultured heart cells. Nature. 1983 ;304:462–464 . doi: 10.1038/304462a0. [DOI] [PubMed] [Google Scholar]
- Chen Y. , Yu N.J. Differential effects of protein kinase C activation on 5-HT1A receptor coupling to Ca2+ and K+ currents in rat serotonergic neurones. J. Physiol. 1994 ;496:129–137 . doi: 10.1113/jphysiol.1996.sp021670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S.H. , Rich D.P. , Marshall J. , Gregory R.J. , Welsh M.W. , Smith A.E. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 1991 ;66:1027–1036 . doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]
- Corey S. , Krapivinsky G. , Krapivinsky L. , Clapham D.E. Number and stoichiometry of subunits in the native atrial G-protein–gated K+ channel, IKACh . J. Biol. Chem. 1998 ;273:5271–5278 . doi: 10.1074/jbc.273.9.5271. [DOI] [PubMed] [Google Scholar]
- Daaka Y. , Luttrell L.M. , Lefkowitz R.J. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997 ;390:88–91 . doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- Dascal N. Signalling via the G protein-activated K+ channels. Cell. Signal. 1997 ;9:551–573 . doi: 10.1016/s0898-6568(97)00095-8. [DOI] [PubMed] [Google Scholar]
- Dascal N. , Schreibmayer W. , Lim N.F. , Weizhen W. , Chavkin C. , DiMagno L. , Labarca C. , Kieffer B.L. , Gaveriaux-Ruff C. , Trollinger D. Atrial G protein–activated K+ channel expression cloning and molecular properties. Proc. Natl. Acad. Sci. USA. 1993 ;90:10235–10239 . doi: 10.1073/pnas.90.21.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascal N. , Lotan I. Expression of exogenous ion channels and neurotransmitter receptors in RNA-injected Xenopus oocytes. In: Longstaff A. , Revest P. , editors. Methods in Molecular Neurobiology. Vol. 13. Humana Press; Totowa, NJ : 1992 . pp. 205–225 . [Google Scholar]
- Fidler-Lim N. , Dascal N. , Labarca C. , Davidson N. , Lester H.A. A G protein–gated K channel is activated via β2 -adrenergic receptors and Gβγ subunits in Xenopus oocytes. J. Gen. Physiol. 1995 ;105:421–439 . doi: 10.1085/jgp.105.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohnwieser B. , Chen L.-Q. , Schreibmayer W. , Kallen R.G. Modulation of the human cardiac sodium channel α-subunit by cAMP and the responsible sequence domain. J. Physiol. 1997 ;498:309–318 . doi: 10.1113/jphysiol.1997.sp021859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedin K.E. , Lim N.F. , Clapham D.E. Cloning of a Xenopus laevis inwardly rectifying K+ channel subunit that permits GIRK1 expression of IKACh currents in oocytes. Neuron. 1996 ;16:423–429 . doi: 10.1016/s0896-6273(00)80060-4. [DOI] [PubMed] [Google Scholar]
- Huang C.-L. , Jan Y.N. , Jan L.Y. Binding of the G protein βγ subunit to multiple regions of G protein–gated inward-rectifying K+ channels. FEBS Lett. 1997 ;405:291–298 . doi: 10.1016/s0014-5793(97)00197-x. [DOI] [PubMed] [Google Scholar]
- Jing J. , Chikvashvili D. , Singer-Lahat D. , Thornhill W.B. , Reuveny E. , Lotan I. Fast inactivation of a brain K+ channel composed of Kv1.1 and Kvβ1.1 subunits modulated by G protein βγ subunits . EMBO (Eur. Mol. Biol. Organ.) J. 1999 ;18:1245–1256 . doi: 10.1093/emboj/18.5.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. β-Adrenergic regulation of the muscarinic-gated K+ channel via cyclic AMP-dependent protein kinase in atrial cells. Circ. Res. 1990 ;67:1292–1298 . doi: 10.1161/01.res.67.5.1292. [DOI] [PubMed] [Google Scholar]
- Liman E.R. , Tytgat J. , Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992 ;9:861–871 . doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- Luchian T. , Dascal N. , Dessauer C. , Platzer D. , Davidson N. , Lester H.A. , Schreibmayer W. A C-terminal peptide of the GIRK1 subunit directly blocks the G protein–activated K+ channel (GIRK) expressed in Xenopus oocytes. J. Physiol. 1997 ;505:13–22 . doi: 10.1111/j.1469-7793.1997.013bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray R. , Spiegel E. Theory and Problems of Statistics 1961 . McGraw-Hill Book Co; New York, NY: pp. 167–184 [Google Scholar]
- Nargeot J. , Nerbonne J.M. , Engels J. , Lester H.A. Time course of the increase in myocardial slow inward current after photochemically generated concentration jump of intracellular cyclic AMP . Proc. Natl. Acad. Sci. USA. 1983 ;80:2395–2399 . doi: 10.1073/pnas.80.8.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher J.A. , Freedman N.J. , Lefkowitz R.J. G protein–coupled receptor kinases. Annu. Rev. Biochem. 1998 ;67:653–692 . doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- Osterrieder W. , Brum G. , Hescheler J. , Trautwein W. , Flockerzi V. , Hofmann F. Injection of subunits of cyclic AMP-dependent protein kinase into cardiac myocytes modulates Ca2+ current. Nature. 1982 ;298:576–578 . doi: 10.1038/298576a0. [DOI] [PubMed] [Google Scholar]
- Perets T. , Blumenstein Y. , Shistik E. , Lotan I. , Dascal N. A potential site of functional modulation by proteinkinase A in the cardiac Ca2+ channel α1C subunit. FEBS Lett. 1996 ;384:189–192 . doi: 10.1016/0014-5793(96)00303-1. [DOI] [PubMed] [Google Scholar]
- Sambrook J. , Fritsch E.F. , Maniatis T. Molecular CloningA Laboratory Manual. 2nd ed. CSH Press; New York, NY : 1989 . [Google Scholar]
- Schreibmayer W. , Dessauer C.W. , Vorobiov D. , Gilman A.G. , Lester H.A. , Davidson N. , Dascal N. Inhibition of an inwardly rectifying K+ channel by G-protein alpha-subunits. Nature. 1996 ;380:624–627 . doi: 10.1038/380624a0. [DOI] [PubMed] [Google Scholar]
- Schreibmayer W. , Lester H.A. , Dascal N. Voltage clamp of Xenopus laevis oocytes utilizing agarose-cushion electrodes. Pflügers Arch. 1994 ;426:453–458 . doi: 10.1007/BF00388310. [DOI] [PubMed] [Google Scholar]
- Sharon D. , Vorobiov D. , Dascal N. Positive and negative coupling of the metabotropic glutamate receptors to a G protein–activated K+ channel, GIRK, in Xenopus oocytes. J. Gen. Physiol. 1997 ;109:477–490 . doi: 10.1085/jgp.109.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman S.K. , Lester H.A. , Dougherty D.A. Subunit stoichiometry of a heteromultimeric GIRK K+ channel. J. Biol. Chem. 1996 ;271:30524–30528 . doi: 10.1074/jbc.271.48.30524. [DOI] [PubMed] [Google Scholar]
- Sorota S. , Rybina I. , Yamamoto A. , Du X.-Y. Isoprenaline can activate the acetylcholine-induced K+ current in canine atrial myocytes via Gs-derived bg subunits . J. Physiol. 1999 ;514:413–423 . doi: 10.1111/j.1469-7793.1999.413ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautwein W. , Taniguchi J. , Noma A. The effect of intracellular cyclic nucleotides and calcium on the action potential and acetylcholine response of isolated cardiac cells . Pflügers Arch. 1982 ;392:307–314 . doi: 10.1007/BF00581624. [DOI] [PubMed] [Google Scholar]
- Uezono Y. , Bradley J. , Min C. , McCarty N.A. , Quick M. , Riordan J.R. , Chavkin C. , Zinn K. , Lester H.A. , Davidson N. Receptors that couple to 2 classes of G proteins increase cAMP and activate CFTR expressed in Xenopus oocytes. Receptors Channels. 1993 ;1:233–241 . [PubMed] [Google Scholar]
- Wang Y.G. , Lipsius S.L. β-Adrenergic stimulation induces acetylcholine to activate ATP-sensitive K+ current in cat atrial myocytes. Circ. Res. 1995 ;77:565–574 . doi: 10.1161/01.res.77.3.565. [DOI] [PubMed] [Google Scholar]
- Wellner-Kienitz M.C. , Bender K. , Brandts B. , Meyer T. , Pott L. Antisense oligonucleotides against receptor kinase GRK2 disrupt target selectivity of β-adrenergic receptors in atrial myocytes . FEBS Lett. 1999 ;451:279–283 . doi: 10.1016/s0014-5793(99)00594-3. [DOI] [PubMed] [Google Scholar]