

Abstract
TRPV6 (CaT1/ECaC2), a highly Ca2+-selective member of the TRP superfamily of cation channels, becomes permeable to monovalent cations in the absence of extracellular divalent cations. The monovalent currents display characteristic voltage-dependent gating and almost absolute inward rectification. Here, we show that these two features are dependent on the voltage-dependent block/unblock of the channel by intracellular Mg2+. Mg2+ blocks the channel by binding to a site within the transmembrane electrical field where it interacts with permeant cations. The block is relieved at positive potentials, indicating that under these conditions Mg2+ is able to permeate the selectivity filter of the channel. Although sizeable outward monovalent currents were recorded in the absence of intracellular Mg2+, outward conductance is still ∼10 times lower than inward conductance under symmetric, divalent-free ionic conditions. This Mg2+-independent rectification was preserved in inside-out patches and not altered by high intracellular concentrations of spermine, indicating that TRPV6 displays intrinsic rectification. Neutralization of a single aspartate residue within the putative pore loop abolished the Mg2+ sensitivity of the channel, yielding voltage-independent, moderately inwardly rectifying monovalent currents in the presence of intracellular Mg2+. The effects of intracellular Mg2+ on TRPV6 are partially reminiscent of the gating mechanism of inwardly rectifying K+ channels and may represent a novel regulatory mechanism for TRPV6 function in vivo.
Keywords: voltage-dependent gating, pore block, calcium channel, TRP channel, CRAC channel
INTRODUCTION
The TRP superfamily consists of cation channels with six transmembrane segments that are related to the product of the Drosophila trp (for transient receptor potential) gene (Clapham et al., 2001). Based on sequence homology, the 21 mammalian TRPs can be subdivided into three subfamilies: members of TRPC subfamily (the C stands for canonical) display the highest homology to Drosophila TRP, members of the TRPV subfamily are most homologous to the vanilloid receptor 1 (VR1, now TRPV1) and members of TRPM subfamily show the highest homology with melastatin (TRPM1). TRP channels vary significantly in their biophysical properties and gating mechanisms, and the exact regulation and function of most TRPs is still unclear.
TRPV6 (previously known as CaT1 and ECaC2) and the homologous TRPV5 (ECaC1) are the two members of the TRP superfamily with the highest known Ca2+ selectivity (PCa/PNa 100), and are supposed to be involved in Ca2+ reabsorption in kidney and intestine (Hoenderop et al., 1999, 2001; Peng et al., 1999; Vennekens et al., 2000; Voets et al., 2001; Yue et al., 2001). Like all other Ca2+-selective channels (Hille, 2001), they become permeable to monovalent cations when all extracellular divalent cations are removed (Vennekens et al., 2000; Yue et al., 2001). In the case of TRPV5 it has been demonstrated that high Ca2+ selectivity and sensitivity to extracellular divalents depends on a single aspartate residue in the pore region between transmembrane domains 5 and 6 (Nilius et al., 2001). Monovalent TRPV6 (and TRPV5) currents are characterized by voltage-dependent opening at negative potentials and extremely strong inward rectification (Voets et al., 2001). These properties are unique within the TRP superfamily, since all other studied members show weak rectification in both inward and outward directions and lack voltage dependence (Clapham et al., 2001). Moreover, these distinct properties can be used to discriminate between TRPV6 and CRAC, the Ca2+ release–activated Ca2+ channel that has been equaled to TRPV6 (Yue et al., 2001), but lacks its voltage dependence and high degree of inward rectification (Voets et al., 2001). The mechanisms underlying the strong inward rectification and voltage-dependent gating of TRPV6 are, however, not well understood.
Rectification of inwardly rectifying K+ (Kir) channels arises from a voltage-dependent block by intracellular Mg2+ and polyamines, which occlude the pore and thereby inhibit outward K+ flux (Vandenberg, 1987; Ficker et al., 1994; Lopatin et al., 1994; Fakler et al., 1995). Upon hyperpolarization, these blocking particles are expelled from the pore, which is observed as a time-dependent opening of the channel (Nichols and Lopatin, 1997). It has been shown that two negative charges in the second transmembrane domain and the COOH-terminal cytoplasmic domain of strongly inwardly rectifying K+ channels are required for the high affinity binding of Mg2+ and polyamines, and that neutralization of these residues leads to weakly rectifying channels (Stanfield et al., 1994; Wible et al., 1994; Yang et al., 1995). We have shown recently that the voltage-dependent gating of TRPV6 depends on intracellular Mg2+ (Voets et al., 2001), raising the question of whether the mechanism of strong inward rectification of this TRP channel is similar to that of Kir channels. In the present work we present data demonstrating that Mg2+-dependent gating indeed contributes to strong inward rectification of TRPV6 and that Mg2+ ions act as a permeant pore blocker.
MATERIALS AND METHODS
Cell Culture and Transfection
HEK-293 cells were grown in DMEM containing 10% (vol/vol) fetal calf serum, 2 mM L-glutamine, 2 U/ml penicillin, and 2 mg/ml streptomycin at 37°C in a humidity-controlled incubator with 10% CO2. They were transiently transfected with the pCINeo/IRES-GFP/mCaT1 vector (Hoenderop et al., 2001) using methods described previously (Trouet et al., 1997) and electrophysiological recordings were performed between 8 and 24 h after transfection.
Single amino acids in the pore region of TRPV6 were mutated using the standard PCR overlap extension technique, and the nucleotide sequences of all mutants were verified by sequencing of the corresponding cDNAs.
Electrophysiology
Transfected cells were identified by their green fluorescence when illuminated at 480 nm using the polychrome IV monochromator (T.I.L.L. Photonics, GmbH). Patch-clamp experiments were performed in the tight-seal whole-cell configuration at room temperature (20–25°C) using an EPC-9 patch-clamp amplifier and Pulse software (HEKA Electronics). Patch pipettes had DC resistances of 2–4 MΩ when filled with the different intracellular solutions. Series resistances were between 3 and 10 MΩ, and were compensated for 60–80%. Residual voltage errors due to uncompensated series resistances were generally <10 mV, and cells with larger voltage errors were omitted from the analysis. Currents were sampled at 10 kHz and filtered at 2.9 kHz using an eight-pole Bessel filter.
Most solutions used in this study were used both as pipette and as extracellular solution. The standard divalent cation-free (DVF) solution contained (in mM): 150 NaCl, 10 EDTA, and 10 HEPES, titrated to pH 7.4 with NaOH. To test the effect of the permeant cation, all NaCl in the DVF solution was either replaced by 150 mM LiCl, CsCl, or KCl, or by 100 mM NMDG and 30 mM BaCl2. In these cases, titration to pH 7.4 was done using the corresponding hydroxide or HCl. Solutions with free Mg2+ concentrations <500 μM were obtained by adding the appropriate amount of MgCl2 to the DVF solution. To obtain solutions containing free Mg2+ concentrations >500 μM, the appropriate amount of MgCl2 was added to a solution containing 10 mM EGTA instead of EDTA. The free Mg2+ concentration of these solutions was calculated using the CaBuf software (ftp://ftp.cc.kuleuven.ac.be/pub/droogmans/cabuf.zip). The intracellular solution used in the Mg2+ uncaging experiments contained (in mM): 150 NaCl, 10 HEPES, 10 EGTA, 5 DM-nitrophen (Molecular Probes, Inc.) and 3 MgCl2, titrated to pH 7.4 with NaOH.
HEK-293 cells endogenously express TRPM7, and TRPM7-like currents have been described in HEK-293 cells dialyzed with Mg2+-free solution (Nadler et al., 2001). To assess the contribution of this channel to the whole-cell currents in HEK-293 cells transfected with TRPV6, we routinely measure currents after switching to an extracellular solution containing 2 mM free Ca2+. Under this condition, TRPV6 carries virtually no outward current, whereas the TRPM7-like current is observed as a strongly outwardly rectifying current (Nadler et al., 2001; Runnels et al., 2001). We found that in most batches of HEK-293 cells, the contribution of these TRPM7-like currents was negligible compared with the robust TRPV6-mediated currents. In batches of cells where the contribution of the TRPM7-like currents was relatively large, we added 100 μM GTPγS to the pipette solution, which has been shown previously to effectively inhibit a TRPM7-like current in RBL cells (Hermosura et al., 2002).
Data analysis and display were done using Microcal Origin software version 7.0 (OriginLab Corporation). Unless noted otherwise, averaged data are shown as mean ± SEM from at least four cells.
RESULTS
HEK-293 cells expressing TRPV6 display large cation currents that are active immediately after breaking into the cell with a NaCl-based pipette solution containing 10 mM EGTA and 2 mM Mg2+. In the presence of physiological concentrations of Ca2+ in the extracellular medium, TRPV6 is highly selective for Ca2+, resulting in currents that reverse at potentials more positive than 50 mV. However, omitting all divalents from the extracellular medium renders the channel permeable to monovalent cations, resulting in large, inwardly rectifying currents that reverse close to 0 mV and display voltage-dependent activation at negative potentials (Hoenderop et al., 2001; Voets et al., 2001; Yue et al., 2001). In general, the TRPV6-mediated currents further increase in amplitude in the first minutes after break-in. We attribute this increase in amplitude to the decrease of the intracellular-free Ca2+ concentration by the wash-in of Ca2+ buffer, which relieves TRPV6 from slow Ca2+-dependent inhibition (Hoenderop et al., 2001; Voets et al., 2001). Unless noted otherwise, all experiments described in this work were performed after the current reached a steady-state level, which was normally achieved within 200 s.
Mg2+-dependent Gating and Strong Inward Rectification of TRPV6
Fig. 1 A displays a TRPV6 I-V relation in the absence of extracellular divalents obtained from a 200-ms linear voltage ramp from −150 to 100 mV. Two typical features of this current trace have been put forward as the TRPV6 fingerprint (Schindl et al., 2002): the negative slope conductance in the voltage region more negative than −100 mV, which reflects time-dependent activation of the current when the voltage is stepped from holding potential (0 mV) to −150 mV, and the extreme inward rectification. However, the shape of the I-V relation obtained from such voltage-ramps depends strongly on the speed and voltage range of the ramp, making this protocol less well suited for quantitative analysis of the rectification and gating behavior of TRPV6. The time dependence of current activation at negative potentials can be better appreciated from the step protocol in Fig. 1 B. Stepping to potentials more negative than −40 mV from a holding potential of 0 mV gives rise to inward current showing time-dependent activation, whereas only very small outward currents are measured at positive potentials (Fig. 1 B).
Figure 1.

Strong inward rectification and voltage-dependent gating of monovalent currents through TRPV6. Extracellular solution contained 150 mM Na+ and 10 mM EDTA; intracellular solution contained 150 mM Na+, 10 mM EGTA, and 2 mM Mg+. (A) I-V relation obtained during a 200-ms linear voltage-ramp from −150 to 100 mV. (B) Currents measured during 100-ms voltage steps ranging from −180 to 100 mV from a holding potential of 0 mV.
We have shown recently that the time-dependent activation of TRPV6 is only observed in the presence of intracellular Mg2+ (Voets et al., 2001). To assess the impact of the Mg2+-dependent gating on inward rectification, we employed the voltage protocol shown in Fig. 2 C. In this protocol, TRPV6 is maximally activated by a hyperpolarizing prepulse to −100 mV, before stepping to test potentials ranging between −160 and 100 mV. The amplitude of the current immediately after the prepulse (Iini) represents the current through fully activated TRPV6, whereas that at the end of the test pulse gives the steady-state current (Iss). In the presence of 1 mM intracellular free Mg2+, clear deactivation of the channel can be observed during the test pulses to potentials more positive than −60 mV (Fig. 2 A). As a result, the I-V relation for Iss normalized to the current at −100 mV is clearly much more inwardly rectifying than for Iini (Fig. 2, D and E). The rectification score, which we defined as the ratio of the slope conductances at −80 and 80 mV and used as a quantitative measure of inward rectification, was 32.1 ± 5.4 for Iss (n = 7) versus 11.1 ± 4.2 for Iini (n = 7). In contrast, the currents in the absence of intracellular Mg2+ show no noticeable time dependence (Fig. 2 B) and consequently Iss and Iini are virtually identical. The shape of the I-V relation for Iss (Fig. 2, D and E) and the slope conductance ratio (10.9 ± 1.2; n = 9) are not different from that of Iini in the presence of intracellular Mg2+. Together, these data indicate that TRPV6 shows clear inward rectification in the absence of intracellular Mg2+, and that deactivation in the presence of 1 mM intracellular Mg2+ increases the degree of steady-state inward rectification by a factor of three.
Figure 2.

Voltage-dependent gating requires intracellular Mg2+ and contributes to strong inward rectification. (A and B) Currents measured in response to the voltage protocol shown in C, obtained with an intracellular solution containing 1 mM free intracellular Mg2+ (A) or 10 mM EDTA and no Mg2+ (B). (C) Voltage protocol used in A and B and subsequent figures. The time-points at which the initial (Iini) and steady-state (Iss) currents and the apparent open probability (Po) were measured are indicated. (D) Average I-V relations obtained from this step protocol and normalized to the current at −100 mV. Shown are Iini and Iss obtained with 1 mM free intracellular Mg2+ and Iss obtained in the absence of intracellular Mg2+. (E) Magnification of the boxed area in D illustrating the effect of Mg2+-dependent gating on the steady-state outward currents. (F) Voltage dependence of the apparent open probability (Po) in the absence and presence of 1 mM intracellular Mg2+. Apparent Po was determined as the inward current measured immediately after the 100-ms test pulses normalized to the maximal inward current.
The voltage protocol shown in Fig. 2, A–C, also allows quantification of the voltage dependence of Mg2+-dependent deactivation. We assessed the apparent open probability (Po) (Fig. 2 F) from the instantaneous inward current during a step to −100 mV following the 100-ms test pulses and by normalizing this value to the maximal inward current. With 1 mM intracellular Mg2+, the apparent Po steeply decreases between −80 and −20 mV, reaches a minimum of ∼0.25 at −20 mV and then slightly increases at more positive potentials, whereas in the absence of intracellular Mg2+ it remains virtually constant at a value close to 1.
Fig. 3, A–C , illustrates the concentration dependence of the effect of intracellular Mg2+ on inward rectification and voltage-dependent gating of TRPV6. Increasing [Mg2+]i from 8 μM to 5 mM leads to a more pronounced time dependence of the currents and a significant reduction of the steady-state outward currents. The rectification score of Iss increased from around 10 at low [Mg2+]i to ∼50 with 5 mM [Mg2+]i (Fig. 3 D), and half maximal reduction of the relative outward conductance, as estimated from fitting a Hill equation to the data points, occurred at a concentration of 154 ± 28 μM. Fig. 3 E shows the voltage dependence of the apparent Po for different [Mg2+]i. Increasing [Mg2+]i causes a leftward shift of the deactivation curve and leads to a decrease of the minimal apparent Po. The Mg2+ concentration at which the apparent Po at 0 mV was half-maximally reduced was 57 ± 14 μM (Fig. 3 F). Note that for all tested intracellular Mg2+ concentrations the apparent Po goes through a nonzero minimum around 0 mV and then increases at more positive potentials, an effect that was most clear at submillimolar [Mg2+]i (for example, see Fig. 3 B, inset).
Figure 3.

Concentration dependence of the effect of intracellular Mg2+ on inward rectification and voltage-dependent gating. (A–C) Current traces with three different intracellular free Mg2+ concentrations obtained using the protocol shown in Fig. 2 C, except that test pulses ranged from −120 to 100 mV. (D) Mg2+ dependence of inward rectification. The rectification score of Iss was determined as the ratio of the slope conductances at −80 and 80 mV. The solid line represent a Hill function fitted to the data. (E) Voltage dependence of the apparent Po for the different intracellular Mg2+ concentrations. (F) Mg2+ dependence of the apparent PO at 0 mV. The solid line represent a Hill-function fitted to the data.
Interaction between Mg2+ and Permeant Cations within the Channel Pore
The most straightforward explanation for the Mg2+-dependent rectification and gating of TRPV6 is that intracellular Mg2+ binds to one or more sites in the channel pore and within the transmembrane electrical field, and thereby blocks permeation of other cations in a voltage-dependent manner. However, it cannot be fully excluded that Mg2+ allosterically modulates an intrinsic gating mechanism. To discriminate between these possibilities we tested whether Mg2+-dependent gating is modulated by the permeant cation.
As shown in Fig. 4 , reducing the extracellular Na+ concentration from 150 to 70 or 30 mM (isomolar substitution by the impermeant cation NMDG+) not only reduces the inward current (Fig. 4, A and B) but also shifts the voltage dependence of the apparent Po to more negative potentials (Fig. 4 C). Similarly, isomolar substitution of extracellular Na+ by the less permeant monovalent cations K+ or Cs+ causes a leftward shift of the voltage dependence of the apparent Po (Fig. 5, A and C) , whereas with 30 mM Ba2+ as the sole charge carrier the voltage dependence is shifted to more positive potentials (Fig. 5, B and C), consistent with the higher relative permeability of Ba2+ compared with Na+. As shown in Fig. 6 , there is a linear relation between the measured reversal potential for the different extracellular solutions and the potential for 50% deactivation (VPo = 0.5). Together, these data demonstrate that permeant cations interact with Mg2+-dependent gating, consistent with direct binding of Mg2+ within the channel pore. Unfortunately, we were not able to directly measure the effects of intracellular Mg2+ on Ca2+ currents through TRPV6. Ca2+ currents undergo rapid and slow Ca2+-dependent inactivation (Hoenderop et al., 2001; Yue et al., 2001), which makes it impossible to accurately measure the apparent Po and the effects of intracellular Mg2+ on thereon.
Figure 4.
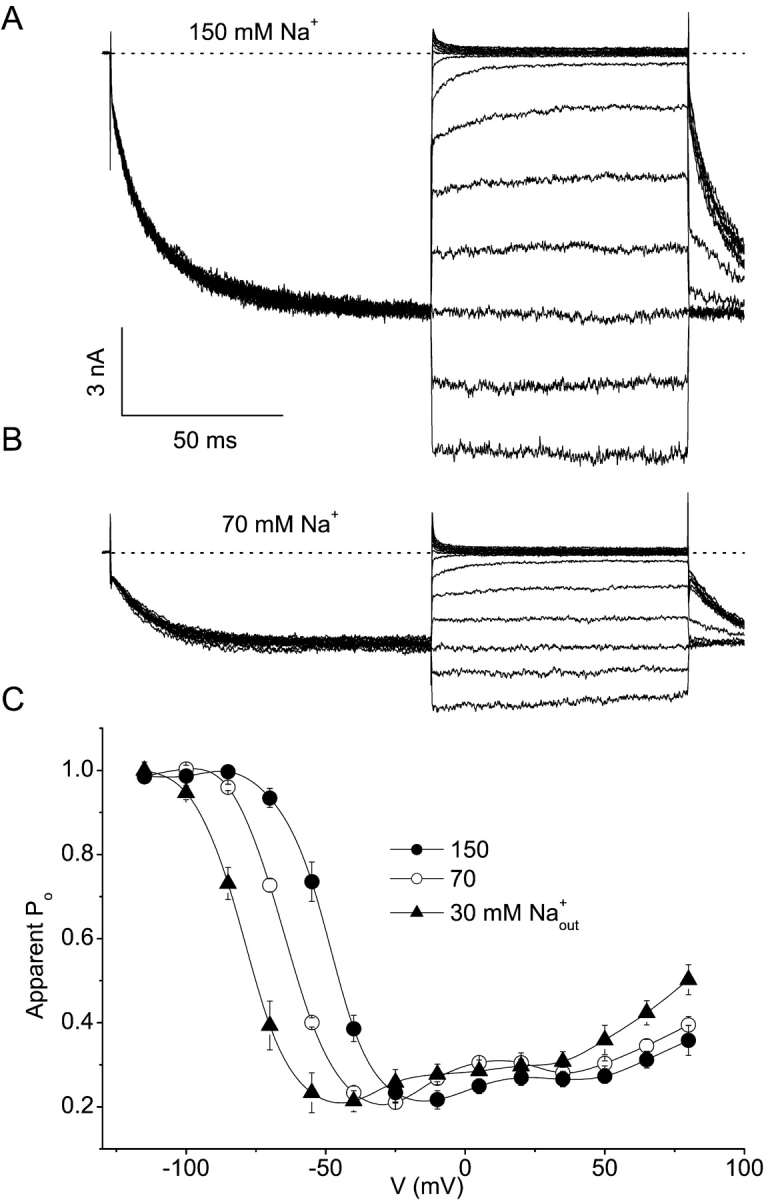
Modulation of the voltage dependence of TRPV6 by the extracellular Na+ concentration. Intracellular solution contained 150 mM Na+, 10 mM EGTA and 2 mM Mg+. (A and B) Current traces in 150 mM (A) or 70 mM extracellular Na+ obtained using the protocol shown in Fig. 2 C, except that test pulses ranged from −140 to 100 mV. (C) Voltage dependence of the apparent Po for three different extracellular Na+ concentrations.
Figure 5.

Modulation of the voltage dependence of TRPV6 by the permeant cation. (A) Current traces with extracellular solutions containing 150 mM of the indicated monovalent cation as the sole inward charge carrier obtained using the protocol shown in Fig. 2 C. (B) Same as A, but using an extracellular solution containing 100 mM NMDG and 30 mM Ba2+. Test pulses ranged from −100 to 80 mV. (C) Voltage dependence of the apparent Po for the different permeant cations.
Figure 6.

Correlation between reversal potential and the potential at which the apparent Po is reduced to 0.5 for the different extracellular cation conditions illustrated in Figs. 4 and 5. All values were corrected for liquid junction potentials.
Mg2+ Acts as a Bidirectional Permeant Blocker
As noted above, the apparent Po reaches a minimum around 0 mV and increases at more positive potentials (see Fig. 3, B and F). This relief of block at positive potentials suggests that under these conditions Mg2+ can be expelled from its binding site(s) and permeate through the channel into the extracellular space. If this were the case, it is expected that the same binding site(s) within the channel pore can also be occupied in a voltage-dependent manner by extracellular Mg2+ ions.
As shown in Fig. 7, A–C , submillimolar concentrations of extracellular Mg2+ ions indeed cause a voltage-dependent block of TRPV6 currents. The voltage dependence of block by extracellular Mg2+ mirrors that by intracellular Mg2+: the apparent Po is maximal at very positive potentials, decreases to a minimum around −40 mV, and increases again at more negative values (Fig. 7 D). The extracellular Mg2+ concentration at which the apparent Po at 0 mV was half-maximally reduced was estimated to be 75 ± 7 μM, comparable to the value of 57 ± 14 μM obtained for intracellular Mg2+ (see Fig. 3 F). Together, these data indicate that Mg2+ can block the TRPV6 pore from either side of the membrane, and permeate the channel if sufficient voltage is applied.
Figure 7.

Voltage-dependent block of TRPV6 by extracellular Mg2+. Intracellular solution contained 150 mM Na+, 10 mM EDTA and no added Mg+. (A) Current traces obtained in the absence of extracellular Mg2+. The voltage was stepped from −100 mV to potentials ranging from −140 to 120 mV before stepping back to −100 mV. (B and C) Same as A, but in the presence of the indicated concentration of free extracellular Mg2+ ions. Note that the voltage was stepped to 60 mV before the test steps, in order to relieve most of the Mg2+ block. (D) Voltage dependence of the apparent open probability (Po) for the different extracellular Mg2+ concentrations. Apparent Po was determined, analogous to Fig. 2 F, as the inward current measured immediately after the 100-ms test pulses normalized to the maximal inward current.
Yue et al. (2001) observed that voltage-dependent activation of TRPV6 at negative potentials only occurs in the full absence of extracellular Mg2+. At 1 mM intracellular Mg2+ and with extracellular Mg2+ buffered at 3 μM, they observed slow inactivation of TRPV6 currents at negative potentials. Under these conditions, the activation at negative potentials due to unblock of intracellular Mg2+ is probably masked by a rapid block of the pore by extracellular Mg2+.
Kinetics of Mg2+ Binding and Unbinding
Fig. 8 presents a quantitative analysis of the kinetics of Mg2+ block and unblock from either side of the membrane. Fig. 8, A–C, show results that were obtained in a divalent-free extracellular solution and with 1 mM intracellular Mg2+. In this condition, maximal block of the channel by the intracellular Mg2+ ions is achieved around 0 mV, whereas the apparent Po is maximal at −100 mV (see Fig. 2 F). Stepping from 0 mV to negative potentials, as illustrated in Fig. 8 A, results in time-dependent opening of TRPV6, which we interpret as the time course of Mg2+ release from its binding site(s) in the channel pore. In the same way, we interpret the negative slope of the I-V relation obtained from linear voltage ramps (e.g., Fig. 1 A) as the time-dependent unblocking of Mg2+ during the hyperpolarized part of the ramp protocol. Conversely, stepping from −100 mV to more depolarized potentials (Fig. 8 B) results in time-dependent closing of the channel, which mainly represents the time course of binding of intracellular Mg2+ into the channel pore. From the superimposed fits of the current traces it can be seen that neither block nor unblock are monoexponential and that two exponential terms are required for an adequate fit. Over the examined voltage range, the two time constants differed roughly by one order of magnitude (Fig. 8 C).
Figure 8.

Analysis of the kinetics of Mg2+ block and unblock. (A and B) Current traces obtained with a divalent-free extracellular solution and with 1 mM free intracellular Mg2+ in response to the indicated voltage step. Single (solid gray) and double (dotted gray) exponential fits are superimposed. (C) Fast (squares) and slow (circles) time constants obtained from double exponential fits as in A and B. The open symbols were obtained from the fits to currents in response to a hyperpolarizing step from a holding potential of 0 mV, as in A. The filled symbols were obtained from the fits to currents in response to a depolarizing step from a holding potential of −100 mV, as in B. (D and E) Current traces obtained with a divalent–free intracellular solution and with 500 μM free extracellular Mg2+ in response to the indicated voltage step. Single (solid gray) and double (dotted gray) exponential fits are superimposed. (F) Fast (squares) and slow (circles) time constants obtained from single or double exponential fits as in (D and E). The open symbols were obtained from the fits to currents in response to a hyperpolarizing step from a holding potential of 80 mV, as in D. The filled symbols were obtained from the fits to currents in response to a depolarizing step from a holding potential of −100 mV, as in E.
Fig. 8, D–F, illustrates a kinetic analysis of current traces obtained with a divalent cation–free intracellular solution and with 500 μM extracellular Mg2+. In this condition, maximal block of the channel by extracellular Mg2+ ions is achieved below −20 mV, whereas block is relieved at more positive potentials (see Fig. 7 B). Stepping from 80 mV to negative potentials (Fig. 8 D) results in a time-dependent block of TRPV6 by extracellular Mg2+, whereas stepping from −100 mV to more depolarized potentials (Fig. 8 E) results in time-dependent release of bound Mg2+ to the extracellular medium. The time course of extracellular Mg2+ block at hyperpolarized potentials (−80 mV and below) again required two exponential terms for an acceptable fit (Fig. 8 D), yielding two time constants that differed by approximately one order of magnitude (Fig. 8 F). In contrast, block at less negative potentials and unblock at positive potentials (Fig. 8, E and F) were well described by a monoexponential function. However, due to the relatively short length of the test pulses (100 or 200 ms), a second slow exponential term with time-constants >100 ms may have escaped detection.
Mg2+-independent Inward Rectification Is Intrinsic
As shown above (Figs. 2 and 3) TRPV6 still displays inward rectification in the absence of intracellular Mg2+ and with identical solutions on either side of the plasma membrane. Inward rectification of Kir channels in the absence of intracellular Mg2+ is due to a pore block by intracellular polyamines such as spermine, spermidine, and putrescine (Nichols and Lopatin, 1997), which is slowly lost in inside-out patches due to the washout of these cytoplasmic gating molecules (Lopatin et al., 1994). To investigate whether a similar mechanism causes rectification of TRPV6 in the absence of intracellular Mg2+, we first measured whole-cell currents using a Mg2+-free intracellular solution containing 5 mM spermine. Currents measured under these conditions were time-independent, displayed moderate inward rectification (rectification score = 10.1 ± 0.7; n = 4) and were indiscernible from currents measured in the absence of intracellular spermine and Mg2+ (unpublished data).
Additionally, we tested whether rectification was sensitive to washout of cytoplasmic factors in inside-out patches. As shown in Fig. 9 , inside-out patches excised in Ca2+- and Mg2+-free medium display macroscopic, inwardly rectifying currents. The amplitude of these currents increased significantly during the first ∼100 s after excision (Fig. 9, A and B). We attribute this current increase mainly to the relief of the tonic inhibition of TRPV6 by intracellular Ca2+ and Mg2+ ions, which have both been shown to exert an inhibitory effect on TRPV6 activity (see below and in Hoenderop et al., 2001; Voets et al., 2001). Addition of 200 μM Mg2+ to the cytoplasmic side of the patch reversibly induced voltage-dependent gating, similar to what is observed in whole-cell currents (Fig. 9 C). Most importantly, channel activity was generally stable for at least 1000 s, without significant changes in rectification (Fig. 9 D). The average rectification score 1,000 s after patch-excision amounted to 9.8 ± 1.4 (n = 4). Together, our data demonstrate that Mg2+-independent rectification does not depend on block by polyamines or other soluble cytoplasmic factors, suggesting that TRPV6 displays intrinsic rectification.
Figure 9.

Inward rectification is preserved in cell-free inside-out patches. The extracellular (pipette) solution contained 150 mM NaCl and 1 mM EDTA. Currents were measured during 400-ms voltage ramps from −150 to 100 mV. Shown are I-V relations representing the average of five subsequent traces obtained within 10 s. (A) I-V relation obtained in the cell-attached mode, immediately before excision of the patch. (B) I-V relation obtained 100 s after patch excision in a divalent-free solution. At this time point, the TRPV6 current had maximally developed. (C) Addition of 200 μM free Mg2+ to the intracellular side of the patch caused a reduction of the inward current and time-dependent opening at negative potentials, comparable with the behavior of the whole-cell current in Fig. 1 A. (D) The effect of intracellular Mg2+ was rapidly reversible, and inward rectification was preserved up to 1,000 s after patch excision.
Asp541 within the Pore Region Is Required for Mg2+ Binding
In the strong inward rectifier K+ channel Kir2.1 (IRK1), high-affinity binding of intracellular Mg2+- and polyamines is strongly affected by neutralizing a single negatively charged aspartate residue (Asp172) in the second transmembrane domain (M2) (Stanfield et al., 1994). The corresponding sixth transmembrane domain (TM6) of TRPV6 does not contain any negatively charged residue, but four negative residues (one glutamate and three aspartates) are present in the pore region between TM5 and TM6. To investigate their possible involvement in Mg2+ binding, we replaced these negatively charged residues one by one by neutral amino acids. All four mutants expressed as functional channels, and three of them (E534A, D547S, and D549A) were indiscernible from WT TRPV6 with respect to Mg2+-dependent gating and inward rectification (Fig. 10, A, B, E, and F) . In contrast, the mutant D541A showed no obvious voltage-dependent gating and only moderate inward rectification with 1 mM free intracellular Mg2+ (Fig. 10, C and F). Moreover, omitting Mg2+ from the intracellular solution did not affect the inward rectification and time-independent nature of the currents through this mutant (Fig. 10 E and unpublished data). Interestingly, the D541A mutation also affected the shape of the I-V relation in the absence of intracellular Mg2+, passing significantly more outward current than WT TRPV6 at potentials >50 mV (Fig. 10 D). Similar to the effect of mutating the corresponding amino acid in TRV5 (Nilius et al., 2001), the D541A mutant channel was also insensitive to application of 1 mM Mg2+ to the extracellular solution and only poorly permeable for Ca2+ (unpublished data). Thus, Asp541 is a crucial determinant of the Mg2+ affinity of TRPV6, indicating that it may form part of a Mg2+ binding site within the channel pore.
Figure 10.

Asp541 within the pore region is required for Mg2+ binding. (A–C) Current traces obtained with 1 mM intracellular Mg2+ for the indicated pore mutants using the protocol shown in Fig. 2 C, except that test pulses ranged from −120 to 80 mV. (D) Average normalized I-V relations of Iss for WT TRPV6 and the D541A mutant in the absence of intracellular Mg2+. (E) Comparison of the rectification scores for WT TRPV6 and four pore mutants in the presence and absence of 1 mM intracellular Mg2+. The rectification of Iss was calculated as described in Fig. 3 D. (F) Voltage dependence of the apparent Po with 1 mM intracellular Mg2+ for the four pore mutants. The solid line represents the data for WT TRPV6.
Mg2+ Uncaging Reveals a Second, Slower Inhibitory Action of Intracellular Mg2+
During the course of the present experiments we consistently observed that the TRPV6 current density was inversely correlated to the intracellular Mg2+ concentration, in line with previous findings (Hoenderop et al., 2001). In a specific set of experiments, the average inward Na+ current density at the end of a 100-ms step to −100 mV measured 500 s after establishment of the whole-cell configuration was 762 ± 245 pA/pF (n = 14) with 1 mM free intracellular Mg2+ versus 1712 ± 384 pA/pF (n = 14) in the absence of intracellular Mg2+ (P < 0.05; unpaired t test). To test whether the effect of Mg2+ on overall current density can be separated from the above-described voltage-dependent pore block, we performed experiments using the photolabile Mg2+ chelator DM-nitrophen. Fig. 11 shows an experiment in which a TRPV6-expressing cell dialyzed with a pipette solution to which DM-nitrophen 60% loaded with Mg2+ was added (calculated free Mg2+ concentration of 3.75 μM) was held at 20 mV and stepped to −100 mV for 100 ms every 3.2 s. In Fig. 11 A, the current amplitude at the beginning and end of this step are plotted as a function of time, with on top representative current traces corresponding to the indicated time points. As expected, dialyzing with this low Mg2+ solution led to a rapid washout of the Mg2+-dependent gating, resulting in time-independent inwardly rectifying currents (Fig. 11, A and C). Moreover, the amplitude of the current grew significantly during the first minutes after establishment of the whole-cell configuration. Subsequently, the cell was illuminated with pulses of UV light from the Polychrome IV monochromator (380 nm; 100-ms pulses at 1 Hz during 20 s) in order to partially photolyze the DM-nitrophen. The resulting increase of the free intracellular Mg2+ concentration, which we estimated to reach ∼1 mM, had a biphasic effect on the TRPV6 current. Initially, the inward currents became time dependent, but the steady-state inward current at the end of the 100-ms step was not affected. However, after ∼10 s of increased intracellular Mg2+, the amplitude of the steady-state current also started to decrease (Fig. 11, A and C). Fig. 11 B displays the ratio of initial over steady-state current, as a quantitative measure of voltage-dependent gating. By comparing the time course of this ratio with that of the steady-state current, the different timing of the two Mg2+-dependent processes can be appreciated: the effect on gating reached its maximum at the end of the UV illumination period (Fig. 11 B), whereas the decrease of the steady-state current was maximal ∼60 s later (Fig. 11 A). The effect of UV illumination was only slowly and partially reversible (Fig. 11 A). We attribute this to the fact that not only the cell but also the tip of the patch-pipette had been illuminated with UV light, such that the increase in intracellular Mg2+ outlasted the period of UV illumination for several hundreds of seconds. To exclude the possibility that photoproducts of the DM-nitrophen photolysis contributed to the dual effects of UV illumination, we performed similar experiments using a DM-nitrophen-containing pipette devoid of Mg2+. Under this condition, UV illumination had no effect on the voltage-dependent properties or steady-state amplitude of TRPV6 currents (unpublished data).
Figure 11.

Separation of two effects of intracellular Mg2+ using photolysis of caged Mg2+. Intracellular solution contained 5 mM DM-nitrophen and 3 mM Mg2+. (A) Time course of the inward current measured at the beginning (Iini) and end (Iss) of a 100-ms step to −100 mV from a holding of 20 mV. The traces on top were recorded at the time points indicated by numbers 1–5. (B) Time course of the ratio of Iini over Iss as a measure of the voltage-dependent block by intracellular Mg2+. (C) Current traces obtained at the time points indicated by an arrow in A using the protocol shown in Fig. 2 C, except that test pulses ranged from −120 to 80 mV.
Finally, we investigated whether this slower inhibition is conserved in cell-free inside-out patches (Fig. 12) . Fig. 12 A shows TRPV6 current traces measured after maximal development of the current in an inside-out patch with the cytoplasmic side exposed to a Ca2+- and Mg2+-free solution (10 EDTA). Subsequently, the patch was rapidly exposed to a solution containing 1 mM free Mg2+ (10 EGTA and 2 mM MgCl2). As expected from the above, this leads to a voltage-dependent block of TRPV6, which is observed within 5 s as a reduction of outward currents and a negative slope conductance at potentials below −120 mV (Fig. 12 B; see also Figs. 9 C and 1 A). After this virtually immediate, voltage-dependent effect of intracellular Mg2+, the currents through the patch continued to decrease during the next 20–30 s (Fig. 12 C). This second, slower effect of Mg2+ did not show clear voltage dependence, as evidenced by the close match of the superimposed, normalized I-V relations obtained 5 and 30 s after addition of Mg2+ (Fig. 12 D). On average, the current decrease between 5 and 60 s after addition of 1 mM Mg2+ to the patch amounted to 55 ± 13% at 100 mV and 53 ± 7% at −100 mV (n = 4). Together, these data demonstrate that increasing intracellular Mg2+ has two distinct effects on TRPV6: it causes a direct voltage-dependent block of the channel pore and a slower, voltage-independent inhibition of the channel. Moreover, both effects are preserved in cell-free inside-out patches, indicating that the actions of Mg2+ do not require any soluble cytosolic factors.
Figure 12.

Both effects of intracellular Mg2+ on TRPV6 are preserved in cell-free inside-out patches. The extracellular (pipette) solution contained 150 mM NaCl and 1 mM EDTA. Currents were measured during 400-ms voltage ramps from −150 to 100 mV. (A) I-V relation obtained 200 s after patch excision in a divalent-free solution. At this time point, the TRPV6 current had maximally developed. (B) I-V relation obtained 5 s after addition of 1 mM Mg2+ to the intracellular side of the patch. (C) I-V relation obtained 30 s after addition of 1 mM Mg2+ to the intracellular side of the patch. (D) Overlay of the I-V relations shown in B and D after normalization to the current at 100 mV, illustrating the lack of voltage dependence of the slow Mg2+-dependent inhibition of TRPV6.
DISCUSSION
In this study, we have systematically studied the effects of intracellular Mg2+ on the inward rectification and voltage-dependent gating of the cation channel TRPV6. Our results demonstrate that submillimolar concentrations of intracellular Mg2+ cause voltage-dependent gating of the channel, which contributes to the strong inward rectification of monovalent cation currents through TRPV6. We found that the voltage dependence varies in parallel with the concentration and relative permeability of the main permeant cation, consistent with a direct interaction between the blocking particle Mg2+ and permeant cations within the channel pore. Block is relieved at positive potentials, indicative a “punch through” mechanism whereby Mg2+ escapes from its binding site toward the extracellular space. Moreover, we show that neutralizing a single negatively charged amino acid within the putative selectivity filter fully abolishes the effect of intracellular Mg2+ on inward rectification and voltage dependence of TRPV6. Thus, Mg2+ acts as a voltage-dependent gate for TRPV6 that directly interacts with the selectivity filter and permeates the channel if the driving force is sufficiently large.
It is well known that Mg2+ acts as a gating particle in other types of cation channels (Hille, 2001). In NMDA-type glutamate receptors, block of the channel pore by extracellular Mg2+ causes outward rectification and inhibits glutamate-induced currents at hyperpolarized potentials (Nowak et al., 1984). Conversely, block by intracellular Mg2+ of outward K+ fluxes causes rectification of Kir channels (Nichols and Lopatin, 1997). From our study, a number of parallels between the effects of intracellular Mg2+ on inward rectification of TRPV6 and Kir channels become apparent. First, the interaction of intracellular Mg2+ with both channel types manifests itself as a time-dependent activation upon hyperpolarization and deactivation upon depolarization. Second, the affinity of TRPV6 and Kir channels for Mg2+ is voltage-dependent with apparent KD values at depolarized potentials in the micromolar to low millimolar range (Vandenberg, 1987). Third, the voltage dependence of Mg2+ block of Kir channels and TRPV6 depends on the nature and extracellular concentration of the permeant cation (Horie et al., 1987), indicating that in both channels the permeant cation can knock Mg2+ off its binding site within the pore.
However, there are also some clear differences between the mechanisms underlying inward rectification in Kir channels and TRPV6. First, we show that intracellular Mg2+ can be “punched” through the channel pore, which relieves block at more depolarized potentials, whereas such a relief of block by intracellular Mg2+ has never been observed in Kir channels (Nichols and Lopatin, 1997). Second, inward rectification of Kir channels in the absence of intracellular Mg2+ reflects channel block by polyamines such as spermine and spermidine (Ficker et al., 1994; Lopatin et al., 1994; Fakler et al., 1995). Although an intrinsic gating mechanism has been proposed for Kir channels (Lee et al., 1999), complete washout of Mg2+ and polyamines in inside-out patches, which is typically achieved within ∼100 s (Lopatin et al., 1994), produces intrinsically nonrectifying K+ channels (Guo and Lu, 2002). In contrast, inward rectification of TRPV6 is preserved in inside-out patches after perfusion of the cytoplasmic side of the patch for >1,000 s with Mg2+- and polyamine-free solution, suggesting that TRPV6 displays intrinsic rectification. Finally, high Mg2+ affinity in Kir channels depends on at least two negatively charged residues: one in TM2 (corresponding to TM6 of TRP channels) and one in the COOH-terminal cytoplasmic domain (Stanfield et al., 1994; Wible et al., 1994; Yang et al., 1995). In contrast, we found that block of TRPV6 by intra- and extracellular Mg2+ is abolished after neutralization of a single aspartate (Asp541) that is located in the pore loop between TM5 and TM6. Assuming that the overall structure of the pore of TRP channels is similar to that of K+ channels (Doyle et al., 1998), this finding indicates that the Mg2+ binding site in TRPV6 is located in the selectivity filter, whereas binding of Mg2+ (and polyamines) in Kir channels is thought to occur in the wider channel vestibule closer to the cytoplasmic side of the channel.
The simplest model for voltage-dependent block by a permeant blocker consists of a single binding site in the channel pore that can be reached by Mg2+ from either side of the membrane and from which Mg2+ can be released to either side. Such a simple model fits the steady-state open probability in function of TRPV6 in function of voltage and Mg2+ reasonably well (unpublished data), but cannot explain the experimentally observed double exponential time dependence of the currents, for which more complicated models should be considered. For example, biexponential time dependence can be modeled assuming two distinct binding sites for Mg2+ within the channel pore. Alternatively, one can envisage that the rate of Mg2+ binding/unbinding to a single binding site can vary, for example depending on the transmembrane voltage or the presence of permeant cations in the channel pore. A more detailed structural analysis of the TRPV6 pore would be needed to assess the validity of such more complex models.
When comparing the rectification properties of TRPV6 (and the highly homologous TRPV5) with the two other known types of highly Ca2+-selective channels, namely the superfamily of voltage-gated Ca2+ channels (VGCCs) and the calcium release–activated Ca2+ channel (CRAC), some interesting differences become apparent. TRPV6, VGCCs and CRAC all display anomalous mole fraction behavior in Na+/Ca2+ mixtures, and conduct large monovalent cation currents in the absence of divalent cations (Hess and Tsien, 1984; Hoth and Penner, 1993; Lepple-Wienhues and Cahalan, 1996; Hoenderop et al., 2001; Vennekens et al., 2001). However, monovalent cation currents through TRPV6 and CRAC are inwardly rectifying, even under symmetrical ionic conditions (Hoth and Penner, 1993; Voets et al., 2001; Yue et al., 2001; Bakowski and Parekh, 2002), whereas instantaneous monovalent cation currents through VGCCs are linear (Lux et al., 1990). Additionally, intracellular Mg2+ further increases the degree of inward rectification of TRPV6, whereas it has no significant effect on the I-V relation of CRAC (Hermosura et al., 2002; Kozak et al., 2002; Prakriya and Lewis, 2002). Thus, although the mechanism of selecting between Ca2+ and monovalent cations appears to be similar in TRPV6, CRAC, and VGCCs, the structural elements that underlie rectification and Mg2+ sensitivity clearly differ among these Ca2+-selective channels. Mg2+ sensitivity and inward rectification are therefore instrumental criteria for the comparison of currents though endogenous and cloned channels.
The origin of the “intrinsic” inward rectification of TRPV6 that remains in inside-out patches in the absence of cytoplasmic Mg2+ is presently not known. TRPV6 may be endowed with an intrinsic, voltage-dependent gating mechanism that closes the channel at positive potentials. Alternatively, rectification could originate from the rapid voltage-dependent block of outward current by some unknown cytosolic factor that withstands perfusion of an inside-out patch for more than 1,000 s or by contaminants in the intracellular recording solution. For example, impurities in typical constituents of recording solutions such as HEPES and EDTA have been shown to cause a moderate degree of inward rectification of Kir channels (Guo and Lu, 2002). However, any voltage-dependent gating mechanism would need to be extremely fast (time constant <100 μs), since with our sampling rate we were unable to resolve time-dependent current inactivation at positive potentials in the absence of intracellular Mg2+. Finally and probably more likely, rectification could be an intrinsic property of the channel pore, and reflect the asymmetric distribution of energy barriers along the channel conduction pathway (Hille, 2001). The finding that the D541A mutation also alters the shape of the I-V relation in the absence of Mg2+ could point to an alteration of the energy barriers in the selectivity filter, although alternative explanations are certainly possible. Studying the properties of additional pore mutations may help to further elucidate the origin of the Mg2+-independent rectification of TRPV6.
Our results confirm previous results showing that intracellular Mg2+ has a slow inhibitory action on steady-state TRPV6 current (Hoenderop et al., 2001). By using the photolysable Mg2+ cage DM-nitrophen, we show that this inhibitory effect of Mg2+ can be separated from the voltage-dependent pore block: the voltage-dependent pore block occurs immediately upon Mg2+-uncaging, whereas the slow inhibitory effect develops over ∼60 s after Mg2+ uncaging. The molecular mechanism of this slower inhibitory effect of intracellular Mg2+ is presently unknown, but its voltage independence and slow development argues against a direct binding of Mg2+ in the channel pore. Our finding that the slow inhibition is preserved in inside-out patches points out that no soluble cytosolic factors are required for this Mg2+-dependent effect. Note that voltage-independent inhibition of a TRP channel by intracellular Mg2+ is not unprecedented: TRPM7, a member of the TRPM branch of the TRP superfamily, is inhibited by intracellular Mg2+ ions with a half-maximal inhibition at ∼0.5 mM (Nadler et al., 2001). This inhibition by Mg2+ is regulated by intracellular nucleotides, and it has been hypothesized that this mechanism couples the activity of TRPM7 to the metabolic state of the cell (Nadler et al., 2001). Interestingly, the activity of CRAC channels, which were proposed to be mediated by TRPV6 (Yue et al., 2001; but see Voets et al., 2001; Prakriya and Lewis, 2002), is not significantly modulated by the intracellular Mg2+ concentration (Kozak et al., 2002; Hermosura et al., 2002; Prakriya and Lewis, 2002).
In this study, voltage-dependent block of TRPV6 by intracellular Mg2+ was studied in the complete absence of extracellular divalents or with extracellular Ba2+ as the sole charge carrier. In the presence of millimolar concentrations of extracellular Ca2+, in which case currents through TRPV6 are almost solely carried by Ca2+, an accurate measurement of the extent of block by intracellular Mg2+ was impossible due to the prominent Ca2+-dependent inactivation of the channel (Hoenderop et al., 2001; Yue et al., 2001). At present, the mechanism of Ca2+-dependent inactivation of TRPV6 is still unresolved, although a calmodulin-binding site in the COOH terminus and residues in the intracellular loop between TM2 and TM3 of TRPV6 have been implicated in the process (Niemeyer et al., 2001; Nilius et al., 2002). However, mutations affecting these sites only slow down the inactivation of TRPV6 without fully abolishing it (Niemeyer et al., 2001; Nilius et al., 2002). Therefore, the actual impact of the block by intracellular Mg2+ on Ca2+ influx through TRPV6 and its physiological implications remain unclear. If the linear relation between reversal potential and VPo = 0.5 (Fig. 6) holds true for Ca2+ currents, one would expect that TRPV6 currents at 1 mM extracellular Ca2+, which reverse around 30 mV (Hoenderop et al., 2001; Yue et al., 2001), would be half maximally blocked by 1 mM intracellular Mg2+ in the voltage range between 0 and 20 mV. TRPV6 is prominently expressed in the apical membrane of enterocytes, and has been proposed to function as the major gatekeeper for Ca2+ absorption in the intestine (Peng et al., 1999). In conditions where the diet contains very low amounts of Ca2+ and/or high concentrations of Ca2+ chelating molecules, the free Ca2+ concentration in the intestinal lumen may drop sufficiently to allow significant monovalent TRPV6 currents (Mupanomunda et al., 1999). Under such conditions, the voltage-dependent block of TRPV6 by intracellular Mg2+ ions could be important to prevent depolarization of the apical membrane and massive Na+ influx into the enterocytes.
In conclusion, our data demonstrate that intracellular Mg2+ acts as a permeant pore blocker of TRPV6 and contributes to the strong inward rectification of this channel, and that Mg2+-binding requires an aspartate in the putative selectivity filter of the pore. Additionally, high intracellular Mg2+ also induces a slow, voltage-independent inhibition of TRPV6. These Mg2+-dependent effects may contribute to the regulation of TRPV6 function in vivo and represent instrumental criteria for the molecular identification of TRPV6-like channels in native cells.
Acknowledgments
We thank Karel Talavera for helpful discussions.
T. Voets is a postdoctoral Fellow of the Fund for Scientific Research-Flanders (Belgium) (FWO-Vlaanderen). This work was supported by the Belgian Federal Government, the Flemish Government, and the Onderzoeksraad KULeuven (F.W.O. G.0214.99, F.W.O. G.0136.00; F.W.O. G.0118.00, F.W.O. G.0172.03, and the Interuniversity Poles of Attraction Program, Prime Ministers Office IUAP).
Olaf S. Andersen served as editor.
References
- Bakowski, D., and A.B. Parekh. 2002. Monovalent cation permeability and Ca2+ block of the store-operated Ca2+ current ICRAC in rat basophilic leukemia cells. Pflugers Arch. 443:892–902. [DOI] [PubMed] [Google Scholar]
- Clapham, D.E., L.W. Runnels, and C. Strubing. 2001. The TRP ion channel family. Nat. Rev. Neurosci. 2:387–396. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., J. Morais Cabral, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Fakler, B., U. Brandle, E. Glowatzki, S. Weidemann, H.P. Zenner, and J.P. Ruppersberg. 1995. Strong voltage-dependent inward rectification of inward rectifier K+ channels is caused by intracellular spermine. Cell. 80:149–154. [DOI] [PubMed] [Google Scholar]
- Ficker, E., M. Taglialatela, B.A. Wible, C.M. Henley, and A.M. Brown. 1994. Spermine and spermidine as gating molecules for inward rectifier K+ channels. Science. 266:1068–1072. [DOI] [PubMed] [Google Scholar]
- Guo, D., and Z. Lu. 2002. IRK1 Inward Rectifier K+ Channels Exhibit No Intrinsic Rectification. J. Gen. Physiol. 120:539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermosura, M.C., M.K. Monteilh-Zoller, A.M. Scharenberg, R. Penner, and A. Fleig. 2002. Dissociation of the store-operated calcium current ICRAC and the Mg-nucleotide-regulated metal ion current MagNuM. J. Physiol. 539:445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, P., and R.W. Tsien. 1984. Mechanism of Ion Permeation through Calcium Channels. Nature (Lond.). 309:453–456. [DOI] [PubMed] [Google Scholar]
- Hille, B. 2001. Ion Channels of Excitable Membranes. Sinauer Associates, Sunderland, MA.
- Hoenderop, J.G., A.W. van der Kemp, A. Hartog, S.F. van de Graaf, C.H. van Os, P.H. Willems, and R.J. Bindels. 1999. Molecular identification of the apical Ca2+ channel in 1, 25-dihydroxyvitamin D3-responsive epithelia. J. Biol. Chem. 274:8375–8378. [DOI] [PubMed] [Google Scholar]
- Hoenderop, J.G., R. Vennekens, D. Müller, J. Prenen, G. Droogmans, R.J. Bindels, and B. Nilius. 2001. Function and expression of the epithelial Ca2+ channel family: comparison of the epithelial Ca2+ channel 1 and 2. J. Physiol. 537:747–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie, M., H. Irisawa, and A. Noma. 1987. Voltage-dependent magnesium block of adenosine-triphosphate-sensitive potassium channel in guinea-pig ventricular cells. J. Physiol. 387:251–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth, M., and R. Penner. 1993. Calcium release-activated calcium current in rat mast cells. J. Physiol. 465:359–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak, J.A., H.H. Kerschbaum, and M.D. Cahalan. 2002. Distinct properties of CRAC and MIC channels in RBL cells. J. Gen. Physiol. 120:221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.K., S.A. John, and J.N. Weiss. 1999. Novel gating mechanism of polyamine block in the strong inward rectifier K channel Kir2.1. J. Gen. Physiol. 113:555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepple-Wienhues, A., and M.D. Cahalan. 1996. Conductance and permeation of monovalent cations through depletion-activated Ca2+ channels (ICRAC) in Jurkat T cells. Biophys. J. 71:787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopatin, A.N., E.N. Makhina, and C.G. Nichols. 1994. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature (Lond.). 372:366–369. [DOI] [PubMed] [Google Scholar]
- Lux, H.D., E. Carbone, and H. Zucker. 1990. Na+ currents through low-voltage-activated Ca2+ channels of chick sensory neurones: block by external Ca2+ and Mg2+. J. Physiol. 430:159–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mupanomunda, M.M., N. Ishioka, and R.D. Bukoski. 1999. Interstitial Ca2+ undergoes dynamic changes sufficient to stimulate nerve-dependent Ca2+-induced relaxation. Am. J. Physiol. 276:H1035–H1042. [DOI] [PubMed] [Google Scholar]
- Nadler, M.J., M.C. Hermosura, K. Inabe, A.L. Perraud, Q. Zhu, A.J. Stokes, T. Kurosaki, J.P. Kinet, R. Penner, A.M. Scharenberg, and A. Fleig. 2001. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature (Lond.). 411:590–595. [DOI] [PubMed] [Google Scholar]
- Nichols, C.G., and A.N. Lopatin. 1997. Inward rectifier potassium channels. Annu. Rev. Physiol. 59:171–191. [DOI] [PubMed] [Google Scholar]
- Niemeyer, B.A., C. Bergs, U. Wissenbach, V. Flockerzi, and C. Trost. 2001. Competitive regulation of CaT-like-mediated Ca2+ entry by protein kinase C and calmodulin. Proc. Natl. Acad. Sci. USA. 98:3600–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius, B., J. Prenen, J.G. Hoenderop, R. Vennekens, S. Hoefs, A.F. Weidema, G. Droogmans, and R.J. Bindels. 2002. Fast and slow inactivation kinetics of the Ca2+ channels ECaC1 and ECaC2 (TRPV5 and TRPV6). Role of the intracellular loop located between transmembrane segments 2 and 3. J. Biol. Chem. 277:30852–30858. [DOI] [PubMed] [Google Scholar]
- Nilius, B., R. Vennekens, J. Prenen, J.G. Hoenderop, G. Droogmans, and R.J. Bindels. 2001. The single pore residue Asp542 determines Ca2+ permeation and Mg2+ block of the epithelial Ca2+ channel. J. Biol. Chem. 276:1020–1025. [DOI] [PubMed] [Google Scholar]
- Nowak, L., P. Bregestovski, P. Ascher, A. Herbet, and A. Prochiantz. 1984. Magnesium gates glutamate-activated channels in mouse central neurones. Nature (Lond.). 307:462–465. [DOI] [PubMed] [Google Scholar]
- Peng, J.B., X.Z. Chen, U.V. Berger, P.M. Vassilev, H. Tsukaguchi, E.M. Brown, and M.A. Hediger. 1999. Molecular cloning and characterization of a channel-like transporter mediating intestinal calcium absorption. J. Biol. Chem. 274:22739–22746. [DOI] [PubMed] [Google Scholar]
- Prakriya, M., and R.S. Lewis. 2002. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. J. Gen. Physiol. 119:487–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runnels, L.W., L. Yue, and D.E. Clapham. 2001. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 291:1043–1047. [DOI] [PubMed] [Google Scholar]
- Schindl, R., H. Kahr, I. Graz, K. Groschner, and C. Romanin. 2002. Store depletion-activated CaT1 currents in rat basophilic leukemia mast cells are inhibited by 2-aminoethoxydiphenyl borate. Evidence for a regulatory component that controls activation of both CaT1 and CRAC (Ca2+ release-activated Ca2+ channel) channels. J. Biol. Chem. 277:26950–26958. [DOI] [PubMed] [Google Scholar]
- Stanfield, P.R., N.W. Davies, P.A. Shelton, M.J. Sutcliffe, I.A. Khan, W.J. Brammar, and E.C. Conley. 1994. A single aspartate residue is involved in both intrinsic gating and blockage by Mg2+ of the inward rectifier, IRK1. J. Physiol. 478:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trouet, D., B. Nilius, T. Voets, G. Droogmans, and J. Eggermont. 1997. Use of a bicistronic GFP-expression vector to characterize ion channels after transfection in mammalian cells. Pflugers Arch. 434:632–638. [DOI] [PubMed] [Google Scholar]
- Vandenberg, C.A. 1987. Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc. Natl. Acad. Sci. USA. 84:2560–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennekens, R., J.G. Hoenderop, J. Prenen, M. Stuiver, P.H. Willems, G. Droogmans, B. Nilius, and R.J. Bindels. 2000. Permeation and gating properties of the novel epithelial Ca2+ channel. J. Biol. Chem. 275:3963–3969. [DOI] [PubMed] [Google Scholar]
- Vennekens, R., J. Prenen, J.G. Hoenderop, R.J. Bindels, G. Droogmans, and B. Nilius. 2001. Pore properties and ionic block of the rabbit epithelial calcium channel expressed in HEK 293 cells. J. Physiol. 530:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets, T., J. Prenen, A. Fleig, R. Vennekens, H. Watanabe, J.G. Hoenderop, R.J. Bindels, G. Droogmans, R. Penner, and B. Nilius. 2001. CaT1 and the calcium-release activated calcium channel manifest distinct pore properties. J. Biol. Chem. 276:47767–47770. [DOI] [PubMed] [Google Scholar]
- Wible, B.A., M. Taglialatela, E. Ficker, and A.M. Brown. 1994. Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature (Lond.). 371:246–249. [DOI] [PubMed] [Google Scholar]
- Yang, J., Y.N. Jan, and L.Y. Jan. 1995. Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+ channel. Neuron. 14:1047–1054. [DOI] [PubMed] [Google Scholar]
- Yue, L., J.B. Peng, M.A. Hediger, and D.E. Clapham. 2001. CaT1 manifests the pore properties of the calcium-release-activated calcium channel. Nature (Lond.). 410:705–709. [DOI] [PubMed] [Google Scholar]