

Abstract
Inhibition of N- (Cav2.2) and P/Q-type (Cav2.1) calcium channels by G-proteins contribute importantly to presynaptic inhibition as well as to the effects of opiates and cannabinoids. Accordingly, elucidating the molecular mechanisms underlying G-protein inhibition of voltage-gated calcium channels has been a major research focus. So far, inhibition is thought to result from the interaction of multiple proposed sites with the Gβγ complex (Gβγ). Far less is known about the important interaction sites on Gβγ itself. Here, we developed a novel electrophysiological paradigm, “compound-state willing-reluctant analysis,” to describe Gβγ interaction with N- and P/Q-type channels, and to provide a sensitive and efficient screen for changes in modulatory behavior over a broad range of potentials. The analysis confirmed that the apparent (un)binding kinetics of Gβγ with N-type are twofold slower than with P/Q-type at the voltage extremes, and emphasized that the kinetic discrepancy increases up to ten-fold in the mid-voltage range. To further investigate apparent differences in modulatory behavior, we screened both channels for the effects of single point alanine mutations within four regions of Gβ1, at residues known to interact with Gα. These residues might thereby be expected to interact with channel effectors. Of eight mutations studied, six affected G-protein modulation of both N- and P/Q-type channels to varying degrees, and one had no appreciable effect on either channel. The remaining mutation was remarkable for selective attenuation of effects on P/Q-, but not N-type channels. Surprisingly, this mutation decreased the (un)binding rates without affecting its overall affinity. The latter mutation suggests that the binding surface on Gβγ for N- and P/Q-type channels are different. Also, the manner in which this last mutation affected P/Q-type channels suggests that some residues may be important for “steering” or guiding the protein into the binding pocket, whereas others are important for simply binding to the channel.
Keywords: α1A and α1B, channel modulation, voltage-dependent regulation, mathematical modeling, G proteins
INTRODUCTION
N- (Cav2.2) and P/Q-type (Cav2.1) calcium channels are the dominant triggers of neurotransmitter release throughout the nervous system (Luebke et al., 1993; Takahashi and Momiyama, 1993; Wheeler et al., 1994; Wu and Saggau, 1994; Dunlap et al., 1995). Their inhibition by G proteins therefore figures critically in presynaptic inhibition (Lipscombe et al., 1989; Miller, 1990; Wu and Saggau, 1994, 1997; Patil et al., 1996; Brody et al., 1997), as well as in the effects of opiates (Wilding et al., 1995; Bourinet et al., 1996; Kaneko et al., 1997) and cannabinoids (Mackie and Hille, 1992; Twitchell et al., 1997; Wilson and Nicoll, 2002). Moreover, the transient relief of such inhibition seen in many systems by repetitive channel activation (Brody et al., 1997; Artim and Meriney, 2000; Brody and Yue, 2000; Currie and Fox, 2002) may support forms of short-term synaptic plasticity (Elmslie et al., 1990; Miller, 1990; Wu and Saggau, 1994; Brody et al., 1997; Brody and Yue, 2000), and hold important implications for the neurocomputational properties of the brain (Markram and Tsodyks, 1996). For these reasons, understanding the molecular mechanisms underlying G-protein inhibition of voltage-gated calcium channels has been a major focus of channel investigation.
Progress along this research direction promises numerous benefits. From the standpoint of general principles of channel modulation, G-protein inhibition represents a prototypic modulatory mechanism that extends not only across α1A,B,E (Cav2.1–3) calcium channels, but possibly to other classes of channels, such as muscarinic K+ channels (Logothetis et al., 1987). In addition, structure-function research of G-protein modulation may yield custom channels and/or custom G-proteins that afford selective modulation of channel types, and provide valuable tools for exploring physiological questions concerning the differential roles of diverse channel targets. Finally, engineered G-proteins demonstrating selective modulation of certain channel types or splice variants could provide critical structural guidelines for the design of novel pharmaceutical agents of like selectivity, a goal with enormous therapeutic potential (Dickenson et al., 2002).
In the past several years, there has been considerable progress in understanding the molecular basis of G-protein inhibition. A dominant form of calcium channel inhibition is thought to result from direct binding of the G-protein βγ complex (Gβγ)* to the pore-forming α1 calcium channel subunit (Ikeda, 1996; Herlitze et al., 1997). Multiple studies have sought to define the channel surface that presumably interacts with Gβγ to produce channel modulation, and the following channel α1 subunit locations have been implicated in such interaction: domain I (Zhang et al., 1996; Page et al., 1997), the loop connecting domains I and II (De Waard et al., 1997; Herlitze et al., 1997; Zamponi et al., 1997; Garcia et al., 1998), the NH2 terminus (Page et al., 1998; Simen and Miller, 2000; Canti et al., 1999), and the COOH terminus (Qin et al., 1997; Furukawa et al., 1998a,b). However, there remains some controversy over the relative importance of these various sites for G-protein modulation. While some studies emphasize the importance of the I-II loop (De Waard et al., 1997; Herlitze et al., 1997; Zamponi et al., 1997), others report persistence of modulation despite presumed elimination of Gβγ binding to the I-II loop (Zhang et al., 1996; Page et al., 1997; Canti et al., 1999; Furukawa et al., 1998a,b). Trying to pool together the past studies, using data scattered among different channel types (i.e., N-type, P/Q-type, and R-type), may lead to false conclusions. Kinetic differences found between N- and P/Q-type modulation (Mintz and Bean, 1993; Bourinet et al., 1996; Zhang et al., 1996; Currie and Fox, 1997; Furukawa et al., 1998a,b; Colecraft et al., 2000, 2001), as well as fundamental differences in the prevalence of “reluctant gating” (Colecraft et al., 2000, 2001; Kinoshita et al., 2001), may suggest that the actual binding is rather different for each channel type. Hence, the complexities of defining channel interaction sites with Gβγ may be further augmented by the possibility of channel-specific customization of sites. It also may be that multiple regions of the channel contribute to an overall binding pocket for Gβγ, much as regions of both NH2 and COOH termini on GIRK channels are important for binding Gβγ (He et al., 2002) and as multiple ligands may orchestrate apoCaM binding to various targets (Jurado et al., 1999; Erickson et al., 2001).
By contrast, mapping interaction sites on the Gβ subunit may prove more tractable at the present time, now that the crystal structure of Gβ1γ2 is known, and the locations of Gβ residues interacting with Gα are determined (Wall et al., 1995; Lambright et al., 1996). Presuming that Gβγ/effector binding may involve a similar pattern of interaction, interest has focused on identifying Gβ residues important for association with its effectors (Ford et al., 1998). Therefore, the structure of Gβ1γ2 became an obvious target for us to manipulate in regard to calcium channel modulation, with the goal of defining which residues may be important in binding to the channel α1 subunit. Thus far, previous work to determine the important residues has focused solely on N-type calcium channels (Ford et al., 1998) and not other calcium channel types subject to G-protein modulation. Given the relative feasibility of mapping Gβγ interaction sites, and sensitized to the channel-isoform–specific variation in G-protein regulation, we here examine Gβ1 interaction with both N- and P/Q-type channels, and identify Gβ1 residues with selective importance for N- versus P/Q-type calcium channels.
MATERIALS AND METHODS
Transfection of Human Embryonic Kidney 293 Cells
Human embryonic kidney (HEK 293) cells were cultured and transiently transfected using a calcium phosphate precipitate protocol. We introduced 8 μg of cDNA of the following channel subunits: rat brain α1A (Stea et al., 1994) or human α1B (Williams et al., 1992), rat brain β2a (Perez-Reyes et al., 1992), and rat brain α2δ (Tomlinson et al., 1993). Gβγ subunits were also overexpressed by introducing 4 μg of cDNA of Gβ1 (Sugimoto et al., 1985) or the appropriate Gβ1 mutant, and 4 μg of Gγ2 (Gautam et al., 1990). Cells were maintained as described previously (Brody et al., 1997).
Electrophysiology
Whole-cell voltage clamp recordings were made 2–4 d after transfection using glass pipettes with a resistance of 1–2.5 MΩ, when filled with an internal solution containing: 135 mM CsMeSO4, 5 mM CsCl, 10 mM EGTA, 10 mM HEPES, 1 mM MgCl2, and 4 mM ATP. Cells were continually perfused with: 150 mM TEA-MeSO4, 10 mM HEPES, 2 mM CaCl2, and 1 mM MgCl2. Standard patch clamp techniques were used with an Axopatch 200A (Axon Instruments, Inc.). Series resistance was compensated 60–70% and currents were filtered at 2 kHz. Leak currents and capacitance transients were subtracted using a P/8 protocol. All analysis was preformed using custom written MATLAB software (MathWorks) and Microsoft Excel. T-tests were performed to establish statistical significance, which was set at level of P < 0.05. All errors are given in ± SEM.
Construction of Gβ1 Mutants
Gβ1 wild-type, Gβ1L55A, Gβ1I80A, and Gβ1W322A were a gift of Dr. H.E. Hamm, Vanderbilt University. All constructs were shuttled out of their original plasmids using HindIII and XbaI and placed in pcDNA3 to allow uniform expression. All other mutations were made using QuikChange (Stratagene). All mutations were verified by sequencing.
Western Blots
HEK 293 cells were harvested and lysed two days post transfection. Cells were collected and washed with PBS, then pelleted. Total protein concentration was determined using a standard BCA protein assay (Pierce Chemical Co.). 10-μg samples were loaded per lane, and resolved on 12.5% SDS-PAGE. Afterwards, the gel was electroblotted to nitrocellulose membrane. Proteins were detected by using a primary Gβ1 polyclonal antibody (Santa Cruz Biotechnology, Inc.) applied at a 1:10,000 dilution in 10 ml TBS with 5% milk, and incubated overnight at 4°C. Unbound antibody was washed off. For detection a secondary anti-rabbit HRP (Amersham Biosciences) was applied at a 1:10,000 dilution in 15 ml TBS with 5% milk, and incubated at room temperature for 1 h. The membrane was then incubated at room temperature for 5 min in 3 ml ECL reagent (Amersham Biosciences), and visualized with chemiluminescence using the VersaDoc imaging system (Bio-Rad Laboratories).
RESULTS
Compound-state Description of G-protein Interaction with Calcium Channels
A prerequisite for undertaking in-depth structure-function analysis of G-protein βγ (Gβγ) subunit interaction with calcium channels is a quantitative description of channel modulation by G proteins. A leading mechanistic framework for such modulation is the “willing-reluctant” model of voltage-dependent G-protein inhibition (Bean, 1989; Elmslie et al., 1990). Here, uninhibited channels (without Gβγ bound) gate according to a “willing” mode in which depolarization readily drives channels from fully closed (C) to open (O) states (Fig. 1 A, right, top mode). By contrast, inhibited channels (with Gβγ bound) gate according to a “reluctant” mode where prolonged and/or larger depolarizations are required to drive channels from closed (C′)to open (O′) states (Fig. 1 A, right, bottom mode). The characteristic, voltage-dependent relief of Gβγ inhibition (Elmslie et al., 1990) can be explained by state-dependent affinity of Gβγ binding, where Gβγ associates tightly with channels residing in deep closed conformations toward the left of each mode, while Gβγ binding is weak or almost nonexistent near or in the open states. Such state-dependent affinity is represented schematically by the relative length of vertical arrows linking states in willing and reluctant modes (Fig. 1 A, right). Hence, changes in voltage cause channels to adopt various positions along the activation pathway, thereby redistributing the fraction of channels in willing versus reluctant modes.
Figure 1.

Gβγ modulation of N-type calcium channels. (A, left) Variable duration reinhibition protocol elicited by facilitating all channels with a depolarizing prepulse to 100 mV, followed by repolarization to −100 mV for variable durations, and then measuring the test-pulse current (TP) at 0 mV (5 ms into the trace). Tail and outward currents were clipped here and throughout this figure for clarity. (A, middle) Normalized test-pulse currents plotted versus duration of the repolarization step. Single exponential fit to determine reinhibition time constant and steady-state W(∞,V), according to the equation I n,rein. = W(∞,−100) + (1 − W(∞,−100))exp[−t/τ(V)], (τ = 20.4 ms). Protocols were repeated for interpulse voltage values of −40, −60, and −80 mV (not depicted). (A, right) State diagram depicting key features of “willing-reluctant” model of G-protein modulation of calcium channels. Highlighted arrows depict vertical rate constants most relevant to reinhibition protocol. (B, left) Variable-duration prepulse protocol used to determine time course of facilitation and elicited currents are shown. (B, middle) Normalized test pulse currents were measured 5 ms into the test pulse (0 mV) and plotted versus duration of prepulse (100 mV). Single exponential fits were determined using a least squares fit method to obtain a time constant and W(∞,V) values using the following equation I n,facil. = W(∞,100) + [W(∞,−100) − W(∞,100)]exp[−t/τ(V)], (τ = 8.3 ms). Protocols were repeated for prepulse voltage values of 40, 60, and 80 mV (not depicted). (B, right) State diagram with highlighted vertical arrows reflecting rates being measured by facilitation protocol. (C, left) Step protocol, with and without prepulse, used to determine time course and steady-state level of binding for given test pulse. Test pulse in exemplar is 0 mV. Step protocols were obtained from −30 to 30 mV. (C, middle) Dual fits of traces with and without a prepulse were fit using a least squares method: I +pre = I max[W(∞,V) + (1 − W(∞,V)) e−t/ τ(V)] e−t/τinact. I -pre = I max[W(∞,−100) + (W(∞,V) − W(∞,−100)) (1 − e−t/ τ(V))] e−t/τinact; (τ = 57.0 ms). (C, right) State diagram modeling G-protein modulation with highlighted arrows depicting rates measured.
This willing-reluctant mechanism is frequently explored using two common electrophysiological protocols, illustrated in Fig. 1 by records of recombinant N-type channels coexpressed with Gβγ subunits. In the first paradigm, termed a “reinhibition” protocol (Fig. 1 A, left), nearly all channels are initially driven to the willing mode by a strong 100-mV prepulse (Colecraft et al., 2000, 2001). Then, a variable-duration interpulse (here, to −100 mV) is interposed before eliciting a test-pulse (TP) current at 0 mV. After the briefest interpulse duration (1 ms in the marked exemplar trace), the test-pulse current thus activates according to a large and uniform phase, as expected of channels entirely in the willing mode. At longer interpulse durations, test-pulse currents adopt a biphasic activation profile, with a rapid activation component mainly reflective of willing channels, and a slower activation component indicative of channel conversion from reluctant to willing modes during the test pulse. Because channels would reside in deep closed conformations during the hyperpolarized interpulse period (Fig. 1 A, right), lengthening of the interpulse period should increase the fraction of channels in the reluctant mode at the onset of the test pulse. This prediction is confirmed by the progressive decline of the rapid activation component with increasing interpulse duration (Fig. 1 A, left). The time course of willing-to-reluctant conversion (at −100 mV) can then be quantified by plotting the normalized amplitude of the rapid activation component as a function of interpulse duration (Fig. 1 A, middle). Because this time course pertains to periods of strong hyperpolarization, the reinhibition protocol mainly reflects the vertical rate constants toward the left end of the modes (Fig. 1 A, right, thick arrows).
Conversion in the opposite direction, from reluctant to willing modes, can be probed by a “facilitation” protocol (Fig. 1 B, left). Here, a strong, variable-duration prepulse is introduced before a test pulse to 0 mV. Without a prepulse, the test-pulse current shows only a small rapid activation component, consistent with most channels residing in the reluctant mode at the onset of the test pulse. During the strong prepulse, channels would be driven toward the open states with corresponding low Gβγ affinity (Fig. 1 B, right), fitting nicely with the enhancement of the rapid activation component with prolongation of the prepulse duration (Fig. 1 B, left). Plots of the rapid-activation amplitude as a function of prepulse duration (Fig. 1 B, middle) thus quantify the kinetics of reluctant-to-willing conversion (at 100 mV) and, in this case, mainly reflects the vertical rate constants nearer the open states (Fig. 1 B, right, thick arrows).
A less-well scrutinized, but telling experimental paradigm concerns reluctant-willing exchange via vertical transitions between the central regions of modes (Fig. 1 C, thick arrows). In the step-equilibration protocol, we apply 300-ms test pulses to intermediate voltages, where channels would populate conformations with intermediate affinity for Gβγ. Accordingly, in the absence of a prepulse, test-pulse currents slowly activate toward a plateau level, presumably specified by steady-state partitioning of channels between reluctant and willing modes at intermediate voltages. If correct, then the same plateau level of current should be reached, regardless of the initial fraction of channels in willing or reluctant modes. In agreement with this prediction, test-pulse currents after a strong prepulse to 100 mV achieve the same steady-state level (Fig. 1 C, left), despite initial rapid activation to a large current amplitude. The response after a prepulse is consistent with an initial predominance of willing channels, followed by rebinding of Gβγ and attainment of the same steady-state partitioning of channels between modes. Expanded plots of test-pulse responses (Fig. 1 C, middle) thus provide high-resolution representations of the time course of willing-reluctant interchange at intermediate voltages. Collectively, these three protocols provide a rather complete kinetic signature of presumed Gβγ binding to channels, sampled over the entire spectrum of functionally relevant voltages.
Full-bore implementation of the state-diagram model for the willing-reluctant mechanism (Boland and Bean, 1993) can represent the complete repertoire of behaviors with impressive precision. However, such implementation constitutes a substantial computational endeavor and requires specification of a rather large number of parameters, many of which may not be uniquely constrained. Hence, such an extensive model may be impractical for understanding the effects of large numbers of channel/Gβγ mutations on structure-function relations. Moreover, such modelling, even if implemented in each case, may not provide easy visualization of dominant trends. Accordingly, we developed a simpler quantitative description of Gβγ interaction with voltage-gated calcium channels, exploiting a critical simplifying feature of the observed kinetics—that the time course of willing-reluctant mode equilibration is approximately single-exponential in form, as viewed across the entire voltage range (Figs. 1, A–C, middle, smooth curve fits). This implies that, at a given voltage, channels achieve “rapid equilibrium” within each mode before appreciable exchange between the willing and reluctant modes (Neher and Steinbach, 1978). Thus, at each voltage V, willing-reluctant interchange can be specified by a single downward (k on(V)) and single upward (k off(V)) effective rate constant (Fig. 2 A), corresponding to presumed bimolecular Gβγ binding and unbinding reactions (Zamponi and Snutch, 1998). These assumptions are the foundation for our compound-state willing-reluctant analysis.
Figure 2.
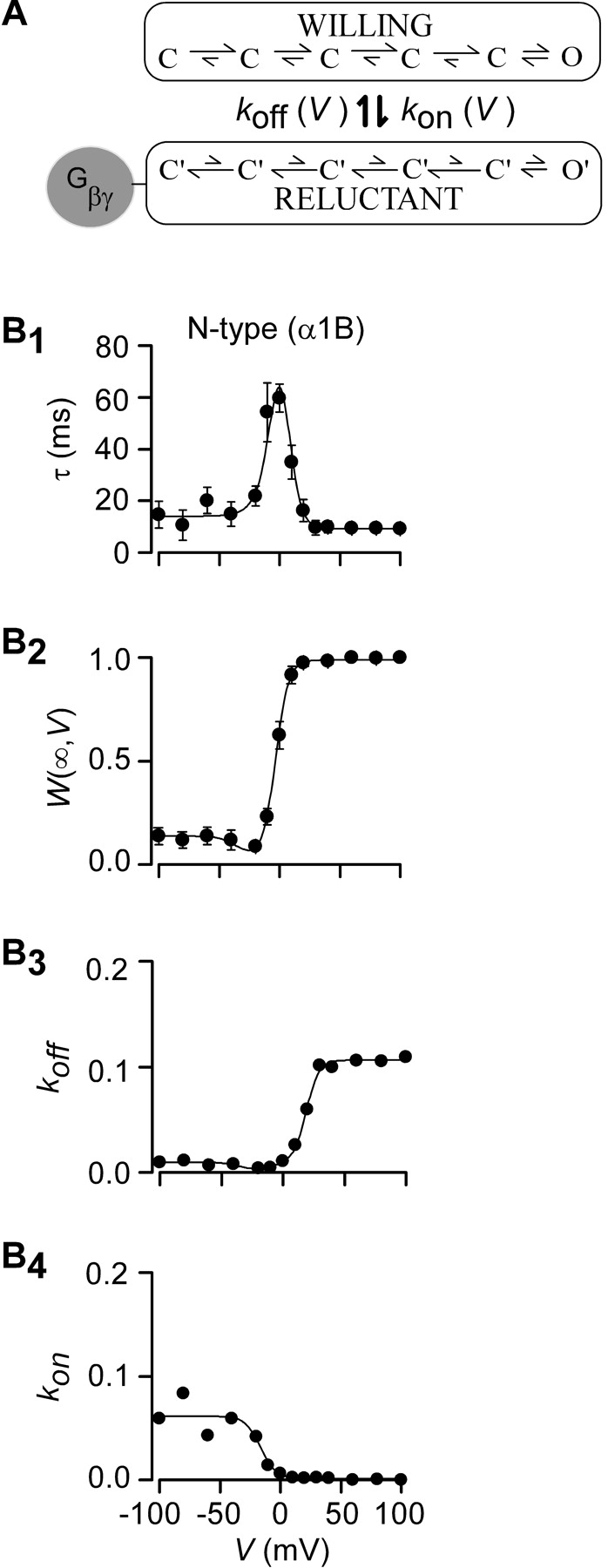
Measured parameters and fits for recombinant N-type channels (α1B/β2a/α2δ). (A) For a particular voltage, willing-reluctant interchange is modeled as a single upward and downward rate. (B1 and B2) Time constants, τ(V), and fraction of channels in the willing mode, W(∞,V), were collected using protocols described in Fig. 1. Fits were obtained using Eqs. 1–5 and simultaneously fitting both parameters using least squares protocol. (B3) Off rates, k off(V), were determined by combining Eqs. 1 and 2 such that: k off (V) = W(∞,V)/τ(V). The smooth fit was obtained by plotting Eq. 5 using parameters determined with fits from parts B1 and B2. (B4) The on rates, k on(V), were also determined by combining Eqs. 1 and 2 where: k on(V) = (1 − W(∞,V))/τ(V). Fits were obtained by inserting the values determined in B1 and B2 into Eq. 4. All concentration values, [Gβγ], for this figure and all following figures are set to one, assuming that concentration on average is equal.
Kinetic Properties of Gβγ Modulation on N-type Channels
We first applied compound-state willing-reluctant analysis to recombinant N-type (α1B/β2a/α2δ) channels (Cav2.2) expressed in HEK 293 cells along with Gβ1γ2. Time constants, τ(V), were determined for voltages ranging from –100 mV to +100 mV (Fig. 2 B1), based on exponential fits to data from all three protocols (Fig. 1, middle). In relation to our simplified model,
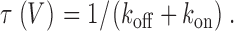 |
(1) |
Hence, the striking, bell-shaped voltage dependence of τ (Fig. 2 B1) is fully consistent with predictions that would be drawn from moderately sized k on and k off magnitudes sampled by partially activated channels, and large k on or k off amplitudes sampled at extreme negative and positive voltages, respectively. To obtain further model constraints, we sought to experimentally gauge the steady-state fraction of channels in the willing mode at a given voltage (W(∞,V)). This could be accomplished because strong depolarizing prepulses result in near exclusive occupancy of the willing mode in our system (Colecraft et al., 2000, 2001). Hence, after normalization of test-pulse currents by the peak current amplitude observed immediately following a strong +100 mV prepulse, the steady-state amplitude of currents (Fig. 1, A–C, middle) gives an estimate of W(∞,V). This estimate presumes that reluctant channels are electrically silent at the times and voltages at which steady-state determinations are made, an assumption supported by previous biophysical analysis (Colecraft et al., 2000, 2001). Given this assumption, our simplified model predicts
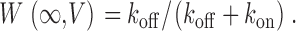 |
(2) |
Hence, determination of W(∞,V) as shown in Fig. 2 B2, together with specification of τ(V), fully specifies k on(V) and k off(V) (Fig. 2, B3 and B4, symbols).
As a convenient means of interpolating data to achieve a continuous representation of k on(V) and k off(V) as a function of voltage, we empirically assumed that these rates could be described by a voltage-dependent Boltzmann factor added to a voltage-independent offset.
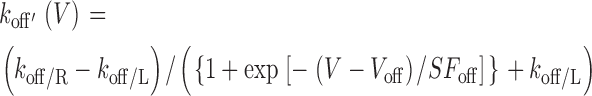 |
(3) |
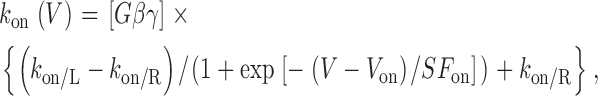 |
(4) |
where V off and V on are midpoint voltages, SF off and SF on are slope factors, and [Gβγ] is the local Gβγ concentration near the cytoplasmic face of the channel. This form ensured that k on(V) and k off(V) would achieve finite assymptotic values at voltage extremes, corresponding to vertical transitions at the extreme right and left ends of the state diagram for the willing-reluctant model. In particular, k off/R and k off/L correspond to upward vertical transitions at the extreme right and left of the willing-reluctant model state diagram (Fig. 1 A, right), and k on/R and k on/L pertain to the analogous downward vertical transitions. Experimental data showed a slight dip in the calculated k off values (W(∞,V)/τ) that could not be fit to satisfaction with a simple Boltzmann equation. A Gaussian curve was added to the k off equation to produce a more precise fit.
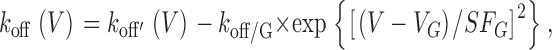 |
(5) |
where k off/G represents the magnitude of the Gaussian, V G is the half maximal voltage, and SF G is the slope factor. The fact that k off does not behave like a simple Boltzmann is not surprising, due to the complex nature of the N-type channel activation (Jones et al., 1997). The close concordance of smooth curve fits (Eqs. 4 and 5) with the time-constant and W(∞,V) data (Fig. 2, B1 and B2) supports the appropriateness of these functional forms. The fits also provide constrained estimates of k on and k off, shown as continuous functions of voltage in Fig. 2, B3 and B4.
Fundamental Differences in the Kinetics of P/Q-type Channel Modulation
Application of the compound-state analysis to recombinant P/Q-type (α1A, β2a, α2δ) channels (Cav2.1) coexpressed with Gβγ revealed striking differences in the kinetics of willing-reluctant exchange (Fig. 3) . In agreement with previous studies (Arnot et al., 2000; Colecraft et al., 2000), time constants from facilitation protocols were consistently faster than those of N-type channels (Fig. 3 D1), fitting with larger k off values at extreme depolarization (Fig. 3 D3). Similarly, time constants from reinhibition protocols were also consistently faster than those of N-type channels (Zhang et al., 1996), consistent with larger k on values at hyperpolarized potentials. New insight comes from the behavior of k on and k off at intermediate voltages: these adopt a voltage-dependent profile that minimizes the bell-shaped contour of τ(V) (Fig. 3 D1). Hence, P/Q-type channels retain very fast reluctant-willing interchange across the entire voltage range, whereas N-type channels feature marked slowing of such exchange over voltages spanned by action-potential spikes.
Figure 3.

Exemplar traces and parameter fits shown for P/Q-type channels. (A, left) Voltage reinhibition protocol as shown in Fig. 1 with a test pulse voltage of −10 mV and exemplar traces collected at −100 mV. (A, right) Peak currents from traces shown on left were normalized and shown versus duration of interpulse to −100 mV. Fits were obtained by using the same equations as in Fig. 1 (τ = 6.3 ms). (B, left) Voltage facilitation protocol and exemplar traces shown for P/Q-type channels at 100 mV. (B, right) Peak currents were normalized and shown versus length of the prepulse. Fits were obtained using a single exponential (τ = 5.2 ms). (C, left) Step protocol and exemplar traces shown for P/Q-type channels at −10 mV. (C, right) Dual fits are shown using traces with and without a prepulse (τ(−10) = 17.9 ms, W(∞,−10) = 0.789). (D1 and D2) Time constants, τ(V), and the fraction of channels in willing mode, W(∞,V), collected are shown along with fits using same equations that were used in Fig. 2. (D3 and D4) Fits for on and off rates (k on(V) and k off(V)) determined are shown along with calculated k on(V) and k off(V). Dashed lines represent N-type channel fits.
These qualitative differences in the kinetic behavior of the two channel types raised the possibility of potentially important distinctions in the interaction surface of Gβγ with N- versus P/Q-type channels. The quantitative fingerprint afforded by compound-state analysis provided a sensitive and efficient screen for such distinctions, which could manifest as differential effects of Gβ point mutations at plausible Gβ–channel interaction loci. These sites were chosen based on the crystallographically determined interaction surface between Gβ and Gα (Ford et al., 1998), under the assumption that a similar surface would pertain to the articulation of Gβ with different calcium channels. In the original crystallography studies the residues interacting with Gα were classified into two groups based on the area of Gα they bound. Six of the residues we mutated: K57, M101, L117, D186, D228, and W332, were bound to Gα on the “switch interface” and the other two residues examined: L55 and I80A, were bound to the “NH2-terminal interface” (Wall et al., 1995; Lambright et al., 1996). Due to their geographical location on the surface of Gβ, we grouped the residues according to four dominant regions of interaction (Fig. 4) .
Figure 4.
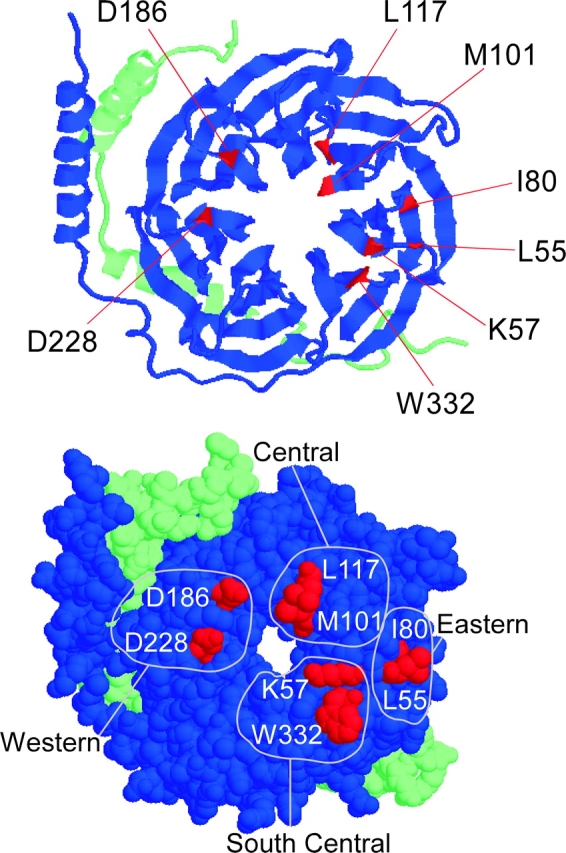
Crystal structure of Gβγ with mutated residues highlighted. (Top) Ribbon model of Gβγ is shown with its Gα binding surface fully exposed. The green strand is Gγ and the blue is Gβ. The NH2 termini of each are in the upper left hand corner. The COOH terminus of Gβ is located on the end of the β stand next to the W332 residue. The COOH terminus of Gγ is in the lower right-hand corner. Residues studied are highlighted in red. (Bottom) Space fill model of Gβγ. Residues are divided into four regions based on their location according to the full set of residues that interact with Gα.
Central Region of Gβ Interaction Surface
The geometric center of the Gβ interaction surface with Gα enfolds amino acids M101 and L117 (Fig. 4). Presuming a similar centrality of this region for channel interaction, one might predict that mutations at either of these positions would strongly affect G-protein association with both channel types. In fact, alanine substitutions at either of these positions (Gβ[M101A], Gβ[L117A]) produced clear-cut changes in G-protein modulation of both channels. In regard to N-type channels, both mutations increased unbinding rates at depolarized voltages (Fig. 5 , A3 and B3), and reduced the degree of steady-state G-protein inhibition at negative potentials (Fig. 5, A2 and B2). For ease of visual comparison, fits for wild-type Gβ behavior are reproduced as dashed curves, here and throughout. The mutations showed similar effects in regard to P/Q-type channels, though the reduction in steady-state G-protein inhibition at hyperpolarized potentials was somewhat greater (Fig. 5, C2 and D2). Hence, the central region appears important for Gβ articulation of both channel types.
Figure 5.

Measured parameters and fits for Gβ1 mutations M101A and L117A. Time constants, τ(V), fraction of channels in willing mode, W(∞,V), and on and off rates shown along with fits for two mutations found on the central region of Gβ1, M101A, and L177A. N-type channel data is shown in A and B; P/Q-type channel data is shown in C and D. Dashed lines represent wild-type Gβ1 data for each particular channel type. Format identical to Fig. 3, D1–D4.
South-central Locus of Interaction
Almost as centrally situated in the presumed interaction surface is the south-central locus of amino acids (Fig. 4), featuring residues K57 and W332. Fitting with the suspected centrality, Gβ[K57A] and Gβ[W332A] mutants also resulted in substantial blunting of G-protein modulation of both channel types. All of the pairings showed clear acceleration of the off rates (Fig. 6 , A3–D3), with the effects of both mutations being particularly strong for P/Q-type channels. Even at hyperpolarized potentials, less than half the P/Q-type channels appear to have Gβ bound at steady-state (Fig. 6, C2 and D2). These effects were among the strongest observed over all mutant G proteins and channels. Overall, the south-central locus of interaction also harbors critical residues for interaction with both channel types.
Figure 6.

Measured parameters and fits for Gβ1 mutations K57A and W332A. Time constants, τ(V), fraction of channels in willing mode, W(∞,V), and on and off rates are shown along with fits for two mutations found on south-central locus of Gβ1, K57A and W332A. N-type channel data is shown in A and B; P/Q-type channel data is shown in C and D. Dashed lines represent wild-type Gβ1 data for each particular channel type. Format identical to Fig. 3, D1–D4.
Eastern Zone of Gβ Interaction
The eastern zone of Gβ (Fig. 4), being more peripherally situated on the interaction surface with Gα and binding to a separate region on Gα than the other regions studied, might be expected to have variable importance for binding to different effector molecules. Moderate distinctions in the total area or center of effector footprints on Gβ would predict such variability. In fact, the Gβ[L55A] mutations showed clear changes in G-protein modulation of P/Q-type channels (Fig. 7 C), while this same mutation demonstrated essentially wild-type G-protein modulatory properties in the context of N-type channels (Fig. 7 A). The L55A mutation, though not as potent as those in the central or south-central loci, produced statistically significant slowing of P/Q-type channel time constants over the middle and lower voltage ranges (Fig. 7 C1), representing a decrease in the on and off rates (Fig. 7 C3), but appeared to have a wild-type W(∞,V) curve. Mutating this residue seems to affect the ability of Gβγ to get to and from its binding site, in a manner corresponding to elevation of an energy barrier that must be overcome when Gβγ enters or exits the channel binding site (Atkins, 1998). Since the mutation leaves the W(∞,V) curve unchanged, the actual binding affinity (dictated by the energy difference between bound and unbound states) would be unaffected. Physically speaking, Gβγ binding and unbinding to a channel may occur like a skeleton key that needs to be oriented correctly to be inserted or removed from a lock. L55A acts as a “steering” mechanism that orients the key to fit through the keyhole and into the lock; mutations at this position entail more stochastic tumbling before the key (Gβγ) can slip into and out of the lock. The I80A mutation when expressed with P/Q-type channels showed slower time constants in the middle range similar to the effects of the L55A mutation and a wild-type W(∞,V) curve, but upon further statistical analysis this difference was not significant (Fig. 7, D1 and D2). I80A also expresses wild-type behavior when expressed with N-type channels (Fig. 7, B1–B4). Most importantly, the selective effect of L55A raises the possibility that certain Gβ “ligands” may be specific for one channel type over the other.
Figure 7.

Measured parameters and fits for Gβ1 mutations L55A and I80A. Time constants, τ(V), fraction of channels in willing mode, W(∞,V), and on and off rates are shown along with fits for two mutations found on the eastern zone of Gβ1, L55A and I80A. N-type channel data is shown in A and B; P/Q-type channel data is shown in C and D. Dashed lines represent wild-type Gβ1 data for each particular channel type. Format identical to Fig. 3, D1–D4.
Western Region of Interaction
In hopes of identifying Gβ foci with converse selectivity for N-type channels, we examined the effects of mutations at the other peripheral region of the interaction surface, the Western locus of Gβ (Fig. 4). However, Gβ[D186A] and Gβ[D228A] mutants showed unmistakable changes in G-protein modulation of both channel types (Fig. 8) . Thus, while peripheral regions may be more likely to have differing importance for interaction with various effectors, in this case N- and P/Q-type channels both appear to rely upon interactions with the western region.
Figure 8.

Measured parameters and fits for Gβ1 mutations D186A and D228A. Time constants, τ(V), fraction of channels in willing mode, W(∞,V), and on and off rates are shown along with fits for two mutations found on the western region of Gβ1, D186A and D228A. N-type channel data is shown in A and B; P/Q-type channel data is shown in C and D. Dashed lines represent wild-type Gβ1 data for each particular channel type. Format identical to Fig. 3, D1–D4.
Comparable Expression Levels of Mutant Gβ Constructs
One caveat to comparative interpretation of the efficacy of different Gβ molecules was the assumption that expression levels were consistent across constructs. While k off is independent of Gβγ concentration, our estimates of k on incorporate this concentration (Eq. 4); hence, variations in expression among different G-protein constructs could itself produce differences in the voltage-dependent profile of k on, without intrinsic changes in channel modulatory properties. Expression of recombinant Gβγ provides us with a way to produce consistent and robust levels of Gβγ among the different G-protein constructs, in a manner that might be difficult to reliably achieve with receptor activated modulation. Western blot analysis of expression levels for the different Gβ constructs argues against such concentration variance (Fig. 9) . Each lane reports Gβ expression from cells cotransfected with the indicated channel subtype, and all mutants produced bands of comparable density, well within a factor of two by quantitative densitometry (see Fig. 9 legend). Blots of serial dilutions of recombinant Gβ protein further established that all bands were imaged in a linear range of responsiveness. These results suggest that average expression levels, taken over many cells, were quite similar. Reassuringly, even if we were to make the liberal supposition that Gβ expression levels of mutant constructs were to fluctuate from 50–200% of wild-type levels, sensitivity analysis (Fig. 9, C and D) indicates that very little change in τ(V) and W(∞,V) relations would result. In this analysis, k off(V) and k on(V)/[Gβγ] are held constant at the fitted values determined for wild-type Gβ data (Figs. 2 and 3), while [Gβγ] is halved or doubled from corresponding mean values. The graphs demonstrate that even these liberal changes in Gβγ concentration would not account for the properties of any of the mutant constructs that appeared to alter modulatory behavior. Furthermore, many of the effects reported above concern distinct alterations of k off, which would be independent of Gβγ concentration. It is also worth emphasizing that all Gβ constructs have been shown previously to fold correctly by gel-filtration assays (Ford et al., 1998). Finally, a remaining presumption of our experiments was that levels of endogenous Gβ were negligible compared with that of recombinant Gβ. The Western blot analysis also confirms this presumption. Overall, the distinctions in G-protein modulation reported in this study are likely to reflect intrinsic alterations in channel–Gβγ interaction.
Figure 9.
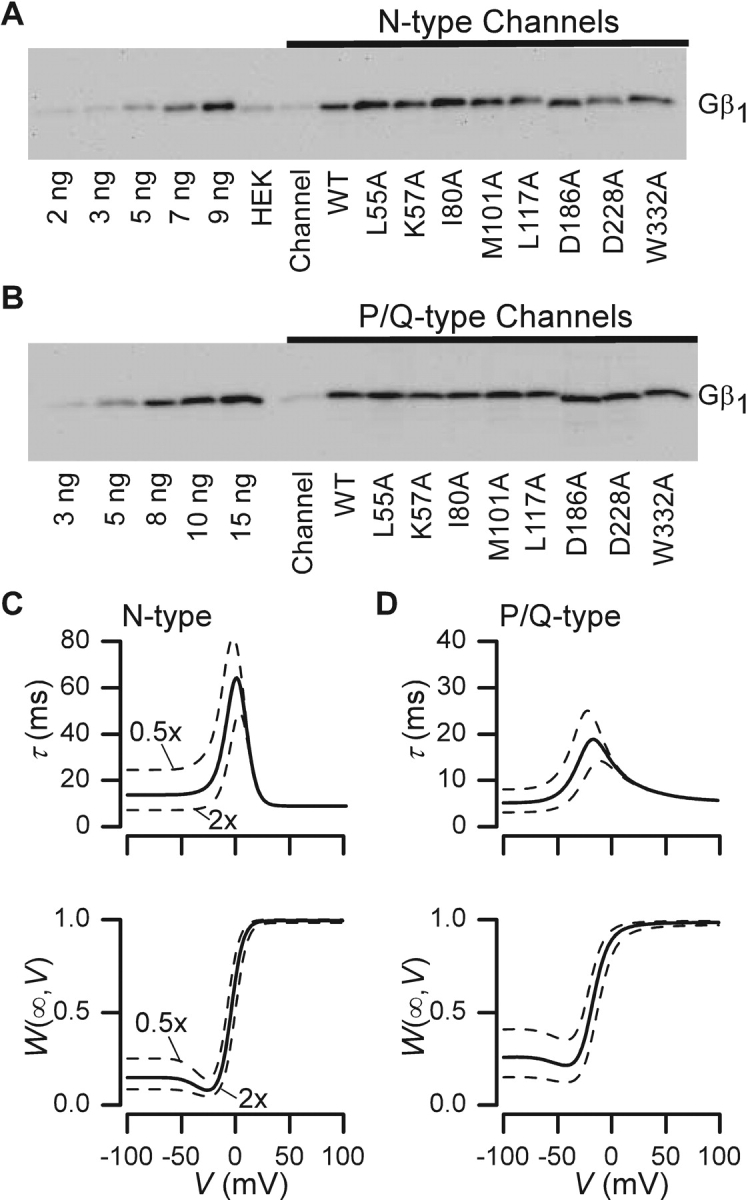
Western blots assaying Gβ expression. (A) Blots shown are for N-type calcium channel (α1B/β2a/α2δ) expressed alone, or coexpressed with wild-type or mutant Gβ1. Samples were obtained from the membrane fraction of cells. The soluble fraction gave far weaker signals. The first five lanes contain varying levels of purified, bovine Gβ1 to demonstrate that samples are in the linear range. “HEK” is HEK 293 cell lysates that have undergone calcium phosphate transfection using only cDNA encoding T antigen. “Channel” identifies extracts from cells transfected with only channel subunits. All other lanes are N-type channel subunits expressed with Gγ2 and either wild-type or mutant Gβ1, as labeled. Mutations D186A and D228A traveled slightly faster than other Gβs expressed, due to the change in charge that occurs with alanine mutation. All lanes were quantified based on band density and compared with the controls. Lanes with overexpressed Gβ (wild-type or mutant) averaged 9.15 ng with a standard deviation of 2.34. (B) Blots for wild-type and mutant Gβ1 coexpressed with P/Q-type calcium channels. Again, various levels of purified Gβ1 are shown in the first five lanes. The “channel” lane shows lysates from cells transfected with only the P/Q-type channel subunits (α1A/β2a/α2δ). Lanes with overexpressed Gβ (wild-type or mutant) averaged 10.63 ng with a standard deviation of 1.56. (C) Solid curves reproduce fits for modulation of N-type channels by wild-type Gβγ, taken from Fig. 2, B1 and B2. Dashed lines represent the simulated changes that would occur when G-protein concentration is increased or decreased by a factor of two, holding all other parameters fixed. (D) Analogous sensitivity analysis for modulation of P/Q-type channels by wild-type Gβγ, with solid curve fits reproduced from Fig. 3, D1 and D2, and format identical to that in C. Again, dashed lines represent the simulated changes that would occur when G-protein concentration is increased or decreased by a factor of two, holding all other parameters constant.
DISCUSSION
We devised a new kinetic approach to characterize G-protein modulation of N- and P/Q-type channels over a wide voltage range, termed compound-state willing-reluctant analysis. This approach revealed surprisingly large differences in the G-protein modulation of these two channel types. Whereas it has been well known that N-type channels facilitate and reinhibit about twofold slower than P/Q-type channels at the extreme voltages (Zhang et al., 1996; Arnot et al., 2000; Colecraft et al., 2000), our compound-state analysis now shows that the apparent equilibration rate of Gβγ interacting with these channels is almost an order of magnitude slower for N-type channels at potentials near 0 mV. Previous work from our lab suggested the presence of such a phenomenon (Colecraft et al., 2000), but the behavior over the entire range has not been shown until now. This stark quantitative difference actually reflects a more general distinction: N-type channels exhibit a profound bell-shaped voltage dependence of Gβγ kinetics, while P/Q-type channels exhibit only a hint of this voltage dependence. Motivated by the possibility that such contrasting functional profiles might reflect differences in channel/G-protein contact surfaces, we undertook alanine-scanning mutagenesis of Gβ and probed for changes in channel modulation using compound-state analysis. This approach identified a number of important residues for functional modulation, resolving effects at loci where previous screens had failed to detect perturbations. Mutations in the three regions that interact with the “switch interface” of Gα all disrupted Gβγ modulation on both channel types. However, the L55A mutation on the eastern zone, which interacts with the “NH2-terminal interface” of Gα, had no effect on N-type channels while considerably slowing the time constants of equilibration in the mid-voltage range with P/Q-type channels. This finding suggests a “selective” difference in Gβ interaction surfaces with the two types of channels, and identification of the full suite of such “selective” residues may ultimately furnish a structural framework for understanding the differing modulatory behaviors.
These findings raise three major points for discussion. First, what are the advantages of the compound-state analysis compared with more common characterization schemes? Second, do the in-depth analyses of G-protein modulation of N- and P/Q-type channels hint at general structure-function principles of modulation? Third, despite a common Gβ interaction surface with effectors, do distinctive contacts at the periphery of the surface afford differential modulation of these different effectors?
Appraisal of Compound-state Analysis for Characterizing G-protein Interaction
Past studies have often focused on characterizing isolated features of G-protein modulation: kinetic slowing, prepulse facilitation, and prepulse reinhibition. In this study, we have developed compound-state analysis, in which all of these aspects are integrated simultaneously, within a simple analytical framework. The result is a “kinetic fingerprint” that extends over a wide voltage range. The virtues of such an approach become apparent by considering how a narrow focus on one modulatory property can be misleading. For example, focusing on a common metric—the degree of facilitation taken from prepulse facilitation protocols (Fig. 10, A and B) —would suggest that while most of the mutations affect G-protein modulation, L55A had no effect. In reality, the L55A mutation exerts unmistakable effects on the kinetics of G-protein modulation of P/Q-type calcium channels (Fig. 7 A). However, the changes caused by this mutation are only apparent upon scrutinizing kinetic slowing at intermediate potentials (Fig. 7 C1). Furthermore, measuring the steady-state fraction of channels in the willing mode (W(∞,V)) over a wide range of voltages complements the kinetic information, and enables estimation of underlying changes of on/off rates that result from Gβ mutations.
Figure 10.
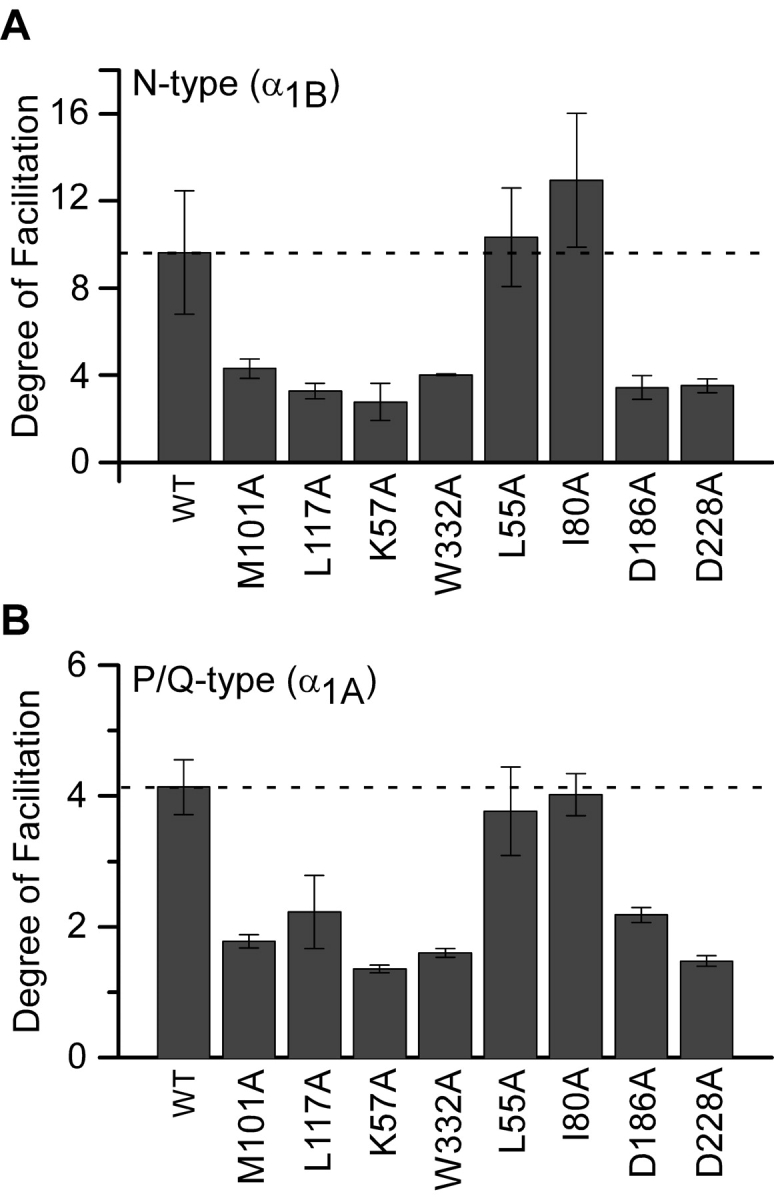
Degree of facilitation measured for N-and P/Q-type channels. (A) The degree of facilitation calculated (1/W(∞,−100 mV)), for N-type channels expressed with Gβ1 wild-type and with each mutant. (B) The degree of facilitation, calculated in same manner as A, for P/Q-type channels expressed with wild-type Gβ1 and with each mutant.
Another important feature of our approach was to characterize channel modulatory properties during consistently strong and uniform expression of various Gβ constructs (Fig. 9). This feature may help to explain why our N-type channel findings (Fig. 10 A) differ considerably from previously published data using the same Gβ1 mutants (Ford et al., 1998). In that study, wild-type Gβ1 showed a weak degree of facilitation (∼1.5) comparable to that of most Gβ1 mutants, except for L55A and I80A. The latter constructs were reported to be “gain of function” mutants that demonstrated markedly enhanced G-protein modulation compared with wild-type Gβ1. By contrast, our wild-type Gβ1 showed very strong modulation, with a degree of facilitation comparable to that of L55A and I80A (Fig. 10 A). At the same time, we reproduced the relative modulatory profile reported for the other Gβ1 mutants. The end result was that we arrive at a nearly inverse picture of Gβ1 mutant effects on modulation. Rather than L55A and I80A showing enhanced modulation, these mutants were the only ones to spare wild-type Gβ1 regulatory activity. All the other Gβ1 mutants were seen to impair modulation compared with wild-type. The basis for the differing findings is unclear, though it appears to arise from unusually weak expression of wild type Gβ1 in the earlier study (Ford et al., 1998).
An important concern with our approach was that the strong, constitutive presence of Gβγ could modulate channels via ancillary signaling pathways, unrelated to the direct binding of Gβγ to channels as modeled by compound-state analysis. In particular, Gβγ could well activate PLC and adenylate cyclase (AC) (Sunahara et al., 1996; Rhee and Bae, 1997), and these events might in principle affect our results. Concerning PLC, its activation leads to cleavage of phosphatidylinositol 4,5-biphosphate (PIP2), triggering a signaling cascade that includes activation of PKC. As many of the mutations in Gβ investigated here also disrupt Gβγ interaction with PLC (Ford et al., 1998), PIP2 levels in these cases would have been higher and PKC activation lower, in comparison to cells expressing wild-type Gβγ. Since PIP2 can inhibit P/Q-type channels in a voltage-dependent manner mimicking G-protein inhibition (Wu et al., 2002), higher PIP2 levels might be mistaken for increased G-protein inhibition. Likewise, since PKC antagonizes G-protein inhibition (Barrett and Rittenhouse, 2000; Cooper et al., 2000), diminished PKC activation could also appear as increased G-protein inhibition. The important point, however, is that increased G-protein modulation was not seen with any of our Gβ mutations, arguing against a major contribution of these ancillary regulatory pathways in the results. Regarding AC, its activation by Gβγ could also modulate N- and P/Q-type channels via activation of protein kinase A (PKA), which has been shown to produce modest facilitation of these calcium currents (Fukuda et al., 1996). Importantly, however, PKA up-regulation of channels occurs in a voltage-independent manner, and would not be expected to affect the voltage-dependent parameters characterized in this study. Finally, an important feature of our results is that constitutive expression of wild-type Gβγ produced modulatory properties that were quantitatively similar to those that we characterized previously for very transient, receptor-mediated activation (Fig. 10 in Colecraft et al., 2000). Since brief, receptor-mediated activation of G-proteins seems less likely to recruit ancillary signaling pathways, the quantitative agreement of effects produced by constitutive expression of Gβγ is reassuring. Overall, while it remains important to recognize the possibility of secondary effects of constitutive Gβγ expression, the pattern of our results provides no clear evidence for such crosstalk.
Emergent Structure-function Themes of Channel Modulation by G-proteins
Our data raises the intriguing possibility of at least two classes of Gβ residues, those involved in binding to the channel, and those involved in steering the Gβ subunit to the binding site. Interestingly, this division seems to mirror the classification of the Gβ residues according to their interaction with either the “switch interface” or “NH2-terminal interface” of Gα (Lambright et al., 1996). Specifically, in the setting of P/Q-type channels, the L55A mutation (“NH2-terminal interface” residues) features an essentially wild-type W(∞,V) curve, but clearly slowed time constants. This constellation of effects fits nicely with a scenario where this residue selectively mediates steering of Gβ to and from a binding site, rather than supporting binding to the channel per se. Disruption of such a steering mechanism would elevate the transition energy barrier that must be overcome for Gβγ to (un)bind to the channel, while sparing the relative free energies of (un)bound channels (Atkins, 1998). By contrast, all mutations on the “switch” interface resulted in clear changes in W(∞,V) curves, consistent with direct changes in binding affinity.
Consideration of such a steering mechanism raises an attractive hypothesis for rationalizing the faster time constants of P/Q- versus N-type channels across all voltages (Fig. 3). The P/Q-type channels may contain extra sites that interact with steering residues to guide the Gβγ subunit more efficiently to and from its main binding site, thus yielding faster kinetics of (un)binding. Therefore, disruption of steering residues would slow down the (un)binding process while sparing the affinity, just as seen with the L55A mutant. Interestingly, Gβ5, the only other Gβ subunit that does not conserve a leucine in the 55 position, also produces much slower facilitation and reinhibition time constants with P/Q-type channels compared with those induced by Gβ1 (Arnot et al., 2000). N-type channels may not show the same effect because they either do not possess areas that interact with analogous steering sites, or there may be more dominant rate-limiting factors that slow down the kinetics of modulation.
By contrast to all Gβ residues examined in this study, yet another class of interacting Gβ sites could involve those that do not form van der Waals contacts with Gα. At first glance, the existence of such residues might appear unlikely from a biological perspective, because a substantial number of such Gβ sites would permit constitutive channel modulation in the absence of explicit G-protein activation (which results in dissociation of Gα/Gβγ). However, the crystal structure of Gαt bound to Gβ1γ2 (Wall et al., 1995; Lambright et al., 1996) shows that, when Gαt is bound to Gβ1γ2, steric constraints created by the sheer size of Gαt would protect (from potential effectors) many Gβ1γ2 sites that are not explicitly involved in van der Waals contacts. This set of sterically protected Gβ1γ2 sites could then serve to interact with effectors, in a manner that would preserve gating by G-protein activation. In fact, peripheral Gβ1 residues outside of the van der Waals contact surface with Gα have proven to be important for interaction with other channel types such as GIRK (Albsoul-Younes et al., 2001; Mirshahi et al., 2002b). Indeed, Mirshahi et al. (2002a) have recently identified three Gβ1 residues that are not van der Waals contacts, but nonetheless affect both GIRK activation and N-type calcium channel modulation (Mirshahi et al., 2002a). It will be interesting to determine whether residues that are not van der Waals contact points affect Gβ steering, binding affinity, and/or other dimensions of interaction.
Custom G-proteins with Selective Modulation
Engineered Gβ subunits with selective modulation of certain types of calcium channels would provide important tools for dissecting physiological questions, and could furnish critical structural guidelines for designing novel therapeutic compounds of like selectivity. Toward this end, the L55A mutation is interesting because it selectively affected P/Q- versus N-type channel modulation. Though the attenuation of P/Q-type channel modulation was incomplete for this mutation, it is possible that substitution of more obtrusive amino acids than alanine could produce much stronger effects while sparing N-type channel regulation.
Beyond possible identification of particular residues with selective importance for one type of channel over another, this study also suggests a general structural theme concerning G-protein specificity. It appears that a core group of Gβ residues, located centrally on the “switch” interface, seems more likely to be of common importance for a number of different effectors, while specificity may be determined by residues nearer the periphery of this interface. In particular, all mutations in the central and south-central regions (Figs. 5–6) disrupted modulation for both N- and P/Q-type channels. By contrast, mutations on the eastern edge of the Gα interaction surface, L55A and I80A, had channel specific or no effects. The L55A mutant only displayed specificity for P/Q-type channels, and the I80A mutation had no significant affect on either channel type. Furthermore, examination of other effector targets lends further support to the idea that specificity may be determined by residues on the periphery. For example, L55A and I80A show very diverse actions across different effectors: both mutations show weakened activation of GIRK channels (Ford et al., 1998; Mirshahi et al., 2002b), and L55A eliminates activation of adenyl cyclase 2 while I80A spares it (Ford et al., 1998). Fitting with the common importance of proximal locations, all central and south-central Gβ mutations tested in this study strongly disrupted activation of adenyl cyclase 2 (Ford et al., 1998). Though there are certainly exceptions to peripheral specificity, this theme may prove to be a useful overall guideline.
Regarding therapeutics, the development of pharmaceuticals that mimic the action of calcium-channel–specific Gβ subunits remains an intriguing clinical prospect (Dickenson et al., 2002; Saegusa et al., 2002). Certainly, N-type calcium channels figure crucially in conducting peripheral pain impulses (Vanegas and Schaible, 2000). Moreover, selective N-type channel toxins such as ω-conotoxin MVIIA (SNX-111 or Zinconotide) and ω-conotoxin CVID (AM-336) attenuate pain in animal models and humans (McIntosh and Jones, 2001; Scott et al., 2002). Much of the action of endorphins and opiates is produced via Gβγ inhibition of calcium channels (Pertwee, 1997), and therein may lie some of the particular advantages of this modality of analgesia. Rather than producing complete tonic blockade of calcium channels, Gβγ inhibition allows low probability opening of N-type channels (Colecraft et al., 2001), and repetitive channel activation can transiently relieve the inhibition of calcium channels (Brody et al., 1997; Brody and Yue, 2000). Introducing pharmaceutics that mimic the action of Gβγ subunits with channel selectivity may thus customize the qualities of analgesia for particular clinical contexts, since different calcium channel isoforms may play different roles in pain perception (Knight et al., 2002; Saegusa et al., 2002). In the nearer term, engineered Gβ subunits with selectivity for certain calcium channels may provide important investigative tools for deepening our fundamental understanding of pain perception, thereby refining the desired activity profile of designer pharmaceutics.
Acknowledgments
We thank Heidi Hamm for donating several of the Gβ constructs and for helpful advice on Gβ Western blots; and Badr Alseikhan for technical assistance and discussion on Western blots.
This work was supported by a National Science Foundation Training Fellowship (H.L. Agler), a National Institutes of Health Training Grant (H.L. Agler), and RO1 grants from the National Institutes of Health (D.T. Yue).
Olaf S. Andersen served as editor.
Footnotes
Abbreviation used in this paper: Gβγ, G-protein βγ complex.
References
- Albsoul-Younes, A.M., P.M. Sternweis, P. Zhao, H. Nakata, S. Nakajima, Y. Nakajima, and T. Kozasa. 2001. Interaction sites of the G protein β subunit with brain G protein-coupled inward rectifier K+ channel. J. Biol. Chem. 276:12712–12717. [DOI] [PubMed] [Google Scholar]
- Arnot, M.I., S.C. Stotz, S.E. Jarvis, and G.W. Zamponi. 2000. Differential modulation of N-type 1B and P/Q-type 1A calcium channels by different G protein subunit isoforms. J. Physiol. 527:203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artim, D.E., and S.D. Meriney. 2000. G-protein-modulated Ca(2+) current with slowed activation does not alter the kinetics of action potential-evoked Ca(2+) current. J. Neurophysiol. 84:2417–2425. [DOI] [PubMed] [Google Scholar]
- Atkins, P. 1998. Chapter 27: Molecular reaction dynamics. Physical Chemistry. W.H. Freeman and Company, New York. 830–843.
- Barrett, C.F., and A.R. Rittenhouse. 2000. Modulation of N-type calcium channel activity by G-proteins and protein kinase C. J. Gen. Physiol. 115:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean, B.P. 1989. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 340:153–156. [DOI] [PubMed] [Google Scholar]
- Boland, L.M., and B.P. Bean. 1993. Modulation of N-type calcium channels in bullfrog sympathetic neurons by luteinizing hormone-releasing hormone: kinetics and voltage dependence. J. Neurosci. 13:516–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet, E., T.W. Soong, A. Stea, and T.P. Snutch. 1996. Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proc. Natl. Acad. Sci. USA. 93:1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody, D.L., P.G. Patil, J.G. Mulle, T.P. Snutch, and D.T. Yue. 1997. Bursts of action potential waveforms relieve G-protein inhibition of recombinant P/Q-type Ca2+ channels in HEK 293 cells. J. Physiol. 499:637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody, D.L., and D.T. Yue. 2000. Relief of G-protein inhibition of calcium channels and short-term synaptic facilitation in cultured hippocampal neurons. J. Neurosci. 20:889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canti, C., K.M. Page, G.J. Stephens, and A.C. Dolphin. 1999. Identification of residues in the N terminus of α1B critical for inhibition of the voltage-dependent calcium channel by Gβ γ. J. Neurosci. 19:6855–6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colecraft, H.M., D.L. Brody, and D.T. Yue. 2001. G-protein inhibition of N- and P/Q-type calcium channels: distinctive elementary mechanisms and their functional impact. J. Neurosci. 21:1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colecraft, H.M., P.G. Patil, and D.T. Yue. 2000. Differential occurrence of reluctant openings in G-protein-inhibited N- and P/Q-type calcium channels. J. Gen. Physiol. 115:175–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, C.B., M.I. Arnot, Z.P. Feng, S.E. Jarvis, J. Hamid, and G.W. Zamponi. 2000. Cross-talk between G-protein and protein kinase C modulation of N-type calcium channels is dependent on the G-protein β subunit isoform. J. Biol. Chem. 275:40777–40781. [DOI] [PubMed] [Google Scholar]
- Currie, K.P., and A.P. Fox. 1997. Comparison of N- and P/Q-type voltage-gated calcium channel current inhibition. J. Neurosci. 17:4570–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie, K.P., and A.P. Fox. 2002. Differential facilitation of N- and P/Q-type calcium channels during trains of action potential-like waveforms. J. Physiol. 539:419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Waard, M., H. Liu, D. Walker, V.E. Scott, C.A. Gurnett, and K.P. Campbell. 1997. Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature. 385:446–450. [DOI] [PubMed] [Google Scholar]
- Dickenson, A.H., E.A. Matthews, and R. Suzuki. 2002. Neurobiology of neuropathic pain: mode of action of anticonvulsants. Eur. J. Pain. 6:51–60. [DOI] [PubMed] [Google Scholar]
- Dunlap, K., J.I. Luebke, and T.J. Turner. 1995. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 18:89–98. [PubMed] [Google Scholar]
- Elmslie, K.S., W. Zhou, and S.W. Jones. 1990. LHRH and GTP-γ-S modify calcium current activation in bullfrog sympathetic neurons. Neuron. 5:75–80. [DOI] [PubMed] [Google Scholar]
- Erickson, M.G., B.A. Alseikhan, B.Z. Peterson, and D.T. Yue. 2001. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 31:973–985. [DOI] [PubMed] [Google Scholar]
- Ford, C.E., N.P. Skiba, H. Bae, Y. Daaka, E. Reuveny, L.R. Shekter, R. Rosal, G. Weng, C.S. Yang, R. Iyengar, et al. 1998. Molecular basis for interactions of G protein βγ subunits with effectors. Science. 280:1271–1274. [DOI] [PubMed] [Google Scholar]
- Fukuda, K., S. Kaneko, N. Yada, M. Kikuwaka, A. Akaike, and M. Satoh. 1996. Cyclic AMP-dependent modulation of N- and Q-type Ca2+ channels expressed in Xenopus oocytes. Neurosci. Lett. 217:13–16. [DOI] [PubMed] [Google Scholar]
- Furukawa, T., R. Miura, Y. Mori, M. Strobeck, K. Suzuki, Y. Ogihara, T. Asano, R. Morishita, M. Hashii, H. Higashida, M. Yoshii, and T. Nukada. 1998. a. Differential interactions of the C terminus and the cytoplasmic I-II loop of neuronal Ca2+ channels with G-protein α and β γ subunits. II. Evidence for direct binding. J. Biol. Chem. 273:17595–17603. [DOI] [PubMed] [Google Scholar]
- Furukawa, T., T. Nukada, Y. Mori, M. Wakamori, Y. Fujita, H. Ishida, K. Fukuda, S. Kato, and M. Yoshii. 1998. b. Differential interactions of the C terminus and the cytoplasmic I-II loop of neuronal Ca2+ channels with G-protein α and β γ subunits. I. Molecular determination. J. Biol. Chem. 273:17585–17594. [DOI] [PubMed] [Google Scholar]
- Garcia, D.E., B. Li, R.E. Garcia-Ferreiro, E.O. Hernandez-Ochoa, K. Yan, N. Gautam, W.A. Catterall, K. Mackie, and B. Hille. 1998. G-protein β-subunit specificity in the fast membrane-delimited inhibition of Ca2+ channels. J. Neurosci. 18:9163–9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam, N., J. Northup, H. Tamir, and M.I. Simon. 1990. G protein diversity is increased by associations with a variety of γ subunits. Proc. Natl. Acad. Sci. USA. 87:7973–7977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, C., X. Yan, H. Zhang, T. Mirshahi, T. Jin, A. Huang, and D.E. Logothetis. 2002. Identification of critical residues controlling G protein-gated inwardly rectifying K+ channel activity through interactions with the β γ subunits of G proteins. J. Biol. Chem. 277:6088–6096. [DOI] [PubMed] [Google Scholar]
- Herlitze, S., G.H. Hockerman, T. Scheuer, and W.A. Catterall. 1997. Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel α1A subunit. Proc. Natl. Acad. Sci. USA. 94:1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, S.R. 1996. Voltage-dependent modulation of N-type calcium channels by G-protein β-γ subunits. Nature. 380:255–258. [DOI] [PubMed] [Google Scholar]
- Jones, L.P., P.G. Patil, T.P. Snutch, and D.T. Yue. 1997. G-protein modulation of N-type calcium channel gating current in human embryonic kidney cells (HEK 293). J. Physiol. 498:601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado, L.A., P.S. Chockalingam, and H.W. Jarrett. 1999. Apocalmodulin. Physiol. Rev. 79:661–682. [DOI] [PubMed] [Google Scholar]
- Kaneko, S., N. Yada, K. Fukuda, M. Kikuwaka, A. Akaike, and M. Satoh. 1997. Inhibition of Ca2+ channel current by mu- and kappa-opioid receptors coexpressed in Xenopus oocytes: desensitization dependence on Ca2+ channel α 1 subunits. Br. J. Pharmacol. 121:806–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, M., T. Nukada, T. Asano, Y. Mori, A. Akaike, M. Satoh, and S. Kaneko. 2001. Binding of G α(o) N terminus is responsible for the voltage-resistant inhibition of α1A (P/Q-type, Ca(v)2.1) Ca2+ channels. J. Biol. Chem. 276:28731–28738. [DOI] [PubMed] [Google Scholar]
- Knight, Y.E., T. Bartsch, H. Kaube, and P.J. Goadsby. 2002. P/Q-type calcium-channel blockade in the periaqueductal gray facilitates trigeminal nociception: a functional genetic link for migraine? J. Neurosci. 22:RC213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambright, D.G., J. Sondek, A. Bohm, N.P. Skiba, H.E. Hamm, and P.B. Sigler. 1996. The 2.0 A crystal structure of a heterotrimeric G protein. Nature. 379:311–319. [DOI] [PubMed] [Google Scholar]
- Lipscombe, D., S. Kongsamut, and R. Tsien. 1989. Alpha-adrenergic inhibition of sympathetic neurotransmitter release mediated by modulation of N-type calcium-channel gating. Nature. 340:639–642. [DOI] [PubMed] [Google Scholar]
- Logothetis, D.E., Y. Kurachi, J. Galper, E.J. Neer, and D.E. Clapham. 1987. The β γ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 325:321–326. [DOI] [PubMed] [Google Scholar]
- Luebke, J.I., K. Dunlap, and T. Turner. 1993. Multiple calcium channel types control glutamatergic synaptic transmission in the hippocampus. Neuron. 11:895–902. [DOI] [PubMed] [Google Scholar]
- Mackie, K., and B. Hille. 1992. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc. Natl. Acad. Sci. USA. 89:3825–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram, H., and M. Tsodyks. 1996. Redistribution of synaptic efficacy between neocortical pyramidal neurons. Nature. 382:807–810. [DOI] [PubMed] [Google Scholar]
- McIntosh, J.M., and R.M. Jones. 2001. Cone venom–from accidental stings to deliberate injection. Toxicon. 39:1447–1451. [DOI] [PubMed] [Google Scholar]
- Miller, R.J. 1990. Receptor-mediated regulation of calcium channels and neurotransmitter release. FASEB J. 4:3291–3299. [PubMed] [Google Scholar]
- Mintz, I.M., and B.P. Bean. 1993. GABAB receptor inhibition of P-type Ca2+ channels in central neurons. Neuron. 10:889–898. [DOI] [PubMed] [Google Scholar]
- Mirshahi, T., V. Mittal, H. Zhang, M.E. Linder, and D.E. Logothetis. 2002. a. Distinct sites on G protein βγ subunits regulate different effector functions. J. Biol. Chem. 277:36345–36350. [DOI] [PubMed] [Google Scholar]
- Mirshahi, T., L. Robillard, H. Zhang, T.E. Hebert, and D.E. Logothetis. 2002. b. Gβ residues that do not interact with Gα underlie agonist-independent activity of K+ channels. J. Biol. Chem. 277:7348–7355. [DOI] [PubMed] [Google Scholar]
- Neher, E., and J.H. Steinbach. 1978. Local anaesthetics transiently block currents through single acetylcholine-receptor channels. J. Physiol. 277:153–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, K.M., G.J. Stephens, N.S. Berrow, and A.C. Dolphin. 1997. The intracellular loop between domains I and II of the B-type calcium channel confers aspects of G-protein sensitivity to the E-type calcium channel. J. Neurosci. 17:1330–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, K.M., C. Canti, G.J. Stephens, N.S. Berrow, and A.C. Dolphin. 1998. Identification of the amino terminus of neuronal Ca2+ channel α1 subunits α1B and α1E as an essential determinant of G-protein modulation. J. Neurosci. 18:4815–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil, P.G., M. de Leon, R.R. Reed, S. Dubel, T.P. Snutch, and D.T. Yue. 1996. Elementary events underlying voltage-dependent G-protein inhibition of N-type calcium channels. Biophys. J. 71:2509–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes, E., A. Castellano, H.S. Kim, P. Bertrand, E. Baggstrom, A.E. Lacerda, X.Y. Wei, and L. Birnbaumer. 1992. Cloning and expression of a cardiac/brain β subunit of the L-type calcium channel. J. Biol. Chem. 267:1792–1797. [PubMed] [Google Scholar]
- Pertwee, R.G. 1997. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 74:129–180. [DOI] [PubMed] [Google Scholar]
- Qin, N., D. Platano, R. Olcese, E. Stefani, and L. Birnbaumer. 1997. Direct interaction of gβγ with a C-terminal gβγ-binding domain of the Ca2+ channel α1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proc. Natl. Acad. Sci. USA. 94:8866–8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee, S.G., and Y.S. Bae. 1997. Regulation of phosphoinositide-specific phospholipase C isozymes. J. Biol. Chem. 272:15045–15048. [DOI] [PubMed] [Google Scholar]
- Saegusa, H., Y. Matsuda, and T. Tanabe. 2002. Effects of ablation of N- and R-type Ca2+ channels on pain transmission. Neurosci. Res. 43:1–7. [DOI] [PubMed] [Google Scholar]
- Scott, D.A., C.E. Wright, and J.A. Angus. 2002. Actions of intrathecal omega-conotoxins CVID, GVIA, MVIIA, and morphine in acute and neuropathic pain in the rat. Eur. J. Pharmacol. 451:279–286. [DOI] [PubMed] [Google Scholar]
- Simen, A.A., and R.J. Miller. 2000. Involvement of regions in domain I in the opioid receptor sensitivity of α1B Ca2+ channels. Mol. Pharmacol. 57:1064–1074. [PubMed] [Google Scholar]
- Stea, A., W.J. Tomlinson, T.W. Soong, E. Bourinet, S.J. Dubel, S.R. Vincent, and T.P. Snutch. 1994. Localization and functional properties of a rat brain α 1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proc. Natl. Acad. Sci. USA. 91:10576–10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto, K., T. Nukada, T. Tanabe, H. Takahashi, M. Noda, N. Minamino, K. Kangawa, H. Matsuo, T. Hirose, S. Inayama, et al. 1985. Primary structure of the β-subunit of bovine transducin deduced from the cDNA sequence. FEBS Lett. 191:235–240. [DOI] [PubMed] [Google Scholar]
- Sunahara, R.K., C.W. Dessauer, and A.G. Gilman. 1996. Complexity and diversity of mammalian adenylyl cyclases. Annu. Rev. Pharmacol. Toxicol. 36:461–480. [DOI] [PubMed] [Google Scholar]
- Takahashi, T., and A. Momiyama. 1993. Different types of calcium channels mediate central synaptic transmission. Nature. 366:156–158. [DOI] [PubMed] [Google Scholar]
- Tomlinson, W.J., A. Stea, E. Bourinet, P. Charnet, J. Nargeot, and T.P. Snutch. 1993. Functional properties of a neuronal class C L-type calcium channel. Neuropharmacology. 32:1117–1126. [DOI] [PubMed] [Google Scholar]
- Twitchell, W., S. Brown, and K. Mackie. 1997. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 78:43–50. [DOI] [PubMed] [Google Scholar]
- Vanegas, H., and H. Schaible. 2000. Effects of antagonists to high-threshold calcium channels upon spinal mechanisms of pain, hyperalgesia and allodynia. Pain. 85:9–18. [DOI] [PubMed] [Google Scholar]
- Wall, M.A., D.E. Coleman, E. Lee, J.A. Iniguez-Lluhi, B.A. Posner, A.G. Gilman, and S.R. Sprang. 1995. The structure of the G protein heterotrimer Gi α1 β1 γ2. Cell. 83:1047–1058. [DOI] [PubMed] [Google Scholar]
- Wheeler, D.B., A. Randall, and R.W. Tsien. 1994. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 264:107–111. [DOI] [PubMed] [Google Scholar]
- Wilding, T.J., M.D. Womack, and E.W. McCleskey. 1995. Fast, local signal transduction between the mu opioid receptor and Ca2+ channels. J. Neurosci. 15:4124–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, M.E., P.F. Brust, D.H. Feldman, S. Patthi, S. Simerson, A. Maroufi, A.F. McCue, G. Velicelebi, S.B. Ellis, and M.M. Harpold. 1992. Structure and functional expression of an omega-conotoxin-sensitive human N-type calcium channel. Science. 257:389–395. [DOI] [PubMed] [Google Scholar]
- Wilson, R.I., and R.A. Nicoll. 2002. Endocannabinoid signaling in the brain. Science. 296:678–682. [DOI] [PubMed] [Google Scholar]
- Wu, L., C.S. Bauer, X.G. Zhen, C. Xie, and J. Yang. 2002. Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature. 419:947–952. [DOI] [PubMed] [Google Scholar]
- Wu, L.G., and P. Saggau. 1994. Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron. 12:1139–1148. [DOI] [PubMed] [Google Scholar]
- Wu, L.G., and P. Saggau. 1997. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 20:204–212. [DOI] [PubMed] [Google Scholar]
- Zamponi, G.W., E. Bourinet, D. Nelson, J. Nargeot, and T.P. Snutch. 1997. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 385:442–446. [DOI] [PubMed] [Google Scholar]
- Zamponi, G.W., and T.P. Snutch. 1998. Decay of prepulse facilitation of N type calcium channels during G protein inhibition is consistent with binding of a single Gβ subunit. Proc. Natl. Acad. Sci. USA. 95:4035–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J.F., P.T. Ellinor, R.W. Aldrich, and R.W. Tsien. 1996. Multiple structural elements in voltage-dependent Ca2+ channels support their inhibition by G proteins. Neuron. 17:991–1003. [DOI] [PubMed] [Google Scholar]