

Abstract
Titin, a giant protein spanning half the sarcomere, is responsible for passive and restoring forces in cardiac myofilaments during sarcomere elongation and compression, respectively. In addition, titin has been implicated in the length-dependent activation that occurs in the stretched sarcomere, during the transition from diastole to systole. The purpose of this study was to investigate the role of titin in the length-dependent deactivation that occurs during early diastole, when the myocyte is shortened below slack length. We developed a novel in vitro assay to assess myocyte restoring force (RF) by measuring the velocity of recoil in Triton-permeabilized, unloaded rat cardiomyocytes after rigor-induced sarcomere length (SL) contractions. We compared rigor-induced SL shortening to that following calcium-induced (pCa) contractions. The RF–SL relationship was linearly correlated, and the SL-pCa curve displayed a characteristic sigmoidal curve. The role of titin was defined by treating myocytes with a low concentration of trypsin, which we show selectively degrades titin using mass spectroscopic analysis. Trypsin treatment reduced myocyte RF as shown by a decrease in the slope of the RF-SL relationship, and this was accompanied by a downward and leftward shift of the SL-pCa curve, indicative of sensitization of the myofilaments to calcium. In addition, trypsin digestion did not alter the relationship between SL and interfilament spacing (assessed by cell width) after calcium activation. These data suggest that as the sarcomere shortens below slack length, titin-based restoring forces act to desensitize the myofilaments. Furthermore, in contrast to length-dependent activation at long SLs, length-dependent deactivation does not depend on interfilament spacing. This study demonstrates for the first time the importance of titin-based restoring force in length-dependent deactivation during the early phase of diastole.
Keywords: connectin, restoring force, skinned myocyte, length-dependent deactivation, titin
INTRODUCTION
Length-dependent activation of cardiac myocytes has been recognized for some time (Fabiato and Fabiato, 1975) and is thought to be an important contributor to the Frank-Starling relation of the heart. Although many components resulting in length-dependent activation have been identified, such as increased cooperativity of the regulatory proteins or reduced interfilament spacing, mechanistically this phenomena is still poorly understood (for reviews see Allen and Kentish, 1985; Gordon et al., 2000; Goldstein et al., 1991). Recently, it has been recognized that the giant myofilament protein titin may play a role in this phenomenon. Titin is the only molecule that spans the sarcomere and integrates the thick and thin filament system (for reviews see Trinick, 1994; Labeit et al., 1997; Maruyama, 1997; Gregorio et al., 1999; Granzier and Labeit, 2002). Experiments at the single molecule level (Kellermayer et al., 1997; Rief et al., 1997; Tskhovrebova et al., 1997) and in intact myocytes (Wang et al., 1993; Trombitas et al., 1995; Helmes et al., 1999; Watanabe et al., 2002) have shown that the I-band segment of titin is extensible and behaves as an entropic spring. The extensibility of titin allows it to impart passive or restoring forces on the myocyte when stretched above or shortened below resting sarcomere length (SL),* respectively. Passive and restoring forces operate in opposite directions, both working to return the myocyte to resting length. In rat myocytes, at SLs from 1.6 to 2.1 μm, titin is responsible for ∼90% of the passive force during stretch, and at least 60% of the restoring force during shortening of sarcomeres (Granzier and Irving, 1995; Helmes et al., 1996).
Several studies have demonstrated that titin regulates the calcium sensitivity of myocytes. These studies examined the relationship between passive and calcium-activated tension using skinned myocardium or isolated cardiac myocytes that have been stretched (Cazorla et al., 1999, 2001; Fukuda et al., 2001; Muhle-Goll et al., 2001). In all studies, the degradation of titin with a weak trypsin solution decreased both passive and active tension generated at high SLs. These changes occur in concert with decreases in interfilament spacing, and can be mimicked by osmotic compression of myofilaments, leading to a model whereby radial force generated by titin on myofilament proteins increases the probability of cross-bridge formation and therefore active tension (Cazorla et al., 1999, 2001; Fukuda et al., 2001; Muhle-Goll et al., 2001).
In the this study, we tested the hypothesis that titin contributes to the length-dependent deactivation that occurs at short SLs, a process that initiates the systolic–diastolic transition in the intact heart. Although a relationship between titin-based restoring force and length-dependent deactivation might be expected at short SLs, to our knowledge this has not been examined, in part due to the technical limitations of measuring restoring force and calcium sensitivity during sarcomere compression. A system is described to measure the relationship between SL and cell width in dynamic contractions induced by calcium or rigor in isolated skinned unloaded myocytes. Furthermore, comparison of the velocity and acceleration of relaxation after readdition of ATP after rigor contractions supports the use of the velocity of sarcomere relengthening as a surrogate measure of restoring force. We show that degradation of titin reduces this measure of restoring force, and increases the calcium sensitivity of myofilaments, supporting the hypothesis that titin plays a role in the length-dependent deactivation during sarcomere compression. Changes in interfilament spacing estimated by following cell width show that interfilament spacing is of minor importance in the deactivation observed at short lengths.
MATERIALS AND METHODS
Myocyte Isolation and Permeabilization
The present study was performed in accordance with the guidelines of the Animal Care and Use Committee of Boston University School of Medicine and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Left ventricular (LV) myocytes were isolated and permeabilized according to a previously described protocol (Lim et al., 2001). Male Wistar rats, weighing between 200 and 250 g, were anesthetized with sodium pentobarbital (50 mg/kg i.p.) and heparinized (200 IU i.v.). The hearts were rapidly excised and arrested in ice-cold cardioplegic Krebs Bulbring (KB) solution (in mmol/L: 85 KOH, 30 KCl, 30 KH2PO4, 3 MgSO4, 0.5 EGTA, 10 HEPES, 50 L-glutamic acid, 20 taurine, 10 2,3-butanediaonemonoxime, and 10 glucose). The hearts were cannulated and retrogradely perfused via the aortic root with 1.2 mmol/L Ca2+ Tyrode solution (in mmol/L: 137 NaCl, 5.4 KCl, 1.8 CaCl2, 0.5 MgCl2, 10 HEPES, 10 glucose, pH 7.4), followed by Ca2+-free Tyrode solution until the hearts ceased beating. Hearts were then perfused with a digestion solution containing 0.08% collagenase A (Boehringer) and 0.02% protease XIV (Sigma-Aldrich). All perfusion solutions were equilibrated with 100% oxygen and maintained at 37°C throughout the isolation process. The digestion solution was eventually washed out with KB solution, and the hearts removed from the cannula. The LV was separated, minced, and gently agitated allowing the myocytes to be dispersed in KB solution. The myocyte suspension was subsequently filtered through a nylon mesh (Tetko, Inc.), resulting in a typical yield of >90% rod-shaped cells.
Myocytes were washed four times with calcium-free KB solution containing 10 mmol/L BDM, 40 μmol/L leupeptin, and 0.5 mmol/L PMSF, resulting in an ∼104-fold dilution of the remaining proteases and calcium. Myocytes were then permeabilized in relaxing solution (mmol/L: 10 EGTA, 5.9 MgAc, 5.9 Na2ATP, 10 creatine phosphate, 40 imidazole, 70 K+-proprionate, 5 NaN3 and, 1 DTT, 50 U/ml creatine phosphokinase, 0.5 PMSF, and 0.04 leupeptin, pH 7.0) containing 1% Triton (Sigma-Aldrich). Cells were kept on ice throughout the skinning procedure. After 50 min, the cells were centrifuged at 10 g and 4°C and resuspended in relaxing solution. This procedure was repeated twice to wash out any remaining Triton, and the skinned cells were kept on ice until further use.
Experimental Solutions
Calcium-free relaxing solution used during the experimental protocol contained in mmol/L: 10 EGTA, 5.9 MgAc, 5.9 Na2ATP, 10 creatine phosphate, 40 imidazole, 70 K+-proprionate, 5 NaN3 and, 1 DTT. Activating solutions ranging from pCa = 7.0 to 5.1 were made by appropriately adding Ca2+-proprionate to the relaxing solution according to the computer program of Fabiato (1988). Rigor solution for calcium-independent shortening was prepared by omitting the Na2ATP from the relaxing solution. For all solutions, the pH was adjusted to 7.0 at 20°C and ionic strength was adjusted with K+-proprionate to 0.2 mol/L. Prior to the start of the experimental protocol, 4 μmol/L leupeptin was added to prevent myofilament protein degradation. When measuring the SL-pCa curve, 50 U/ml creatine phosphokinase was added to prevent the decline in ATP supply. Creatine phosphokinase was not added to the rigor solutions.
Mechanical Setup
The experimental apparatus used to measure myocyte mechanics has been described previously in detail (Lim et al., 2001). Skinned myocytes were placed in a custom-made chamber, which was mounted on an inverted microscope (Nikon Diaphot epifluorescence microscope), and the myocytes were visualized using a Nikon 40 (1.3 numerical aperture) oil-immersion fluorescence objective lens. The mechanical setup consisted of a 3-barrel pipette, a fast-step solution switcher (SF-77B, Warner Instrument Corp.), and an 8-channel gravity perfusion system (Cell MicroControls). The 3-barrel pipette and fast-step switcher were attached to micromanipulators, which allowed for precise 3-way positioning of the pipette opening over the myocyte. The myocyte was exclusively perfused by a single barrel at a constant flow of 250 μl/min. The fast-step motor, allowed for rapid switching (in 20 ms) to an adjacent barrel, effectively changing the perfusion solution to the myocyte. By linking the 8-channel valve controlled perfusion system via manifolds to the fast-step solution switcher, and hence the 3-barrel pipette, it was possible to perfuse a single myocyte with up to 9 different solutions in one experiment. When comparing the effect of changing calcium levels, care was taken to equalize pCa solution levels in the syringes of the gravity perfusion system, and to use identical lengths of tubing to assure negligible differences in perfusion pressure.
SL Measurement
Cells were imaged using a variable frame rate (60–240 Hz) CCD-camera, and the images were digitized and displayed on a computer screen at a sampling speed of 240 Hz. Camera, acquisition, and analysis software for SL measurements, were obtained from IonOptix Corp. The algorithm for measuring sarcomere spacing has been described in detail (Lim et al., 2001). A user-defined region of interest (ROI) is selected within the myocyte (typically 50–60% of the total cell surface area). The video lines were averaged vertically, and the program performs a Fast-fourrier transform (FFT) on the averaged density trace and the peak of the resulting power spectra for the area of interest is obtained. The spectral peak represents the sarcomere spacing, which is ultimately converted to SL. With this algorithm, we were able to resolve changes in sarcomere spacing as small as 2–3 nm.
Cell-width Measurement
Measuring cell width gives an indication of the interfilament lattice spacing of the myocyte. Cell-width measurement was performed in real-time using off-axis edge detection techniques on the digitized cell image (IonOptix Corp.). The video images were also stored on video-tape for subsequent analysis of SL, as described earlier. To be able to synchronize the cell-width and sarcomere-length measurements, a TTL pulse associated with the perfusion switch was recorded in the data trace of cell width measurements and on the audio channel of the VCR used to tape the contraction. With the subsequent analysis of the videotape for sarcomere spacing, the recorded TTL pulse again was registered on the data trace. This allowed us to correlate the cell width with sarcomere spacing within one video frame, or 4 ms.
Experimental Protocol
SL-pCa curves
Skinned myocytes were plated on a coverslip affixed with vacuum grease (Dow Corning) to the myocyte chamber and superfused with relaxing solution (pCa 9.0). After 5 min of stabilization, the relaxing solution was switched to a calcium-containing solution, and SL was measured when the myocyte shortened to a new steady-state (∼5 s). The myocyte was subsequently switched to relaxing solution until recovery of slack SL was obtained. This process was repeated for increasing calcium-containing solutions up to pCa 5.1. At calcium concentrations greater than pCa 5.1 the unloaded myocyte would irreversibly hypercontract, precluding the measurement of maximum calcium activation and generation of Hill plots. The pCa-SL itself, however, was extremely reproducible, as shown in this (see Fig. 5 B) and a previous study, allowing us to directly compare nonnormalized data (Lim et al., 2001).
Figure 5.

The effect of trypsin digestion on titin, resting length, and restoring force. (A) SL tracing of a myocyte exposed briefly to ATP-free solution before and after trypsin digestion. The decrease in resting length during trypsin digestion is observed in 80% of the cells. (B) The relationship between dL·dt−1 max and SLPC before and after 3 and 9 min of trypsin digestion. With increasing trypsin digestion time, there was a progressive decrease in the slope of the dL·dt−1 max versus SL relation.
Novel method for determining restoring force in skinned myocytes
Measuring diastolic properties in skinned cardiac myocytes using currently available methods is technically demanding, requiring expensive equipment, and inherently difficult due to buckling of myocytes at short SLs. We developed a new assay which estimated restoring force by measuring the velocity of recoil in permeabilized myocytes after rigor-induced contractions. Using the fast-step switcher, the skinned myocyte was made to contract by perfusion with ATP-free rigor solution, which leads to calcium-independent activation. Switching back to ATP-containing relaxing solution led to a rapid recoil or relengthening of the myocyte back to slack length. To assess the restoring force, the speed (dL·dt−1) and acceleration (dL·dt−2) of the relengthening myocyte were calculated off-line. The data were smoothed using a low-pass Butterworth filter and the first derivative, dL·dt−1, was calculated (IonOptix). The resulting dataset was used to calculate the second derivative, Δ dL·dt−1/Δt, for every data point. Each myocyte was subjected to multiple contractions to varying SLs (as in Fig. 3) and the dL·dt−1 max and dL·dt−2 max were plotted against the SL at peak contraction (SLPC) for the various contractions to obtain a relation between SLPC and speed and acceleration of relengthening. We found that consistency in switching the perfusate back to ATP-containing solution, which determines the onset of relaxation, is important for reproducible measurements. Therefore, within an experiment the same two-barrel combination was always used to switch from ATP-free to ATP-containing solution, to avoid bias in barrel-position or switching.
Figure 3.

(A) SL tracing of a single myocyte showing a gradual increase in rigor induced contractions followed by relaxation, and (B) the corresponding first derivative. These figures show a strong correlation between the amplitude of sarcomere shortening (difference between resting and the shortest SL achieved during a contraction, defined as SLPC) and the maximum speed reached during relengthening. (C) The SLPC was graphed versus peak acceleration (dL·dt−2 max, closed circles) and peak velocity (dL·dt−1 max, open triangles) on the left and right hand side of the y-axis value, respectively, and shows a strong correlation between the maximum acceleration and the maximum velocity. (D) The relationship between the dL·dt−1 max and SLPC for 11 myocytes. For the range of SLs studied, the relation is approximately linear. Resting length and regression coefficient are normally distributed.
Trypsin perfusion of myocytes
Mild trypsin digestion was used to specifically degrade titin in skinned myocytes, as described (Higuchi 1992). In single myocytes, the previously described mechanical measurements were first determined in the absence of trypsin treatment. The perfusion solution was subsequently switched from leupeptin-containing relaxing solution to leupeptin-free relaxing solution containing trypsin (2.5 U/ml, Sigma-Aldrich). Myocytes were exposed to trypsin for 3, 6, or 9 min, after which the perfusion solution was switched back to trypsin-free, leupeptin-containing relaxing solution. The mechanical measurements were then repeated in trypsin-digested myocytes.
Trypsin digestion and gel electrophoresis
Mild trypsin digestion was used to specifically degrade titin in skinned myocytes using a modified protocol described previously (Granzier and Irving, 1995). Briefly, after the skinning and washing procedures, the cells were washed five times with leupeptin-free relaxing solution. Trypsin was made up in leupeptin-free relaxing solution at a concentration of 0.5 μg/ml (5 U/ml). Trypsin digestion of skinned myocytes was initiated by adding one volume of trypsin to one volume of cell suspension (final trypsin concentration 0.25 μg/ml or 2.5 U/ml) and cells were trypsinized at room temperature. Trypsin digestion was terminated at 3, 6, 9, and 30 min by adding ice-cold relaxing solution containing 0.25 mmol/L leupeptin and 0.5 mmol/L PMSF to the trypsinized cells. Cells were subsequently solubilized by adding one volume of cell suspension to nine volumes of solubilization buffer (50 mM Tris-Cl, 5% SDS, 10% glycerol, 80 mM DTT, pH 6.8 at 25°C) preheated to 90–95°C. The samples were solubilized for 60 s and allowed to cool down to room temperature. Equal amounts of protein (100 μg) were electrophoresed on a 12.5% acrylamide gel, or specifically analyzed for titin on a 2% polyacrylamide gel (Tatsumi and Hattori, 1995).
Protein preparation and mass spectroscopy
To identify proteins released into the media after trypsin digestion, an aliquot of the trypsinized samples was pelleted by centrifugation and 50 μl of supernatant were loaded on a 10% acrylamide gel, separated by SDS-PAGE, and silver stained. Specific bands of protein were excised from the gel, and processed for in gel digestion as described (Rosenfeld et al., 1992; Wilm and Mann, 1996). Briefly, the gel was cut into small but uniform pieces. The gel pieces were dehydrated using acetonitrile and then rehydrated in 100 mM ammonium bicarbonate. To protect peptides from oxidation, DTT (10 mM) was added in 100 mM ammonium bicarbonate and incubated at 56°C for 1 h. To protect the amino terminus, blocking samples were treated with 10 mM iodoacetamide in 100 mM ammonium bicarbonate. The gel pieces were washed with ammonium bicarbonate and dried by acetonitrile, twice. After completely dehydrating with acetonitrile gel pieces were digested in trypsin (12.5 ng/μl in 50 mM ammonium bicarbonate) at 37°C for 10–12 h. The peptides were extracted from the gel in 50% acetonitrile and 5% formic acid. The extract was concentrated in a speed vacuum and for matrix-assisted LASER desorption and ionization/time of flight/mass spectroscopy (MALDI-TOF/MS) analysis directly spotted on the plate using α-Cyanol as matrix. The samples were analyzed using Perceptive/Voyager MALDI-TOF/MS with reflection mode setting. The protein was identified by mass fingerprinting using the protein-prospector database.
Statistics
Values were reported as the mean ± standard deviation. All SL versus dL·dt max −1 and dL·dt−2 max data were analyzed using linear regression analysis. Groups were compared using a Student's t test, or two-way ANOVA as described. P < 0.05 was considered statistically significant.
RESULTS
Calcium-activated Versus Rigor Contractions: Effect on Cell Width
Fig. 1 A shows typical tracings of SL, and Fig. 1 B the corresponding cell width in a skinned myocyte exposed to increasing calcium solutions. Calcium-activated shortening resulted in an immediate increase in cell width. The percentage increase in cell width was highly variable, in part because a one-dimensional measurement does not take into account the differences in cross-sectional area or orientation of the cell. The SL–cell width relation, however, was very linear and reproducible within a cell. Linear regression showed an r2 > 0.95 (n = 3) looking at the endpoints of SL and cell width for contractions induced by pCa 6.2, 6.1, 6.0, and 5.9. When reperfused with relaxing solution, the sarcomere rapidly returned to slack length (Fig. 1 A), accompanied by an immediate reduction in cell width to resting levels (Fig. 1 B).
Figure 1.

SL and cell-width tracings of contracting, permeabilized myocytes. Calcium activation. (A) The top step line denotes perfusion switches from relaxing solution (pCa 9) to calcium activating solutions (step-up) back to relaxing solution (step-down). Upon exposure to calcium, sarcomere shortening is observed accompanied by an increase in cell width, except at pCa 7, where there is no shortening but an increase in cell width. (B) ATP-free rigor activation. (C) The top black bars and bottom white bars indicate switching between rigor perfusion and relaxing perfusion, respectively. Rigor activation initially leads to a narrowing of the cell, without sarcomere shortening. After on average 3.3% narrowing of the cell, sarcomere shortening commences. This sarcomere shortening is then accompanied by an increase in cell width (D). For clarity, individual SL and cell width tracings were enlarged for calcium activation (E) and rigor activation (F), respectively. Calcium-induced shortening followed by relaxation shows a direct positive correlation between SL and cell width. Rigor activation shows that a reduction in cell width (interfilament spacing) is necessary (a) before active sarcomere shortening, accompanied by an increase cell width, can take place (b). Relaxation following “rigor-induced” contractions shows a rapid phase of relengthening, which is associated with a rapid decrease in cell width (c), followed by a gradual return to resting length and width (d).
The relationship between cell width and SL was profoundly different for calcium activation versus rigor (ATP-free) activation (Fig. 1, C and D). This is clearly illustrated in Fig. 1, E (calcium activation) and F (rigor activation), where individual SL and cell-width tracings were enlarged for clarity. When switching from relaxing to rigor solution, a lag phase (anywhere from 1 to 30 s) was observed before the initiation of sarcomere shortening (Fig. 1 F, a). During this lag phase, the cell gets narrower (Fig. 1 F, a) by an average of 3.7 ± 3.2% (n = 4). Interestingly, virtually no sarcomere shortening was observed until this reduction in cell width was achieved. Once sarcomere shortening commenced there was an increase in cell width with a slope similar to that observed for calcium-activated contraction (Fig. 1 F, b). When switched from rigor to relaxing solution, a clear two-phase process, very different from the calcium-induced data, can be observed (compare Fig. 1, E and F). The fast phase of relengthening is associated with a rapid decrease in cell width to the level just before contraction (Fig. 1 F, c). This is followed by a slow phase with a gradual return of cell width to resting levels (Fig. 1 F, d).
Restoring Force Measurements
We examined the dynamics of relengthening of the sarcomere, by addition of ATP after a rigor-induced contraction, as a potential estimate of restoring force. Fig. 2 A shows a typical tracing of SL after exposure of the skinned myocytes to rigor and relaxing solutions. The myocyte in relaxing solution had a SL of ∼1.9 μm. As shown in Fig. 1, a rapid change to rigor solution initially has no effect on SL as ATP is being washed away. After a lag, the cell begins to contract as shown by a gradual reduction in SL. Upon rapidly switching back to relaxing solution (as indicated by arrow), ATP is reintroduced to the myofilaments. It is interesting that there is an ∼100 ms delay from the readdition of ATP until the initiation of relaxation, during which there is further contraction, with an increase in the velocity of shortening (Fig. 2 B). This is similar to what has been seen in rigor contractions in intact myocytes, where relaxation was initiated by photolytic activation of caged ATP (Niggli and Lederer, 1991), and likely represents the acceleration of contraction during the time period when [ATP] is close to that optimal for rigor (3–10 μM). Upon the initiation of relaxation, the myocyte rapidly recoils to its resting SL. Fig. 2, B and C, show the corresponding velocity and acceleration, first and second derivatives of the SL tracing, respectively.
Figure 2.
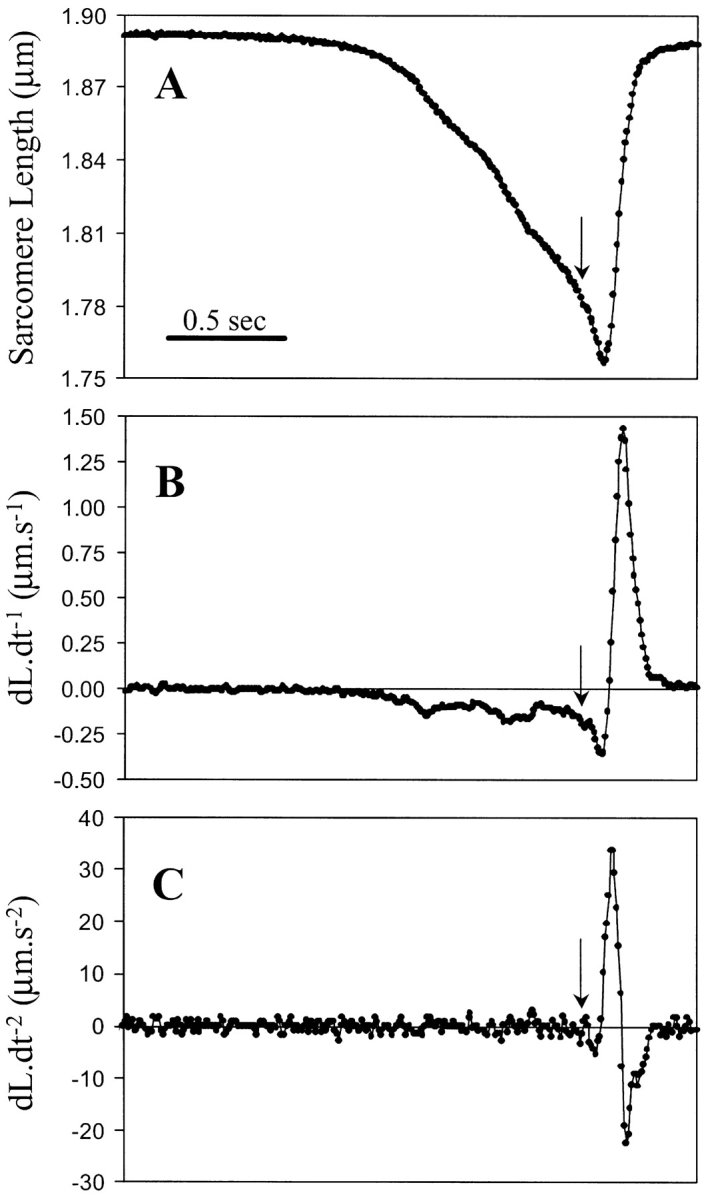
Changes in cell length, and the corresponding first and second derivative of the changes in cell length for a rigor induced contraction and relaxation. (A) At a given time, the perfusate is switched to ATP-free rigor solution, and while the cell is shortening the perfusate is switched back to ATP containing solution (arrow). After ∼150 ms the myocyte starts to relengthen, indicating completion of the perfusate switch and detachment of cross-bridges. (B) First derivative (dL·dt−1) of the tracing shown in A, indicating velocity of shortening and relengthening. (C) Second derivative (dL·dt−2) of the tracing shown in A, indicating acceleration and deceleration of the sarcomere. All tracings were acquired at a sampling rate of 120 Hz.
The acceleration of myocyte relengthening is a direct indicator of the restoring force acting on the myocyte. The acceleration is linearly related to the net force acting on the system and is described by Newton's second law: F = m × a (force equals the product of mass and acceleration of the sarcomere). The net force is the restoring force minus the viscous drag that the myocyte experiences during relengthening. As illustrated in Fig. 2, B and C, at maximum velocity, i.e., dL·dt−1 max, both forces are equal and dL·dt−2 max is zero. Where the acceleration becomes negative, the viscous drag is greater than the restoring force of the myocyte. The acceleration drops exponentially with increasing velocity due to increasing viscous drag. Thus, only at zero velocity, when viscous drag is also zero, does a direct relation exists between the acceleration and the restoring force. In fact, we found that the maximum acceleration (dL·dt−2 max) did occur when the velocity was zero (0.05 ± 0.08 μm/s, n = 7). Plotting dL·dt−2 max versus the SLPC gives an approximation of the relation between SL and restoring force during shortening.
Using dL·dt−2 max to approximate restoring force, however, has one significant disadvantage in that the second derivative is more susceptible to noise when using the raw data tracings (Fig. 2 C). We therefore examined whether the first derivative of the length tracing, i.e., dL·dt−1 max, could be used as an alternative for approximating restoring force. We compared the relationships between SLPC versus dL·dt-1 max and dL·dt−2 max. Fig. 3 A shows a typical tracing of SL and the corresponding dL·dt−1 (Fig. 3 B) in a skinned myocyte exposed to varying degrees of rigor contractions. Fig. 3 C plots dL·dt−2 max and dL·dt−1 max against SLPC, resulting in the relation dL·dt−1 max= m dL·dt−2 max, with an r2 of 0.999. Fig. 3 D shows individual tracings for a number of contractions of differing SLPC with the resulting dL·dt−1 max. As can be seen, the greater the SLPC, the higher the dL·dt−1 max during relengthening, and the relation of SLPC and dL·dt−1 max is approximately linear. For a given cell, the relationship between the SLPC and dL·dt−1 max is very reproducible and a linear regression fit with in an r2 = 0.98 ± 0.01 (n = 4). While the relationship between dL·dt−2 max and SLPC was also linear, the noise in the measure of dL·dt-2 max gave greater variability and resulted in r2 = 0.92 ± 0.04 (n = 4) (e.g., Fig. 3 C). Thus, dL·dt−1 max proved to be as reliable, and less noisy than dL·dt−2 max. We therefore used dL·dt−1 max as an indicator of restoring force in the remainder of the results section.
Trypsin Selectively Digests Titin
Proteolytic digestion of skinned myofibers with weak solutions of trypsin (2.5 U/ml) are reported to degrade titin (Higuchi, 1992). We examined the ability of this protocol to selectively degrade titin using mass spectroscopy protein “fingerprinting”. Myocyte total cell lysates run on SDS-PAGE before and after trypsin digestion are shown in Fig. 4 and show degradation of titin (Fig. 4 A), as has been reported without apparent degradation of other myofilament proteins (Fig. 4 B) (Higuchi, 1992; Astier et al., 1993; Granzier and Irving, 1995; Helmes et al., 1996; Cazorla et al., 1999, 2001; Fukuda et al., 2001). Media from the same conditions run on SDS-PAGE shows the appearance of many bands, consistent with the proteolytic release of peptides from the skinned cells (Fig. 4 C). Eight bands, as outlined, were isolated from the sample treated for 30 min with trypsin and processed, further digested, and analyzed by MALDI-TOF/MS. Comparison of the mass spectroscopy peptide masses with the protein-prospector database identified only titin in six of eight protein bands with very high score. Moreover, most of the peptide fragments identified represent fragments of titin (Fig. 4 C). Of note, the most prominent band appearing in the media with trypsin digestion (labeled as eight on the gel) is titin. The signals from the two remaining bands were too weak to be identified with confidence. As we did not evaluate every band appearing in the supernatant, we cannot completely exclude the possibility that other proteins are also degraded to some degree. However, these data show that there is fairly selective proteolytic degradation of titin with the trypsin protocol as used, consistent with prior investigators' conclusions (Higuchi, 1992; Astier et al., 1993; Granzier and Irving, 1995; Helmes et al., 1996; Cazorla et al., 1999, 2001; Fukuda et al., 2001).
Figure 4.

The effect of trypsin digestion of skinned myocytes on titin. Permeabilized myocytes were trypsinized according to the protocol in materials and methods, and equal amounts of protein (100 μg) were loaded on a 2% acrylamide/ 1.5% agarose gel (A) or a 12.5% acrylamide gel (B) and separated by electrophoresis. An aliquot of the trypsinized samples were pelleted by centrifugation and equal amounts of volume (50 μl) from the supernatant were loaded on a 10% acrylamide gel and separated by electrophoresis (C). From the supernatant samples, eight protein bands (labeled S1 to S8), which appeared at 3 min of trypsinization and increased in density over time, were excised from the gel and analyzed by mass spectroscopy. Of the eight proteins, six were identified as titin fragments (shown in bold), while the remaining two were not identifiable. The mass spec pattern of S5 is shown (D), peaks corresponding to titin fragments are indicated with an asterix.
Effect of Titin Degradation on the Restoring Force
To examine the role of titin in the restoring force measured with this protocol, skinned myocytes were exposed to trypsin (2.5 U/ml) for various durations. Fig. 5 A shows the restoring force measured before and after 12 min of trypsin digestion. After trypsin treatment, the restoring force was clearly decreased as shown by the prolonged sarcomere relengthening. Fig. 5 B demonstrates the relationship between SL and dL·dt−1 max. With increasing duration of trypsin exposure, dL·dt−1 max or the restoring force, gradually decreased over the entire SL range. This was a consistent finding. When we tested six myocytes treated for 6 min with trypsin, the slope of the regression line relating SLPC and dL·dt−1 max decreased by 59 ± 17% (P < 0.05). Interestingly, trypsin treatment usually also led to a gradual reduction in the resting SL in the myocytes (e.g., Fig. 5 A, or Fig. 6, A and B), though this was variable and did not correlate clearly with the changes in restoring force.
Figure 6.

The effect of trypsin digestion on calcium induced sarcomere shortening. (A) Shown in black are the SL tracings of a cardiac myocytes exposed to a range of pCa concentrations. The gray line shows the shortening for the same cell and the same calcium concentrations after a 6 min trypsin digestion. (B) The SLPC-pCa relation of skinned cardiac myocytes before and after a 6 min trypsin digestion (n = 6). Values are significantly different for every calcium concentration. (C) Images of a skinned cardiac myocyte after mild trypsin digestion at 0, 4, and 8 s after exposure to calcium-containing solution (pCa 5.4).
Effect of Titin Degradation on the SL-pCa Relationship
We examined the role of titin in the SL-pCa relationship by selectively degrading titin with trypsin. Fig. 6 A shows a representative SL tracing of a skinned myocyte exposed to increasing calcium concentrations, and superimposed, the same myocyte after 6 min of mild trypsin digestion (2.5 U/ml). The SL-pCa relationships for untreated and trypsin-treated cells (all treated 6 min) are shown in Fig. 6 B. The SL-pCa curve shifted downward following titin degradation. At physiological pCa (≥6) there was a statistically significant interaction between trypsin digestion and pCa (P = 0.025 by 2-way ANOVA), consistent with an increase in calcium sensitivity after titin degradation. This increased calcium sensitivity was only seen at low calcium concentrations. At higher calcium concentrations (pCa < 6), the difference in the SLPC between trypsin-treated and untreated cells remained constant. In many instances, mild trypsin digestion of skinned myocytes followed by exposure to high calcium led to a rapid and irreversible hypercontracture of the myocyte, as shown in Fig. 6 C.
We examined the effect of trypsin digestion on the SL–cell width relationship during Ca-induced contractions. Serial measures of SL and cell width were made in the same cell during contractions induced by change to pCa6.1 and pCa 5.5 before trypsin, and pCa6.1 after 6 min trypsin digestion. After trypsin digestion, pCa 6.1 resulted in a greater reduction in SL, with an identical change in cell width for any degree of sarcomere shortening as before trypsin. Thus, trypsin digestion did not affect the SL–cell width relation (Fig. 7 A).
Figure 7.

Relationship between restoring force and cell width and Ca+2 sensitivity. (A) The relationship between cell width and SL following activation at pCa 6.1 (before: black, and after: light gray, 6 min trypsin) and pCa5.5 (gray). Cell width was normalized to resting cell width. The predicted cell width, assuming filament spacing based deactivation, at pCa 6.1 after 6 min of trypsin digestion is shown (triangle). (B) The relation between dL·dt−1 max and SLPC and the SLPC-pCa relation from the same myocyte were obtained before and after 6 min of trypsin digestion. Values were then extrapolated from these two figures, to generate the dL·dt−1 max-pCa relationship for this cell (C).
We found that the velocity of relaxation after release from Ca-induced contraction was slower than for rigor-induced contractions, making the use of these velocities an inadequate measure of restoring force. Therefore, to examine the effect of trypsin digestion on the restoring force–pCa relationship in a single myocyte we derived the force-pCa curve in the same cell before and after trypsin digestion. To do this, we measured restoring force using rigor contractions, followed by generation of pCa curves before and after trypsin digestion (Fig. 7, B and C). For any given SL in the pCa curves, the restoring force is derived from the rigor contractions. The curves show that the relationship between pCa and restoring force is superimposable before and after tryspsin digestion. These results support the conclusion that the calcium sensitivity of the myofibrils is a function of titin-based restoring force, and not titin per se.
DISCUSSION
In this study, we used a novel method to examine the role of titin in the restoring force, calcium sensitivity, and length-dependent deactivation of isolated cardiac myocytes. Previous studies have shown that for cardiac myocytes, titin contributes to the Frank-Starling effect at long SLs during the transition from diastole to systole (Cazorla et al., 1999, 2001; Le Guennec et al., 2000; Fukuda et al., 2001). Here, we show that titin may also be responsible for the deactivation that takes place when the myocyte is shortened below resting length, i.e., during the transition from systole to diastole. A picture is starting to emerge where titin plays a critical role not only through the development of passive and restoring forces that move the sarcomere toward resting length, but also through regulation of calcium activation/deactivation during the cardiac cycle.
Novel Method to Assess Restoring Force in Skinned Cardiomyocytes
Numerous studies have looked at the passive mechanical properties of the sarcomere by stretching whole myocytes or myofibrils to SLs greater than resting length, which correlates to the late phase of diastole (Allen and Kentish, 1985; Gordon et al., 2000). Studies of the mechanical properties, in particular the restoring force, during the early phase of diastole when the sarcomere is shortened below resting length, are inherently problematic because myocytes are difficult to compress in vitro, as they will buckle (Fabiato and Fabiato, 1978; Nichols and Lederer, 1989; Helmes et al., 1996). In this study we used a novel approach to assess myocyte restoring force, by measuring the velocity of recoil in isolated unloaded skinned myocytes after ATP-free rigor-induced contractions. A rigor-induced contraction essentially loads the springs responsible for the restoring force, and the relaxation upon restitution of ATP in the system triggers the relengthening (Niggli and Lederer, 1991). We found that the velocity and acceleration of sarcomere relengthening are tightly correlated with the amplitude of the sarcomere shortening, as expected for a simple spring. There are limitations of both acceleration and velocity of relengthening as surrogates for restoring force. Maximum acceleration occurs when velocity is 0, and therefore eliminates viscous drag. However, relaxation is initiated by wash-in of ATP, myosin-actin cross-bridges may continue to form, limiting both maximal acceleration as well as velocity. While the acceleration seems a priori a more robust measure, it's accurate determination requires higher sampling frequencies than velocity, which likely accounts for the tighter correlation seen for the velocity/SLPC relationship (Fig. 3 C).
Previous investigations have concluded that in isolated myocytes, titin is responsible for the majority of the passive and restoring force generated at physiological SLs (SL 1.6–2.2 μm) (Granzier and Irving, 1995; Helmes et al., 1996, 1999). The method used has been to expose skinned preparations to low concentrations of trypsin, resulting in titin proteolysis without significant degradation of other myofilament proteins (Higuchi, 1992; Astier et al., 1993; Granzier and Irving, 1995; Helmes et al., 1996; Cazorla et al., 1999, 2001; Fukuda et al., 2001). We have expanded on these findings by using mass spectroscopy to identify proteolyzed peptides released into the media, and found that titin is selectively degraded with the trypsin protocol used, in agreement with previous reports. In this study, we used a quick switching system to control trypsin exposure. That the restoring force is so closely linked to the duration of trypsin digestion is consistent with the notion that the entropic spring properties of titin power the relengthening of the myocyte. Thus, we believe that this method can provide the fidelity to test the effect of treatments on the mechanical properties of the myofilaments in the early phase of diastole.
Calcium and Rigor Activation Differentially Affect Cell Width
The quick switching method allowed us to carefully compare the effects of calcium and rigor activation on cell-width dynamics. The simultaneous measurements of cell width and SL for both calcium and rigor-activated shortening led to some surprising findings. With calcium-induced shortening, there was a corresponding increase in cell width, except at very low [Ca2+], where we observed slight changes in cell width without corresponding sarcomere shortening. The mechanism for these changes in cell width at low [Ca2+] is not clear. We found that the relation between cell width and SL is independent of both calcium and titin, as illustrated in Fig. 7 A. Potential mechanisms for the change in cell width could be expansion of the myofilament lattice, with overlap/buckling of thin filaments and/or bending of thick filaments at very short SLs (Robinson and Winegrad, 1979; Krueger et al., 1980; Krueger and London, 1984; Trombitas et al., 1995).
Contrary to calcium activation, rigor caused early narrowing of the cell, which preceded sarcomere shortening. The cell narrowing is a slow process, taking multiple seconds. Rigor-induced sarcomere shortening started after the cell narrowed by ∼3–5%. A possible explanation of this phenomenon is that the initial formation of rigor bridges causes activation insufficient for contraction, but sufficient to pull the thin filaments toward the thick filament. This is consistent with other investigators' observations that during isometric rigor contractions there are measurable decreases in interfilament spacing (e.g., Millman and Irving, 1988). With time this can cause a visco-elastic compression of the Z-band and/or M-line, leading to a reduction in filament spacing, reducing the threshold for activation. Once sarcomere shortening commences, cell widening is observed, and happens presumably due to expansion of the filament lattice spacing as observed for calcium activation. Relengthening is associated with an equally rapid reduction in cell width to the narrowed state from just before shortening started. Like the initial cell narrowing, the late rewidening is slow, with a complete return in cell width not being achieved until after several minutes. During this slow phase of increasing cell width there is also a slow increase in SL. To what extent these dynamic changes in cell width during calcium- and rigor-induced contractions can be explained by changes in interfilament spacing warrants further investigation.
Titin Regulation of Active Contraction at the Systolic-Diastolic Transition
The steep relationship between force production and SL in cardiac muscle has been well-characterized, and points to the existence of a so-called length-sensor responsible for the length dependence of the calcium sensitivity of the cardiac myofilaments (for review see Allen and Kentish, 1985). Several recent studies have shown a role for titin in length-dependent regulation of contraction at long SLs (1.9–2.3 μm) (Cazorla et al., 2001; Fukuda et al., 2001). These studies showed that titin increases the calcium-sensitivity at long SLs. Our present findings complement those studies and complete the description of titin as a length-sensor by showing that at short SLs the restoring force generated by titin decreases calcium sensitivity. In this context, we are defining calcium sensitivity as the SL attained at a given pCa, and not the traditional pCa50. Thus, titin contributes to the Frank-Starling relationship at both long and short myofilament lengths. Collectively, these observations show that titin has a role beyond its function as a passive entropic spring, and provides critical regulation of active contraction throughout the cardiac cycle.
The length dependence of calcium sensitivity in cardiac muscle is believed to arise in large part from the changes in interfilament lattice spacing that occur with changes in SL (McDonald and Moss, 1995). As shown by Cazorla et al. (2001) the passive force generated by titin can regulate the interfilament spacing, and this may explain the effect of titin on length dependent activation at the diastolic-systolic transition. They showed that reducing interfilament spacing using osmotic compression increases the calcium sensitivity of actin and myosin to a similar extent as filament stretch. Similar conclusions were reached by Fukuda et al. (2001) who used measures of muscle diameter as a surrogate for interfilament spacing. Controversy, however, still persists regarding the role of interfilament spacing and myofilament calcium sensitivity as recent studies would suggest (Konhilas et al., 2002a,b). Beyond interfilament spacing, it has been suggested that the titin-derived force pulling on the myosin filament could affect the organization of the myosin heads and thus affect the force generation by regulating the population of myosin heads available for force generation (Granzier and Wang, 1993; Cazorla et al., 2001; Fukuda et al., 2001). All these studies concentrated on length dependent activation at long lengths, i.e., above resting length (1.9–1.95 μm).
Length dependence, however, is even stronger below resting length (Hibberd and Jewell, 1982; Allen and Kentish, 1985). A widely presented argument has been that the increase in filament spacing with sarcomere shortening effectively stops contraction by uncoupling the actin–myosin interactions. The contractile force would then be so reduced that the restoring force, often referred to as “internal resistance” is sufficient to stop shortening. Our experiments do not support this mechanism. We measured the relationship between cell width and sarcomere shortening in cells before and after trypsin digestion. A prediction of the interfilament spacing argument is that a decrease in myofilament length-dependent deactivation (as occurs with trypsin digestion) would change the cell-width/SL relationship, decreasing the relative change in cell width for any change in SL (Fig. 7 A). Counter to this prediction, however, the SL/CW relation is identical after trypsin digestion. The SL-pCa relation is completely independent of CW in these experiments.
This leaves the question by what mechanism the restoring force modulates the contractile state of the myofilaments. Based on ultrastructural and recent biochemical data, there are at least two possible pathways. Mechanically, titin-based restoring force may act directly on the tip of the myosin filament during sarcomere shortening, leading to buckling of the myosin filament as can be seen in high magnification micrographs (Trombitas et al., 1995). This conformational disorder may reduce the reactivity of the myosin heads, thereby leading to deactivation at short SLs. Due to the high cooperativity of myosin binding to actin, a small reduction in the population of available myosin heads will have a strong impact on the force generation of the contractile apparatus. Recent data suggest that the A-band fibronectin-like (FN) domains of titin that are bound to the myosin filaments play a regulatory role in the length dependence of active contraction (Muhle-Goll et al., 2001). It is believed that the FN domains of titin interact with and keep the myosin heads close to the thick filament stalk, thus preventing acto–myosin interaction. Clipping the tension bearing part of titin in the I-band by trypsin could possibly translate in a slight conformational change in these FN domains, leading to a more favorable configuration for acto–myosin binding. Alternatively, the clipped I-band titin fragment could interact with the myosin filament, and as such disrupt the proposed inhibitory effect of the FN domains. Clearly more experiments are needed to elucidate the mechanism by which titin and the restoring force regulate contractility.
Physiological and Pathophysiological Significance
A picture is now emerging that reveals how cardiac titin is not simply a template for sarcomere assembly during myocardial genesis, but is a bidirectional spring that actively regulates myofilament calcium sensitivity during the cardiac cycle. At the end of diastole, during sarcomere stretch, titin increases calcium sensitivity that presumably promotes the rapid onset of contraction once calcium is released. One might speculate that together with the passive force imparted by its stretched elastic domain, titin improves the efficiency of ventricular systole. Similarly, titin appears to contribute to the deactivation at short SLs. That is, at the end of systole, a minimal decrease in intracellular calcium concentration is sufficient to stop contraction and to start relengthening, fueled at least in part by the titin-based restoring force. The high speed relengthening of the myocyte will preclude reattachment of strong cross-bridges, and relengthening can take place unhampered by the intracellular calcium still present in the cell, ensuring rapid diastolic filling. Thus, we speculate that titin plays a central role in the hysteresis of the relationship between myocyte length and cytosolic calcium during contraction and relaxation, as has been observed in isolated single myocytes (Spurgeon et al., 1992). Titin degradation has been observed in the setting of heart failure (Morano et al., 1994), and perhaps the reduced levels of intact myofilament titin may contribute to the diastolic dysfunction as well as decreased myocardial efficiency that occurs in this and other clinical settings. More work in animal models of heart failure, as well as measurements in failing human myocardium, is necessary to fully address this possibility.
Acknowledgments
We thank William Lehman and Carl Apstein for helpful discussions.
Supported by grants 0150549N (D.B. Sawyer) from the American Heart Association, National Affiliate and HL03878 (D.B. Sawyer) from the National Heart Lung and Blood Institute. C.C. Lim is supported by National Institutes of Health postdoctoral training grant T32 HL07224-27.
M.H. Helmes and C.C. Lim contributed equally to this work.
Footnotes
Abbreviation used in this paper: SL, sarcomere length.
References
- Allen, D.G., and J.C. Kentish. 1985. The cellular basis of the length-tension relation in cardiac muscle. J. Mol. Cell. Cardiol. 17:821–840. [DOI] [PubMed] [Google Scholar]
- Astier, C., J.P. Labbe, C. Roustan, and Y. Benyamin. 1993. Effects of different enzymic treatments on the release of titin fragments from rabbit skeletal myofibrils. Purification of an 800 kDa titin polypeptide. Biochem. J. 290:731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazorla, O., G. Vassort, D. Garnier, and J.Y. Le Guennec. 1999. Length modulation of active force in rat cardiac myocytes: is titin the sensor? J. Mol. Cell. Cardiol. 31:1215–1227. [DOI] [PubMed] [Google Scholar]
- Cazorla, O., Y. Wu, T.C. Irving, and H. Granzier. 2001. Titin-based modulation of calcium sensitivity of active tension in mouse skinned cardiac myocytes. Circ. Res. 88:1028–1035. [DOI] [PubMed] [Google Scholar]
- Fabiato, A. 1988. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 157:378–417. [DOI] [PubMed] [Google Scholar]
- Fabiato, A., and F. Fabiato. 1975. Dependence of the contractile activation of skinned cardiac cells on the sarcomere length. Nature. 256:54–56. [DOI] [PubMed] [Google Scholar]
- Fabiato, A., and F. Fabiato. 1978. Myofilament-generated tension oscillations during partial calcium activation and activation dependence of the sarcomere length-tension relation of skinned cardiac cells. J. Gen. Physiol. 72:667–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda, N., D. Sasaki, S. Ishiwata, and S. Kurihara. 2001. Length dependence of tension generation in rat skinned cardiac muscle: role of titin in the Frank-Starling mechanism of the heart. Circulation. 104:1639–1645. [DOI] [PubMed] [Google Scholar]
- Goldstein, M.A., J.P. Schroeter, and L.H. Michael. 1991. Role of the Z band in the mechanical properties of the heart. FASEB J. 5:2167–2174. [DOI] [PubMed] [Google Scholar]
- Gordon, A.M., E. Homsher, and M. Regnier. 2000. Regulation of contraction in striated muscle. Physiol. Rev. 80:853–924. [DOI] [PubMed] [Google Scholar]
- Granzier, H., and S. Labeit. 2002. Cardiac titin: an adjustable multi-functional spring. J. Physiol. 541:335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzier, H.L., and T.C. Irving. 1995. Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys. J. 68:1027–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzier, H.L., and K. Wang. 1993. Interplay between passive tension and strong and weak binding cross- bridges in insect indirect flight muscle. A functional dissection by gelsolin-mediated thin filament removal. J. Gen. Physiol. 101:235–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio, C.C., H. Granzier, H. Sorimachi, and S. Labeit. 1999. Muscle assembly: a titanic achievement? Curr. Opin. Cell Biol. 11:18–25. [DOI] [PubMed] [Google Scholar]
- Helmes, M., K. Trombitas, T. Centner, M. Kellermayer, S. Labeit, W.A. Linke, and H. Granzier. 1999. Mechanically driven contour-length adjustment in rat cardiac titin's unique N2B sequence: titin is an adjustable spring. Circ. Res. 84:1339–1352. [DOI] [PubMed] [Google Scholar]
- Helmes, M., K. Trombitas, and H. Granzier. 1996. Titin develops restoring force in rat cardiac myocytes. Circ. Res. 79:619–626. [DOI] [PubMed] [Google Scholar]
- Hibberd, M.G., and B.R. Jewell. 1982. Calcium- and length-dependent force production in rat ventricular muscle. J. Physiol. 329:527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi, H. 1992. Changes in contractile properties with selective digestion of connectin (titin) in skinned fibers of frog skeletal muscle. J. Biochem. (Tokyo). 111:291–295. [DOI] [PubMed] [Google Scholar]
- Kellermayer, M.S., S.B. Smith, H.L. Granzier, and C. Bustamante. 1997. Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science. 276:1112–1116. [DOI] [PubMed] [Google Scholar]
- Konhilas, J.P., T.C. Irving, and P.P. de Tombe. 2002. a. Myofilament calcium sensitivity in skinned rat cardiac trabeculae: role of interfilament spacing. Circ. Res. 90:59–65. [DOI] [PubMed] [Google Scholar]
- Konhilas, J.P., T.C. Irving, and P.P. de Tombe. 2002. b. Length-dependent activation in three striated muscle types of the rat. J. Physiol. 544:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger, J.W., D. Forletti, and B.A. Wittenberg. 1980. Uniform sarcomere shortening behavior in isolated cardiac muscle cells. J. Gen. Physiol. 76:587–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger, J.W., and B. London. 1984. Contraction bands: differences between physiologically vs. maximally activated single heart muscle cells. Adv. Exp. Med. Biol. 170:119–134. [DOI] [PubMed] [Google Scholar]
- Labeit, S., B. Kolmerer, and W.A. Linke. 1997. The giant protein titin. Emerging roles in physiology and pathophysiology. Circ. Res. 80:290–294. [DOI] [PubMed] [Google Scholar]
- Le Guennec, J.Y., O. Cazorla, A. Lacampagne, and G. Vassort. 2000. Is titin the length sensor in cardiac muscle? Physiological and physiopathological perspectives. Adv. Exp. Med. Biol. 481:337–348. [DOI] [PubMed] [Google Scholar]
- Lim, C.C., M.H. Helmes, D.B. Sawyer, M. Jain, and R. Liao. 2001. High-throughput assessment of calcium sensitivity in skinned cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 281:H969–H974. [DOI] [PubMed] [Google Scholar]
- Maruyama, K. 1997. Connectin/titin, giant elastic protein of muscle. FASEB J. 11:341–345. [DOI] [PubMed] [Google Scholar]
- McDonald, K.S., and R.L. Moss. 1995. Osmotic compression of single cardiac myocytes eliminates the reduction in Ca2+ sensitivity of tension at short sarcomere length. Circ. Res. 77:199–205. [DOI] [PubMed] [Google Scholar]
- Millman, B.M., and T.C. Irving. 1988. Filament lattice spacing of frog striated muscle. Radial forces, lattice stability, and filament compression in the A-band of relaxed and rigor muscle. Biophys. J. 54:437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morano, I., K. Hadicke, S. Grom, A. Koch, R.H. Schwinger, M. Bohm, S. Bartel, E. Erdmann, and E.G. Krause. 1994. Titin, myosin light chains and C-protein in the developing and failing human heart. J. Mol. Cell. Cardiol. 26:361–368. [DOI] [PubMed] [Google Scholar]
- Muhle-Goll, C., M. Habeck, O. Cazorla, M. Nilges, S. Labeit, and H. Granzier. 2001. Structural and functional studies of titin's fn3 modules reveal conserved surface patterns and binding to myosin S1–a possible role in the Frank-Starling mechanism of the heart. J. Mol. Biol. 313:431–447. [DOI] [PubMed] [Google Scholar]
- Nichols, C., and W. Lederer. 1989. The role of ATP in energy-deprivation contractures in unloaded rat venticular myocytes. Can. J. Physiol. Pharmacol. 68:183–194. [DOI] [PubMed] [Google Scholar]
- Niggli, E., and W.J. Lederer. 1991. Restoring forces in cardiac myocytes. Insight from relaxations induced by photolysis of caged ATP. Biophys. J. 59:1123–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rief, M., M. Gautel, F. Oesterhelt, J.M. Fernandez, and H.E. Gaub. 1997. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 276:1109–1112. [DOI] [PubMed] [Google Scholar]
- Robinson, T.F., and S. Winegrad. 1979. The measurement and dynamic implications of thin filament lengths in heart muscle. J. Physiol. 286:607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld, J., J. Capdevielle, J.C. Guillemot, and P. Ferrara. 1992. In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal. Biochem. 203:173–179. [DOI] [PubMed] [Google Scholar]
- Spurgeon, H.A., W.H. duBell, M.D. Stern, S.J. Sollott, B.D. Ziman, H.S. Silverman, M.C. Capogrossi, A. Talo, and E.G. Lakatta. 1992. Cytosolic calcium and myofilaments in single rat cardiac myocytes achieve a dynamic equilibrium during twitch relaxation. J. Physiol. 447:83–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumi, R., and A. Hattori. 1995. Detection of giant myofibrillar proteins connectin and nebulin by electrophoresis in 2% polyacrylamide slab gels strengthened with agarose. Anal. Biochem. 224:28–31. [DOI] [PubMed] [Google Scholar]
- Trinick, J. 1994. Titin and nebulin: protein rulers in muscle? Trends Biochem. Sci. 19:405–409. [DOI] [PubMed] [Google Scholar]
- Trombitas, K., J.P. Jin, and H. Granzier. 1995. The mechanically active domain of titin in cardiac muscle. Circ. Res. 77:856–861. [DOI] [PubMed] [Google Scholar]
- Tskhovrebova, L., J. Trinick, J.A. Sleep, and R.M. Simmons. 1997. Elasticity and unfolding of single molecules of the giant muscle protein titin. Nature. 387:308–312. [DOI] [PubMed] [Google Scholar]
- Wang, K., R. McCarter, J. Wright, J. Beverly, and R. Ramirez-Mitchell. 1993. Viscoelasticity of the sarcomere matrix of skeletal muscles. The titin- myosin composite filament is a dual-stage molecular spring. Biophys. J. 64:1161–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, K., P. Nair, D. Labeit, M.S. Kellermayer, M. Greaser, S. Labeit, and H. Granzier. 2002. Molecular mechanics of cardiac titin's PEVK and N2B spring elements. J. Biol. Chem. 277:11549–11558. [DOI] [PubMed] [Google Scholar]
- Wilm, M., and M. Mann. 1996. Analytical properties of the nanoelectrospray ion source. Anal. Chem. 68:1–8. [DOI] [PubMed] [Google Scholar]