

Abstract
The selectivity filter of all known T-type Ca2+ channels is built by an arrangement of two glutamate and two aspartate residues, each one located in the P-loops of domains I–IV of the α1 subunit (EEDD locus). The mutations of the aspartate residues to glutamate induce changes in the conduction properties, enhance Cd2+ and proton affinities, and modify the activation curve of the channel. Here we further analyze the role of the selectivity filter in the gating mechanisms of T-type channels by comparing the kinetic properties of the α1G subunit (CaV3.1) to those of pore mutants containing aspartate-to-glutamate substitution in domains III (EEED) or IV (EEDE). The change of the extracellular pH induced similar effects on the activation properties of α1G and both pore mutants, indicating that the larger affinity of the mutant channels for protons is not the cause of the gating modifications. Both mutants showed alterations in several gating properties with respect to α1G, i.e., faster macroscopic inactivation in the voltage range from −10 to 50 mV, positive voltage shift and decrease in the voltage sensitivity of the time constants of activation and deactivation, decrease of the voltage sensitivity of the steady-state inactivation, and faster recovery from inactivation for long repolarization periods. Kinetic modeling suggests that aspartate-to-glutamate mutations in the EEDD locus of α1G modify the movement of the gating charges and alter the rate of several gating transitions. These changes are independent of the alterations of the selectivity properties and channel protonation.
Keywords: pH, activation, selectivity filter, pore mutant, CaV3.1
INTRODUCTION
One of the corner stones of the classical description of ion channels is the uncoupling between the mechanisms of gating and the ionic conduction through the pore. A large number of structure-function studies in many channel types have supported this idea by showing that special protein structures like the S4 segments account for voltage sensing that controls channel activation and that the pore loops between S5 and S6 segments determine the ionic conduction. However, a usual concomitant outcome is that the pore structure influences the gating processes (Hille, 2001). Mutations of pore residues modify the gating properties of potassium channels (Taglialatela et al., 1992; Lopez-Barneo et al., 1993; Heginbotham et al., 1994; Becker et al., 1996; Molina et al., 1998; Kiss et al., 1999; Proks et al., 2001; Yellen, 2002), sodium channels (Tomaselli et al., 1995; Balser et al., 1996; Townsend and Horn, 1999; Hilber et al., 2001; Sheng et al., 2001; Kuhn and Greeff, 2002), and CNG channels (Flynn et al., 2001). To our knowledge, although there are a large number of studies regarding the structural determinants of ionic permeation and selectivity in high voltage–activated (HVA)* Ca2+ channels (Ugarte et al., 1998; McCleskey, 1999; Varadi et al., 1999), there is only one report addressing the relationship between pore structure and gating mechanisms in these channels. Yatani et al. (1994) showed that the replacement of the P-loop segments of repeats III or IV of the L-type Ca2+ channel with the corresponding sequence of the brain (BI-2) Ca2+ channel induced changes in the kinetics of macroscopic activation and inactivation.
Recently, we have studied permeation properties of the T-type Ca2+ channel α1G, in comparison with pore mutants containing aspartate-to-glutamate substitutions in the pore loops of domains III and IV (called EEED, EEDE and EEEE) (Talavera et al., 2001). We showed that the aspartate residues of the characteristic EEDD pore locus of α1G control several of its conduction properties, i.e., the Ca2+/Ba2+ selectivity, the absence of anomalous mole fraction effect between Ca2+ and Ba2+, the low Cd2+ sensitivity and the proton block. In addition, and unexpectedly, all pore mutants showed alterations in the activation curves with respect to α1G, i.e., a positive shift in the voltage for half-maximal activation and an increase in the slope factor of the activation. These modifications were similar to those promoted by low extracellular pH (pHe) in the T-type Ca2+ channel α1H (Delisle and Satin, 2000). Thus, we suggested that the alterations in the activation curves induced by the aspartate-to-glutamate mutations could be due to the enhanced proton sensitivity of the mutant channels. In the companion paper we prove that the mechanisms of proton block in the T-type Ca2+ channels α1G and α1H are qualitatively similar, and give additional evidence that the EEDD pore locus is a target for protonation. The purpose of the present experiments was to investigate the mechanism by which the structure of the selectivity filter influences the activation curve. To this end we have tested the protonation hypothesis described above against the possible modification of the activation process as a direct result of the changed structure of the selectivity filter. We found that the structure of the selectivity filter influences the gating of α1G independently of proton effects. The differential gating properties of the pore mutants with respect to α1G can be explained by kinetic models that predict a redistribution of the gating charges associated with each gating step and the modification of the rate of several channel transitions.
MATERIALS AND METHODS
Solutions
Before current recordings, cells were rinsed with Krebs solution containing: 150 mM NaCl, 6 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 10 mM glucose, 10 mM HEPES, and titrated to pH 7.4 with 1 N NaOH. The standard extracellular solution contained 20 mM CaCl2, 120 mM NMDG, 5 mM CsCl, 5 mM glucose, and 10 mM HEPES. HEPES was substituted by a mixture of MES, HEPES and TAPS (5 mM each) when studying the effect of pHe to expand the buffering range from pHe 5.5 to 9.1. All test solutions were titrated with HCl and were kept Mg2+ free to avoid extracellular Mg2+ block (Serrano et al., 2000). The intracellular (pipette) solution contained 102 mM CsCl, 10 mM HEPES, 5 mM MgCl2, 5 mM Na2-ATP, 10 mM TEA-Cl, 10 mM EGTA, titrated to pH 7.4 with 1 N CsOH. All chemicals were purchased from Sigma-Aldrich.
Electrophysiology
We used human embryonic kidney cells (HEK293) transfected with the wild-type α1G or the EEED and EEDE pore mutants as in previous works (Staes et al., 2001; Talavera et al., 2001). Currents were recorded in the whole-cell configuration of the patch-clamp technique using an EPC-7 (LIST Electronics) patch-clamp amplifier and filtered with an eight-pole Bessel-filter (Kemo). For control of voltage-clamp protocols and data acquisition, we used an IBM-compatible PC with a TL-1 DMA interface (Axon Instruments, Inc.) and the software pCLAMP (Axon Instruments, Inc.). Bath solutions were perfused by gravity via a multibarreled pipette. Patch pipettes were pulled from Vitrex capillary tubes (Modulohm) using a DMZ-Universal puller (Zeitz-instruments). An Ag-AgCl wire was used as reference electrode. Adequate voltage control was achieved by using low pipette resistances (1–2.5 MΩ) and series resistance compensation to the maximum extent possible (50–70%). Membrane capacitive transients were electronically compensated and the linear background components were digitally subtracted before data analysis. Current traces were filtered at 2.5–5 kHz and digitized at 5–10 kHz. All experiments were performed at room temperature (22–25°C).
Stimulation Protocols and Data Analysis
I-V curves were obtained from the peak amplitude of currents evoked by the application 200-ms voltage steps from −90 to 60 mV. I-V curves of the EEED and the EEDE pore mutants the were fitted to:
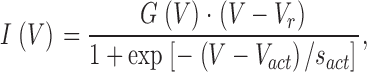 |
(1) |
where I is the measured peak current, V the step potential, V act the potential of half-maximal activation, s act the slope parameter of activation, and G(V) the conductance, which is voltage dependent. For the wild-type α1G, the steady-state activation was best described by the sum of two Boltzmann components as described in the accompanying paper, Talavera et al. (2003)(this issue). To account for the strong inward rectification, we have approximated G(V) from a fit of the amplitudes of the tail current after a depolarizing step to 100 mV in the voltage range from −70 to 110 mV for α1G and from −70 to 70 mV for the pore mutants to the equation:
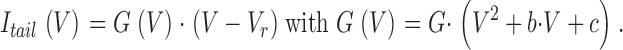 |
(2) |
The average values of the fitted parameters b and c for each experimental condition were then used to fit the I-V curves to Eq. 1.
The time constants of inactivation (τinac) were determined from single-exponential fit of the decaying phase of the current traces. Because of the relatively small amplitude of the currents through the mutant channels, the direct determination of the time constant of activation was sometimes impaired by the presence of residual capacitive transients. Thus, the time constant of activation (τact) was estimated from the time to the peak (t p) and τinac, by solving iteratively the following transcendent equation, based on an m 2h model of gating:
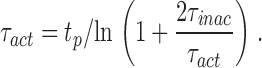 |
(3) |
The decaying phase of the voltage dependence of τact was fitted with an exponential function of the form:
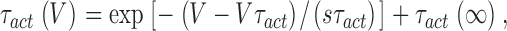 |
(4) |
where sτact is the voltage sensitivity, τact(∞) is the asymptotic value at positive potentials and Vτact is the voltage at which τact is equal to 1 + τact(∞).
Steady-state inactivation (h ∞) was determined from the peak current recorded during a 160-ms test pulse to 0 mV after a 5,120-ms lasting prepulse to potentials between −100 and −25 mV. These peak currents were normalized to that following the prepulse to −100 mV and the voltage dependence was fitted by the equation:
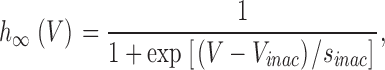 |
(5) |
where V inac is the potential of half-maximal inactivation and s inac the slope factor for the inactivation.
Tail currents were recorded during voltage steps between −140 and 110 mV after a 7.5-ms lasting depolarization to 100 mV. At each voltage, we determined the amplitude I tail and time constant of current decay τdecay from a single-exponential fit of the time course of the tail currents. τdecay corresponds to the time constant of deactivation (τdeac) at potentials negative to the activation threshold and to the time constant of the macroscopic inactivation (τinac) at more positive potentials. The voltage dependence of τdecay can be expressed by:
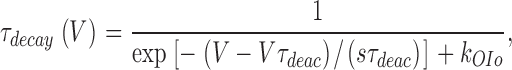 |
(6) |
where V is the repolarisation potential, Vτdeac the voltage at which τdeac is equal to 1 ms, sτdeac the voltage sensitivity of the time constant of deactivation, and k OIo is the rate constant of the transition between the open state (O) and the closest inactivated state (Io) (see Talavera et al., 2003, in this issue, for details).
Reactivation kinetics at −100 mV were determined using a standard double pulse protocol to 0 mV. Reactivation was calculated as a function of the interpulse duration by the ratio between the current amplitude in each pulse (I(pulse2)/I(pulse1)) and was fit by a double exponential function of the form:
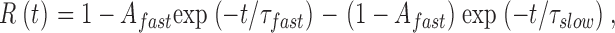 |
(7) |
where τslow and τfast are the time constants describing slow and fast reactivation components, respectively. A fast is the probability for the channels to reactivate via the fast component.
In all voltage protocols the holding potential was −100 mV and the stimulation frequency 0.5 Hz, with the exception of the inactivation protocol in which it was 0.2 Hz. The data were analyzed using WinASCD (ftp://ftp.cc.kuleuven.ac.be/pub/droogmans/winascd.zip; G. Droogmans, Laboratory of Physiology, KU Leuven). For all measurements, pooled data are given as mean ± SEM. We used Student's t test, taking P < 0.05 or P < 0.01 as the level of significance.
Data Simulation
We used MATLAB (MathWorks) to solve a Markov model for the gating of the EEED and EEDE mutants in the presence of 20 mM extracellular [Ca2+] and pHe 7.4. Parameter optimization and numerical solution of the differential equations were performed with the built-in functions fmin and expm, respectively.
RESULTS
Aspartate-to-glutamate Mutations in the Selectivity Filter Affect Macroscopic Kinetics of α1G
We have shown previously that exchanging the aspartate residues (D1487 and/or D1810) in the selectivity filter of α1G for glutamate shifted the activation curve of the channel to more positive potentials and decreased its voltage sensitivity (Talavera et al., 2001). In this paper we extended the kinetic characterization of the EEED and the EEDE mutants in comparison with the wild-type α1G. The shift of the activation curve of these mutants can be noticed from the current traces shown in Fig. 1 A. At pHe 7.4 step depolarization to −40 mV induced a current through α1G but not through the mutant channels, and maximal inward current was attained near −20 mV in α1G but close to 0 mV in both mutants. Panel B shows that the times to the peak (t p) were not significantly different between mutant channels and α1G. However, the time constant of macroscopic inactivation of the mutants (τinac) were significantly larger at −30 mV but about twofold smaller at potentials between −10 and 50 mV (Fig. 1 C) compared with the corresponding values of α1G. Fig. 1 D shows that the estimated time constant of activation (τact) was larger for both mutant channels than for α1G in the voltage range from −10 to 30 mV. The fit of Eq. 4 to the voltage dependence of τact for the different channels yields that there is a significant shift to positive potentials and decreased voltage sensitivity in the mutant channels with respect to α1G (Fig. 1 D, inset). However, the saturating values of τact at very positive potentials were not different among these channels: τact(∞) was 0.53 ± 0.04 ms, 0.51 ± 0.05 ms, and 0.5 ± 0.05 ms for α1G and the EEED and EEDE mutants, respectively.
Figure 1.

Comparison of activation and inactivation kinetics for α1G and the pore mutants. (A) Current traces recorded from typical cells expressing α1G or the EEED or EEDE pore mutants during voltage steps to the potentials indicated by the labels. B and C show the voltage dependences of the average time to peak (t p) and the average time constant of macroscopic inactivation (τinac), respectively, for α1G (▪, N = 11) and the EEED (○, N = 8) and EEDE (▵, N = 6) pore mutants. Continuous lines are the voltage dependences of t p and τinac predicted by the kinetic models for α1G and the pore mutants (see discussion). (D) Estimated time constant of activation as function of test potential; solid lines are the fit of experimental data with Eq. 4 from which the average Vτact and sτact shown in the inset were obtained (α1G, filled bars; EEED, dashed bars; and EEDE, empty bars). The double asterisks denote significant difference from the values obtained for α1G with P < 0.01.
The Effects of Aspartate-to-glutamate Mutations in the EEDD Pore Locus on the Activation Mechanism of α1G Are Not Related to the Proton Effects
Alterations in the activation curves of the EEED and EEDE pore mutants could be due to an enhanced effect of protons on gating caused by their increased binding of protons to the selectivity filter (Talavera et al., 2001). To test this hypothesis we compared the effects of changing the pHe on the I-V curves of these mutants and α1G (Fig. 2 , A–C). A pHe change from 9.1 to 7.4 did not affect the I-V curve of α1G. Acidification to pHe 6.8 decreased the inward current, whereas a stronger acidification also shifted the peak inward current to more positive potentials and decreased the activation slope of the I-V curve. Both mutant channels were more sensitive to pHe changes: a decrease of pHe from 9.1 to 8.2 already reduced the peak inward current. However, the shift of the I-V curve and the reduction of the activation slope were only clear at pHe 6.2 or below, similar to the observations from the wild-type channel. Extracellular acidification to pH 5.5 dramatically shifted the reversal potential of the EEED mutant to less positive values and allowed a robust flux of outward currents (Fig. 2 B), whereas little outward current was observed in α1G and the EEDE mutant.
Figure 2.

Effects of extracellular protons on the I-V curves of α1G and the pore mutants. (A–C) I-V curves obtained from typical cells expressing α1G or the EEED or the EEDE pore mutants, respectively, at different pHe (9.1, ▪; 8.2, □; 7.4, ○; 6.8, ▵; 6.2, ▿; 5.5, ⋄). (D) Activation curves of α1G (▪, n = 7), EEED (○, n = 8) and EEDE (▵, n = 6) at pHe 9.1. (E and F) pHe dependence of the voltage for half-maximal activation (V act) and the slope factor of the activation (s act), respectively (same legend as in D).
A change of pHe in the range from 9.1 to 6.8 did not significantly alter the activation properties of α1G, while acidification below 6.2 significantly shifted the voltage for half-maximal activation (V act) toward positive potentials and increased the slope factor (s act) of the steepest component of the activation (see Talavera et al., 2003, in this issue). If the modifications of activation in the mutant channels were due to their larger proton affinity, it would be expected the pHe dependence of V act and s act were shifted along the pHe axis in the alkaline direction with respect to the wild-type channel. Accordingly, the activation curves of the mutants would resemble that of α1G at high pHe. However, this does not occur, as the difference in activation between the mutants and the wild-type channel persist at pHe as high as 9.1 (Fig. 2 D). Furthermore, the variations of V act and s act with pHe followed the same qualitative pattern as in α1G (Fig. 2, E and F). The activation curves of both mutant channels showed an ∼20-mV positive shift and more than twofold increase in s act with respect to α1G, independently from changes in pHe between 9.1 and 5.5. These results indicate that the changes in the activation process of α1G due to the aspartate-to-glutamate mutations are not related to proton effects on channel function, but to the modification of the structure of the selectivity filter itself. Therefore, we have also compared other gating properties of the pore mutants and α1G. Given that the pHe effects in α1G and the mutant channels were similar at all pHe values, all further experiments were conducted at pHe 7.4.
Steady-state Inactivation of α1G and the Pore Mutants
Because T-type channel inactivation is coupled to the activation process (Droogmans and Nilius, 1989; Chen and Hess, 1990), it was interesting to test whether the modifications in activation kinetics induced by the mutations in the selectivity filter were accompanied by changes in steady-state inactivation. Fig. 3 A shows typical current traces recorded when applying the inactivation protocol to cells expressing α1G or the mutant channels. Comparison of average inactivation curves shown in Fig. 3 B indicate that the aspartate-to-glutamate mutations induced some changes in the steady-state inactivation of α1G. The voltage for half-maximal inactivation (V inac) was not different between α1G (−52.4 ± 1.2 mV), the EEED mutant (−54.2 ± 0.7 mV), and the EEDE mutant (−50.2 ± 1.0 mV) (Fig. 6 A). The slope factor (s inac) was significantly different from that of wild-type (3.8 ± 0.2 mV) in both EEED (5.96 ± 0.16 mV, P < 0.01) and EEDE (4.76 ± 0.15, P < 0.01) mutants (Fig. 6 B).
Figure 3.

Voltage dependence of steady-state inactivation of α1G and the pore mutants. (A) Typical current traces recorded during the application of the steady-state inactivation protocol in cells expressing α1G or the EEED or EEDE pore mutants. The dotted traces correspond to the prepulse to −50 mV. Average voltage dependence of the steady-state inactivation of α1G (▪, n = 6), EEED (○, n = 5) and EEDE (▵, n = 10). Continuous lines are the inactivation curves calculated from Eq. 5 using the average values of V inac and s inac determined for each channels type.
Figure 6.

Summary of gating properties of the EEED and EEDE mutants compared with the α1G channel. (A) Average voltage for half-maximal inactivation (V inac). (B) Slope factor of the voltage dependence of the steady-state inactivation (s inac). (C) Voltage at which τdeac is equal to 1 ms (Vτdeac). (D) Voltage sensitivity of the time constant of deactivation (sτdeac). (E) Average fast (τfast, dashed bars) and slow (τslow, empty bars) time constants of recovery from inactivation. (F) Probability for the channels to reactivate via the fast component, A fast. (**), denote significant difference respect to the values obtained for the wild-type α1G with P < 0.01.
Kinetics of Deactivation and Macroscopic Inactivation Are Faster in the EEED and EEDE Mutants
In the accompanying paper we demonstrated that deactivation of the EEED mutant is faster than that of α1G at pHe values of 9.1 and 6.2. In this work we extend this comparison to the EEDE mutant at pHe 7.4. Fig. 4 A shows typical tail current traces through α1G and both pore mutants, recorded during the repolarization to −80 or 0 mV after a 7.5-ms lasting prepulse to 100 mV. The decay of the tail currents, which were clearly faster in the mutants than in the wild-type channel, could always be described by a single exponential function. Fig. 4 B shows the voltage dependence of the average time constant of current decay (τdecay) as a function of the repolarization potential for the various channels. At potentials negative to the activation threshold, τdecay corresponds to the time constant of deactivation τdeac, which was clearly smaller for the pore mutants than for α1G in the voltage range studied. The fit of the experimental data with Eq. 6 allows determining the properties of the voltage dependence of τdeac. The value of voltage at which τdeac was equal to 1 ms (Vτdeac) was used as an index for the position along the voltage axis and was −95.5 ± 1.4 mV and −90.1 ± 1.6 mV for the EEED and the EEDE, respectively, which are less negative than the value −116 ± 2 mV for α1G (Fig. 6 C). In addition, the voltage sensitivity of the deactivation was significantly smaller for the EEED (sτdeac = 35.6 ± 1.4 mV, P < 0.01) and the EEDE mutant (sτdeac = 35.5 ± 1.5 mV, P < 0.01), than for α1G (sτdeac = 24.0 ± 0.6 mV) (Fig. 6 D). At potentials around the activation threshold the decay of tail currents is determined by both deactivation and inactivation processes. Interestingly, the faster decay of tail currents in the mutants compared with the wild-type in the voltage range from −10 to 50 mV is in line with the faster macroscopic inactivation during step depolarizations (Fig. 1 C). Note that the differences in the rate of the tail current decay tend to disappear at very positive potentials.
Figure 4.

Aspartate-to-glutamate mutations in the selectivity filter of α1G induced faster deactivation kinetics. (A) Typical tail current traces recorded at −80 mV (left) or at 0 mV (right) from cells expressing α1G or the EEED or EEDE mutant channels. (B) Voltage dependence of average time constant of the decay (τdecay) of tail currents recorded after 7.5-ms depolarization to 100 mV for α1G (▪, n = 14) and the EEED (○, n = 11) and EEDE (▵, n = 12) mutants. Continuous lines are functions of the form of Eq. 6 calculated using the average values of Vτdeac and sτdeac determined for each channel type.
Aspartate-to-glutamate Mutations Induce Changes in the Reactivation Kinetics
Given that the aspartate-to-glutamate mutations induced changes in the inactivation properties, it was interesting to examine if kinetics of the recovery from inactivation were altered as well by these pore mutations. Fig. 5 A shows current traces elicited by the application of the reactivation protocol in three cells expressing α1G or the pore mutants. Average reactivation curves for each channel type are shown in Fig. 5 B. Both pore mutants displayed a slower recovery for short repolarization periods but a faster recovery for intervals >200 ms. To characterize the reactivation kinetics we fitted the experimental data with Eq. 7, which yielded two time constants (τfast and τslow) and the fraction of reactivation (A fast) that occurs via the fast process. τfast was larger in both mutant channels (τfast = 150 ± 11 ms and 132 ± 10 ms for EEED and EEDE, respectively), accounting for their slower reactivation for short reactivation periods compared with α1G (τfast = 70 ± 3 ms). However, τslow was not significantly altered by the pore mutations (Fig. 6 E). The faster reactivation in the mutant channels for long repolarization periods is accounted for by the larger fraction of recovery that occurs through the fast process in the mutants; A fast = 0.83 ± 0.08 and 0.66 ± 0.05, for EEED and EEDE, respectively, compared with 0.46 ± 0.01 for the wild-type channel (Fig. 6 F).
Figure 5.

Reactivation kinetics of α1G and the pore mutants. (A) current traces recorded during the application of the reactivation protocol to typical cells expressing α1G or the EEED or EEDE pore mutants. (B) Average reactivation curves of α1G (▪, n = 6) and the EEED (○, n = 8) and EEDE (▵, n = 8) mutants. Continuous lines are the fit of the experimental points to Eq. 7.
DISCUSSION
The segregation of the ionic permeation from the gating processes is a comfortable hypothesis in the analysis of ionic channel function. It allows the description of ionic currents as the multiplication of the parameters describing the state of the channel gates by the amplitude of the single channel current (Hille, 2001). Although special structures determine the gating or the conduction properties of the channels (e.g., S4 segments and P-loops), recent studies involving the mutation of residues in the pore loops of several types of ion channels suggest that ionic transport and pore structure are related to the gating mechanisms (see introduction). This work presents the first analysis of the gating alterations induced by the modification of the pore structure of T-type Ca2+ channels.
The Modification of the Activation Curve by Mutations in the Selectivity Filter Is Not Related to Channel Protonation
We have shown previously that at pHe 7.4, the mutation of the pore residues D1487 and D1810 for glutamate induce a positive shift in the voltage for half-maximal activation and an increase in the slope factor of the activation of the T-type channel α1G (Talavera et al., 2001). It could be hypothesized that these modifications are due to an increased proton sensitivity of the mutants, such that at pHe 7.4 these channels undergo proton-induced modifications in channel gating (Tytgat et al., 1990; Delisle and Satin, 2000), which can only be observed at lower pHe values in the wild-type α1G. We tested this hypothesis by comparing the effects of extracellular protons on the activation properties of α1G and the EEED and EEDE pore mutants. Although there are marked differences in the voltage of half-maximal activation (V act) and the slope factor (s act) between the mutants and α1G, the pHe dependency of these parameters in range from 9.1 to 5.5 is similar for the wild-type and the mutated channels. These results indicate that the differences in the activation properties of the pore mutants are not related to channel protonation and that the proton effects on T-type channel activation are not controlled by the protonation of the selectivity filter.
Possible Mechanisms of Gating Modification by Alterations in the Pore Structure
The alteration of gating properties by mutations in the pore may result from the modification of the substrate for ions that modulate channel gating. Evidence from Shaker K+ channels indicates that K+ permeation, TEA+ block and pore mutations that alter selectivity for K+ influence the C-type inactivation, which consists in a constriction of the outer mouth of the pore (Yellen, 1998; Kiss et al., 1999). Similarly, the reduction of the extracellular concentration of permeant ions decreases the open probability of cardiac Na+ channels at positive potentials, with this effect being susceptible to mutations in the selectivity filter (Townsend et al., 1997; Townsend and Horn, 1999).
Shuba et al. (1991) showed that the type and concentration of the permeant ion affect the mean open time of the T-type Ca2+ channel in mouse neuroblastoma cells and proposed a relationship between the ion permeation rate and the local dielectric permeability affecting the voltage sensing. They hypothesized that ion-specific interactions with the selectivity filter are the basis of the particular kinetic modulation by each permeant ion and suggested that channel closure involves a narrowing of the selectivity filter. However, we find some limitations in the interpretation given by these authors since they did not address how important factors like the screening of surface charges or proton effects could contribute to the diverse gating properties observed in the presence of different permeant ions. Recently, Alvarez et al. (2000) proposed that T-type channel reopening after strong depolarizing prepulses depends on a Na+-Ca2+ competition for intrapore binding sites. In addition, we have obtained evidence that the speed of macroscopic inactivation depends on whether Ca2+ or Ba2+ is the charge carrier (Klugbauer et al., 1999; Staes et al., 2001) and that Ca2+ modulates the gating of the T-type Ca2+ channel α1G by counteracting proton effects on activation (Talavera et al., 2003, in this issue).
Both EEED and EEDE mutants show a decreased Ca2+/monovalent cation selectivity with respect to α1G, as apparent from the reversal potentials. Thus, it could be envisaged that the modification of the activation properties in the mutant channels are due to the decrease of the Ca2+ permeation. However, this is in contrast with the fact that when proton modulation is avoided, the voltage sensitivity of the activation of α1G is not Ca2+ dependent and is larger than that of the pore mutants (Talavera et al., 2003, in this issue, and present results). The aspartate-to-glutamate mutations induced a decrease in the voltage sensitivity of the time constant of deactivation. Yet, the changes in extracellular proton, Ca2+ or Na+ concentrations do not modify this parameter in α1G (Talavera et al., 2003, in this issue). In summary, this evidence suggests that the gating changes induced by the aspartate-to-glutamate mutations are not related to the modification of pore occupancy but to the change of the selectivity filter structure itself.
Modification of Channel Kinetics Induced by the Aspartate-to-glutamate Mutations. Mechanistic Insights from Kinetic Modeling
The study of changes of specific gating transitions induced by the alteration of the structure of the selectivity filter may allow unraveling the role of pore structures in the gating process. To quantitatively account for these gating modifications we developed a kinetic model for each mutant in comparison to that obtained for the wild-type channel in the accompanying paper (pHe 7.4 and 20 mM [Ca2+]e). Fig. 7 A shows the kinetic scheme of the Markov model used. 13 out of the 20 rate constants defining the model are markedly (more than twofold) different in the mutants respect to the wild-type (Table I) , reflecting that the mutation of D1487 or D1810 to glutamate altered basically all gating properties of α1G.
Figure 7.

Simulation of the gating of α1G and the EEED and EEDE pore mutants. (A) Markov model used to describe the gating properties of the wild-type and both pore mutants. The thick arrows represent voltage-dependent transitions and the dotted arrows denote the transitions that are significantly different in the mutants with respect to α1G. (B) predicted time course of the open probability of α1G and the EEDE mutant during voltage steps in the range of −60 to 90 mV from a holding potential of −100 mV. The asterisks denote that the EEDE mutant shows faster macroscopic inactivation than the wild type in the voltage range from −10 to 50 mV. The same occurred for the EEED mutant (Fig. 1 C). C–F show the fit of average activation, steady-state inactivation, voltage dependence of τdecay and reactivation curves, respectively, for α1G (▪) and the EEED (○) and EEDE (▵) pore mutants. The inset in E shows simulated tail currents corresponding to repolarization potentials of −80 (continuous lines) or 0 mV (dashed lines) for α1G (thick lines) and EEDE (thin lines) pore mutant.
TABLE I.
Model Parameters Optimized to Fit the Experimental Data Obtained for α1G and the EEED and EEDE Pore Mutants at pHe 7.4 in 20 mM [Ca2+]e
| Parameter | Value | Parameter | Value | ||||
|---|---|---|---|---|---|---|---|
| α1G | EEED | EEDE | α1G | EEED | EEDE | ||
| q1 | 1.22 | 0.343 | 0.392 | δ1 | 0.819 | 0.0115 | 9.90 × 10−3 |
| q2 | 4.95 | 5.36 | 5.31 | δ2 | 0.128 | 0.598 | 0.607 |
| q3 | 0.980 | 1.40 | 1.38 | δ3 | 0 | 0 | 0 |
| KC1C2 | 2.85 | 1.51 | 1.81 | KC2C1 | 1.11 | 36.1 | 35.0 |
| KC2C3 | 17.9 | 50.3 | 53.8 | KC3C2 | 0.105 | 0.320 | 0.359 |
| kC3O = kI3Io | 2.35 | 2.30 | 2.30 | KOC3 = KIoI3 | 9.66 × 10−3 | 0.0684 | 0.0762 |
| KI1I2 | 0.0240 | 1.93 × 10−3 | 1.10 × 10−3 | KI2I1 | 4.35 × 10−3 | 8.02 × 10−4 | 8.23 × 10−4 |
| KI2I3 | 0.573 | 1.72 | 1.52 | KI3I2 | 2.50 × 10−5 | 2.60 × 10−5 | 2.80 × 10−5 |
| kC1I1 | 9.93 × 10−4 | 1.98 × 10−4 | 3.12 × 10−4 | kI1C1 | 3.31 × 10−4 | 1.98 × 10−4 | 3.12 × 10−4 |
| kC2I2 | 0.0120 | 0.210 | 0.104 | kI2C2 | 1.86 × 10−3 | 3.65 × 10−3 | 4.00 × 10−3 |
| kC3I3 | 0.0900 | 50.1 | 50.1 | kI3C3 | 1.04 × 10−4 | 2.07 × 10−3 | 5.34 × 10−3 |
| kOIo | 0.0900 | 0.105 | 0.105 | kIoO | 1.04 × 10−4 | 4.34 × 10−6 | 1.12 × 10−6 |
Both mutant channels show a positive shift and an increase in the slope factor of the voltage dependence of the time constant of activation (τact) with respect to α1G, in accordance with the changes observed in the activation curve. However, the asymptotic value of τact at positive potentials did not differ between these channels, indicating that initial transitions between closed states, but not the transition to the open state from the immediate closed state of the activation pathway, are affected by the aspartate-to-glutamate mutations. Accordingly, the predicted values for k C3O and k I3Io are very similar for the three channels. The modification of the activation properties by mutations in the selectivity filter may arise from alterations in the mechanism of voltage sensing and/or in the coupling of the movement of voltage sensor and the channel gates as it has been shown that S4 segments may interact with pore domains in HERG (Gandhi et al., 2000) and Shaker K+ channels (Li-Smerin et al., 2000; Elinder et al., 2001). The results of the kinetic modeling indicate that several voltage-dependent rate constants of the activation pathway are modified by the mutations. Notably, the model predicts that a fraction of the gating charge associated with the first box (q 1) in α1G moves during the transitions in the second and third boxes in the mutant channels, keeping the total gating charge constant (∼7.1). Interestingly, the coupling of the local electric potential sensed by q 1 with the membrane potential (δ1) is smaller in the mutant channels with respect to that of the wild-type. In contrast, δ2 is larger in the pore mutants than in α1G. The invariance of the total gating charge is consistent with the fact that the pore mutations are not likely to alter the total size of the gating charge, but to change its movement during voltage sensing. These results also strengthen the idea that gating differences between the pore mutants and α1G are not due to differential proton affinities, since the simulation of the gating modifications due to channel protonation predict a reduction of the total gating charge (Talavera et al., 2003, in this issue). Fig. 7, B and C, show that the models accurately reproduce the distinctive activation properties of the mutant channels: less negative potential for half-maximal activation and smaller voltage sensitivity than α1G. The maximal open probability (POmax) is predicted to be ∼10-fold smaller in the mutant channels when compared with that of the wild-type.
The mutation of the aspartate residues did not affect the voltage of half-maximal inactivation, but reduced significantly the voltage sensitivity of the steady-state inactivation. Considering that the macroscopic inactivation is coupled to channel activation (Droogmans and Nilius, 1989; Chen and Hess, 1990; Serrano et al., 1999; Burgess et al., 2002), it is reasonable to think that the modification of the voltage dependence of the steady-state inactivation arises from the slowing of initial steps of the activation. As shown in Fig. 7 D the kinetic models give an appropriate description of the experimental data.
Interestingly, the time constant of the macroscopic inactivation (τinac) is nearly halved in the mutant channels with respect to the wild-type in the voltage range from −10 to 50 mV, contrary to what would be expected from a slowing of initial steps of channel activation and the activation-inactivation coupling. This apparent contradiction is solved when considering the effects of the mutations on the deactivation kinetics. The macroscopic inactivation is the result of the transitions from the open state (O) to the inactivated state (IO, strict inactivation), and to the adjacent noninactivated closed state preceding the open state in the activation pathway (C3, strict deactivation). The differences in τinac between the mutant channels and α1G tend to disappear at very positive potentials (Fig. 4 and Fig. 12 of Talavera et al., 2003, in this issue), indicating that the transition between the open state and the inactivated state is not significantly altered by the pore mutations. Thus, the drastic increase in the rate of deactivation in both mutant channels should account for the increase in the rate of the macroscopic inactivation of these channels with respect to α1G. As shown in Fig. 7 E, the models accurately predict that the decay of the tail currents is indeed faster for the mutant channels in the voltage range from the activation threshold to ∼50 mV. This is reflected in an increase of the rate of macroscopic inactivation of the mutants in this voltage range, as noted by the asterisks in Fig. 7 B and clearly observed in Fig. 1 C. It should be stressed, however, that given the direct description of the voltage dependence of τdecay by Eq. 6, an accurate description of τdecay in the mutant channels is only obtained when the rate constant k C3I3 is much larger for the mutant channels than for α1G (see Table I). This condition ensures a low probability of state C3 (see materials and methods in the accompanying paper, Talavera et al., 2003, in this issue).
It has been shown that pore loops are flexible and able to undergo measurable motions during the gating of sodium (Benitah et al., 1997, 1999), potassium (Zheng and Sigworth, 1997, 1998; Yellen, 1998; Kiss et al., 1999) and calcium channels (Cloues and Sather, 2000). Proks et al. (2001) found that mutations within the P-loop altered the intraburst kinetics of ATP-sensitive potassium channel and suggested that the selectivity filter constitutes a fast gate of this channel. Thus, the fact that the substitution of D1487 and D1810 of α1G by a larger amino acid increase of the closing rate suggests that the selectivity filter forms part of a gate controlling the lifetime of the open state.
The recovery from inactivation was also altered by the aspartate-to-glutamate mutations. The reactivation of T-type Ca2+ channels is rather complex and consists of at least two exponential components. T-type channel reactivation has been shown to be preceded by the deactivation (Kuo and Yang, 2001; Burgess et al., 2002). So, it is tempting to explain the increased probability of reactivation via the fast component in the mutant channels by the increased deactivation rate with respect to α1G. However, this is not a plausible interpretation since the differences in the course of reactivation of the pore mutants with respect to α1G are demonstrated at a potential (−100 mV) at which the speed of the reactivation is saturated and no longer voltage dependent (Burgess et al., 2002). Alternatively, aspartate-to-glutamate mutations might modify the interactions between the P-loops and S6 segments that had been shown to control the reactivation of α1G (Marksteiner et al., 2001). Model simulations indicate that many parameters influence the time course of reactivation (Fig. 7 F), and that the main difference between the pore mutants is given by the variations of q 1 and k C1I1.
General Remarks
The present work is the first addressing the modifications of T-type channel gating by mutations in the selectivity filter. We show that modification of the side chain of residues 1487 and 1810 in the P-loops of domains III and IV, respectively, induces dramatic changes in the activation and deactivation processes of the T-type Ca2+ channel subunit α1G, with the consequent modification of the coupled processes of inactivation and recovery from inactivation. The fact that the EEED and the EEDE mutants partially mimic the structure of the selectivity filter of HVA Ca2+ channels raises the question of a possible correlation between some of the gating properties of voltage-dependent Ca2+ channels with their pore structure. Notably, all T-type Ca2+ channels, which show the characteristic low-voltage threshold of activation and slow deactivation kinetics, have an EEDD pore locus, in contrast with the conserved EEEE pore locus of the fast-deactivating HVA Ca2+ channels (Hofmann et al., 1999). Although large efforts have been devoted to the study of the conduction properties of many pore mutants of HVA Ca2+ channels, very little or nothing is known about possible gating modifications in these mutants with respect to the wild-type parents. Thus, it would be very interesting to address this point in order to achieve a better understanding of the structure-function relationship in other voltage-dependent Ca2+ channels.
Acknowledgments
We thank Dr. F. Hofmann for the α1G clone and Dr. M. Staes for the help in the construction of the pore mutants. We are also grateful to Drs. T. Voets, J.L. Alvarez, and G. Vassort for helpful discussions. The expert technical assistance of M. Crabbé, H. Van Weijenbergh, S. de Swaef, and M. Schuermans is greatly acknowledged. We also thank Professor V. Flockerzi for providing the vector pCAGGSM2.
This work was supported by the Belgian Federal Government, the Flemish Government and the Onderzoeksraad KU Leuven (GOA 99/07, F.W.O. G.0237.95, F.W.O. G.0214.99, F.W.O. G. 0136.00; Interuniversity Poles of Attraction Program, Prime Ministers Office IUAP Nr.3P4/23, and C.O.F./96/22-A069), and by “Levenslijn” (7.0021.99).
Olaf S. Andersen served as editor.
Karel Talavera's permanent address is Instituto de Cardiologia y Cirusia Cardiovascular, 10400 Lattabana, Cuba.
Footnotes
Abbreviation used in this paper: HVA, high voltage–activated.
References
- Alvarez, J.L., A. Artiles, K. Talavera, and G. Vassort. 2000. Modulation of voltage-dependent facilitation of the T-type calcium current by sodium ion in isolated frog atrial cells. Pflugers Arch. 441:39–48. [DOI] [PubMed] [Google Scholar]
- Balser, J.R., H.B. Nuss, N. Chiamvimonvat, M.T. Perez-Garcia, E. Marban, and G.F. Tomaselli. 1996. External pore residue mediates slow inactivation in μ1 rat skeletal muscle sodium channels. J. Physiol. 494:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, D., I. Dreyer, S. Hoth, J.D. Reid, H. Busch, M. Lehnen, K. Palme, and R. Hedrich. 1996. Changes in voltage activation, Cs+ sensitivity, and ion permeability in H5 mutants of the plant K+ channel KAT1. Proc. Natl. Acad. Sci. USA. 93:8123–8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitah, J.P., Z. Chen, J.R. Balser, G.F. Tomaselli, and E. Marban. 1999. Molecular dynamics of the sodium channel pore vary with gating: interactions between P-segment motions and inactivation. J. Neurosci. 19:1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitah, J.P., R. Ranjan, T. Yamagishi, M. Janecki, G.F. Tomaselli, and E. Marban. 1997. Molecular motions within the pore of voltage-dependent sodium channels. Biophys. J. 73:603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess, D.E., O. Crawford, B.P. Delisle, and J. Satin. 2002. Mechanism of inactivation gating of human T-type (low-voltage activated) calcium channels. Biophys. J. 82:1894–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C.F., and P. Hess. 1990. Mechanism of gating of T-type calcium channels. J. Gen. Physiol. 96:603–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloues, R.K., and W.A. Sather. 2000. Permeant ion binding affinity in subconductance states of an L-type Ca2+ channel expressed in Xenopus laevis oocytes. J. Physiol. 524:19–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delisle, B.P., and J. Satin. 2000. pH modification of human T-type calcium channel gating. Biophys. J. 78:1895–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droogmans, G., and B. Nilius. 1989. Kinetic properties of the cardiac T-type calcium channel in the guinea- pig. J. Physiol. 419:627–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinder, F., R. Mannikko, and H.P. Larsson. 2001. S4 charges move close to residues in the pore domain during activation in a K channel. J. Gen. Physiol. 118:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn, G.E., J.P. Johnson, Jr., and W.N. Zagotta. 2001. Cyclic nucleotide-gated channels: shedding light on the opening of a channel pore. Nat. Rev. Neurosci. 2:643–651. [DOI] [PubMed] [Google Scholar]
- Gandhi, C.S., E. Loots, and E.Y. Isacoff. 2000. Reconstructing voltage sensor-pore interaction from a fluorescence scan of a voltage-gated K+ channel. Neuron. 27:585–595. [DOI] [PubMed] [Google Scholar]
- Heginbotham, L., Z. Lu, T. Abramson, and R. MacKinnon. 1994. Mutations in the K+ channel signature sequence. Biophys. J. 66:1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilber, K., W. Sandtner, O. Kudlacek, I.W. Glaaser, E. Weisz, J.W. Kyle, R.J. French, H.A. Fozzard, S.C. Dudley, and H. Todt. 2001. The selectivity filter of the voltage-gated sodium channel is involved in channel activation. J. Biol. Chem. 276:27831–27839. [DOI] [PubMed] [Google Scholar]
- Hille, B. 2001. Ion Channels of Excitable Membranes. 3rd edition. Sinauer Associates, Inc., Sunderland, MA. 646–662.
- Hofmann, F., L. Lacinova, and N. Klugbauer. 1999. Voltage-dependent calcium channels: from structure to function. Rev. Physiol. Biochem. Pharmacol. 139:33–87. [DOI] [PubMed] [Google Scholar]
- Kiss, L., J. LoTurco, and S.J. Korn. 1999. Contribution of the selectivity filter to inactivation in potassium channels. Biophys. J. 76:253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer, N., E. Marais, L. Lacinova, and F. Hofmann. 1999. A T-type calcium channel from mouse brain. Pflugers Arch. 437:710–715. [DOI] [PubMed] [Google Scholar]
- Kuhn, F.J., and N.G. Greeff. 2002. Mutation D384N alters recovery of the immobilized gating charge in rat brain IIA sodium channels. J. Membr. Biol. 185:145–155. [DOI] [PubMed] [Google Scholar]
- Kuo, C.C., and S. Yang. 2001. Recovery from inactivation of t-type Ca2+ channels in rat thalamic neurons. J. Neurosci. 21:1884–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin, Y., D.H. Hackos, and K.J. Swartz. 2000. A localized interaction surface for voltage-sensing domains on the pore domain of a K+ channel. Neuron. 25:411–423. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo, J., T. Hoshi, S.H. Heinemann, and R.W. Aldrich. 1993. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1:61–71. [PubMed] [Google Scholar]
- Marksteiner, R., P. Schurr, S. Berjukow, E. Margreiter, E. Perez-Reyes, and S. Hering. 2001. Inactivation determinants in segment IIIS6 of Ca(v)3.1. J. Physiol. 537:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleskey, E.W. 1999. Calcium channel permeation: A field in flux. J. Gen. Physiol. 113:765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina, A., P. Ortega-Saenz, and J. Lopez-Barneo. 1998. Pore mutations alter closing and opening kinetics in Shaker K+ channels. J. Physiol. 509:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proks, P., C.E. Capener, P. Jones, and F.M. Ashcroft. 2001. Mutations within the P-loop of Kir6.2 modulate the intraburst kinetics of the ATP-sensitive potassium channel. J. Gen. Physiol. 118:341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano, J.R., S.R. Dashti, E. Perez-Reyes, and S.W. Jones. 2000. Mg2+ block unmasks Ca2+/Ba2+ selectivity of alpha1G T-type calcium channels. Biophys. J. 79:3052–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano, J.R., E. Perez-Reyes, and S.W. Jones. 1999. State-dependent inactivation of the alpha1G T-type calcium channel. J. Gen. Physiol. 114:185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, S., J. Li, K.A. McNulty, T. Kieber-Emmons, and T.R. Kleyman. 2001. Epithelial sodium channel pore region. Structure and role in gating. J. Biol. Chem. 276:1326–1334. [DOI] [PubMed] [Google Scholar]
- Shuba, Y.M., V.I. Teslenko, A.N. Savchenko, and N.H. Pogorelaya. 1991. The effect of permeant ions on single calcium channel activation in mouse neuroblastoma cells: ion-channel interaction. J. Physiol. 443:25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staes, M., K. Talavera, N. Klugbauer, J. Prenen, L. Lacinova, G. Droogmans, F. Hofmann, and B. Nilius. 2001. The amino side of the C-terminus determines fast inactivation of the T- type calcium channel alpha1G. J. Physiol. 530:35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela, M., G.E. Kirsch, A.M. VanDongen, J.A. Drewe, H.A. Hartmann, R.H. Joho, E. Stefani, and A.M. Brown. 1992. Gating currents from a delayed rectifier K+ channel with altered pore structure and function. Biophys. J. 62:34–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera, K., M. Staes, A. Janssens, N. Klugbauer, G. Droogmans, F. Hofmann, and B. Nilius. 2001. Aspartate residues of the Glu-Glu-Asp-Asp (EEDD) pore locus control selectivity and permeation of the T-type Ca2+ channel alpha(1G). J. Biol. Chem. 276:45628–45635. [DOI] [PubMed] [Google Scholar]
- Talavera, K., A. Janssens, N. Klugbauer, G. Droogmans, and B. Nilius. 2003. Extracellular Ca2+ modulates the effects of protons on gating and conduction properties of the T-type Ca2+ channel α1G (CaV3.1). J. Gen. Physiol. 121:511–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaselli, G.F., N. Chiamvimonvat, H.B. Nuss, J.R. Balser, M.T. Perez-Garcia, R.H. Xu, D.W. Orias, P.H. Backx, and E. Marban. 1995. A mutation in the pore of the sodium channel alters gating. Biophys. J. 68:1814–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend, C., H.A. Hartmann, and R. Horn. 1997. Anomalous effect of permeant ion concentration on peak open probability of cardiac Na+ channels. J. Gen. Physiol. 110:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend, C., and R. Horn. 1999. Interaction between the pore and a fast gate of the cardiac sodium channel. J. Gen. Physiol. 113:321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tytgat, J., B. Nilius, and E. Carmeliet. 1990. Modulation of the T-type cardiac Ca channel by changes in proton concentration. J. Gen. Physiol. 96:973–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarte, G., F. Perez, and R. Latorre. 1998. How do calcium channels transport calcium ions? Biol. Res. 31:17–32. [PubMed] [Google Scholar]
- Varadi, G., M. Strobeck, S. Koch, L. Caglioti, C. Zucchi, and G. Palyi. 1999. Molecular elements of ion permeation and selectivity within calcium channels. Crit. Rev. Biochem. Mol. Biol. 34:181–214. [DOI] [PubMed] [Google Scholar]
- Yatani, A., A. Bahinski, M. Wakamori, S. Tang, Y. Mori, T. Kobayashi, and A. Schwartz. 1994. Alteration of channel characteristics by exchange of pore-forming regions between two structurally related Ca2+ channels. Mol. Cell. Biochem. 140:93–102. [DOI] [PubMed] [Google Scholar]
- Yellen, G. 1998. The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 31:239–295. [DOI] [PubMed] [Google Scholar]
- Yellen, G. 2002. The voltage-gated potassium channels and their relatives. Nature. 419:35–42. [DOI] [PubMed] [Google Scholar]
- Zheng, J., and F.J. Sigworth. 1997. Selectivity changes during activation of mutant Shaker potassium channels. J. Gen. Physiol. 110:101–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, J., and F.J. Sigworth. 1998. Intermediate conductances during deactivation of heteromultimeric Shaker potassium channels. J. Gen. Physiol. 112:457–474. [DOI] [PMC free article] [PubMed] [Google Scholar]