

Abstract
Measurements of membrane capacitance were applied to dissect the cellular mechanisms underlying PKA-dependent and -independent stimulation of insulin secretion by cyclic AMP. Whereas the PKA-independent (Rp-cAMPS–insensitive) component correlated with a rapid increase in membrane capacitance of ∼80 fF that plateaued within ∼200 ms, the PKA-dependent component became prominent during depolarizations >450 ms. The PKA-dependent and -independent components of cAMP-stimulated exocytosis differed with regard to cAMP concentration dependence; the K d values were 6 and 29 μM for the PKA-dependent and -independent mechanisms, respectively. The ability of cAMP to elicit exocytosis independently of PKA activation was mimicked by the selective cAMP-GEFII agonist 8CPT-2Me-cAMP. Moreover, treatment of B-cells with antisense oligodeoxynucleotides against cAMP-GEFII resulted in partial (50%) suppression of PKA-independent exocytosis. Surprisingly, B-cells in islets isolated from SUR1-deficient mice (SUR1−/− mice) lacked the PKA-independent component of exocytosis. Measurements of insulin release in response to GLP-1 stimulation in isolated islets from SUR1−/− mice confirmed the complete loss of the PKA-independent component. This was not attributable to a reduced capacity of GLP-1 to elevate intracellular cAMP but instead associated with the inability of cAMP to stimulate influx of Cl− into the granules, a step important for granule priming. We conclude that the role of SUR1 in the B cell extends beyond being a subunit of the plasma membrane KATP-channel and that it also plays an unexpected but important role in the cAMP-dependent regulation of Ca2+-induced exocytosis.
Keywords: insulin, Ca2+, cAMP, cAMP-GEFII, SUR1
INTRODUCTION
Insulin secretion from the pancreatic B-cells is controlled by metabolic fuels, neurotransmitters released from intraislet nerve endings, paracrine mechanisms, and circulating hormones (Ashcroft and Rorsman, 1989; Satin and Kinard, 1998). Several modulators of insulin secretion act by activation of protein kinases and phosphatases (Nesher et al., 2002). For example, the capacity of glucagon, GLP-1, and GIP to enhance insulin secretion involves stimulation of adenylate cyclase with resultant increase in the intracellular cAMP concentration (Gromada et al., 1998). Cyclic AMP initiates several processes that culminate in enhancement of insulin secretion. These include stimulation of electrical activity by a reduction of KATP-channel activity (Holz et al., 1993), increase of the L-type Ca2+-current (Ämmälä et al., 1993; Kanno et al., 1998), activation of depolarizing cation-permeable ion channels (Leech and Habener, 1997), mobilization of Ca2+ from intracellular stores (Gromada et al., 1995; Kang et al., 2001), and an effect on the release process itself (Jones et al., 1988). Many of these effects are likely to be mediated by activation of PKA. However, there is increasing evidence that PKA-independent mechanisms are also significant. For example, the ability of cAMP to mobilize Ca2+ from intracellular stores is mediated by the cAMP receptor protein cAMP-GEFII (Kang et al., 2001) and we and others have demonstrated previously that the effects of cAMP on secretion involves both PKA-dependent and -independent mechanisms (Renström et al., 1997; Ozaki et al., 2000). The latter pathway appears to be mediated by cAMP-GEFII and this protein has been suggested to play a prominent role in incretin-induced insulin secretion (Nakazaki et al., 2002). Exactly how cAMP, via binding to cAMP-GEFII, promotes exocytosis remains unestablished but the effect may involve Rim2 (Ozaki et al., 2000). RIM proteins have recently been shown to promote priming of granules for release in neurons (Wang et al., 1997; Schoch et al., 2002) and it is tempting to speculate that they fulfil a similar function in the B cell.
Here we have studied the PKA-dependent and -independent effects of cAMP on insulin secretion in B-cells taken from wild-type and SUR1 knockout mice. We demonstrate that the PKA-independent pathway of cAMP accounts for a rapid component of release that appears particularly significant for incretin-stimulated insulin secretion. We finally propose a working model for the regulation of exocytosis in the B cell that incorporates Rim2, cAMP-GEFII, and SUR1 in granule priming and release.
MATERIALS AND METHODS
Animals, Isolation of Islets, and Preparation and Culture of B-cells
Most experiments were performed on B-cells isolated from NMRI-mice (Bomholtgaard). The mice lacking the SUR1 receptor (SUR1−/− mice; 6–12 mo of age) used in this study were obtained from Dr. M. Magnuson (Shiota et al., 2002) and bred at the Karolinska Institute in Stockholm. The mice were killed by cervical dislocation, the pancreas was quickly removed, and islets were isolated by collagenase digestion. The procedures for sacrificing the animals were approved by the ethical committee at Lund University and by local authorities in Hamburg.
For electrophysiology, the islets were dissociated into single cells using a Ca2+-free solution. The resulting cell suspension was plated on Corning petri dishes or glass coverslips (for confocal microscopy) and maintained in tissue culture for 6–30 h in RPMI 1640 medium containing 10% (vol/vol) fetal calf serum, 100 μg/ml streptomycin, and 100 i.u./ml penicillin.
The influence of the cAMP-binding protein cAMP-GEFII, which mediates cAMP-dependent but PKA-independent exocytosis (Ozaki et al., 2000), was tested by culturing intact islets from NMRI-mice in the presence of 4 μM of antisense phosphorothioate–substituted oligodeoxynucleotides (ODNs)* against mouse cAMP-GEFII (5′-CAACGGCCTTTTATCC-3′) or control ODNs (5′-ACCTACGTGACTACGT-3′) (provided by S. Seino, Chiba University, Japan) for 96 h. The control ODNs were made such that chemical properties were similar to that of antisense ODN (Ozaki et al., 2000). These islets were then dissociated into single cells and cultured as described above. Most cells prepared from islets cultured for 4 d had small exocytotic responses. The analysis of the effects of treatment with antisense cAMP-GEFII or the control ODN was therefore confined to the five cells in each group displaying the largest capacitance increases (the total number of cells analyzed was 114). Because of the difficulty maintaining secretion in primary B-cells, some experiments (Fig. 4, C–E) were instead performed using mouse insulinoma MIN6 cells (Miyazaki et al., 1990). These were either treated with the control and antisense ODNs specified above for 96 h or were subjected to transient transfection using mutant cAMP-GEF-II expression vector pSRα-cAMP-GEFII (G114E/G422D; provided by S. Seino) reconstituted in effectene transfection reagent (QIAGEN). The cells were cotransfected with eGFP (CLONTECH Laboratories, Inc.) To facilitate the identification of the transfected cells. Electrophysiological experiments were performed ≥36 h after transfection.
Figure 4.

Antisense cAMP-GEFII abolishes cAMP- and sulfonylurea-induced secretion. (A and B) Increase in cell capacitance (ΔCm, lower trace) elicited by a 500-ms depolarization from −70 to 0 mV. The experiments were conducted on B-cells prepared from islet pretreated for 72 h with 4 μM control (A) or antisense cAMP-EGFII ODNs (B). Experiments were conducted in the presence of 10 μM Rp-cAMPs alone (left) and in the simultaneous presence of 2–10 μM forskolin and 10 μM Rp-cAMPS (right). (C) Capacitance increases recorded from MIN6-cells during standard whole-cell recordings when the cells were infused with a buffer containing ∼1.5 μM free Ca2+ under control conditions (black trace) and in presence of 0.1 mM cAMP (red trace) or 0.1 mM cAMP and 0.1 mM tolbutamide (blue trace). The cells had been cultured for 96 h with control oligodeoxynucleotides. (D) As in C but cells had been cultured for 96 h with antisense cAMP-GEFII. (E) As in C but the cells had been transfected with the cAMP-GEFII mutant G114E/G422D ≥36 h before the experiments. (F) Distribution of insulin (red) and cAMP-GEFII (EPAC) immunoreactivity (green) in MIN-6 and primary B-cells under control conditions and when the cells had been cultured with anti-sense cAMP-GEFII. Bar, 10 μm.
Solutions
The extracellular solution consisted of (in mM) 118 NaCl, 20 tetraethylammonium-chloride (TEA-Cl), 5.6 KCl, 1.2 MgCl2, 2.6 CaCl2, 5 D-glucose, and 5 HEPES (pH 7.4 with NaOH). In the perforated patch experiments, forskolin (2–10 μM; from Sigma-Aldrich) or GLP-1 (10 nM; Peninsula Laboratories) was added to increase intracellular cAMP levels and enhance exocytosis (Ämmälä et al., 1993). PKA was inhibited by treating the cells with the membrane-permeant cAMP antagonists Rp-cAMPS or Rp-8-Br-cAMPS (Biologic) for ≥5 min. The pipette solution used for the perforated patch measurements contained (in mM) 76 Cs2SO4, 10 NaCl, 10 KCl, 1 MgCl2, and 5 HEPES (pH 7.35 with CsOH). The pipette solution in the standard whole-cell measurements in which the cells were stimulated with voltage-clamp depolarizations consisted of (in mM) 125 Cs-glutamate, 10 CsCl, 10 NaCl, 1 MgCl2, 0.05 EGTA, 3 Mg-ATP, 5 HEPES (pH 7.1 using CsOH; intracellular solution I). Cyclic AMP or the selective cAMP-GEFII-agonist 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8CPT-2Me-cAMP; Enserink et al., 2002) was added to the latter solution at the indicated concentrations. To block PKA, Rp-cAMPS was included at a concentration of 0.5 mM. When exocytosis was stimulated by infusion of a buffer with elevated [Ca2+]i (Figs. 4, C–E, 9, and 10), the pipette solution contained (in mM) 125 K-glutamate, 10 KCl, 10 NaCl, 1 MgCl2, 3 Mg-ATP, 5 HEPES (pH 7.15 with KOH), 5 or 9 CaCl2, and 10 EGTA (intracellular solution II). The free Ca2+ concentrations were estimated to be 0.17 and 1.5 μM using the binding constants of Martell and Smith (1974).
Electrophysiology
Patch electrodes were made from borosilicate glass capillaries coated with Sylgard close to the tips and fire-polished. The pipette resistance ranged between 2 and 4 MΩ when the pipettes were filled with the intracellular solutions specified above. The zero-current potential of the pipette was adjusted in the bath before giga-seal formation. Either the perforated patch whole-cell technique (Figs. 1 and 4, A and B), in which the cells remain metabolically intact, or the standard whole-cell configuration (Figs. 2, 3, 4, C–E, 5, 7, 9, and 10), which allows intracellular application of reagents simply by including them in the pipette solution, was used. All measurements were conducted using EPC7 or EPC9 patch-clamp amplifiers and the Pulse software (version 8.30 or later; Heka Elektronik).
Figure 1.

GLP-1 potentiates exocytosis in B-cells. Increases in cell capacitance (ΔCm, bottom trace) elicited by progressively longer (5, 10, 15, 30, 50, 100, 150, 250, 350, 450, and 850 ms) depolarizations from −70 to 0 mV (V, top trace) under (A) control conditions and (B) 4 min after the inclusion of 10 nM GLP-1 in the extracellular medium. The interval between two successive depolarizations was 15 s for pulses ≤50 ms and 30 s for longer pulses. For clarity, only responses to the 30-, 100-, 250-, and 450-ms depolarizations are shown. The recording was performed using the perforated patch configuration. (C) Relationship between pulse duration (t) and increase in cell capacitance (ΔCm) under control conditions and in the presence of 10 nM GLP-1 as indicated. The curves were derived by fitting Eq. 3 to the data points for depolarizations ≤350 ms. The dotted lines represent linear fits to the values measured in response to the two longest depolarizations. Data are mean values ± SEM of seven paired experiments. *P < 0.05 and **P < 0.01.
Figure 2.
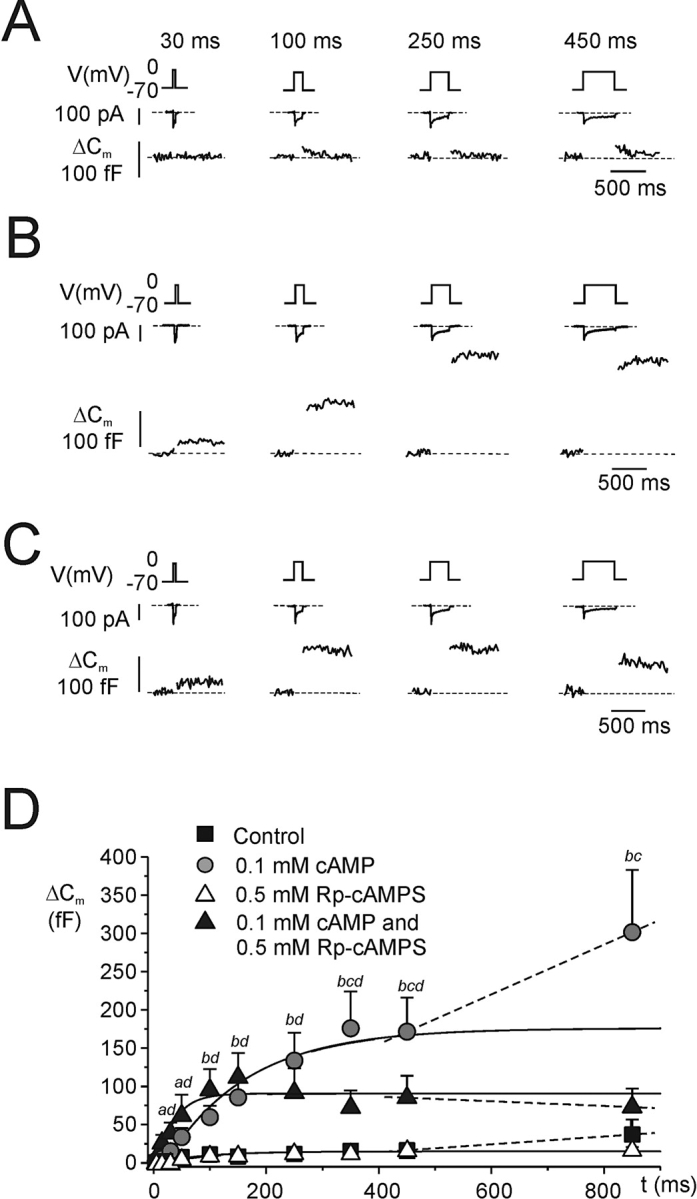
Kinetics of cAMP-stimulated exocytosis. (A) As in Fig. 1 but recordings were performed using the standard whole-cell configuration and the cells were dialyzed with standard cAMP-free intracellular solution. The recording commenced ≥1 min after establishment of the whole-cell configuration to allow complete equilibration of the pipette solution with the cytoplasm. (B) As in A but cAMP was included at a concentration of 0.1 mM. (C) As in B but 0.5 mM of the PKA-inhibitor Rp-cAMPS was added to the pipette solution. (D) Relationship between pulse duration (t) and increase in cell capacitance (ΔCm) under control conditions, in the presence of 0.1 mM cAMP, in the simultaneous presence of 0.1 mM cAMP and 0.5 mM Rp-cAMPS and in the presence of 0.5 mM Rp-cAMPS alone as indicated. Data are mean values ± SEM of 5–10 experiments. aP < 0.05 and bP < 0.01 for comparison between cAMP and control. cP < 0.05 for comparison of cAMP alone with the combination of cAMP and Rp-cAMPS; dP < 0.05 for comparison of control with the combination of 0.1 mM cAMP and 0.5 mM Rp-cAMPS. Note that intracellular cAMP enhances both an early and a late component of exocytosis and that Rp-cAMPS principally affects the late component.
Figure 3.

Concentration dependence of cAMP-induced stimulation of secretion. (A) As in Fig. 2 but the cells were infused with intracellular medium containing 0 (white square), 1 (light gray triangle), 10 (light gray circle), 50 (gray diamond), 100 (dark gray square), and 500 μM cAMP (black triangle). The curves were obtained by approximating Eq. 3 to the data points. Data are mean values ± SEM of 5–9 experiments. (B) Normalized size of cAMP-induced increase in IRP ([ΔCm − ΔCm,basal]/[ΔCm,500 − ΔCm,basal], where ΔCm,basal, ΔCm, and ΔCm,500 are the calculated values of IRP in the absence of cAMP at a given cAMP concentration and in the presence of 500 μM cAMP, respectively (left axis), and the absolute values of IRP (right axis) displayed against the intracellular [cAMP]. The curve shown was derived by approximating Eq. 4 to the normalized mean values. The open circles represents the amplitude of IRP observed in the simultaneous presence of 0.1 mM cAMP and 0.5 mM Rp-cAMPS. (C) Exocytotic responses elicited by trains consisting of ten 500-ms depolarization from −70 to 0 mV in the presence of 0, 1, 10, 50, 100, and 500 μM cAMP. (D) As in B but the summed increase in cell capacitance evoked by the last nine pulses ( ) of the train was analyzed. The open circle represents
) of the train was analyzed. The open circle represents  in the simultaneous presence of 0.1 mM cAMP and 0.5 mM Rp-cAMPS. Data are mean values ± SEM of 4–6 experiments.
in the simultaneous presence of 0.1 mM cAMP and 0.5 mM Rp-cAMPS. Data are mean values ± SEM of 4–6 experiments.
Exocytosis was detected as changes in cell capacitance, which was estimated by the Lindau-Neher technique (Gillis, 1995), implementing the “Sine + DC”-feature of the lock-in module (40 mV peak-to-peak and a frequency of 500–1,000 Hz). Secretion was elicited by single voltage-clamp depolarizations from −70 to 0 mV of variable duration or trains of 10 500-ms depolarizations (1-Hz stimulation frequency) to 0 mV. During the experiments, the cells were continuously superfused with the extracellular medium at a rate of <2 ml/min and the volume of the chamber reduced to 0.5 ml by a plastic insert. All electrophysiological measurements were performed at ≈33°C.
Immunocytochemistry
Primary B-cells and MIN-6 cells were fixed in 3% paraformaldehyde in K-PIPES (Sigma-Aldrich) and permeabilized with 0.1% Triton X-100. After blocking of nonspecific sites with 5% normal donkey serum, the cells were incubated for 2 h in the presence of goat-raised anti–cAMP-GEFII/anti-EPACII (C-19, 1:50, Santa Cruz Biotechnology, Inc.) and a guinea-pig polyclonal antiinsulin antibody (B 65–1, 1:500, Euro-diagnostica). Anti–goat Cy3 and anti–guineapig Cy5 (Jackson ImmunoResearch Laboratories) were then used to label the detected sites. Images were acquired using the 543-nm (Cy3) and 633-nm (Cy5) lines of a ZEISS 510 LSM confocal microscope. Emitted light was visualized using a 63×/1.3 NA oil objective and a bandpass filter (565–615 nm; Cy3) or a >650-nm filter (Cy5). The samples were scanned sequentially with the appropriate settings to minimize crosstalk.
Insulin Measurements
Islets were preincubated in Earle's balanced Salt Solution (EBSS; Invitrogen) containing 0.1% BSA and 2.8 mM glucose for 30 min at 37°C. Batches of five islets were subsequently incubated for 90 min at 37°C in EBSS supplemented with 0.1% BSA, 2.8 or 20 mM glucose and the respective test compound added. The incubation was terminated by placing the islets on ice. The supernatant was removed and stored at −20°C pending the insulin measurements (ELISA). Forskolin was included at a concentration of 10 μM in the experiments involving intact islets (rather than the 2 μM used for the single-cell studies) to overcome any diffusion barriers within the intact islet to ensure that exocytosis in B-cells in the islet core is also stimulated.
Measurements of Islet cAMP
Groups of 10 islets were preincubated at 37°C in Krebs-Ringer bicarbonate buffer containing 115 mM NaCl, 4.7 mM KCl, 2.6 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 20 mM NaHCO3, 16 mM HEPES, 2 mg/ml BSA, and 1 mM IBMX for 1 h and incubated for another 15 min in same buffer with or without GLP-1 and forskolin. The reaction was stopped by addition of 50 mM HCl and neutralized with NaOH. Concentration of cAMP was determined by a cAMP [125I] scintillation proximity assay (Amersham Biosciences).
Measurements of Granular Cl− Uptake
Changes in insulin granular Cl−-fluxes or pH (Figs. 9, C–E, and 10) were monitored by confocal imaging of LysoSensorTMGreen DND-189® (LSG; Molecular Probes) fluorescence as described previously (Barg et al., 2001a; Renström et al., 2002b). LSG (1 μM) was added during the last 30 min of cell culture and was included in extracellular buffer that continuously superfused the cells. LSG fluorescence was excited using the 488-nm line of a ZEISS 510 confocal microscope. The emitted light was collected with a 63×/1.3 NA oil objective and a >505-nm filter. Laser scanning was performed with low pixel resolution (128 × 128) and with 6-s intervals to minimize photobleaching. After giga-seal formation, it was ascertained that LSG fluorescence was stable for >30 s before the standard whole-cell configuration was established. The holding potential was set to −70 mV and the temperature held at 20°C to prevent exocytosis (Renström et al., 1996) and concomitant cell swelling. Prior to experiments using Rp-cAMPS, the dishes were pretreated with the membrane-permeant analogue 8-Br-Rp-cAMPS (0.5 mM), which did not interfere with LysoSensorGreen loading. Under the experimental conditions used (0.1 mM of the protonophore CCCP), stimulation of Cl− flux is reflected as a reduction of LSG fluorescence. In Fig. 9, C–E, tolbutamide-induced changes in intragranular pH in intact cells were estimated using the same method. To prevent tolbutamide from eliciting exocytosis, with resultant loss of LSG fluorescence, a Ca2+-free extracellular solution containing (in mM) 138 NaCl, 5.6 KCl, 1.2 MgCl2, 5 HEPES, 5 glucose, and 0.2 μM EGTA (pH 7.4 with NaOH) was used for these experiments.
Figure 9.

Sulfonylureas retain ability to stimulate exocytosis in SUR1−/− B-cells. (A-B) Changes in cell capacitance (ΔCm) under control conditions (ctrl) and in the presence of 0.1 mM tolbutamide (tolb) in SUR1−/− B-cells dialyzed with intracellular solution II with free Ca2+-concentrations of 0.17 μM (A) or 1.5 μM (B), respectively. Cyclic AMP was included at a concentration of 0.1 mM to promote granule mobilization (Renström et al., 1997). Traces shown are representative of 6–11 experiments. (C and D) Tolbutamide-induced granular acidification in wild-type (C) and SUR1−/− B-cells (D). Acidification is estimated as the relative increase (in %) in the initial LSG-signal (ΔF/Fo) after addition of tolbutamide. (E) Relative change in LSG fluorescence in wild-type (wt) and SUR1−/− B-cells 60 s after addition of tolbutamide. Data are mean values ±SEM of 7–8 experiments.
PCR
Total mouse islet RNA from wild-type and SUR1−/− mice was isolated using RNeasy Mini Kit (QIAGEN). 1 μg of total RNA was used for reverse transcription using random hexamer primers and Superscript II (Invitrogen). In a negative control, Superscript II was omitted from the reaction. PCR was performed under the following conditions: 3 min at 94°C followed by 35 cycles of 45 s at 94°C, 45 s at 58°C, and 45 s at 72°C with primers: Rim2 sense: 5′GCTCAAACAAGGTTATGTGGGTG3′; Rim2 antisense: 5′CCTGTCTTGTGAGCCCATGAGC3′; cAMPGEFII sense: 5′GGAGGAGAAAAGGTGGTGCT3′; and cAMPGEFII antisense: 5′CTCTCAAACTCCGCATAGAAC3′.
Data Analysis
The kinetic model used for fitting experimental data (in Figs. 1–3 and 7) considers two pools of granules; the pool of immediately releasable granules (P) and the pool of fused granules (P1). Release can then be described as
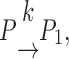 |
(1) |
where k is
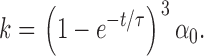 |
(2) |
Figure 7.

SUR1 knockout mice lack PKA-independent component of cAMP-stimulated exocytosis. (A–C) As in Fig. 2, A–C but experiments were performed on B-cells isolated from SUR1 knockout mice. (D) Relationship between pulse duration (t) and increase in cell capacitance (ΔC m) under control conditions, in the presence of 0.1 mM cAMP, and in the simultaneous presence of 0.1 mM cAMP and 0.5 mM Rp-cAMPS. The curves were derived by approximating Eq. 3 to the data points for depolarizations ≤350 ms. The dashed lines connect data point for depolarizations >350 ms and were derived by linear approximation. aP < 0.05 for comparison between cAMP and control. bP < 0.05 for comparison of cAMP alone with the combination of cAMP and Rp-cAMPS. Data are mean values ± SEM of 5–8 experiments. (E) Exocytosis measured in the presence of 0.1 mM cAMP during depolarizations ≤100 ms in wild-type (data taken from Fig. 2 D) and in SUR1−/− mice. Note slow onset of exocytosis in the knockout B-cells. **P < 0.01.
The pool dynamics is described by the differential equation
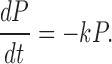 |
(3) |
Solutions to Eq. 3 were fit to the experimental data and the initial size of P (ΔCm,∞), taken to represent the immediately releasable pool (IRP), and the values of τ were thus estimated. The constant α0 describes the decay rate of P, which varies between individual experiments and depended on the initial pool size. A power function was used to account for the fact that the capacitance increase occurred after a delay and is consistent with the idea that the effect of Ca2+ on exocytosis in B-cells (Barg et al., 2001b) as well as other secretory cells (Thomas et al., 1993; Heinemann et al., 1994) exhibits great cooperativity. Tentatively, the time constant τ represent the sequential binding of Ca2+-ions to the Ca2+-sensor of secretion. A third power yielded the best results and suggest that a minimum of three Ca2+ ions are involved.
All data are quoted as mean values ± SEM of indicated number of experiments. Statistical significance was evaluated using Student's t test.
RESULTS
Stimulation of Exocytosis by GLP-1
We have reported previously that the incretin hormone GLP-1 and agents that increase intracellular cAMP levels such as the adenylate cyclase activator forskolin stimulate exocytosis in insulin-secreting mouse B-cells by both PKA-dependent and -independent mechanisms (Ämmälä et al., 1993; Gromada et al., 1997; Renström et al., 1997). Here we explored how GLP-1 affects the kinetics of exocytosis by applying progressively longer depolarizations (5–850 ms) from −70 to 0 mV before (Fig. 1 A) and 4 min after addition of 10 nM of the hormone (Fig. 1 B). The experiments were performed in metabolically intact cells using the perforated patch method. The relationships between pulse length and the amplitude of the exocytotic response in the absence and presence of GLP-1 are summarized in Fig. 1 C. It can be seen that whereas the amplitude of the exocytotic responses increased with pulse length for short depolarizations, the responses plateaued during depolarizations ≥200 ms. A secondary acceleration was observed during long depolarizations in the presence of GLP-1, but not in the absence of the hormone. The solution of Eq. 3 was approximated to the data points to derive the size of the IRP (ΔCm,∞). The mean values of seven paired experiments are given in Table I (lines 1 and 2). It is clear that GLP-1 increased IRP 2.3-fold. This enhancement occurred without any effect on the kinetics of exocytosis and the τ averaged 48 ± 18 ms and 35 ± 17 ms in the absence and presence of GLP-1, respectively. Application of the adenylate cyclase activator forskolin (2 μM) mimicked the effects of GLP-1 on the size of IRP (Table I, line 3).
TABLE 1.
Summary of the Effects of cAMP and PKA Inhibition in B-cells from Wild-type and SUR1−/− Mice
| Line | Condition | ΔCm,∞ (=IRP) | ΔCm/Δt450–850 | n | |
|---|---|---|---|---|---|
| fF | fFs−1 | ||||
| Wild-type B-cells | 1 | Control | 45 ± 7 | 6 ± 8 | 7 |
| Perforated patch | 2 | GLP-1 (10 nM) | 104 ± 15a | 56 ± 17a | 7 |
| 3 | Forskolin (2 mM) | 123 ± 30a | 152 ± 58a | 11 | |
| Wild-type B-cells | 4 | Control | 11 ± 2 | 25 ± 12 | 7 |
| Standard whole-cell | 5 | cAMP (1 μM) | 18 ± 6 | 16 ± 4 | 6 |
| 6 | cAMP (10 μM) | 57 ± 10c | 150 ± 55b | 5 | |
| 7 | cAMP (50 μM) | 83 ± 20c | 94 ± 18b | 5 | |
| 8 | cAMP (100 μM) | 157 ± 27c | 317 ± 119b | 7 | |
| 9 | cAMP (500 μM) | 176 ± 38c | 306 ± 135b | 6 | |
| 10 | cAMP (100 μM) and Rp-cAMPS (0.5 μM) | 96 ± 26 | −39 ± 35d | 7 | |
| SUR1−/− B-cells | 11 | Control | 16 ± 6 | 43 ± 19 | 5 |
| Standard whole-cell | 12 | cAMP (0.1 μM) | 68 ± 18e,f | 77 ± 40 | 8 |
| 13 | cAMP (0.1 μM) and Rp-cAMPS (0.5 μM) | 18 ± 3g | 3 ± 10g | 6 |
The size of IRP was estimated from the approximation of the solution of Eq. 3 to the data points in Figs. 1 C, 2 D, and 7, D and E. ΔCm/Δt450–850 is the rate of capacitance increase occurring between 450- and 850-ms data points. Data are mean values ± SEM of indicated number of cells tested. Statistical significances refer to comparisons within the same column.
P < 0.01 versus line 1; bP < 0.01 versus line 4; cP < 0.05 versus line 4; dP < 0.02 versus line 8; eP < 0.05 versus line 11; fP < 0.05 versus line 8; gP < 0.05 versus line 12.
Effects of cAMP and PKA Inhibition during Standard Whole-cell Recordings
We proceeded to study the effects of cAMP and Rp-cAMPS (a blocker of cAMP-dependent activation of PKA) on exocytosis. The experiments were conducted using the standard whole-cell configuration, in which the electrode solution dialyzes the cell interior allowing control of the composition of the cytosol. Fig. 2 shows exocytotic responses in three different cells elicited by progressively longer voltage-clamp depolarizations under control conditions (A), in the presence of 0.1 mM cAMP (B) and in the simultaneous presence of 0.1 mM cAMP and 0.5 mM of Rp-cAMPS (C). Under control conditions, the exocytotic responses were small and plateaued after a capacitance increase of <20 fF (Fig. 2 A). Inclusion of 0.1 mM cAMP in the intracellular solution exerted a strong stimulatory effect that was detectable already during brief stimuli (Fig. 2, B and C) and longer depolarizations elicited progressively larger exocytotic responses. Interestingly, the effect of cAMP was only partially suppressed by Rp-cAMPS and the capacitance increase elicited by ≤250-ms depolarizations was the same in the absence and presence of the PKA-inhibitor (Fig. 2, C and D). We have demonstrated elsewhere that the effects of Rp-cAMPS are identical to those of the peptide inhibitor PKI (Renström et al., 1997), making it likely that the observed effects of Rp-cAMPS do indeed reflect PKA inhibition.
The relationships between pulse length and the magnitude of the exocytotic responses under control conditions, with an intracellular cAMP concentration of 0.1 mM and in the simultaneous presence of cAMP and Rp-cAMPS are summarized in Fig. 2 D. Solutions to Eq. 3 were approximated to the data points and the average size of IRP determined under the different experimental conditions is presented in Table I (lines 4, 8, and 10). Cyclic AMP produced a 15-fold increase of IRP that was only partially reversed by Rp-cAMPS. The capacitance increase plateaued during depolarizations lasting ≥200 ms with a secondary stimulation being observed during longer depolarizations under both control conditions and in the presence of cAMP (50 fF/s under control conditions and 300 fF/s in the presence of cAMP). The latter component of capacitance increase was highly sensitive to inclusion of Rp-cAMPS and the ΔC/Δt-value in fact assumed a negative value in the presence of the PKA-inhibitor (−30 fF/s; i.e., less than what was observed under control conditions in the absence of cAMP), possibly indicative of endocytosis. These experiments confirm our earlier observation that cAMP stimulates exocytosis by both PKA-dependent and -independent mechanisms and provide the novel observation that the PKA-independent action is particularly prominent during brief depolarizations. Control experiments using 0.5 mM Rp-cAMPS alone revealed that the compound did not exert any stimulatory effect on its own (Fig. 2 D).
Differential cAMP Dependence of PKA-dependent and -independent Stimulation of Exocytosis
We subsequently investigated the cAMP dependence of the PKA-dependent and -independent actions on exocytosis. Cyclic AMP was applied intracellularly at concentrations of 0, 1, 10, 50, 100, and 500 μM. The data are summarized in Fig. 3 A. Solutions of Eq. 3 were approximated to the data points and the values of Cm∞ and ΔC/Δt450–850 are presented in Table I. Fig. 3 B shows the normalized increase in IRP (S) against the cAMP concentration (the increase in IRP at 500 μM of cAMP taken as unity). The Hill equation
 |
(4) |
was fit to the data points to derive the Hill coefficient (n) and the K d value, the concentration of cAMP at which stimulation is half-maximal (K d). In Eq. 4, Smax represent the calculated maximal cAMP-induced stimulation. We thus obtained a K d value of 29 μM. For comparison, the normalized response seen in the presence of 0.1 mM cAMP and 0.5 mM Rp-cAMPS is shown (open circle). It is clear that the early cAMP-dependent component of exocytosis is little affected by PKA-inhibition.
The effects of varying the intracellular concentration of cAMP on exocytosis elicited by trains of 10 500-ms depolarizations from −70 to 0 mV (Fig. 3 C) were analyzed similarly. The total increase in cell capacitance elicited by the train in the absence of cAMP is quite variable but in this series of experiments averaged 31 ± 4 fF (n = 6). A low concentration of cAMP (1 μM) only marginally stimulated exocytosis, but much larger responses were obtained after inclusion of ≥10 μM cAMP in the pipette solution. Fig. 3 D summarizes the amplitude of the capacitance increases elicited by the last nine pulses of the train at the different concentrations of cAMP (1–500 μM). The K d value for this late component was estimated to 6 μM by approximating the data to Eq. 4. Unlike what was observed for the rapid component (Fig. 3 B), the late cAMP-dependent effect was highly sensitive to Rp-cAMPS and in the presence of this antagonist, the increase in cell capacitance elicited by the last nine pulses was in fact less than that observed in the complete absence of cAMP (Fig. 3 D, open circle).
cAMP Mediates its PKA-independent Component via cAMP-GEFII
The cAMP-binding protein cAMP-GEFII binds cAMP at concentrations ≈10 μM and has been reported to mediate PKA-independent effects of the nucleotide on insulin secretion (Ozaki et al., 2000). We next confirmed that this effect also contributes to the cAMP-induced enhancement of exocytosis monitored as increases in membrane capacitance using the perforated patch whole-cell configuration (Fig. 4, A and B) . The PKA-dependent effects were prevented by pretreatment of the cells with 500 μM of the membrane-permeant PKA inhibitor 8-Br-Rp-cAMPS. After pretreatment with control oligonucleotide, exocytosis elicited by a 500-ms depolarization from −70 to 0 mV applied in the presence of forskolin amounted to 91 ± 8 fF (n = 5). This value is about twice that measured in cells pretreated with antisense cAMP-GEFII oligonucleotide, which averaged 48 ± 5 fF (n = 5; P < 0.01). Exocytosis in the presence of Rp-cAMPS alone amounted to 5 ± 5 fF (n = 5) and 10 ± 4 (n = 5) after pretreatment with control and antisense ODNs, respectively. We conclude that cAMP-GEFII mediates >55% of the PKA-independent component of exocytosis (i.e., 100% * {1 − [48 fF − 10 fF]/[91 fF − 5 fF]}).
The cAMP effector protein cAMP-GEFII was originally identified by two-yeast hybrid screening of a MIN6-cell library using the sulfonylurea receptor SUR1 as the bait (Ozaki et al., 2000). We have reported previously that sulfonylureas, such as tolbutamide and glibenclamide, stimulate exocytosis by a late effect on the exocytotic machinery that does not involve closure of plasma membrane KATP channels and that culminates in accelerated priming of the secretory granules by a PKC-dependent mechanism (Eliasson et al., 1996; Barg et al., 1999, 2001a). We analyzed the significance of cAMP-GEFII for sulfonylurea-stimulated exocytosis using clonal MIN-6 cells. In control cells (pretreated with control oligonucleotides or untransfected cells, no difference being observed between the two sets of cells), the capacitance increased steadily after establishment of the whole-cell configuration at a rate (ΔC/Δt) of 9 ± 1 fF/s (n = 17; Fig. 4 C). Inclusion of 0.1 mM cAMP in the pipette solution almost doubled the rate of exocytosis and the ΔC/Δt-value rose to 17 ± 2 fF/s (n = 18, P < 0.001 vs. control). Addition of tolbutamide (0.1 mM) produced a further 30% stimulation of exocytosis and ΔC/Δt amounted to 22 ± 1 fF/s (n = 13; P < 0.05 vs. rate measured with cAMP alone). When the same type of experiment was repeated in cells pretreated with antisense cAMP-GEFII oligonucleotides (Fig. 4 D), ΔC/Δt averaged 12 ± 3 fF/s (n = 12) under control conditions, 16 ± 3 fF/s (n = 14) in the presence of cAMP alone and 14 ± 3 fF/s in the simultaneous presence of cAMP and tolbutamide (n = 14; P < 0.05 vs. rate observed in cells exposed to cAMP and tolbutamide but incubated with the control oligonucleotide). Thus, reduced expression of cAMP-GEFII not only abolished the effect of cAMP on exocytosis, it also interfered with the ability of tolbutamide to stimulate exocytosis. This conclusion is reinforced by experiments performed in MIN-6 cells, which had been transfected with a dominant-negative mutant of cAMP-GEFII (G114/G422D; Ozaki et al., 2000; Fig. 4 E). In this series of experiments, ΔC/Δt averaged 8 ± 2 fF/s (n = 16) under control conditions, 8 ± 1 fF/s (n = 15) in the presence of cAMP alone and 8 ± 2 fF/s in the simultaneous presence of cAMP and tolbutamide (n = 13). The effects of treatment with the antisense oligonucleotide on exocytosis echo those on cAMP-GEFII immunoreactivity (Fig. 4 F). Whereas both MIN6-cells and primary mouse B-cells treated with the control oligonucleotide exhibited clear cAMP-GEFII/EPACII immunoreactivity, that only weakly colocalized with insulin, cells treated with the antisense oligonucleotide showed little cAMP-GEFII immunoreactivity. Notably, the antisense treatment did not affect insulin immunoreactivity.
A Selective cAMP-GEFII Agonist Promotes Rapid Exocytosis
The data of Fig. 4 suggest that cAMP-GEFII is responsible for the PKA-independent component of cAMP-stimulated exocytosis. We next tested the effects of the selective cAMP-GEFII agonist 8CPT-2Me-cAMP on B cell exocytosis. Secretion was elicited by a train of 10 depolarizations from −70 to 0 mV under control conditions, in the presence of 0.1 mM 8CPT-2Me-cAMP alone and in the simultaneous presence of both the agonist and 0.5 mM Rp-cAMPS (Fig. 5 A). The total increase in capacitance elicited by the train averaged 99 ± 14 fF (n = 6) under control conditions, 274 ± 51 fF (n = 6; P < 0.01 vs. control) in the presence of 0.1 mM 8CPT-2Me-cAMP and 178 ± 30 fF (n = 5) in the simultaneous presence of 0.1 mM 8CPT-2Me-cAMP and 0.5 mM Rp-cAMPS. The latter value is statistically different from the control value (P < 0.05) but not from that observed in the presence of 8CPT-2Me-cAMP alone. Fig. 5 B illustrates the increase in cell capacitance elicited by the individual pulses during the train. It is clear that the action of 8CPT-2Me-cAMP is particularly pronounced during the first part of the train and that this effect was unaffected by PKA-inhibition (pulses 1 and 2). Surprisingly, an exocytotic component sensitive to Rp-cAMPS was observed during the middle part of the train (pulses 3–5). We speculate that Ca2+-entry associates with some Ca2+-dependent activation of adenylate cyclase leading to sufficient generation of cAMP to promote granule mobilization by the PKA-dependent mechanism (Fig. 3 D). This scenario would also explain the slow increase in capacitance that occurs in the complete absence of cyclic AMP (Figs. 3 C and 5 A) and that can likewise be suppressed by Rp-cAMPS (Figs. 2 C and 3 D; Renström et al., 1997).
Figure 5.

PKA-independent stimulation of exocytosis by the cAMP-GEFII–selective agonist 8CPT-2Me-cAMP. (A) Capacitance increases (bottom) elicited by trains of 10 depolarizations from −70 to 0 mV (top) under control conditions (left), in the presence 100 μM 8pCPT-2Me-cAMP (middle) and in the simultaneous presence of the agonist and 0.5 mM Rp-cAMPs. (B) Mean increase in membrane capacitance (ΔCm,n − ΔCm,n − 1) during each individual depolarization of the train (n). Data are mean values ± SEM of 5–6 experiments. aP < 0.05, bP < 0.001, and cP < 0.01 for comparison between 8CPT-2Me-cAMP and control. dP < 0.01 and eP < 0.05 for comparison between the combination of 8CPT-2Me-cAMP and Rp-cAMPS and control. fP < 0.05 for comparision between 8CPT-2Me-cAMP and the combination of 8CPT-2Me-cAMP and Rp-cAMPS.
Impaired Insulinotropic Action of Glucose and the Incretin GLP-1 in vitro
It has been reported recently that insulin secretion from SUR1 knockout mice (SUR1−/−) exhibits a much lower sensitivity to intracellular cAMP and appears unaffected by stimulation with incretins (Shiota et al., 2002; Nakazaki et al., 2002). We have repeated these experiments focusing on the ability of GLP-1 and forskolin to stimulate glucose-induced insulin secretion and the effects of inhibiting PKA using Rp-cAMPS in wild-type and SUR1−/− islets (Table II) . Wild-type islets had low basal insulin secretion, which increased five- to ninefold in response to stimulation with 1 μM glibenclamide or 20 mM glucose. GLP-1 and forskolin potentiated the stimulatory action of glucose 2.8- to 3.6-fold. The latter effects were partially (65–70%) counteracted by Rp-8-Br-cAMPS. In islets from SUR1−/− mice, basal secretion was increased >1.4-fold relative to that seen in wild-type islets. As expected, glibenclamide had no effect but glucose retained a 4.5-fold stimulatory action. GLP-1 and forskolin potentiated glucose-induced secretion from SUR1−/− islets 2.2- to 2.8-fold; i.e., not much less than the relative stimulation in wild-type islets. The same observations were made in two separate experimental series. However, unlike the situation in the wild-type islets, the effects of forskolin and GLP-1 were fully (≥90%) antagonized by Rp-cAMPS. The smaller secretory responses in SUR1−/− B-cells are not attributable to degranulation and islet insulin content averaged 280 ± 10 ng insulin/islet (n = 6) and 253 ± 10 ng/islet (n = 6) in wild-type and SUR1−/− mice, respectively. GLP-1 increased cAMP content more than than sevenfold in SUR1−/− islet; similar to the ninefold elevation seen in the wild-type islets (Fig. 6) . The effects of GLP-1 were dose-dependent and bell-shaped in both wild-type and SUR1−/− islets. The maximum cAMP levels were observed at 10 nM and at this concentration the content approached the values observed in response to 10 μM forskolin.
TABLE II.
Insulin Release (ng/islet/h)
| Line | Conditions | Wild-type (SUR1+/+) | Knockout (SUR1−/−) |
|---|---|---|---|
| 1 | 2.8 mM glucose | 0.27 ± 0.04(n = 5) | 0.39 ± 0.06 (n = 5)i |
| 2 | 2.8 mM glucose +1 μM glibenclamide |
1.34 ± 0.10 (n = 5)a | 0.41 ± 0.09 (n = 5)k |
| 3 | 20 mM glucose | 2.53 ± 0.23 (n = 5)b,c | 1.76 ± 0.27 (n = 5)a,c,k |
| 4 | 20 mM glucose 10 nM GLP-1 |
7.01 ± 0.51 (n = 5)d | 3.87 ± 0.37 (n = 5)g,j |
| 5 | 20 mM glucose 10 nM GLP-1 +100 μM Rp-8-Br-cAMPS |
4.11 ± 0.33 (n = 5)e | 1.91 ± 0.31 (n = 5)e,j |
| 6 | 20 mM glucose +10 mM forskolin |
8.97 ± 0.62 (n = 5)d | 4.88 ± 0.49 (n = 5)d,i |
| 7 | 20 mM glucose +10 mM forskolin +100 μM Rp-8-Br-cAMPS |
4.54 ± 0.46 (n = 5)f | 2.07 ± 0.43 (n = 5)h |
Impaired PKA-independent incretin effect in islet from SUR1−/− mice. Secretion of insulin from isolated islets taken from wild-type (SUR1+/+) and SUR1 knockout (SUR1−/−) in the absence and presence of different insulin secretagogues and inhibitors. Incubation was performed with five islets in each tube and at either 2.8 or 20 mmol/l glucose. Data are presented as mean values ± SEM of indicated number of experiments (n).
P < 0.01 versus line 1; bP < 0.001 versus line 1; cP < 0.02 versus line 2; dP < 0.001 versus line 3; eP < 0.01 versus line 4; fP < 0.001 versus line 6; gP < 0.01 versus line 3; hP < 0.01 versus line 6; iP < 0.001; jP < 0.01 versus corresponding value in wild-type islets; kP < 0.05 versus corresponding value in wild-type islets.
Figure 6.

GLP-1–induced cAMP production in wild-type and SUR1−/− B-cells. The amount of cAMP generated was measured in wild-type and SUR1−/− B-cells as indicated at 0–100 nM GLP-1. Values observed in the presence of 10 μM forskolin have been included for comparison. Data are mean values ± SEM of six experiments in each group. All values in the presence of GLP-1 are statistically different from control value (P < 0.001; not shown). *P < 0.05 for the comparison between wild-type and SUR1−/− islets.
Lack of an Early PKA-independent Component in B-cells from SUR −/− Mice
Fig. 7, A–C , shows the effects of intracellular cAMP and PKA inhibition on exocytosis measured as a capacitance increase in B-cells from SUR1−/− mice using the same protocol as in Fig. 2. Under basal conditions (no cAMP, Fig. 7 A), the exocytotic responses were small and ≤20 fF for depolarizations lasting 450 ms. As was the case in wild-type B-cells, inclusion of 0.1 mM cAMP in the intracellular medium resulted in much larger exocytotic responses (Fig. 7 B). However, unlike the situation in wild-type B-cells, the effects of cAMP were fully antagonized by 0.5 mM Rp-cAMPS (compare Figs. 2 C and 7 C). The relationships between pulse length and exocytosis under control conditions, in the presence of cAMP and in the simultaneous presence of both cAMP and Rp-cAMPS are summarized in Fig. 7 D. The size of IRP under the different experimental conditions was determined by fitting Eq. 3 to the observed data points (Table I, lines 11–13). It is apparent that cAMP produced a fourfold increase in pool size but that it is ∼90 fF smaller than in wild-type animal (compare lines 8 and 12 of Table I). The effect of cAMP in B-cells from SUR1−/− mice, unlike what was observed in their wild-type counterparts, was completely inhibited by Rp-cAMPS. Moreover, the fast component of exocytosis (detectable during the first 100 ms) was more prominent in wild-type than in SUR1−/− islets (Fig. 7 E). Thus, the value of τ (see Eq. 2) was 3.5-fold higher in B-cells from SUR1−/− mice than in their wild-type counterparts (45 ± 13 ms vs. 12 ± 4 ms; P < 0.05). We can discard the possibility that the differences between wild-type and SUR1−/− B-cells were due to a reduction of voltage-gated Ca2+-entry. The Ca2+-current amplitude during depolarizations to 0 mV averaged −57 ± 5 pA (n = 12) and −64 ± 8 pA (n = 8) in SUR1−/− and wild-type B-cells, respectively.
cAMP-GEFII and Rim2 are Transcribed in SUR1−/− B-cells
We tested whether the failure of cAMP to exert a PKA-independent stimulatory action on exocytosis in SUR1−/− B-cells results from loss of transcription of the putative effector proteins. However, applying RT-PCR to islets isolated from SUR1−/− mice indicate that both cAMP-GEFII and Rim2 remain expressed in islets from the knockout animals (Fig. 8) .
Figure 8.

RT-PCR analyses of Rim2 and cAMP-GEFII expression in islets from wild-type and SUR1−/− mice. Expression profiles of cAMP-GEFII and Rim2 (+) in wild-type and SUR−/− islets. In a negative control reaction (−), reverse transcriptase was omitted. PCR products were analyzed on 1% agarose gels and bands of the predicted size were obtained from Rim2 (262 bp) and cAMP-GEFII (554 bp), after 35 amplification cycles. Molecular weight standard is shown to the right.
Sulfonylureas Stimulate Exocytosis in B-cells from SUR1−/− Mice
It has been suggested that sulfonylureas stimulate exocytosis in B-cells by a direct effect on the exocytotic machinery (Eliasson et al., 1996). We investigated whether this effect is retained in SUR1−/− B-cells using a Ca2+-infusion protocol (intracellular solution II supplemented with 0.1 mM cAMP). In SUR1−/− B-cells, tolbutamide (0.1 mM) increased the rate of capacitance increase ≥2.5-fold at both 0.17 and 1.5 μM intracellular free Ca2+ (Fig. 9, A and B) ; from 5 ± 1 fF/s (n = 8) to 14 ± 3 fF/s (n = 11; P < 0.05) at low Ca2+ and from 16 ± 3 fF/s (n = 6) to 37 ± 9 fF/s (n = 6; P < 0.05) at the higher intracellular Ca2+-concentration (Fig. 9, A and B). These rates and the extent of stimulation are comparable to effects reported previously (Barg et al., 1999, 2001a). The failure of glibenclamide to stimulate insulin secretion in B-cells from SUR1−/− mice (Table II) we attribute to the glucose concentration in this series of experiments. The direct action of sulfonylureas on exocytosis is particularly pronounced at high glucose, whereas it is without effect at low concentrations of the sugar (unpublished data).
Acidification of the granule interior has been identified as an important reaction in the preparation of the granules for release (Barg et al., 2001a; Renström et al., 2002a,b). As shown in Fig. 9 C, extracellular application of tolbutamide resulted in a pronounced acidification of the granules (detected as an increase in granular LSG fluorescence), an effect that was maintained in B-cells from SUR1−/− mice (Fig. 9 D), although the average effect was reduced by ∼30% (Fig. 9 E). Apparently, the ability of tolbutamide to promote granule priming is largely unperturbed by ablation of SUR1.
Cyclic AMP Stimulates Influx of Cl− by a PKA-independent Mechanism in Wild-type but Not in SUR1−/− B-cells
ClC3 channels have been shown previously to play an important role in granule priming by providing the shunt conductance required for the granules to acidify (Barg et al., 2001a; Renström et al., 2002a). Given that the stimulatory effects of both the PKA-independent mechanism of cAMP and those of the sulfonylureas on exocytosis appear to involve cAMP-GEFII (Fig. 4, C–E), it is tempting to speculate that their actions converge at the level of granular ClC3-channels. In wild-type B-cells, inclusion of cAMP accelerated granule deprotonation (corresponding to stimulation of Cl− influx) fourfold over that seen under control conditions, an effect resistant to PKA inhibition (Fig. 10, A and C) . Interestingly, the ability of cAMP to accelerate Cl− uptake into the granules was almost abolished in B-cells from SUR1−/− mice (Fig. 10, B and C) and the rate of fluorescence decrease was not significantly different from that observed in the absence of cAMP. We are not implying that granules in B-cells from SUR1−/− mice are unable undergo priming. Indeed the ability of Ca2+ alone to acidify the granules was the same in wild-type and SUR1−/− B-cells (Fig. 10 C). It is evident from both the capacitance measurements and insulin release experiments that SUR1−/− B-cells contain a large pool of release-competent granules. We postulate, however, that the ability of cAMP to accelerate priming is much reduced in the SUR1−/− B-cells and that this accounts for the poor incretin effects in these cells.
Figure 10.

Granular Cl− fluxes and pH in wild-type and SUR1−/− B-cells. Changes in insulin granule pH estimated as changes in LysoSensor-Green™ DND-189 (LSG) fluorescence and detected by confocal imaging. (A) Mean changes in LSG fluorescence (in percentage of initial values; F/F0) ± SEM recorded when using a Ca2+-containing pipette solution (n = 6; ctrl), with cAMP (0.1 mM; n = 5; cAMP) added to the pipette solution, and when both cAMP and Rp-cAMPS (0.5 mM; n = 4; Rp+cAMPS) were included. The arrow indicates the time at which the standard whole-cell configuration were established with the onset of wash-in of the pipette solution. The top trace (PB) represents (F/F0) parallel recordings in intact (whole-cell configuration not established) neighboring cells (n = 15), providing an estimate of the extent of photo-bleaching in these experiments. (B) Same as in A but recorded on B-cells isolated from SUR−/− mice under control conditions (n = 4, ctrl) and in the presence of 0.1 mM cAMP (n = 4, cAMP). (C) Mean decreases in LSG fluorescence (in percentage of initial; [F0 − F]/F0) ± SEM, measured 60 s after standard whole-cell establishment, under the respective condition as indicated. All experiments (except the left panel labeled PB) were conducted in the presence of 1.5 μM free intracellular Ca2+. *P < 0.05.
DISCUSSION
The ability of cAMP-increasing agents to stimulate insulin secretion is well established (Wollheim and Sharp, 1981), but the precise cellular processes involved are not fully understood. Here we have combined measurements of insulin release with high-resolution capacitance measurements, antisense oligonucleotide technology, and the use of transgenic animals to dissect the mechanisms by which cAMP accelerates exocytosis. We confirm that cAMP enhances insulin secretion by both PKA-dependent and -independent mechanisms (Renström et al., 1997). The latter pathway involves the cAMP effector protein cAMP-GEFII and, somewhat surprisingly, requires the participation of the sulfonylurea protein SUR1. Here we discuss in turn the properties of PKA-dependent and -independent exocytosis, their respective roles in insulin secretion and the possible interaction of the PKA-independent mechanism of exocytosis with the sulfonylurea receptor protein SUR1.
The PKA-dependent and -independent Mechanisms of cAMP-stimulated Exocytosis Can Be Temporally Separated and Exhibit Distinct Concentration Dependence
Our data indicate that PKA-independent (Rp-cAMPS–insensitive) effects of cAMP on exocytosis accounts for a capacitance increase of ∼100 fF (at 0.1 mM cAMP) that plateaus within ∼100 ms, whereas the PKA-dependent (Rp-cAMPS–sensitive) component became apparent during depolarizations >150 ms. (Figs. 2 D and 3, B and D). We can discard the possibility that the rapid component of capacitance increase reflects release of GABA-containing synaptic-like microvesicles (SLMVs) rather than insulin-containing granules as has previously been proposed for rat B-cells (Takahashi et al., 1997). Using an assay that allows detection of single-vesicle release of GABA, we estimate that exocytosis of SLMVs contribute only ≈1% of the total capacitance increase (Braun et al., 2002).
The PKA-dependent and -independent mechanisms of cAMP-stimulated exocytosis not only differ with regard to speed (Figs. 2–3), they can also be distinguished by differences in their dose–response properties (Fig. 3). Whereas the PKA-dependent pathway exhibits a K d of 6 μM, a fivefold higher concentration is required to activate the PKA-independent mechanism. Previous data argue that the PKA-independent effects are mediated by the cAMP-sensing protein cAMP-GEFII (Ozaki et al., 2000; Kashima et al., 2001). Three observations corroborate this notion. First, treatment of islets with antisense oligonucleotides against cAMP-GEFII resulted in a marked reduction of PKA-independent exocytosis (Fig. 4 A). Second, transfection of MIN6-cells with a dominant mutant of cAMP-GEFII (G114E/G422D; Fig. 4 E) abolished cAMP-stimulated secretion. Third, inclusion of the specific cAMP-GEFII agonist 8CTP-2Me-cAMP in the intracellular solution mimicked the Rp-cAMPS-resistant component of cAMP-induced secretion (Fig. 5).
We propose that the PKA-independent component of exocytosis can be used to estimate the cAMP concentration at the release sites. Under basal conditions, the size of IRP amounted to 45 fF, which rose to ≈100 fF and ≈120 fF after stimulation with 10 nM GLP-1 or 2 μM forskolin (Table I). Using the dose–response curve in Fig. 3 B, these values correspond to a cAMP concentrations of 8, 42, and 70 μM under basal conditions in the presence of GLP-1 and after stimulation with forskolin. Thus, the cAMP concentration sensed by the exocytotic machinery may vary by almost a factor of 10 under different experimental conditions. It is interesting to compare the above concentrations with the reported cAMP levels in the absence and presence of forskolin. In one study performed in the absence of any phosphodiesterase inhibitor, the cAMP content of isolated mouse islets was found to increase from a basal 4 fmol/islet to ≈65 fmol in the presence of 10 μM forskolin (Eddlestone et al., 1985). The above values can be converted to global intracellular cAMP concentrations taking the intracellular water volume to be ≈2 nl in mouse islets (Ashcroft et al., 1970). We thereby estimate that cAMP increases from a basal concentration of ≈2 to ≈30 μM in the presence of forskolin. Since the latter value is only ≈40% of that suggested by the capacitance measurements, it appears that fairly steep intracellular gradients of cAMP may exist within the B cell. This is perhaps not so unexpected given that adenylate cyclase is situated in the plasma membrane, i.e., close to the release site. It is also worthy of note that already the basal concentration (2 μM) is sufficient to allow some limited PKA-dependent stimulation of granule mobilization (Fig. 3 D).
Evidence for PKA-dependent and -independent Components of Insulin Release in vitro in Wild-type and SUR1−/− mice
Although SUR1−/− B-cells lack functional KATP-channels, these islets retain some glucose dependence of insulin secretion (3.5-fold stimulation; Table II). Contrary to what was reported recently by others (Nakazaki et al., 2002), islets from SUR1−/− mice also respond well to GLP-1 and forskolin although the magnitude of the responses is only 50% of those seen in wild-type islets. However, an important difference exists with respect to the effects of PKA inhibition. Whereas 30–35% of insulin secretion elicited by GLP-1 or forskolin is resistant to Rp-cAMPS in wild-type islets, the PKA-independent component is absent in the SUR1−/− islets. Consistent with data reported by others (Nakazaki et al., 2002), this difference cannot be explained by a reduced ability of GLP-1 and forskolin to increase intracellular cAMP, which was only marginally reduced in the knockout islets. Indeed, the lack of a rapid PKA-independent component of insulin secretion was confirmed using capacitance measurements in which B-cells from SUR1−/− mice were dialyzed with 0.1 mM cAMP (Fig. 7 E). These findings therefore raise the interesting alternative possibility that SUR1 somehow is involved in the exocytotic process. It is important to note that both cAMP-GEFII and Rim2 are transcribed in the SUR1−/− B-cells (Fig. 8). Although PCR data cannot be equated to protein levels, these results nevertheless argue that the loss of PKA-independent cAMP-induced exocytosis cannot simply be attributed to the absence of the effector proteins. It may seem surprising that whereas our data indicate that GLP-1 retains a good stimulatory action in isolated islets, in vivo experiments on the same SUR1−/− mouse strain suggest the complete loss of GLP-1–stimulated secretion (Shiota et al., 2002 and unpublished data). Although we are unable to provide a simple explanation to this discrepancy, this observation might indicate that the PKA-independent component of cAMP-induced exocytosis plays a particularly important role for incretin-stimulated insulin secretion in vivo. It should be noted that the PKA-independent action of cAMP is operational at higher intracellular cAMP concentrations than the PKA-dependent mechanism (Fig. 3 D) and one possible explanation is therefore that stimulation with GLP-1 resulted in a larger increase in intracellular cAMP levels in the in vitro experiments than that attained in vivo. This concept would in fact be in accordance with the observations of Nakazaki et al. (2002), who found that whereas GLP-1 failed to stimulate insulin secretion, forskolin retained the ability to potentiate insulin secretion and especillay so when applied in the presence of IBMX.
PKA-independent Effect of cAMP Involves Stimulation of Cl− Influx and Granular Acidification
We and others have demonstrated previously that sulfonylureas stimulate exocytosis in B-cells via an effect exerted on the secretory machinery itself and that is not mediated by closure of plasma membrane KATP-channels (Flatt et al., 1994; Eliasson et al., 1996; Tian et al., 1998; Barg et al., 1999). This effect remained observable in the presence of forskolin but disappeared when PKC was maximally activated by the phorbol ester PMA or inhibited by bisindolylmaleimide (Eliasson et al., 1996). The ability of sulfonylureas to stimulate exocytosis in SUR1−/− B-cells therefore suggests that inactivation of this gene with resultant suppression of the PKA-independent action does not interfere with the ability of PKC activation to stimulate secretion. Indeed, it has been reported recently that the potency of PMA to stimulate insulin secretion is at least as strong in the SUR1−/− islets as in their wild-type counterparts (Nakazaki et al., 2002).
The ability of sulfonylureas to stimulate exocytosis in B-cells may not only be of pharmacological significance. It can be speculated that it contributes to the metabolic regulation of insulin secretion (i.e., the “augmenting” or “KATP channel–independent” effects of glucose on insulin release; Henquin, 2000; Henquin et al., 2002). The importance of the metabolic state for exocytosis is witnessed by the prompt (within a few seconds) inhibition when the cytoplasmic ATP/ADP-ratio is lowered by flash photolysis of caged ADP even if ATP and Ca2+ are present at levels that would otherwise be stimulatory (Barg et al., 2002a). This effect of ADP can be reversed by tolbutamide and in the simultaneous presence of the sulfonylurea and ADP, exocytosis proceeds at the same rate as in the presence of ATP alone (Barg et al., 2001a).
A recent model postulates that the inhibitory and stimulatory effects of ADP and tolbutamide on exocytosis are secondary to changes in granular Cl− fluxes and pH; stimulation of exocytosis occurs upon intragranular acidification and activation of Cl− influx (Barg et al., 2001a; Renström et al., 2002a). We now extend these observations and demonstrate that binding of cAMP to cAMP-GEFII stimulates exocytosis via the same mechanism. We also provide evidence that the poor incretin effect characterizing B-cells from SUR1−/− mice associates with the failure of cAMP to activate these processes (Fig. 10).
Model
In Fig. 11 we outline a hypothetical model that appears to account for our observations. We propose that SUR1, in addition to its well-known role in the formation of functional KATP-channels (see i.e., Ashcroft and Gribble, 1998), also facilitates the interaction between cAMP-GEFII and downstream effector proteins including RIM2. The ability of SUR1 itself to associate with cAMP-GEFII is suggested by the original identification of cAMP-GEFII in insulin-secreting cells by yeast-two-hybrid screening of a mouse cDNA library using SUR1 as the bait (Ozaki et al., 2000). RIM proteins have been proposed to subserve scaffolding roles in neurons (compare Wang et al., 1997; Schoch et al., 2002) and it is easy to imagine how SUR1, Rim2, and cAMP-GEFII can give rise to a molecular net of protein–protein interactions in the B cell that culminates in the priming of the insulin secretory granules. The ability of sulfonylureas to stimulate exocytosis in B-cells from SUR1−/− mice reinforces previous data, indicating that the granular sulfonylurea-binding protein (gSUR) is distinct from SUR1 (Barg et al., 1999; Renström et al., 2002a). We have suggested that gSUR, via interaction with the ClC3 channels and regulation of granular Cl− fluxes, modulates the release competence of the insulin granules (Barg et al., 2001a). We now extend this concept and propose that cAMP-GEFII, by assembling with the gSUR–ClC3 complex, promotes granule priming. A tight association of cAMP-GEFII, gSUR, and ClC3 is indeed suggested by the finding that the stimulatory action of sulfonylureas is lost following down-regulation of cAMP-GEFII using antisense ODNs or a dominant negative construct (Fig. 4). It is unlikely that the sulfonylureas bind directly to cAMP-GEFII because the latter protein has a molecular weight of 110 kD and no sulfonylurea-binding proteins of this molecular weight have been identified. We propose that the binding of sulfonylureas to gSUR stabilizes the interaction between cAMP-GEFII/gSUR and ClC3 thus facilitating granule priming. We acknowledge that this scenario by necessity is speculative because the identity of gSUR remains enigmatic and the interaction with other proteins and effects of sulfonylureas have accordingly not been possible to test.
Figure 11.

Schematic summarizing the interactions between cAMP-GEFII, SUR, and exocytosis-regulating proteins. See text for details. SG, secretory granule; ClC3, ClC3 chloride channel, SUR1, sulfonylurea receptor.
To account for the loss of PKA-independent stimulation of exocytosis in SUR1−/− B-cells, we propose that the cAMP-GEFII–gSUR1 complex fails to associate correctly with the granular ClC3-channel in the absence of SUR1. Accordingly, the ability of cAMP to exert its PKA-independent effect on granular Cl− fluxes (Fig. 10) and exocytosis (Fig. 7) is lost. This concept would account for the observations that the effects of both cAMP and sulfonylureas converge at the level of granular acidification. We finally emphasize that although SUR1, cAMP-GEFII, ClC3, and gSUR are important for granule priming, membrane fusion still depends on the SNARE proteins VAMP2, syntaxin-1, SNAP25, and the Ca2+-sensor synaptotagmin (Jacobsson et al., 1994; Lin and Scheller, 2000; Satin, 2000) and exocytosis can therefore proceed in the absence of cAMP or in SUR1-deficient B-cells, albeit at a lower rate than in wild-type B-cells.
The interactions between cAMP and sulfonylureas and their respective receptors are clearly very complex. However, based on the present findings as well as those published by others (Nakazaki et al., 2002; Shiota et al., 2002) it now seems justifiable to conclude that the role of SUR1 in the B cell extends beyond being a subunit of the KATP-channel and that it also plays an unexpected but important role in the control of the insulin secretory machinery.
Acknowledgments
Lena Eliasson, Xiaosong Ma, and Erik Renström contributed equally to this study and their names appear in alphabetical order. We thank Dr. Mark Magnusson for generating the SUR1−/− mice and Susumo Seino for kindly providing us with the antisense cAMP-GEF II and control ODNs, as well as the dominant negative mutant of cAMP-GEFII. We also express our gratitude to Kristina Borglid and Britt-Marie Nilsson for skilled technical assistance.
Financial support was obtained from the Swedish Research Council (grants 8647, 13147, 4286, 9890, and 12234), the Juvenile Diabetes Research Foundation, the National Institutes of Health (DK58508), the Swedish Diabetes Association, the Novo Nordisk Foundation, the Albert Påhlssons Stiftelse, the Åke Wibergs Stiftelse, the Magnus Bergvalls Stiftelse, the Crafoordska Siftelsen, and the Royal Physiolgraphic Society in Lund.
David C. Gadsby served as editor.
Footnotes
Abbreviations used in this paper: IRP, immediately release pool; ODN, oligodeoxynucleotide.
References
- Ämmälä, C., F.M. Ashcroft, and P. Rorsman. 1993. Calcium-independent potentiation of insulin release by cyclic AMP in single β-cells. Nature. 363:356–358. [DOI] [PubMed] [Google Scholar]
- Ashcroft, F.M., and F.M. Gribble. 1998. Correlating structure and function in ATP-sensitive K+ channels. Trends Neurosci. 21:288–294. [DOI] [PubMed] [Google Scholar]
- Ashcroft, F.M., and P. Rorsman. 1989. Electrophysiology of the pancreatic B-cell. Prog. Biophys. Mol. Biol. 54:87–143. [DOI] [PubMed] [Google Scholar]
- Ashcroft, S.J., C.J. Hedeskov, and P.J. Randle. 1970. Glucose metabolism in mouse pancreatic islets. Biochem. J. 118:143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg, S., E. Renström, P.O. Berggren, A. Bertorello, K. Bokvist, M. Braun, L. Eliasson, W.E. Holmes, M. Köhler, P. Rorsman, and F. Thevenod. 1999. The stimulatory action of tolbutamide on Ca2+-dependent exocytosis in pancreatic β cells is mediated by a 65-kDa mdr-like P-glycoprotein. Proc. Natl. Acad. Sci. USA. 96:5539–5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg, S., P. Huang, L. Eliasson, D.J. Nelson, S. Obermüller, P. Rorsman, F. Thevenod, and E. Renström. 2001. a. Priming of insulin granules for exocytosis by granular Cl− uptake and acidification. J. Cell Sci. 114:2145–2154. [DOI] [PubMed] [Google Scholar]
- Barg, S., X. Ma, L. Eliasson, J. Galvanovskis, S.O. Göpel, S. Obermüller, J. Platzer, E. Renström, M. Trus, D. Atlas, et al. 2001. b. Fast exocytosis with few Ca2+ channels in insulin-secreting mouse pancreatic B cells. Biophys. J. 81:3308–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg, S., L. Eliasson, E. Renström, and P. Rorsman. 2002. a. A subset of secretory granules in close contact with L-type Ca2+-channels accounts for first-phase insulin secretion in mouse β-cells. Diabetes. 51:S74–S82. [DOI] [PubMed] [Google Scholar]
- Braun, M., B. Birnir, J. Broman, L. Eliasson, H. Mulder, and P. Rorsman. 2002. GABA is released from rat pancreatic β-cells by Ca2+-dependent exocytosis of synaptic –like microvesicles. Diabetologia. 45:A163. [Google Scholar]
- Eddlestone, G.T., S.B. Oldham, L.G. Lipson, F.H. Premdas, and P.M. Beigelman. 1985. Electrical activity, cAMP concentration, and insulin release in mouse islets of Langerhans. Am. J. Physiol. 248:C145–C153. [DOI] [PubMed] [Google Scholar]
- Eliasson, L., E. Renström, C. Ämmälä, P.O. Berggren, A.M. Bertorello, K. Bokvist, A. Chibalin, J.T. Deeney, P.R. Flatt, J. Gäbel, et al. 1996. PKC-dependent stimulation of exocytosis by sulfonylureas in pancreatic β cells. Science. 271:813–815. [DOI] [PubMed] [Google Scholar]
- Enserink, J.M., A.E. Christensen, J. de Rooij, M. van Triest, F. Schwede, H.G. Genieser, S.O. Doskeland, J.L. Blank, and J.L. Bos. 2002. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 4:901–906. [DOI] [PubMed] [Google Scholar]
- Flatt, P.R., O. Shibier, J. Szecowka, and P.O. Berggren. 1994. New perspectives on the action of sulfonylureas and hypoglycemic sulfonamides on the pancreatic β-cell. Diabete Metab. 20:157–162. [PubMed] [Google Scholar]
- Gillis, K.D. 1995. Techniques for membrane capacitance measurements. Single Channel Recording. B. Sakmann and E. Neher, editors. Plenum Press, New York. 155–198.
- Gromada, J., S. Dissing, K. Bokvist, E. Renström, J. Frökjaer-Jensen, B.S. Wulff, and P. Rorsman. 1995. Glucagon-like peptide I increases cytoplasmic calcium in insulin-secreting βTC3-cells by enhancement of intracellular calcium mobilization. Diabetes. 44:767–774. [DOI] [PubMed] [Google Scholar]
- Gromada, J., W.G. Ding, S. Barg, E. Renström, and P. Rorsman. 1997. Multisite regulation of insulin secretion by cAMP-increasing agonists: evidence that glucagon-like peptide 1 and glucagon act via distinct receptors. Pflugers Arch. 434:515–524. [DOI] [PubMed] [Google Scholar]
- Gromada, J., J.J. Holst, and P. Rorsman. 1998. Cellular regulation of islet hormon secretion by the incretin hormon glucagon-like peptide 1. Pflugers Arch. 435:583–594. [DOI] [PubMed] [Google Scholar]
- Heinemann, C., R.H. Chow, E. Neher, and R.S. Zucker. 1994. Kinetics of the secretory response in bovine chromaffin cells following flash photolysis of caged Ca2+. Biophys. J. 67:2546–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin, J.C. 2000. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 49:1751–1760. [DOI] [PubMed] [Google Scholar]
- Henquin, J.C., N. Ishiyama, M. Nenquin, M.A. Ravier, and J.C. Jonas. 2002. Signals and pools underlying biphasic insulin secretion. Diabetes. 51:S60–S67. [DOI] [PubMed] [Google Scholar]
- Holz, G.G. IV, W.M. Kuhtreiber, and J.F. Habener. 1993. Pancreatic β-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7-37). Nature. 361:362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsson, G., A.J. Bean, R.H. Scheller, L. Juntti-Berggren, J.T. Deeney, P.O. Berggren, and B. Meister. 1994. Identification of synaptic proteins and their isoform mRNAs in compartments of pancreatic endocrine cells. Proc. Natl. Acad. Sci. USA. 91:12487–12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P.M., D.M. Salmon, and S.L. Howell. 1988. Protein phosphorylation in electrically permeabilized islets of Langerhans. Effects of Ca2+, cyclic AMP, a phorbol ester and noradrenaline. Biochem. J. 254:397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, G., O.G. Chepurny, and G.G. Holz. 2001. cAMP-regulated guanine nucleotide exchange factor II (Epac2) mediates Ca2+-induced Ca2+ release in INS-1 pancreatic β-cells. J. Physiol. 536:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno, T., S. Suga, J. Wu, M. Kimura, and M. Wakui. 1998. Intracellular cAMP potentiates voltage-dependent activation of L-type Ca2+ channels in rat islet β-cells. Pflugers Arch. 435:578–580. [DOI] [PubMed] [Google Scholar]
- Kashima, Y., T. Miki, T. Shibasaki, N. Ozaki, M. Miyazaki, H. Yano, and S. Seino. 2001. Critical role of cAMP-GEFII-Rim2 complex in incretin-potentiated insulin secretion. J. Biol. Chem. 276:46046–46053. [DOI] [PubMed] [Google Scholar]
- Leech, C.A., and J.F. Habener. 1997. Insulinotropic glucagon-like peptide-1-mediated activation of non-selective cation currents in insulinoma cells is mimicked by maitotoxin. J. Biol. Chem. 272:17987–17993. [DOI] [PubMed] [Google Scholar]
- Lin, R.C., and R.H. Scheller. 2000. Mechanisms of synaptic vesicle exocytosis. Annu. Rev. Cell Dev. Biol. 16:19–49. [DOI] [PubMed] [Google Scholar]
- Martell, A.E., and R.M. Smith. 1974. Critically stability constants. Plenum Press, New York.
- Miyazaki, J., K. Araki, E. Yamato, H. Ikegami, T. Asano, Y. Shibasaki, Y. Oka, and K. Yamamura. 1990. Establishment of a pancreatic β cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 127:126–132. [DOI] [PubMed] [Google Scholar]
- Nakazaki, M., A. Crane, M. Hu, V. Seghers, S. Ullrich, L. Aguilar-Bryan, and J. Bryan. 2002. cAMP-activated protein kinase-independent potentiation of insulin secretion by cAMP is impaired in SUR1 null islets. Diabetes. 51:3440–3449. [DOI] [PubMed] [Google Scholar]
- Nesher, R., E. Anteby, M. Ydovizky, N. Warwar, N. Kaiser, and E. Cerasi. 2002. β-Cell protein kinases and the dynamics of the insulin response to glucose. Diabetes. 51:S68–S73. [DOI] [PubMed] [Google Scholar]
- Ozaki, N., T. Shibasaki, Y. Kashima, T. Miki, K. Takahashi, H. Ueno, Y. Sunuga, H. Yano, Y. Matsuura, T. Iwanaga, et al. 2000. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat. Cell Biol. 2:805–811. [DOI] [PubMed] [Google Scholar]
- Renström, E., L. Eliasson, K. Bokvist, and P. Rorsman. 1996. Cooling inhibits exocytosis in single mouse pancreatic B-cells by suppression of granule mobilization. J. Physiol. 494:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renström, E., L. Eliasson, and P. Rorsman. 1997. PKA-dependent and PKA-independent stimulation of exocytosis by cAMP in mouse pancreatic B-cells. J. Physiol. 502:105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renström, E., S. Barg, F. Thévenod, and P. Rorsman. 2002. a. Sulphonylurea-mediated stimulation of insulin exocytosis via an ATP-sensitive K+ channel-independent action. Diabetes. 51:S33–S36. [DOI] [PubMed] [Google Scholar]
- Renström, E., R. Ivarsson, and S. Shears. 2002. b. Inositol 3,4,5,6-tetrakisphosphate inhibits insulin granule acidification and fusogenic potential. J. Biol. Chem. 277:26717–26720. [DOI] [PubMed] [Google Scholar]
- Satin, L.S., and T.A. Kinard. 1998. Neurotransmitters and their receptors in the islets of Langerhans of the pancreas: what messages do acetylcholine, glutamate, and GABA transmit? Endocrine. 8:213–223. [DOI] [PubMed] [Google Scholar]
- Satin, L.S. 2000. Localized calcium influx in pancreatic β-cells: its significance for Ca2+-dependent insulin secretion from the islets of Langerhans. Endocrine. 13:251–262. [DOI] [PubMed] [Google Scholar]
- Schoch, S., P.E. Castillo, T. Jo, K. Mukherjee, M. Geppert, Y. Wang, F. Schmitz, R.C. Malenka, and T.C. Südhof. 2002. RIM1α forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 415:321–326. [DOI] [PubMed] [Google Scholar]
- Shiota, C., O. Larsson, K.D. Shelton, M. Shiota, A.M. Efanov, M. Hoy, J. Lindner, S. Kooptiwut, L. Juntti-Berggren, J. Gromada, et al. 2002. Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J. Biol. Chem. 277:37176–37183. [DOI] [PubMed] [Google Scholar]
- Takahashi, N., T. Kadowaki, Y. Yazaki, Y. Miyashita, and H. Kasai. 1997. Multiple exocytotic pathways in pancreatic β-cells. J. Cell Biol. 138:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, P., J.G. Wong, A.K. Lee, and W. Almers. 1993. A low affinity Ca2+ receptor controls the final steps in peptide secretion from pituitary melanotrophs. Neuron. 11:93–104. [DOI] [PubMed] [Google Scholar]
- Tian, Y.A., G. Johnson, and S.J. Ashcroft. 1998. Sulfonylureas enhance exocytosis from pancreatic β-cells by a mechanism that does not involve direct activation of protein kinase C. Diabetes. 47:1722–1726. [DOI] [PubMed] [Google Scholar]
- Wang, Y., M. Okamoto, F. Schmitz, K. Hofmann, and T.C. Südhof. 1997. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature. 388:593–598. [DOI] [PubMed] [Google Scholar]
- Wollheim, C.B., and G.W. Sharp. 1981. Regulation of insulin release by calcium. Physiol. Rev. 61:914–973. [DOI] [PubMed] [Google Scholar]