

Abstract
Tyrosine side chains participate in several distinct signaling pathways, including phosphorylation and membrane trafficking. A nonsense suppression procedure was used to incorporate a caged tyrosine residue in place of the natural tyrosine at position 242 of the inward rectifier channel Kir2.1 expressed in Xenopus oocytes. When tyrosine kinases were active, flash decaging led both to decreased K+ currents and also to substantial (15–26%) decreases in capacitance, implying net membrane endocytosis. A dominant negative dynamin mutant completely blocked the decaging-induced endocytosis and partially blocked the decaging-induced K+ channel inhibition. Thus, decaging of a single tyrosine residue in a single species of membrane protein leads to massive clathrin-mediated endocytosis; in fact, membrane area equivalent to many clathrin-coated vesicles is withdrawn from the oocyte surface for each Kir2.1 channel inhibited. Oocyte membrane proteins were also labeled with the thiol-reactive fluorophore tetramethylrhodamine-5-maleimide, and manipulations that decreased capacitance also decreased surface membrane fluorescence, confirming the net endocytosis. In single-channel studies, tyrosine kinase activation decreased the membrane density of active Kir2.1 channels per patch but did not change channel conductance or open probability, in agreement with the hypothesis that tyrosine phosphorylation results in endocytosis of Kir2.1 channels. Despite the Kir2.1 inhibition and endocytosis stimulated by tyrosine kinase activation, neither Western blotting nor 32P labeling produced evidence for direct tyrosine phosphorylation of Kir2.1. Therefore, it is likely that tyrosine phosphorylation affects Kir2.1 function indirectly, via interactions between clathrin adaptor proteins and a tyrosine-based sorting motif on Kir2.1 that is revealed by decaging the tyrosine side chain. These interactions inhibit a fraction of the Kir2.1 channels, possibly via direct occlusion of the conduction pathway, and also lead to endocytosis, which further decreases Kir2.1 currents. These data establish that side chain decaging can provide valuable time-resolved data about intracellular signaling systems.
Keywords: unnatural amino acids, ion channel, Kir2.1, clathrin, endocytosis
INTRODUCTION
Ion channel activity can be modulated both by covalent modifications to channel proteins (e.g., phosphorylation) and specific noncovalent protein–protein interactions, typically involving special sequence motifs. These two mechanisms often interact; for instance, tyrosine phosphorylation leads to endocytosis via interactions with other proteins (Glenney et al. 1988; Schlessinger and Ullrich 1992; van der Geer et al. 1994). Tyrosine phosphorylation mechanisms take on added importance because they can also be activated by G protein–coupled receptors (Chen et al. 1994; Dikic et al. 1996).
The inwardly rectifying K+ channel Kir2.1 (Kubo et al. 1993) is one of several K+ channels that shows decreased function under conditions favoring tyrosine phosphorylation (Jonas and Kaczmarek 1996; Levitan 1999). Mutation of the single Kir2.1 tyrosine residue Y242 (Fig. 1) to phenylalanine abolishes this inhibition (Wischmeyer et al. 1998). However, it is not known whether this residue is phosphorylated directly; indeed, only a few studies clearly establish that any tyrosine in an ion channel is phosphorylated directly by a tyrosine kinase (Holmes et al. 1996a,Holmes et al. 1996b; Yu et al. 1997; Felsch et al. 1998; Ling et al. 2000; Rogalski et al. 2000).
Figure 1.

Topology of Kir2.1 from Mus musculus (GenBank accession number X73052). The sequence surrounding the tyrosine-242 contains consensus sequences for both tyrosine phosphorylation and tyrosine-based interaction with clathrin adaptor proteins. Other consensus endocytosis sequences are also shown.
Interestingly, the intracellular COOH-terminal tyrosine (Y242) implicated in Kir2.1 inhibition (Wischmeyer et al. 1998) is also part of a YXXΦ tyrosine–based motif recognized by the μ chains of clathrin adaptor proteins (Fig. 1), where X may be any amino acid and Φ is a hydrophobic amino acid, typically L, F, or M (Kirchhausen et al. 1997). Previous studies show that such a motif regulates the expression of the epithelial sodium channel (Shimkets et al. 1997). Endo- and exocytosis are potentially general and powerful methods for the regulation of channel activity, but the detailed mechanisms that target ion channels and other membrane proteins for these pathways are not yet understood.
We have developed and exploited an experimental technique that enables additional mechanistic insight into signaling pathways involving tyrosine side chains. By nonsense codon suppression (Noren et al. 1989; Nowak et al. 1995), we introduced the caged tyrosine analogue o-nitrobenzyltyrosine (Tyr(ONB)) into Kir2.1 at the critical Y242 position. Because the tyrosyl side chain is protected as the nitrobenzyl ether, this analogue cannot be phosphorylated. Also, crystallographic data suggest that a tyrosine protected in this way would not permit binding by proteins of clathrin-mediated pathways, such as the μ subunit of AP-2 (Owen and Evans 1998). However, UV irradiation of the protein releases the nitrobenzyl protecting group, revealing the wild-type (WT) tyrosine residue and permitting both phosphorylation and/or recognition by the adaptor proteins. Thus, a signal transduction pathway can be primed and arrested at the point where it awaits decaging of a specific side chain in the specific protein of interest. For the study of intracellular signal transduction, caged tyrosine represents a powerful tool.
We used this method to conduct time-resolved functional measurements during and after the decaging. We measured macroscopic currents, single-channel currents, membrane capacitance, and membrane fluorescence. As expected, we find that decaging of the single tyrosine residue leads to inhibition of the K+ current; surprisingly, we find that decaging also leads to substantial endocytosis. Blockade of this endocytosis by a dominant negative dynamin mutant partially suppresses channel inhibition. We find no convincing direct evidence that the target tyrosine itself is phosphorylated after the decaging event. Thus, we suggest that the decaged tyrosine may not be phosphorylated directly but interacts instead with other proteins via the tyrosine-based motif. These interactions lead first to modest inhibition of channel function, and then to general endocytosis.
MATERIALS AND METHODS
Chemical Synthesis
The 4PO-protected amino acid Tyr(ONB) was prepared as reported (Miller et al. 1998). In brief, l-tyrosine was complexed with copper(II) to protect the amino and carboxyl termini. To this complex was added nitrobenzyl chloride under basic conditions, yielding Tyr(ONB). Addition of 4-pentenoic anhydride gave the N-protected 4-PO-Tyr(ONB). The amino acid was coupled to the dinucleotide dCA by established procedures (Nowak et al. 1998). In summary, the carboxyl terminus was converted to the cyanomethyl ester by treatment with chloroacetonitrile. The active ester was condensed with dCA under basic conditions to give the aminoacylated dinucleotide, which was enzymatically ligated to a 74-base tRNA precursor with T4 RNA ligase.
DNA/RNA Constructs
tRNA THG73 was modified from eukaryotic Tetrahymena thermophila tRNAGln(CUA) as described previously (Nowak et al. 1998) and inserted into pUC19 giving the plasmid pTHG73. pTHG73 was linearized by FokI and transcribed in vitro using the MEGAshortscript kit (Ambion).
The tyrosine codon at position 242 of the mouse Kir2.1 cDNA in pcDNA I was mutated to phenylalanine or to the nonsense codon TAG, giving two mutants called Kir2.1(Y242F) and Kir2.1(Y242TAG), respectively. Mutagenesis was carried out using the Quickchange kit (Stratagene). Mutations were verified by sequencing of both strands through the affected regions. The hemagglutinin antigenic sequence YPYDVPDYA was added to the carboxyl terminus of Kir2.1 and to Kir2.1(Y242F) using PCR amplification, giving Kir2.1-HA and Kir2.1(Y242F)-HA. The Kir2.1 plasmids were linearized by NotI.
The cDNA for v-Src kinase (a gift of Dr. I.B. Levitan, University of Pennsylvania, Philadelphia, PA), PyK2 (a gift of Dr. J. Schlessinger, New York University, New York, NY), human WT dynamin I and dynamin-I-K44A (gifts of Dr. Alex van der Bliek and Dr. Thomas Moss, both from University of California, Los Angeles, CA), and rat TrkB (gift of G. Yancoupolis, Regeneron Pharmaceuticals, Tarrytown, NY) were linearized by XbaI, SspI, SalI, and SalI, and XbaI or NotI, respectively. All mRNAs were transcribed in vitro using the SP6 or T7 mMESSAGE mMACHINE kit (Ambion) as appropriate.
Suppression of Kir2.1-Y242TAG with Tyr(ONB) in Xenopus Oocytes
The 4PO-Tyr(ONB)-tRNA in 1 mM sodium acetate, pH 4.5, was deprotected just before injection by mixing with an equal volume of a saturated aqueous solution of iodine (1.2 mM; England et al. 1997) for 10 min at ambient temperature. 10–15 ng of Kir2.1(Y242TAG) mRNA and 20–25 ng of tRNA-Tyr(ONB) in a total volume of 32.2 nl per oocyte were coinjected into stage V and VI oocytes by a Drummond automatic injector. The cRNA for WT Kir2.1 cRNA, the conventional mutant Kir2.1-Y242F, or Kir2.1-HA was injected at 1 ng/oocyte. Oocytes were incubated in 50% L-15 medium supplemented with 7.5 mM HEPES, 0.8 mM of glutamine, and 10 μg/ml of Gentamycin, pH 7.5, at 18–20°C. After incubation for 24–48 h, functional measurements were made.
Electrophysiology
For macroscopic recordings, oocytes were voltage clamped at 0 mV in a high K+ solution (96 mM KCl, 2 mM NaCl, 1 mM MgCl2, 1.5 mM CaCl2 and 5 mM HEPES, pH 7.5) with two electrodes (filled with 3 M KCl, resistance 0.5–3 MΩ) and stepped to a test potential of −80 mV using a GeneClamp 500 (Axon Instruments). Membrane capacitance was measured on oocytes clamped at 0 mV in response to a train of 10-mV pulses delivered at 2.9 Hz with 50% duty cycle. Voltage command signals were generated, and membrane capacitance were measured, by the algorithms in pCLAMP 8.0 software (Axon Instruments). In preliminary experiments, capacitance measurements were verified manually by integrating the response to 10-mV test pulses.
For cell-attached recordings of single-channel recordings, the pipet solution and the bath contained the same high K+ solution. The patch channel of the GeneClamp 500 was used, with 2 kHz filtering. The signals were recorded and analyzed using FETCHEX, FETCHAN, and pSTAT in the pCLAMP 6 suite.
Decaging Optics
The apparatus for Tyr(ONB) decaging was reported previously (Miller et al. 1998). Light from a 300-W Hg arc lamp was filtered through a Schott UG11 filter to provide 300–350 nm light and was focused through a 50-mm quartz lens (Oriel) onto a liquid light guide (Oriel, 1 m long and 3 mm in diameter) connected to the recording chamber. At the chamber, the end of the liquid light guide contacted a Pyrex coverslip placed at the bottom surface of the chamber, upon which the oocyte rested. Miller et al. 1998 reported that 3-s irradiation was sufficient for photolysis of >90% of caged tyrosine on the oocyte surface.
Fluorescence Labeling and Measurements
For nonspecific labeling of the cell surface, oocytes were incubated in ND96 solution (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, and 5 mM Hepes, pH 7.5) containing 10 μM tetramethylrhodamine-5-maleimide for 30 min at 20–22°C. The labeled oocytes were washed five times with ND96 solution to remove unbound fluorescent dye. The labeling intensity was examined at the animal pole. Confocal images were taken with a Bio-Rad MRC 600 microscope with a 10× objective lens, with 10 average scans.
Surface fluorescence from the animal pole of labeled oocytes was measured using an inverted reflected light fluorescence microscope (model IX-70-FLA; Olympus) and other methods previously described (Li et al. 2000). In brief, exciting light was delivered from a stabilized 100-W Hg lamp. We used a 40×, NA 1.3 objective lens. A photomultiplier tube (PMT) attached at the side port recorded the fluorescence. To avoid bleaching, a shutter in the excitation pathway was opened and the fluorescence was measured for only 10–20 s at intervals of 2–2.5 min. The emitted signal (as voltage from the PMT output) was appropriately amplified, filtered, and sent to an Axon Digidata interface for collecting data by pCLAMP 7.
Immunochemical Detection
Oocytes were injected with Kir2.1-HA. Plasma membranes were isolated by physical dissection (Ivanina et al. 1994). The hypotonic solution used to prepare the oocytes for dissection was modified to contain 5 mM phenylarsine oxide (PAO) and 1 mM Na orthovanadate (Huyer et al. 1997), one EDTA-free Boehringer Complete tablet per 40 ml, and 0.08% SDS (which expedites dissection). PAO was dissolved in DMSO and stored at −80°C. Yolk and pigment granules were removed by a 2-min 14,000 rpm spin at 4°C, and the supernatant was dissolved in 15 μl 2× SDS gel loading buffer. Samples were boiled for 5 min or heated to 55°C for 10 min before loading. SDS-PAGE was performed using 10% polyacrylamide (40T:1C) gels or 10% Tris-Cl ReadyGels (BioRad Laboratories).
Proteins were transferred to nitrocellulose overnight at 30 V. Blots were blocked for 1 h in 1× TPBS (PBS containing 0.1% [vol/vol] Tween 20) and 5% (wt/vol) nonfat dried milk (Carnation). Blots were exposed to primary antibody for 1 h in TPBS with 5% milk, washed three times 10 min each in TPBS, and then incubated for 1 h in TPBS containing 5% milk and secondary antibody. Detection used ECL reagents from Amersham.
Primary antibodies included monoclonal HA.11 ascites fluid; monoclonal antiphosphotyrosine antibodies 2G8-D6, 6G9, 1G2, PY20, PY72 (BAbCo), and 4G10 (Upstate Biotechnology); and anti-TrkB antibody C14 (Santa Cruz Biotechnology, Inc.). All BAbCo antibodies and 4G10 were diluted 1:1,000 for Western blotting; C14 was diluted 1:200. Of the phosphotyrosine antibodies, 4G10 had the highest affinity under our conditions. PY72 also performed well. HRP-conjugated secondary antibodies included goat anti–mouse (Jackson Immunologicals) and donkey anti–rabbit (Amersham), diluted 1:5,000 and 1:200, respectively.
Data Analysis
The experimental data were analyzed using ORIGIN (Microcal Software, Inc.) and CLAMPFIT 8 software (Axon Instruments).
Reagents
BDNF was provided by Dr. Andy Welcher (Amgen, Inc.).
RESULTS
Kir2.1 Currents Are Decreased by Tyrosine Kinase
Initially, we verified that the inwardly rectifying potassium channel Kir2.1 from Mus musculus is modulated by tyrosine phosphorylation under the present experimental conditions (Wischmeyer et al. 1998). Xenopus oocytes expressing Kir2.1 were voltage clamped at 0 mV in a recording solution containing 96 mM KCl. Potassium currents elicited by a voltage step to a test potential of −80 mV were recorded. Fig. 2 A shows that oocytes expressing Kir2.1 gave robust (13.3 ± 1.2 μA) whole-cell K+ currents after 24 h. Oocytes treated with the tyrosine phosphatase inhibitor PAO (Webb 1966; Pafford et al. 1995) showed no reduction of current, giving whole-cell K+ currents of 13.1 ± 1.4 μA. However, oocytes coinjected with v-Src gave an average current of 7.8 ± 0.3 μA, a 41% current reduction relative to those injected with Kir2.1 alone. This effect was enhanced by treatment with PAO; oocytes coinjected with v-Src and treated with PAO gave average currents of only 5.7 ± 0.6 μA.
Figure 2.
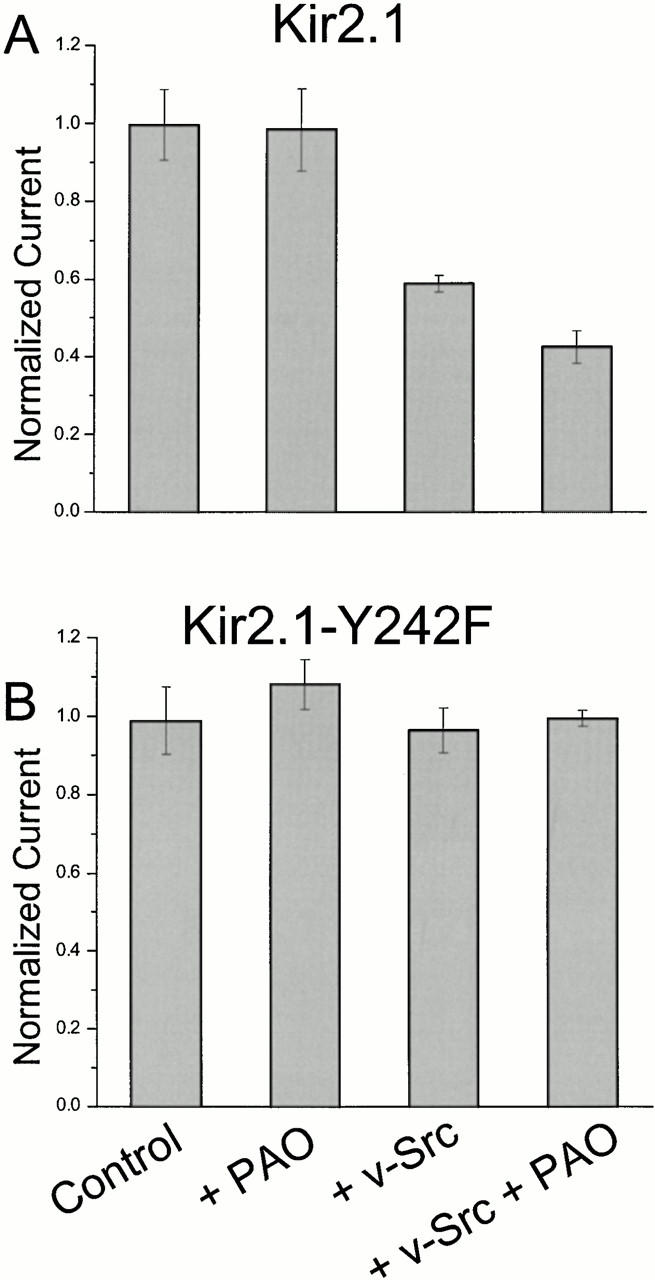
Whole-cell current from oocytes expressing Kir2.1 (A) and Kir2.1-Y242F (B). Data columns represent normalized currents (mean ± SEM, 4–5 oocytes). Where indicated, oocytes were coinjected with cRNA for tyrosine kinases v-Src or PyK2 and incubated with 10 μM PAO for at least 30 min before recording. 100% corresponds to 13.3 μA in A and 31.4 μA in B.
Fig. 2 B confirms that Y242 is critical for the effect of tyrosine kinases (Wischmeyer et al. 1998). The tyrosine residue at position 242 was changed to phenylalanine by site-directed mutagenesis. Oocytes expressing the Kir2.1-Y242F construct were unaffected by treatment with the tyrosine phosphatase inhibitor PAO and/or tyrosine kinases. For instance, untreated oocytes expressing the Y242F construct gave average whole-cell K+ currents of 31.4 ± 2.7 μA.; oocytes coexpressing v-Src and treated with PAO gave currents of 31.2 ± 0.9 μA. The overall expression for the experiment depicted in Fig. 2 was higher than that for the WT Kir2.1, which represents imprecision in mRNA quantitation or batch-dependent expression differences among oocytes. There were no consistent differences in the expression level between the WT and Y242F constructs. In other experiments on Y242F at expression levels comparable to those of Fig. 2 A, we confirmed that no combination of v-Src expression and PAO gave significant decreases for this mutant.
For both constructs, similar but less extensive results on the inhibition of K+ currents were obtained with the tyrosine phosphatase inhibitor pervanadate substituting for PAO. When the v-Src cRNA injection was omitted but oocytes were injected with TrkB (a receptor tyrosine kinase) and exposed to BDNF, reproducible inhibition in the presence of PAO was also observed (data not shown). When the v-Src cRNA injection was omitted but oocytes were injected with the proline-rich tyrosine kinase PyK2, reproducible inhibition in the presence of PAO was also observed (data not shown). These data suggest that several different activators of tyrosine phosphorylation can inhibit the function of Kir2.1.
Tyrosine Phosphorylation Decreases Total Active Kir2.1 Channels per Patch without Changing Conductance or Open Probability
Single-channel experiments were conducted on cell-attached patches at −80 and −100 mV on batches of oocytes that showed at least a 40% decrement in Kir2.1 current between control cells and those coexpressing v-Src and treated with PAO. We found that the v-Src/PAO combination primarily affected the number of active channels per patch (Table ). Fig. 3 presents tracings and all-points histograms for oocytes injected either with Kir2.1 alone and untreated with PAO, or with Kir2.1 and v-Src and treated with PAO. The single-channel conductances for control and v-Src/PAO oocytes were 21 ± 0.3 and 20.4 ± 0.7 pS (mean ± SEM, n = 27 and 20, respectively), indicating no change in single-channel conductance. We found no major conductance substates. Thus, none of the phosphorylation-induced decrease in macroscopic currents derives from a decreased single-channel current.
Table 1.
Single-channel Measurements
| γ, pS | Popen | NPopen | |
|---|---|---|---|
| Control | 21.1 ± 0.3 | 0.53 ± 0.08 | 1.0 ± 0.2 |
| v-Src/PAO | 20.4 ± 0.7 | 0.54 ± 0.08 | 0.54 ± 0.08 |
Single-channel parameters for cell-attached patches from oocytes expressing Kir2.1 alone or coexpressed with v-Src and treated with PAO. γ, single-channel conductance. Data are given as mean ± SEM; number of patches ranged from 10–28.
Figure 3.
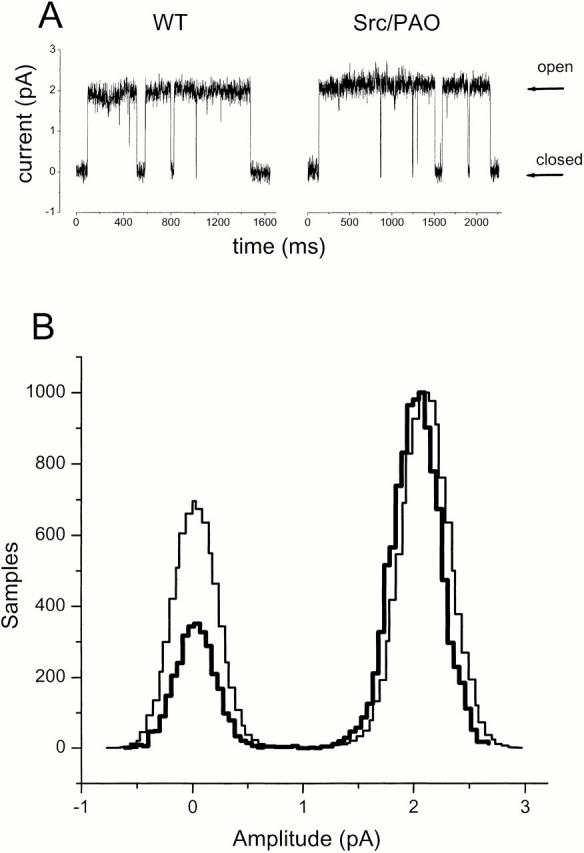
Conditions favoring tyrosine phosphorylation do not change single-channel conductance of WT Kir2.1. (A) Traces showing exemplar single-channel currents from cell-attached patches at −100 mV. The arrows point to the closed states and to the open states for control oocytes (left) and for oocytes coinjected with v-Src and treated with PAO (right). (B) Normalized all-points amplitude histograms for exemplar patches. Data from an oocyte injected with Kir2.1 alone are shown as heavy lines, and data from an oocyte injected with Kir2.1 + v-Src and exposed to PAO are shown as light lines.
In patches that contained only one active channel, we found a wide range in the open probability Popen (0.15–0.86), but neither this range nor the average open probability showed a difference between control and v-Src/PAO patches. However, v-Src/PAO patches consistently displayed fewer active channels. In a series of recordings, we used pipets with <10% variation in resistance and with similar shapes to assure similar tip diameters and therefore similar patch areas. In this series, multichannel patches from v-Src/PAO oocytes displayed NPopen roughly half that of patches from control oocytes (Table ). These data show that the loss of active channels from the membrane accounts for most of the effect of v-Src/PAO on macroscopic currents.
Photolytic Decaging of Tyrosine-242 Leads to a Decrease in Current
To control the state of the tyrosine side chain at position 242, the unnatural amino acid Tyr(ONB) was incorporated into Kir2.1 at position 242 (Nowak et al. 1998). The nonsense codon TAG was introduced using site-directed mutagenesis. mRNA encoding the Kir2.1-Y242TAG construct was generated by in vitro transcription. This message was coinjected into Xenopus oocytes along with the nonsense suppressor tRNA that had been chemically charged with the synthetic amino acid Tyr(ONB). To verify that functional protein was produced by this procedure and to assess the effect of decaging the caged tyrosine residue, oocytes were studied as in the experiments of Fig. 2. As in most unnatural amino acid experiments to date, the current measurements show that expression levels are rather lower (in this case, by a factor of 5–10) than the levels for WT channel or for conventional site-directed mutants (compare the information in the legends of Fig. 2 and Fig. 4). UV light was delivered to oocytes for 3 s (Miller et al. 1998). Fig. 4 (A and B) presents representative data following such irradiation of oocytes expressing Kir2.1-Y242TAG suppressed with Tyr(ONB). In addition, data for a nonirradiated control oocyte are shown. In oocytes that had been coinjected with v-Src and treated with PAO, decaging of the Tyr(ONB) residue had a noticeable effect on the current: there was a decrease over the 30-min time course of our experiments. Fig. 4 C presents normalized current and capacitance values averaged over several oocytes.
Figure 4.
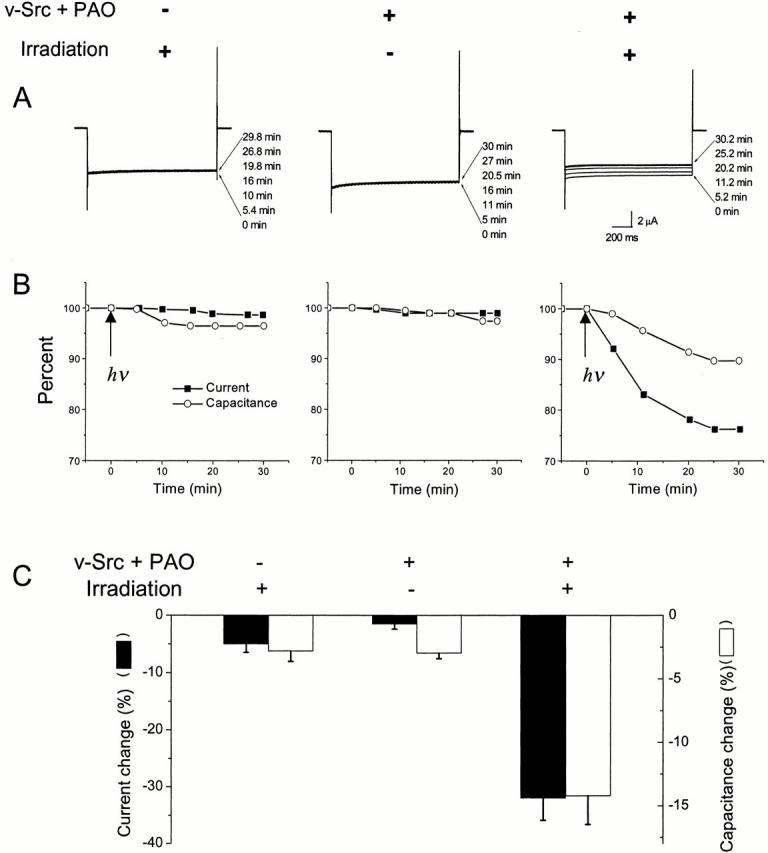
Inhibition of current and decrease of capacitance in oocytes expressing Kir2.1-Y242TAG suppressed with Tyr(ONB). (A) Typical current traces from individual oocytes. The left panel shows the effect of irradiating an oocyte (3 s at arrow) that is neither coexpressing v-Src nor exposed to PAO. The center panel shows the effect of coexpression of v-Src and exposure to PAO, but no irradiation of the oocyte. The right panel shows traces from an oocyte coexpressing v-Src, exposed to PAO, and irradiated. Oocytes were clamped at 0 mV and stepped to −80 mV to elicit an inward current. Bath solutions contained 96 mM KCl. Time after irradiation is shown. (B) Plots of current and capacitance versus time from the individual oocytes in A. (C) Averaged data from batches of oocytes recorded at t = 30 min. The filled bars correspond to the left-hand y-axis, indicating normalized current decrease. Average values (and ranges) for 100% current were 8.7 (5.5–13), 6.5 (4.5–10.4), and 6.6 (1.6–15.5) μA for the three groups (left to right). The hollow bars correspond to the right-hand y-axis, indicating capacitance decrease. Average values (and ranges) for 100% capacitance were 190 (159–227), 187 (167–202), and 194 (132–234) nF for the three groups (left to right). Error bars indicate SEM, n = 4–6 oocytes.
The filled bars and the left-hand y-axis in Fig. 4 C represent current measurements. Control oocytes were injected with Kir2.1-Y242TAG mRNA and tRNA-Tyr(ONB) but not with v-Src mRNA. Current from these oocytes decreased by only 5.0 ± 1.5% over the 30-min interval after irradiation, showing that decaging the tyrosine in the absence of a kinase had only a small effect on Kir2.1 currents. In a second control, whole-cell currents were measured in nonirradiated oocytes coexpressing Kir2.1 containing Tyr(ONB) and v-Src. After 30 min, the average current decrease was only 1.5 ± 0.9%. This is consistent with the supposition that while the nitrobenzyl protecting group remains, the residue at 242 is unavailable as a substrate for interaction with other proteins (either tyrosine kinases or adaptor proteins). Importantly, oocytes coexpressing Kir2.1 with the caged tyrosine and v-Src, treated with PAO, and irradiated showed an average current decrease of 32.0 ± 0.9% 30 min after irradiation. This shows that the decaging of Kir2.1-Y242 under conditions favoring tyrosine phosphorylation leads to a large decrease in current.
Although these data qualitatively agree with the data for the WT and Y242F construct, present limitations of the nonsense suppression technique prevent direct quantitative comparisons. First, suppression is presumably incomplete, leading to a substantial proportion of Kir2.1 subunits truncated at the 242 position (Saks et al. 1996). To control for this effect, we expressed the Y242TAG construct alone; there were no currents. When we coexpressed Y242TAG and WT channels, we saw currents whose modulation properties were like those depicted in the experiment of Fig. 2. Therefore, dominant negative effects because of truncated subunits probably play no role in our experiments. Second, readthrough of the nonsense codon may occur, so that natural residues could be incorporated at position 242 (Saks et al. 1996), although we have no direct evidence that this is occurring in the present system. Therefore, it is possible that some channels would contain one or more Kir2.1 subunits with natural residues at position 242. Although we know that a channel with four Y242F subunits does not respond to v-Src and PAO (Fig. 2), we do not know how modulation would proceed for channels with varying numbers of subunits containing one or more of the other 18 natural amino acid residues. In the unlikely event that readthrough produces a channel with tyrosine at position 242 in one or more subunits, the modulation has presumably occurred during the PAO incubation period (>30 min). We assume that the general effect of any readthrough that may occur would be to decrease the modulatory effect of irradiation.
Decaging Tyrosine-242 Also Causes a Capacitance Decrease
Importantly, Fig. 4 shows that the photochemically induced current decrease in oocytes expressing v-Src and Kir2.1-Y242Tyr(ONB) was accompanied by a significant capacitance decrease. A change in the capacitance is a direct measure of net endocytotic activity of a cell (i.e., exocytosis minus endocytosis). In Fig. 4 A, the representative time course data show that the capacitance decrease accompanied the reduction in the whole-cell current. In both control cases, where irradiation of oocytes caused no current inhibition, the capacitance remained essentially constant over the length of the experiment. Hollow bars and the right-hand y-axis on Fig. 4 C show normalized whole-cell capacitance measurements averaged over batches of five oocytes. Again, the correlation between current decrease and capacitance decrease is clear. Control oocytes that were irradiated without v-Src coexpression showed an overall capacitance decrease of only 2.8 ± 0.8% at 30 min after irradiation. The second control, in which oocytes coexpressing Kir2.1 with caged tyrosine and v-Src were studied without irradiation, also showed little capacitance decrease (3.0 ± 0.4%) over the time course of recording. In contrast, oocytes expressing v-Src and Kir2.1, in which the Tyr(ONB) residue at 242 is uncaged by irradiation, show an overall decrease in membrane capacitance of 14.1 ± 1.9% over 30 min. This decrease suggests that the membrane surface area has decreased as a result of increased endocytotic activity.
In additional controls, we measured little decrease in capacitance (1.9 ± 0.5%, n = 5) when oocytes were injected only with v-Src cRNA, treated with PAO, and irradiated. Therefore, the capacitance change does not arise from possible photosensitive processes involving v-Src. In additional controls, oocytes were injected with t-RNA-Tyr(ONB) and v-Src cRNA, but not with Kir2.1 cRNA, and then treated with PAO. Again, irradiation produced no capacitance change, ruling out a possible suppression of stop codons in endogenous proteins as the source of the photosensitivity. Furthermore, there were no changes in capacitance associated with decaging of tyrosine in our previous study of caged tyrosine at the nicotinic acetylcholine receptor (Miller, 1998). Therefore, tyrosine decaging in general does not lead to membrane endocytosis.
We compared the time course of the current decrease and the capacitance decrease by normalizing to the decrease at 30 min and interpolating the approximate time to half-completion for this decrease. The average values were quite comparable: 11.8 min for the current and 11.1 min for the capacitance. These data indicate that the capacitance change and the current decrease followed similar time courses. Nonetheless, some of our measurements (Fig. 4 B) suggest that the capacitance decrease begins after a delay of several minutes. This possible initial delay has not been investigated systematically.
Dominant Negative Dynamin Eliminates Capacitance Decrease and Partially Eliminates Current Decrease
Capacitance measurements may reflect the contribution of various mechanisms of endocytosis. To test specifically whether clathrin-mediated pathways were involved, experiments were performed with a dominant negative dynamin isoform (Fig. 5). If Kir2.1 inhibition resulted exclusively from clathrin-mediated endocytosis, coinjection of oocytes with dominant negative dynamin (dynamin-K44A; van der Bliek et al. 1993; Damke et al. 1994) at levels capable of blocking endogenous dynamin activity would eliminate both the current change and the capacitance change. Fig. 5 presents data that distinguish between these two events. Capacitance and current data for individual oocytes coinjected with Kir2.1-Y242TAG and v-Src, suppressed with tRNA-Tyr(ONB), and treated with PAO are presented in Fig. 5 A. The left panel shows data for an oocyte without added dynamin; irradiation at time zero induces a decrease in the whole-cell current and an accompanying decrease in capacitance. The center panel shows data for an oocyte coinjected with cRNA for WT dynamin; the current and capacitance changes closely mirror each other. The right panel shows data for an oocyte injected with dynamin-K44A; in response to photolytic decaging, the current decreases but the capacitance remains constant.
Figure 5.

Dynamin expression distinguishes inhibition of the channel from endocytosis. (A) Current and capacitance data from representative oocytes coexpressing v-Src and Kir2.1-Y242TAG suppressed with Tyr(ONB) and treated with PAO. Data were recorded 48 h after injection. (Left) An oocyte without coinjected dynamin cRNA. (Center) An oocyte coinjected with 10 ng WT dynamin cRNA. The capacitance decrease for this cell was unusually large. (Right) An oocyte coinjected with 10 ng dominant negative dynamin-K44A. (B) Data for current and capacitance for oocytes recorded 30 min after irradiation (mean ± SEM, n = 5). Left-hand y-axis and full bars represent normalized current decrease. Average values (and ranges) for 100% were 4.1 (3.15–4.8), 3.1 (2.8–3.7), and 4.1 (3.0–5.7) μA for the three groups (left to right). Right-hand axis corresponds to hollow bars, representing normalized capacitance decrease. The rightmost column shows capacitance change of uninjected control oocytes. Average values (and ranges) for 100% capacitance were 197 (191–205), 225 (191–248), 189 (174–199), and 197 (190–209) nF for the four groups (left to right). Error bars represent SEM.
Fig. 5 B shows the averaged current and capacitance data for three to eight oocytes and also the capacitance change of uninjected control oocytes over the course of the experiment. Current change is indicated by filled bars and the left-hand y-axis. When no dynamin mRNA was coinjected, the average current decrease 30 min after irradiation was 41.1 ± 0.7%. Coexpression with a WT dynamin isoform had an insignificant effect on the current decrease: the average decrease was 46.5 ± 5.7%. Dominant negative dynamin affected the current decrease considerably, but it did not eliminate the effect. The average current decrease was 27.9 ± 1.5% in the presence of dominant negative dynamin (significantly different from both zero dynamin and WT dynamin; P < 10−4). The hollow bars and right-hand axis of Fig. 5 B represent the normalized capacitance decrease. Without added dynamin, the average capacitance change associated with decaging tyrosine on Kir2.1 in the presence of v-Src was 17.7 ± 5.8%. Injection of WT dynamin gave rise to an average capacitance decrease of 26.0 ± 6.6%, not significantly larger than that of oocytes containing only endogenous dynamin. The effect of the dominant negative K44A dynamin isoform, however, was dramatic. Oocytes expressing dominant negative dynamin displayed an average capacitance change of −4.4 ± 0.7% over 30 min. This degree of change is significantly less than the data for zero dynamin and for WT dynamin (P < 10−4) but not significantly different from the baseline capacitance change for uninjected oocytes (−3.3 ± 1.4%).
Although coexpression of WT dynamin produced no significant change in the average decrease of either current or capacitance, we noted two effects that contributed to the increased variability among data for these oocytes. First, as noted in the legend to Fig. 5 B, these oocytes had a larger range of capacitance at the beginning of monitoring, including some values that were clearly larger than the range for the other oocytes in the experiment of Fig. 5. Second, in several such cells coinjected with cRNA for WT dynamin, the current and the capacitance continued to decrease at the end of the 30-min measurement period. This effect has not been systematically investigated.
By combining current and capacitance measurements with the expression of dominant negative dynamin, we have shown that significant current decrease occurs in the absence of appreciable capacitance change. The 28% current reduction in the absence of clathrin-mediated endocytosis is significantly smaller than the 41% reduction seen in cells where the endocytotic machinery has not been perturbed. Thus, clathrin-mediated endocytosis accounts for only a portion of the whole-cell current reduction. In addition, a nonendocytotic mechanism reduces current after the decaging of Tyr242.
Fluorescent Labeling Corroborates Dynamin-mediated Endocytosis
To observe the loss of plasma membrane directly, and to complement the electrophysiological data on current and capacitance change, we labeled and measured surface proteins on the oocyte. Oocytes expressing Kir2.1 with Tyr(ONB) were exposed to the impermeant thiol-reactive fluorophore tetramethylrhodamine-5-maleimide. Since the reagent is not specific for Kir2.1, uninjected oocytes were also labeled as a control. Oocytes treated in this manner displayed fluorescence at the cell surface (Fig. 6 A). Oocytes expressing the channel displayed 65% greater fluorescence intensity than uninjected oocytes, suggesting that one or both of the two extracellular cysteines of Kir2.1 are labeled by the maleimide. After 3 s of irradiation, fluorescence was measured at the animal pole at 2- or 2.5-min intervals over a 30-min period (Li et al. 2000). Fig. 6 B presents the time course of fluorescence for oocytes injected with no dynamin, WT, and dominant negative dynamin, along with controls in which v-Src is not expressed and in which Tyr(ONB) is not uncaged by photolysis. Net membrane internalization is revealed by decreased fluorescence. Corroborating the capacitance measurements, irradiation leads to internalization only in oocytes coexpressing v-Src, caged Kir2.1, and endogenous or added WT dynamin under conditions favoring tyrosine phosphorylation.
Figure 6.
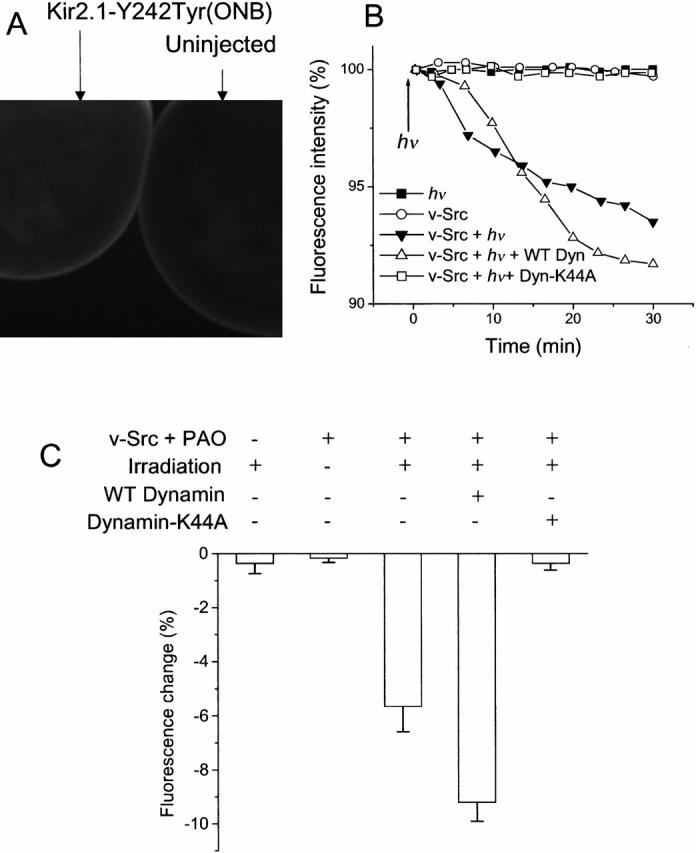
Fluorescence analysis of membrane retrieval. (A) Confocal image of two oocytes labeled with 10 μM tetramethylrhodamine-5-maleimide, a membrane-impermeant thiol-reactive fluorophore. The oocyte at left was injected with v-Src and Kir2.1-Y242TAG suppressed with Tyr(ONB). The oocyte on the right was not injected. (B) Fluorescence measurements for individual oocytes coexpressing v-Src and Kir2.1 Y242Tyr(ONB) and exposed to PAO. Oocytes were labeled with 10 μM tetramethylrhodamine-5-maleimide for 30 min before recording. Fluorescence is measured as PMT output voltage. As indicated, controls included omission of coinjected v-Src and omission of irradiation. (C) Normalized, average fluorescence change at t = 30 min for oocytes expressing Kir2.1-Y242TAG suppressed with Tyr(ONB) and treated with PAO, and then labeled with tetramethylrhodamine maleimide. The first column represents a control in which oocytes were not coinjected with v-Src. In all other columns, oocytes expressed the Kir2.1-Y242Tyr(ONB) along with v-Src. The second column represents a control in which oocytes were not irradiated. In the third column, no exogenous dynamin was added. The fourth column shows oocytes coexpressing WT dynamin. The fifth column represents oocytes coexpressing dynamin-K44A. Each bar represents six or seven oocytes. The average values (and ranges) for 100% fluorescence were 5.76 (3–9.5), 5.74 (3.5–7.2), 6.45 (3.7–9.5), 5.60 (3.93–6.92), and 5.62 (1–9.7) V.
Averaged data from six or seven oocytes are collected in Fig. 6 C. In oocytes expressing Kir2.1 with Tyr(ONB) at Y242 but not v-Src, the fluorescence change at 30 min was similar to that of nonirradiated cells expressing v-Src, −0.4 ± 0.4% and −0.2 ± 0.2%, respectively. Cells that expressed both channel and kinase without added dynamin showed an average fluorescence change of −5.7 ± 0.9% 30 min after irradiation. The coinjection of WT dynamin increased endocytosis markedly: there was a −9.2 ± 0.7% change over 30 min. As before, dominant negative dynamin suppressed membrane internalization to the level of uninjected oocytes. Oocytes coinjected with dynamin-K44A showed only a −0.4 ± 0.2% fluorescence change 30 min after tyrosine decaging. Both capacitance and fluorescence measurements imply that dynamin is required for decaging-induced endocytotic activity in the cell.
There Is No Direct Evidence for Tyrosine Phosphorylation of Kir2.1
Active tyrosine kinases decrease the activity of WT Kir2.1 (Fig. 2), are required for the decreases in both current and membrane area after tyrosine decaging (Fig. 4), and produce decreases in active channel density per patch (Fig. 3). None of these effects is observed for the Y242F mutant channel. The most direct interpretation of these data is that covalent modification of Y242 causes these effects, as proposed in earlier work on this system (Wischmeyer et al. 1998). It is also possible that this tyrosine residue is the interaction partner of proteins that must themselves be tyrosine phosphorylated for full activity, as is the case for many of the proteins in the clathrin-mediated endocytotic pathway.
Therefore, we sought evidence that Kir2.1 is phosphorylated at position 242 in the presence of kinases and phosphatase inhibitors. The primary strategy used to detect phosphorylation of Y242 was Western blotting with antiphosphotyrosine antibodies. For the purposes of immunoprecipitation and immunostaining of the channel, a Kir2.1 construct with the hemagglutinin (HA) epitope at the carboxyl terminus was generated. We verified in electrophysiological experiments that functional expression and inhibition by v-Src/PAO of Kir2.1-HA were similar to WT Kir2.1. In addition, a Y242F-HA construct was made and shown to behave like the Y242F channel.
To establish whether Western blotting with anti-PY antibodies was sufficiently sensitive to detect tyrosine phosphorylation of integral membrane proteins expressed in oocytes, control experiments were performed using the receptor tyrosine kinase TrkB. This kinase undergoes autophosphorylation on tyrosine in response to the binding of its ligand, BDNF (Chao 1992). Plasma membranes from BDNF-stimulated oocytes expressing TrkB were isolated by physical dissection. Western blotting with an anti-TrkB antibody (Santa Cruz C14) shows readily observable staining at the expected molecular mass (Fig. 7 A; Naruse et al. 1998). As expected, uninjected oocytes show no anti-TrkB staining. Blotting with an antiphosphotyrosine antibody (4G10) detects numerous phosphoproteins in the oocyte membrane, which is consistent with the effects of treating the oocytes with a tyrosine phosphatase inhibitor. Oocytes expressing TrkB contain a tyrosine-phosphorylated protein that colocalizes with TrkB and that is not present in uninjected oocytes. Densitometry showed that the phosphorylated TrkB band is roughly as intense as the nearby bands corresponding to endogenous phosphorylated proteins.
Figure 7.

Western blotting shows that Kir2.1 appears not to be phosphorylated on tyrosine, where detection of TrkB phosphorylation serves as a control for the method. The experiment is typical of at least six similar results. (A) Western blot of membranes from oocytes expressing TrkB shows positive staining with both anti-TrkB and anti-PY antibodies. (Lane 1) Uninjected oocytes; and (lane 2) TrkB. Nitrocellulose blot is stained on the left with C14 anti-TrkB antibody, and on the right with 4G10 antiphosphotyrosine antibody. Lane 1 has been loaded with 50% more membrane protein than lane 2 (which received protein from membrane dissected from 21 oocytes), to emphasize the absence of an endogenous phosphorylated band at the position of TrkB. Arrowheads indicate TrkB. (B) Western blot of membranes from oocytes expressing epitope-tagged Kir2.1 shows positive staining with anti-HA antibody, but not with the antiphosphotyrosine antibody 4G10. (Lane 1) Uninjected oocytes; (lane 2) Kir2.1-HA; (lane 3) Kir2.1Y242F-HA; and (lane 4) Kir2.1-HA prepared by in vitro translation from wheat germ extract. Nitrocellulose blot is stained on the left with BAbCo HA.11 antihemagglutinin antibody, and on the right with 4G10 antiphosphotyrosine antibody. Arrows indicate Kir2.1. Lanes 1–3 contain protein from membranes dissected from 21–23 oocytes.
Comparable experiments were carried out with Kir2.1-HA constructs (Fig. 7 B). Kir2.1 constructs are clearly detectable by the anti-HA antibody (HA.11) at the appropriate molecular mass. A protein produced by in vitro translation of Kir2.1-HA mRNA from wheat germ extract confirms the assignment of monomeric Kir2.1 subunits. The stained upper molecular mass bands presumably represent oligomers that are not denatured, even under SDS-PAGE conditions (Ramjeesingh et al. 1999). In contrast, staining Western blots with the antiphosphotyrosine antibody gives no convincing signal corresponding to Kir2.1 monomer or oligomer. Phosphoproteins are clearly present at a variety of other molecular masses, which is consistent with the TrkB results and corroborates that the assay does detect membrane phosphoproteins. That these phosphoproteins are also present in uninjected oocytes treated with PAO confirms that they do not arise from Kir2.1. Any staining in the region corresponding to monomeric Kir2.1 is <10% as intense as that for nearby endogenous phosphorylated proteins.
In addition to these experiments, a number of other immunoprecipitation and immunodetection schemes were examined. Immunoprecipitation of phosphoproteins from oocyte homogenates with anti-PY antibodies followed by detection with anti-HA antibodies was undertaken, as well as the reverse. Also, a panel of antiphosphotyrosine antibodies was examined to optimize sensitivity under the present experimental conditions, with the result that 4G10 gave the highest sensitivity, followed closely by PY72. A completely different assay was also attempted, namely autoradiography. Even though this technique is often considered to be more sensitive than Western blotting, it has the disadvantage of not distinguishing between phosphorylation on tyrosine, serine, or threonine without phosphopeptide mapping. Oocytes were supplied with 32P either by coinjection with mRNA encoding the channel, or by incubation in γ-[32P]ATP. HA-tagged channels were immunoprecipitated from whole-cell membrane preparations and from physically dissected membranes by both anti-HA and anti-PY antibodies and subjected to SDS-PAGE. No differences between the Kir2.1-HA and Kir2.1-Y242F-HA channels were ever observed in 32P autoradiography, and further mapping was not attempted. That we did not detect tyrosine phosphorylation of Kir2.1 leads us to the explanation that other protein(s) are phosphorylated. We conclude that interactions among these proteins, or between these proteins and Kir2.1, lead to downregulation of K+ current and endocytosis after Y242 is decaged.
DISCUSSION
The major new finding is that decaging of a single defined residue in a K+ channel (Y242 in Kir2.1), under conditions favoring tyrosine phosphorylation, leads to substantial clathrin-mediated endocytosis. The endocytosis is detected by decreases in both capacitance (up to 26% with overexpression of WT dynamin) and in surface fluorescence. Because the protein involved is an ion channel, we are also able to compare high resolution physiological assays with the measurements of surface area. The combined data show that decaging also leads to inhibition of Kir2.1 currents via a decreased density of active channels. Interestingly, significant inhibition of the current is observed even when clathrin-mediated endocytosis is blocked by a dominant negative dynamin mutant.
Fig. 8 suggests several alternative sequences of events after irradiation that leads to tyrosine decaging. Our data argue against scheme A, in which the channel itself is directly phosphorylated. Instead, we favor scheme B or C, in which decaging reveals a tyrosine-based interaction motif in Kir2.1. Under conditions favoring tyrosine phosphorylation either of the adapter protein (Fig. 8, pathway B) or of an unspecified upstream protein (Fig. 8, pathway C), the channel protein then interacts with the μ subunit of the adaptor protein AP-1, AP-2, or AP-3, or with a similar molecule (Ohno et al. 1998). The interaction inhibits the channel, possibly via direct occlusion of the conduction pathway. As the interaction proceeds further (perhaps because several Kir2.1 subunits each interact with a μ subunit), this interaction between the adaptor protein and the channel also leads to endocytosis of 15–26% of the membrane, as measured by the capacitance decrease. Thus, the inhibition of channel conductance accompanies, but does not cause, endocytosis. Indeed, the total Kir2.1 current does not decrease to zero, suggesting that only a portion of the channels undergo endocytosis. Our data suggest that the 28% inhibition of macroscopic currents in the absence of clathrin-mediated endocytosis (Fig. 5 B) occurs without detectable membrane retraction. The greater macroscopic inhibition in the presence of WT dynamin (∼46%) would be caused by retraction of [1 − (1 − 0.46)/(1 − 0.28)] = 25% of the Kir2.1 channels. Thus, it is possible that, once tyrosine decaging in Kir2.1 has triggered the endocytotic events, the fraction of channels withdrawn is comparable to the fraction of general membrane withdrawn. That single-channel currents were still recorded in v-Src/PAO patches also suggests that only a fraction of the channels disappeared from the surface membrane; but the single-channel recordings suggest that comparably sized patches contain ∼50% fewer channels during v-Src/PAO treatment. Presumably, part of this decrease occurs because channels are inhibited but remain in the membrane; and part of the decrease occurs because channels are withdrawn.
Figure 8.
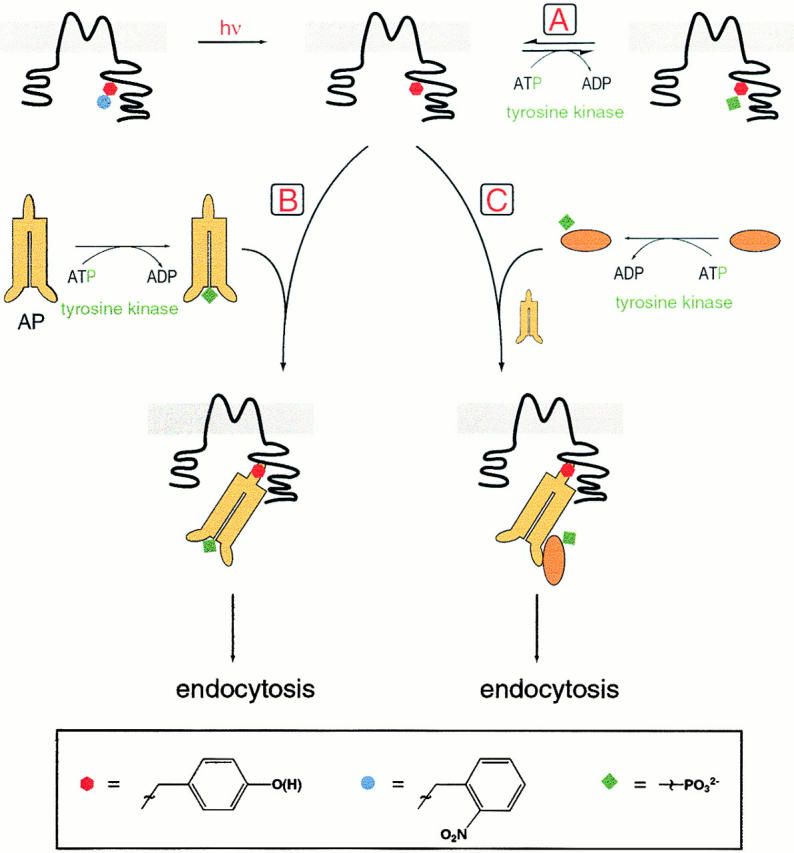
Schematic interpretation of the experiments. Irradiation leads to decaging of Tyr242 of Kir 2.1. Pathway A shows direct phosphorylation of the newly revealed tyrosine, which may be reversible/transient. Failure to observe phosphotyrosine directly makes this pathway unlikely. Pathways B and C involve the μ subunit of AP-1, AP-2, or AP-3 or a similar molecule. In pathway B the adaptor protein (AP) is phosphorylated, which enables it to bind to the now available tyrosine-based interaction motif of Kir 2.1 and inhibit the channel. Endocytosis follows. Pathway C includes phosphorylation of an unspecified protein, which, in combination with the adaptor protein, binds to and inhibits Kir 2.1 and leads to endocytosis.
Our data with phosphotyrosine-specific antibodies and with 32P labeling reveal no evidence for a pathway such as A in Fig. 8, involving direct phosphorylation of Kir2.1. It remains formally possible that Y242 is phosphorylated directly, possibly transiently, and that this phosphorylation aids either the channel inhibition or the endocytosis. Furthermore, because the channel is a homotetramer, an individual channel might contain both phosphorylated and nonphosphorylated Y242 side chains. However if it occurs, this direct phosphorylation has escaped our detection. In support of the idea that Kir2.1 does not undergo direct tyrosine phosphorylation, no such phosphorylation was reported for mammalian cells in the study of Wischmeyer et al. 1998.
Withdrawal of Substantial Membrane Area
Stage 5 and 6 oocytes are capable of extensive membrane trafficking. Membrane area increases by 40% as a result of phosphorylation of CFTR (Takahashi et al. 1996; Peters et al. 1999), and decreases by >35% as a result of C-kinase activation (Vasilets et al. 1990; Bourinet et al. 1992; Quick et al. 1997). Furthermore, IGF-1, which like several other activators of receptor tyrosine kinases induces meiotic oocyte maturation, also induces endocytosis (Taghon and Sadler 1994). In the cases studied, these membrane additions and deletions occur over a time course of minutes to tens of min, as seen here. Furthermore, a similar time course describes the decrease in K+ currents after activation of a receptor tyrosine kinase (Timpe and Fantl 1994). On a slower time scale (measured in days), membrane area increases as a result of the Na+ channel β2 subunit (Isom et al. 1995) and sodium-glucose exchanger (Hirsch et al. 1996) expression. The present study is unique in showing that massive membrane trafficking, 15–26% in 30 min, can be initiated by changing the structure of a single residue in a single species of membrane molecule.
What is the ratio between the amount of membrane area withdrawn and the number of Kir2.1 channels inhibited? For 14 oocytes in the experimental series of Fig. 4 C, the absolute values of current inhibition and capacitance decrease were 1.9 μA and 32 nF, respectively. At −80 mV, the single-channel current is ∼1.7 pA, and the open probability is roughly 50%; therefore, the time-averaged current through a channel is 0.85 pA. Therefore, the average number of electrophysiologically revealed channels inhibited is 2.2 × 106.
These data lead to the conclusion that an amount of membrane with a capacitance of 1.5 × 10−5 nF = 1.5 × 10−2 pF is retrieved per active channel inhibited. For a typical membrane capacitance of 8 × 10−3 pF/μm2, these data suggest that ∼2 μm2 of the net membrane area undergoes endocytosis per electrophysiologically active channel inhibited. This area is >50 times larger than the surface area (0.031 μm2) of the typical 100-nm-diam clathrin-coated vesicle. The ratio of membrane area withdrawn per electrophysiologically active channel inhibited is comparably large for the WT dynamin injected oocytes.
We conclude that the membrane area equal to many clathrin-coated vesicles is retrieved per electrophysiologically active channel inhibited. This conclusion appears at odds with the way YXXΦ motifs typically work: by collecting and concentrating, through direct interaction with the clathrin coat component AP2, YXXΦ-tagged proteins in clathrin-coated pits. We cannot rule out the existence of a large excess of electrophysiologically silent Kir2.1 channels in the membrane that are decaged and also help to induce endocytosis. However, careful comparisons of freeze-fracture data and electrophysiological data on oocytes have shown that, in many cases, there are no electrophysiologically silent channels or transporters (Zampighi et al. 1995; Eskandari et al. 1999). Despite quantitative uncertainties, our data show that decaging of single tyrosine residues under conditions favoring tyrosine phosphorylation is sufficient to organize a significant apparatus for endocytosis.
The ratio between membrane area withdrawn and channels inhibited is not constant: it decreases to essentially zero for the dynamin-K44A–injected oocytes (Fig. 5 B). Therefore, the ratio depends on the state of the endocytotic machinery within the cell. Even with overexpressed WT dynamin, irradiation results in only a 50% decrease of currents. Thus, many channels remain on the surface membrane. Possible reasons include the following: (1) channel retrieval only at the rate of general membrane retrieval, as discussed above; (2) readthrough of the stop codon, resulting in channels with one or more natural residues at position 242; (3) channels localized in regions devoid of endocytotic machinery; and (4) complete membrane recycling, so that channels reappear on the surface membrane.
Fluorescence Measurements of Endocytosis
This paper introduces a fluorescence assay for endocytosis in oocytes. At present, we are not certain of its physical basis. We suggest that the decreased fluorescence occurs because the protein labeled with rhodamine dye enters the low pH quenching environment of vesicles and/or because the exciting and/or emitted light is partially shielded by intracellular pigment granules and other structures. The optical assay of membrane retraction does all of the following: works in real time; does not kill cells; is simple (no special constructs needed); is general (it also works with Gq-coupled receptors; Tong, Y., and M. Li., unpublished observations); and is nearly linear (within a factor of two) with capacitance measurements, which is the gold standard method for measuring membrane traffic.
Modulatory Mechanisms at Kir Channels
Kir2 channels are subject to several regulatory mechanisms. The single-channel conductance of Kir2.3 is decreased by interactions between the channel and PSD-95 via a PDZ domain (Nehring et al. 1999). Kir2.1 channels can also be inhibited by depletion of inositol phosphates (Huang et al. 1998). Kir2.1 channels apparently also display increased open probability when phosphorylated by A-kinase; dephosphorylation by PP2A inhibits function (Ruppersberg and Fakler 1996). Kir3 channels are inhibited via direct phosphorylation of an amino-terminal tyrosine residue (Rogalski et al. 2000) and are facilitated via an unknown pathway involving A-kinase (Müllner et al. 2000). The indirect mechanism that we favor, partially involving membrane retraction (Fig. 8), is probably not unique to Kir2.1; the novel aspect is the scale of membrane retraction stimulated by tyrosine decaging at a single group.
As noted earlier, several tyrosine kinases are able to induce the current inhibition we observed (Wischmeyer et al. 1998). We do not know how many steps intervene between these kinases and the protein that ultimately interacts with Kir2.1. Identification of these steps and these proteins represents a long term goal for studies on the molecular detail of phosphorylation-mediated ion channel regulation. Several targets have been identified for Src and related kinases in Xenopus oocytes (Thorn et al. 1999).
Interactions with Adaptor Proteins
The experiments show that tyrosine decaging leads to dynamin-mediated endocytosis—but only if the tyrosine side chain at Y242 is revealed in a phosphorylation context. Furthermore, the delay observed in some of our experiments suggests that this endocytosis occurs subsequent to the steps that directly cause initial inhibition of the current.
The motif YXXΦ, where X is any amino acid and Φ is a hydrophobic residue, is a recognition signal for adaptor proteins that target polypeptides to clathrin-coated vesicles. Interactions with the μ subunits of AP-1 and AP-3 (μ1 and μ3A, respectively) are especially favored by YXPL, the sequence found in Kir2.1 at positions 242–245 (Ohno et al. 1998; Sanderfoot et al. 1998). Crystallographic evidence confirms that the AP-2 μ subunit is able to bind peptides containing the YXXΦ motif (Owen and Evans 1998). Our data suggest that the AP-2 μ subunit or a similar endogenous molecule in the oocytes does have the very specific property that it signals only while bound to Kir2.1 but blocks signal transduction otherwise. A study of the ENaC channel in Xenopus oocytes has demonstrated that a tyrosine residue in such a motif is functionally important in targeting an ion channel for removal from the plasma membrane (Shimkets et al. 1997).
Thus, a partial explanation for the endocytosis we observed with Kir2.1 would be that caged tyrosine protects Kir2.1 from incorporation into clathrin-coated vesicles (Fig. 8, pathway B or C). The nitrobenzyl group adds steric bulk to the tyrosine side chain and prohibits the tyrosyl hydroxyl from engaging in hydrogen bonding. Once photolysis liberates the WT residue in a tyrosine-based endocytotic motif, a fraction of the channels would be removed from the plasma membrane by a clathrin-mediated pathway. Indeed, other data indicate that the YXXΦ motif cannot interact with adaptor proteins if the tyrosine residue is phosphorylated (Boll et al. 1996; Shiratori et al. 1997). However, because membrane area equivalent to many clathrin-coated vesicles is withdrawn per channel inhibited, a catalytic step of presently unknown nature may participate in the signal transduction pathway between the kinase and an effector required for vesicle formation.
Kir2.1 contains two additional COOH-terminal YXXΦ sequences, YEPV from 326–329 and YSRF, comprising residues 340–344, as well as a potential dileucine endocytosis motif at 231–232. It is now appreciated that tyrosine-based motifs can act cooperatively and allosterically to enhance protein–protein interactions during interactions with other proteins involved in membrane trafficking (Rodriguez-Tarduchy et al. 1996; Sunder-Plassmann et al. 1997; Vely et al. 1997; Haucke and De Camilli 1999). For instance, it is possible that decaging Y242 allows the local conformation of the channel to be folded correctly, so that another endocytotic motif becomes available for endocytosis. Thus, several regions of each Kir2.1 subunit could play a role in the endocytotic event that is triggered by decaging of Y242.
Ion Channel Modulation Studied by Decaging Tyrosine
These data suggest that both function and endocytosis of an inward rectifier K+ channel are regulated directly by a single side chain and indirectly by tyrosine phosphorylation events that are not yet specified. Decreased Kir2.1 function is certainly accompanied by substantial membrane exocytosis but probably not by direct tyrosine phosphorylation of the channel protein. The experiments suggest that membrane trafficking must be considered as a contributory mechanism in any modulatory event at ion channels, even if pharmacology and site-directed mutagenesis suggest that phosphorylation is involved. In addition, these experiments describe a new way to use caged proteins (Pan and Bayley 1997; Curley and Lawrence 1999a,Curley and Lawrence 1999b; Marriott and Walker 1999) for determining the kinetics of cellular events. The caged tyrosine method is applicable to a variety of other studies in signal transduction, and the technique can readily be extended to other caged side chains.
Acknowledgments
We thank Hai-Rong Li for help with the oocytes, Mike Walsh for assistance with instrumentation, Tatiana Ivanina and Abraham Kovoor for advice on phosphorylation assays, and members of our laboratory for much discussion.
This research was supported by grants from the National Institutes of Health (GM29836 and NS34407), by National Research Service Awards to Y. Tong and M. Li, by a fellowship from the Burroughs-Wellcome Foundation Computational Molecular Biology Program to G. Shapovalov, by a Predoctoral Training Award from NIH to E. Slimko, by a Boehringer Fellowship to G.S. Brandt, and by the Alexander von Humboldt Foundation.
Footnotes
Abbreviations used in this paper: HA, hemagglutinin; PAO, phenylarsine oxide; PMT, photomultiplier tube; Tyr(ONB), tyrosine analogue o-nitrobenzyltyrosine; WT, wild-type.
References
- Boll W., Ohno H., Songyang Z., Rapoport I., Cantley L.C., Bonifacino J.S., Kirchhausen T. Sequence requirements for the recognition of tyrosine-based endocytic signals by clathrin AP-2 complexes. EMBO (Eur. Mol. Bio. Organ.) J. 1996;15:5789–5795. [PMC free article] [PubMed] [Google Scholar]
- Bourinet E., Fournier F., Lory P., Charnet P., Nargeot J. Protein kinase C regulation of cardiac calcium channels expressed in Xenopus oocytes. Pflügers Arch. 1992;421:247–255. doi: 10.1007/BF00374834. [DOI] [PubMed] [Google Scholar]
- Chao M.V. Neurotrophin receptorsa window into neuronal differentiation. Neuron. 1992;9:583–593. doi: 10.1016/0896-6273(92)90023-7. [DOI] [PubMed] [Google Scholar]
- Chen Y.H., Pouyssegur J., Courtneidge S.A., Van Obberghen-Schilling E. Activation of Src family kinase activity by the G protein-coupled thrombin receptor in growth-responsive fibroblasts. J. Biol. Chem. 1994;269:27372–27377. [PubMed] [Google Scholar]
- Curley K., Lawrence D.S. Caged regulators of signaling pathways Pharmacol. Ther 82 1999. 347 354a [DOI] [PubMed] [Google Scholar]
- Curley K., Lawrence D.S. Light-activated proteins Curr. Opin. Chem. Biol 3 1999. 84 88b [DOI] [PubMed] [Google Scholar]
- Damke H., Baba T., Warnock D.E., Schmid S.L. Induction of mutant dynamin specifically blocks endocytic-coated vesicle formation. J. Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I., Tokiwa G., Lev S., Courtneidge S.A., Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- England P.M., Lester H.A., Davidson N., Dougherty D.A. Site-specific, photochemical proteolysis applied to ion channels in vivo. Proc. Natl. Acad. Sci. USA. 1997;94:11025–11030. doi: 10.1073/pnas.94.20.11025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskandari S., Snyder P.M., Kreman M., Zampighi G.A., Welsh M.J., Wright E.M. Number of subunits comprising the epithelial sodium channel. J. Biol. Chem. 1999;274:27281–27286. doi: 10.1074/jbc.274.38.27281. [DOI] [PubMed] [Google Scholar]
- Felsch J.S., Cachero T.G., Peralta E.G. Activation of protein tyrosine kinase PYK2 by the m1 muscarinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA. 1998;95:5051–5056. doi: 10.1073/pnas.95.9.5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenney J.R., Jr., Chen W.S., Lazar C.S., Walton G.M., Zokas L.M., Rosenfeld M.G., Gill G.N. Ligand-induced endocytosis of the EGF receptor is blocked by mutational inactivation and by microinjection of anti-phosphotyrosine antibodies. Cell. 1988;52:675–684. doi: 10.1016/0092-8674(88)90405-9. [DOI] [PubMed] [Google Scholar]
- Haucke V., De Camilli P. AP-2 recruitment to synaptotagmin stimulated by tyrosine-based endocytic motifs. Science. 1999;285:1268–1271. doi: 10.1126/science.285.5431.1268. [DOI] [PubMed] [Google Scholar]
- Hirsch J.R., Loo D.D.F., Wright E.M. Regulation of Na+/glucose cotransporter expression by protein kinases in Xenopus laevis oocytes. J. Biol. Chem. 1996;271:14740–14746. doi: 10.1074/jbc.271.25.14740. [DOI] [PubMed] [Google Scholar]
- Holmes T.C., Fadool D.A., Levitan I.B. Tyrosine phosphorylation of the Kv1.3 potassium channel J. Neurosci 16 1996. 1581 1590a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes T.C., Fadool D.A., Ren R., Levitan I.B. Association of Src tyrosine kinase with a human potassium channel mediated by SH3 domain Science 274 1996. 2089 2091b [DOI] [PubMed] [Google Scholar]
- Huang C.L., Feng S., Hilgemann D.W. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. 1998;391:803–806. doi: 10.1038/35882. [DOI] [PubMed] [Google Scholar]
- Huyer G., Liu S., Kelly J., Moffat J., Payette P., Kennedy B., Tsaprailis G., Gresser M.J., Ramachandran C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 1997;272:843–851. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- Isom L.L., Ragsdale D.S., Dejongh K.S., Westenbroek R.E., Reber B.F.X., Scheuer T., Catterall W.A. Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 1995;83:433–442. doi: 10.1016/0092-8674(95)90121-3. [DOI] [PubMed] [Google Scholar]
- Ivanina T., Perets T., Thornhill W.B., Levin G., Dascal N., Lotan I. Phosphorylation by protein kinase A of RCK1 K+ channels expressed in Xenopus oocytes. Biochemistry. 1994;33:8786–8792. doi: 10.1021/bi00195a021. [DOI] [PubMed] [Google Scholar]
- Jonas E., Kaczmarek L. Regulation of potassium channels by protein kinases. Curr. Opin. Neurobiol. 1996;6:318–323. doi: 10.1016/s0959-4388(96)80114-0. [DOI] [PubMed] [Google Scholar]
- Kirchhausen T., Bonifacino J.S., Riezman H. Linking cargo to vesicle formationreceptor tail interactions with coat proteins. Curr. Opin. Cell Biol. 1997;9:488–495. doi: 10.1016/s0955-0674(97)80024-5. [DOI] [PubMed] [Google Scholar]
- Kubo Y., Baldwin T.J., Jan Y.N., Jan L.Y. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- Levitan I.B. Modulation of ion channels by protein phosphorylation. How the brain works. Adv. Second Messenger Phosphoprotein Res. 1999;33:3–22. doi: 10.1016/s1040-7952(99)80003-2. [DOI] [PubMed] [Google Scholar]
- Li M., Farley R., Lester H.A. An intermediate state of the GABA transporter GAT1 revealed by simultaneous voltage clamp and fluorescence. J. Gen. Physiol. 2000;115:491–508. doi: 10.1085/jgp.115.4.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S., Woronuk G., Sy L., Lev S., Braun A.P. Enhanced activity of a large conductance, calcium-sensitive K+ channel in the presence of Src tyrosine kinase. J. Biol. Chem. 2000;275:30683–30689. doi: 10.1074/jbc.M004292200. [DOI] [PubMed] [Google Scholar]
- Marriott G., Walker J.W. Caged peptides and proteinsnew probes to study polypeptide function in complex biological systems. Trends Plant Sci. 1999;4:330–334. doi: 10.1016/s1360-1385(99)01452-1. [DOI] [PubMed] [Google Scholar]
- Miller J.P., Silverman S.K., England P.M., Dougherty D.A., Lester H.A. Flash decaging of tyrosine sidechains in an ion channel. Neuron. 1998;20:619–624. doi: 10.1016/s0896-6273(00)81001-6. [DOI] [PubMed] [Google Scholar]
- Müllner C., Vorobiov D., Bera A.K., Uezono Y., Yakubovich D., Frohnwieser-Steinecker B., Dascal N., Schreibmayer W. Heterologous facilitation of G protein–activated K+ channels by β-adrenergic stimulation via cAMP-dependent protein kinase. J. Gen. Physiol. 2000;115:547–558. doi: 10.1085/jgp.115.5.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruse S., Thinakaran G., Luo J.J., Kusiak J.W., Tomita T., Iwatsubo T., Qian X., Ginty D.D., Price D.L., Borchelt D.R., Wong P.C., Sisodia S.S. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- Nehring R., Wischmeyer E., Döring F., Veh R., Sheng M., Karschin A. Neuronal Kir channels differentially couple to PDZ proteins of the PSD-95/SAP90 family. J. Neurosci. 1999;20:156–162. doi: 10.1523/JNEUROSCI.20-01-00156.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren C.J., Anthony-Cahill S.J., Griffith M.C., Schultz P.G. A general-method for site-specific incorporation of unnatural amino-acids into proteins. Science. 1989;244:182–188. doi: 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- Nowak M.W., Kearney P.C., Sampson J.R., Saks M.E., Labarca C.G., Silverman S.K., Zhong W., Thorson J., Abelson J.N., Davidson N., Schultz P.G., Dougherty D.A., Lester H.A. Nicotinic receptor binding site probed with unnatural amino acid incorporation in intact cells. Science. 1995;268:439–442. doi: 10.1126/science.7716551. [DOI] [PubMed] [Google Scholar]
- Nowak M.W., Gallivan J.P., Silverman S.K., Labarca C.G., Dougherty D.A., Lester H.A. In vivo incorporation of unnatural amino acids into ion channels in a Xenopus oocyte expression system. Methods Enzymol. 1998;293:504–529. doi: 10.1016/s0076-6879(98)93031-2. [DOI] [PubMed] [Google Scholar]
- Ohno H., Aguilar R.C., Yeh D., Taura D., Saito T., Bonifacino J.S. The medium subunits of adaptor complexes recognize distinct but overlapping sets of tyrosine-based sorting signals. J. Biol. Chem. 1998;273:25915–25921. doi: 10.1074/jbc.273.40.25915. [DOI] [PubMed] [Google Scholar]
- Owen D.J., Evans P.R. A structural explanation for the recognition of tyrosine-based endocytotic signals. Science. 1998;282:1327–1332. doi: 10.1126/science.282.5392.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pafford C.M., Simples J.E., Jr., Strong J.A. Effects of the protein tyrosine phosphatase inhibitor phenylarsine oxide on excision-activated calcium channels in Lymnaea neurons. Cell Calcium. 1995;18:400–410. doi: 10.1016/0143-4160(95)90055-1. [DOI] [PubMed] [Google Scholar]
- Pan P., Bayley H. Caged cysteine and thiophosphoryl peptides. FEBS Lett. 1997;405:81–85. doi: 10.1016/s0014-5793(97)00165-8. [DOI] [PubMed] [Google Scholar]
- Peters K.W., Qi J., Watkins S.C., Frizzell R.A. Syntaxin 1A inhibits regulated CFTR trafficking in Xenopus oocytes. Am. J. Physiol. 1999;277:C174–C180. doi: 10.1152/ajpcell.1999.277.1.C174. [DOI] [PubMed] [Google Scholar]
- Quick M.W., Corey J.L., Davidson N., Lester H.A. Second messengers, trafficking-related proteins, and amino acid residues that contribute to the functional regulation of the rat brain GABA transporter GAT1. J. Neurosci. 1997;17:2967–2979. doi: 10.1523/JNEUROSCI.17-09-02967.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramjeesingh M., Huan L.J., Garami E., Bear C.E. Novel method for evaluation of the oligomeric structure of membrane proteins. Biochem. J. 1999;342:119–123. [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Tarduchy G., Sahuquillo A.G., Alarcon B., Bragado R. Apoptosis but not other activation events is inhibited by a mutation in the transmembrane domain of T cell receptor β that impairs CD3ζ association. J. Biol. Chem. 1996;271:30417–30425. doi: 10.1074/jbc.271.48.30417. [DOI] [PubMed] [Google Scholar]
- Rogalski S., Appleyard S., Pattillo A., Terman G., Chavkin C. TrkB activation by BDNF inhibits the G protein gated inward rectifier Kir3 by tyrosine phosphorylation of the channel. J. Biol. Chem. 2000;275:25082–25088. doi: 10.1074/jbc.M000183200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppersberg J.P., Fakler B. Complexity of the regulation of Kir2.1 K+ channels. Neuropharmacology. 1996;35:887–893. doi: 10.1016/0028-3908(96)00092-5. [DOI] [PubMed] [Google Scholar]
- Saks M.E., Sampson J.R., Nowak M.W., Kearney P.C., Du F., Abelson J.N., Lester H.A., Dougherty D.A. An engineered Tetrahymena tRNAGln for in vivo incorporation of unnatural amino acids into proteins by nonsense suppression. J. Biol. Chem. 1996;271:23169–23175. doi: 10.1074/jbc.271.38.23169. [DOI] [PubMed] [Google Scholar]
- Sanderfoot A.A., Ahmed S.U., Marty-Mazars D., Rapoport I., Kirchhausen T., Marty F., Raikhel N.V. A putative vacuolar cargo receptor partially colocalizes with AtPEP12p on a prevacuolar compartment in Arabidopsis roots. Proc. Natl. Acad. Sci. USA. 1998;95:9920–9925. doi: 10.1073/pnas.95.17.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J., Ullrich A. Growth factor signaling by receptor tyrosine kinases. Neuron. 1992;9:383–391. doi: 10.1016/0896-6273(92)90177-f. [DOI] [PubMed] [Google Scholar]
- Shimkets R.A., Lifton R.P., Canessa C.M. The activity of the epithelial sodium channel is regulated by clathrin-mediated endocytosis. J. Biol. Chem. 1997;272:25537–25541. doi: 10.1074/jbc.272.41.25537. [DOI] [PubMed] [Google Scholar]
- Shiratori T., Miyatake S., Ohno H., Nakaseko C., Isono K., Bonifacino J.S., Saito T. Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity. 1997;6:583–589. doi: 10.1016/s1074-7613(00)80346-5. [DOI] [PubMed] [Google Scholar]
- Sunder-Plassmann R., Lialios F., Madsen M., Koyasu S., Reinherz E.L. Functional analysis of immunoreceptor tyrosine-based activation motif (ITAM)-mediated signal transductionthe two YxxL segments within a single CD3ζ-ITAM are functionally distinct. Eur. J. Immunol. 1997;27:2001–2009. doi: 10.1002/eji.1830270826. [DOI] [PubMed] [Google Scholar]
- Taghon M.S., Sadler S.E. Insulin-like growth factor 1 receptor-mediated endocytosis in Xenopus laevis oocytes. A role for receptor tyrosine kinase activity. Dev. Biol. 1994;163:66–74. doi: 10.1006/dbio.1994.1123. [DOI] [PubMed] [Google Scholar]
- Takahashi A., Watkins S.C., Howard M., Frizzell R.A. CFTR-dependent membrane insertion is linked to stimulation of the CFTR chloride conductance. Am. J. Physiol. 1996;271:C1887–C1894. doi: 10.1152/ajpcell.1996.271.6.C1887. [DOI] [PubMed] [Google Scholar]
- Thorn J.M., Armstrong N.A., Cantrell L.A., Kay B.K. Identification and characterisation of Xenopus moesin, a Src substrate in Xenopus laevis oocytes. Zygote. 1999;7:113–122. doi: 10.1017/s0967199499000465. [DOI] [PubMed] [Google Scholar]
- Timpe L.C., Fantl W.J. Modulation of a voltage-activated potassium channel by peptide growth factor receptors. J. Neurosci. 1994;14:1195–1201. doi: 10.1523/JNEUROSCI.14-03-01195.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bliek A.M., Redelmeier T.E., Damke H., Tisdale E.J., Meyerowitz E.M., Schmid S.L. Mutations in human dynamin block an intermediate stage in coated vesicle formation. J. Cell Biol. 1993;122:553–563. doi: 10.1083/jcb.122.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Geer P., Hunter T., Lindberg R.A. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu. Rev. Cell Biol. 1994;10:251–337. doi: 10.1146/annurev.cb.10.110194.001343. [DOI] [PubMed] [Google Scholar]
- Vasilets L.A., Schmalzing G., Madefessel K., Haase W., Schwarz W. Activation of protein kinase C by phorbol ester induces downregulation of the Na+/K+-ATPase in oocytes of Xenopus laevis . J. Membr. Biol. 1990;118:131–142. doi: 10.1007/BF01868470. [DOI] [PubMed] [Google Scholar]
- Vely F., Nunes J.A., Malissen B., Hedgecock C.J. Analysis of immunoreceptor tyrosine-based activation motif (ITAM) binding to ZAP-70 by surface plasmon resonance. Eur. J. Immunol. 1997;27:3010–3014. doi: 10.1002/eji.1830271138. [DOI] [PubMed] [Google Scholar]
- Webb J.L. Enzyme and Metabolic Inhibitors. Vol. 3 1966. Academic Press; New York: pp. 1228 [Google Scholar]
- Wischmeyer E., Doring F., Karschin A. Acute suppression of inwardly rectifying Kir2.1 channels by direct tyrosine kinase phosphorylation. J. Biol. Chem. 1998;273:34063–34068. doi: 10.1074/jbc.273.51.34063. [DOI] [PubMed] [Google Scholar]
- Yu X.M., Askalan R., Keil G.J., II, Salter M.W. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- Zampighi G.A., Kreman M., Boorer K.J., Loo D.D., Bezanilla F., Chandy G., Hall J.E., Wright E.M. A method for determining the unitary functional capacity of cloned channels and transporters expressed in Xenopus laevis oocytes. J. Membr. Biol. 1995;148:65–78. doi: 10.1007/BF00234157. [DOI] [PubMed] [Google Scholar]