

Abstract
Nonmuscle myosin can generate force and shortening in smooth muscle, as revealed by studies of the urinary bladder from mice lacking smooth muscle myosin heavy chain (SM-MHC) but expressing the nonmuscle myosin heavy chains A and B (NM-MHC A and B; Morano, I., G.X. Chai, L.G. Baltas, V. Lamounier-Zepter, G. Lutsch, M. Kott, H. Haase, and M. Bader. 2000. Nat. Cell Biol. 2:371–375). Intracellular calcium was measured in urinary bladders from SM-MHC–deficient and SM-MHC–expressing mice in relaxed and contracted states. Similar intracellular [Ca2+] transients were observed in the two types of preparations, although the contraction of SM-MHC–deficient bladders was slow and lacked an initial peak in force. The difference in contraction kinetics thus do not reflect differences in calcium handling. Thick filaments were identified with electron microscopy in smooth muscle cells of SM-MHC–deficient bladders, showing that NM-MHC can form filaments in smooth muscle cells. Maximal shortening velocity of maximally activated, skinned smooth muscle preparations from SM-MHC–deficient mice was significantly lower and more sensitive to increased MgADP compared with velocity of SM-MHC–expressing preparations. Active force was significantly lower and less inhibited by increased inorganic phosphate. In conclusion, large differences in nucleotide and phosphate binding exist between smooth and nonmuscle myosins. High ADP binding and low phosphate dependence of nonmuscle myosin would influence both velocity of actin translocation and force generation to promote slow motility and economical force maintenance of the cell.
Keywords: nonmuscle myosin, urinary bladder, ATP, ADP, phosphate
INTRODUCTION
Nonmuscle myosins of the myosin II super family are ubiquitously expressed motor proteins in eukaryotic cells and are involved in a large variety of important physiological tasks, including cell migration and cytokinesis (Spudich, 1989). Mutations in a nonmuscle myosin heavy chain gene have been associated with severe disease affecting many organ systems (Kelley et al., 2000; Heath et al., 2001). Nonmuscle myosin expression is altered in several pathophysiological conditions, e.g., atherosclerotic plaque formation (Kuro-o et al., 1991).
The velocity of actin over nonmuscle myosin is slow compared with muscle myosin in vitro (Umemoto and Sellers, 1990), suggesting differences in the kinetics of the cross-bridge interactions. In general, a relationship between the rate of the ADP release from the actin–myosin–ADP complex and the shortening velocity of muscle fibers has been proposed (Siemankowski et al., 1985; Weiss et al., 2001). Consistent with this, smooth muscle myosin has a higher ADP affinity in vitro (Cremo and Geeves, 1998) and in muscles fibers (Arheden and Arner, 1992; Nishiye et al., 1993) compared with the faster skeletal muscle myosin. This correlation is also valid within the smooth muscle group; higher ADP affinity has been demonstrated in slow compared with fast smooth muscle types (Fuglsang et al., 1993; Khromov et al., 1998; Löfgren et al., 2001). At present, information regarding the kinetics of the actin–nonmuscle myosin interaction of the organized contractile system is very sparse.
Recently, a trangenic mouse strain lacking the smooth muscle myosin heavy chain (SM-MHC)* expression has been introduced (Morano et al., 2000). These animals die a few days after birth but their urinary bladder smooth muscle obtained immediately postnatally can contract by the action of the nonmuscle myosins. This contraction has a slow onset in response to stimulation and is characterized by a low shortening velocity. The smooth muscle of these animals provide a unique model for studies of regulation and contractile kinetics of nonmuscle myosin.
In the present study we have used the urinary bladder from SM-MHC–deficient mice to answer the following questions: (a) Is the slow onset of the contraction of intact smooth muscle preparations from the SM-MHC–deficient mice due to altered Ca2+ handling? (b) Can nonmuscle myosins form thick filaments in the smooth muscle cells? (c) What is the shortening velocity of fully activated organized nonmuscle myosin? (d) What are the binding characteristics of substrates and products (MgATP, MgADP, and inorganic phosphate) in the cross-bridge cycle of nonmuscle myosin?
MATERIALS AND METHODS
Animals and Preparations
SM-MHC–deficient mice were generated using gene targeting as described previously (Morano et al., 2000). New-born mice were killed within 24 h of birth and their urinary bladders were dissected. Preparations were used for measurements of intracellular calcium in intact or chemically skinned preparations and used for mechanical measurements. Samples were frozen for gelelectrophoresis and fixed for immunohistochemistry and electronmicroscopy. In some analyses, urinary bladder from 3-wk-old SM-MHC–expressing mice were used.
Homozygous SM-MHC–deficient mice were identified by the thin-walled and distended urinary bladder and the lack of an initial force transient in intact preparations (Morano et al., 2000). This definition of the phenotype was confirmed by gelelectrophoresis showing a low total myosin heavy chain content and a lack of the 205-kD SM1-MHC band in these animals. It has been shown that the myosin content is essentially similar in the mice heterozygous and homozygous for the SM-MHC gene (Morano et al., 2000) and these animals were analyzed as one group denoted SM-MHC expressing.
Measurements of Intracellular Calcium in Intact Preparations
Small strips (2–3 mm long, ∼500 μm wide) were prepared from the urinary bladder wall in the sagittal direction. The preparations were wrapped at each end with aluminum foil, and transferred to an oxygenated HEPES (N-(2-Hydroxyethyl) piperazine-N′-(2-ethanesulfonic acid)) buffered solution containing, in mM: NaCl 118, KCl 5, Na2HPO4 1.2, MgCl2 1.2, HEPES 24 (pH 7.4), glucose 10, and CaCl2 1.5. Loading of Fura-2 was performed for 3–4 h at 22°C in the HEPES-buffered solution containing 10 μM Fura-2-AM (Molecular Probes), 1% DMSO, and 0.02% Pluronic F127. After an ∼30-min wash in the HEPES buffer the preparations were transferred to a 1-ml bath on the stage of an inverted microscope. The muscles were kept in a Krebs-Henseleit solution of the following composition (in mM): NaCl 119, glucose 12.2, KCl 4.6, NaHCO3 25, KH2PO4 1.2, MgSO4 1.2, CaCl2 2 at 37°C, gassed with 95% O2/5% CO2 giving a pH of 7.4. Recordings of force and intracellular free Ca2+ concentration were made in the Krebs-Henseleit solution as described previously (Lucius et al., 1998) in the relaxed state (with and without 2 mM extracellular CaCl2) and after activation with 80 mM KCl. Force and fluorescence signals (at an emission wavelength of 510 nm) at excitation wavelengths 340 and 380 nm were recorded with a time resolution of 4–6 Hz. An internal calibration of the Ca2+ signal was made as described previously (Lucius et al., 1998), using 50 μM ionomycin for permeabilization, 20 mM MnCl2 for quenching, and an apparent dissociation constant of the calcium–fura-2 complex of 224 nM (Grynkiewicz et al., 1985).
Preparation of Permeabilized Tissue
Strips (2–3 mm long, ∼500 μm wide) of urinary bladder were pinned to silica gel and permeabilized with 1% Triton X-100 as described previously (Arner and Hellstrand, 1985), and thereafter kept in a glycerol containing solution at −20°C and used for quick-release experiments and isometric force registrations, as described below, within a month. All experiments on skinned muscle preparations were performed at room temperature (22°C).
Quick-release Experiments
Maximal unloaded shortening velocity (Vmax) at maximal activation was examined with the isotonic quick-release method (compare Arner and Hellstrand, 1985). The preparations were attached with aluminum clips between a force transducer (AE 801; SensoNor) and an isotonic lever arm. The experimental procedure was executed as described previously (Löfgren et al., 2001). The preparations were kept maximally activated using repeated incubation with ATP-γ-S (Arheden et al., 1988). Contractions were elicited by transfer to an ATP-containing solution (see below) and at the plateau of each contraction, a series of 15–25 releases to different afterloads was performed. After each release, the length and afterload were recorded for 1 s. Velocity was determined 100 ms after release. Vmax was calculated by fitting the Hill equation (Hill, 1938; Eq. 1) to the force-velocity data and extrapolating to zero afterload:
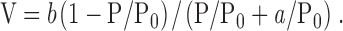 |
(1) |
In this equation, V is the velocity, Po is isometric force, P is afterload, and a and b are constants. Two series of experiments were performed: (a) varied [MgATP] in the presence of an ATP-regenerating system and (b) varied [MgADP] at constant [MgATP] in the absence of an ATP-regenerating system. The ATP and ADP concentrations were applied at random order. At the end of the experiments in the first series, a contraction in 3.2 mM MgATP was followed by a 40 min long incubation in rigor solution (0 MgATP and 0 phosphocreatine and creatine kinase with 50 U/ml hexokinase and 10 mM glucose) before a series of releases was performed, in order to determine the apparent Vmax in rigor (Löfgren et al., 2001).
Measurements of Isometric Force
Skinned preparations were mounted between a force transducer (AE 801; SensoNor) and a stainless steel pin and maximally activated using ATP-γ-S (Arheden et al., 1988). In the series of experiments where the effects of inorganic phosphate on isometric force was determined, the inorganic phosphate was introduced in the ATP-containing activating solution and the different concentrations of inorganic phosphate were applied at random order. In a separate series of experiments, the maximal force generation at 3.2 mM MgATP was determined. These preparations were fixed in 1% glutaraldehyde, embedded in Epon, and sectioned for light microscopy to determine preparation cross-sectional area.
Solutions Used in Experiments on Skinned Preparations
For the experiments on skinned preparations, a solution of the following composition was used: 30 mM TES, 4 mM EGTA, and 2 mM free Mg2+. The ionic strength and the pH were adjusted to 150 mM and 6.9, respectively, using KCl and KOH. The standard ATP-containing solution contained 3.2 mM MgATP and an ATP-regenerating system with 12 mM of phosphocreatine and 0.5 mg/ml of creatine kinase. In the ADP-containing solutions, the ATP-regenerating system was not used and 0.2 mM of the myokinase inhibitor AP5A was added (Feldhaus et al., 1975).
Western Blotting and Immunohistochemistry
Pieces of whole bladder wall were quickly frozen in liquid N2 and kept at −80°C. The samples and SDS gels were prepared essentially as described by Wede et al. (2002). Samples were loaded on three gels. One gel was stained with Coomassie blue and the other two were used for Western blot using a polyclonal rabbit antibody against nonmuscle myosin heavy chain A (NM-MHC-A), a gift from Dr. R. Adelstein (Kelley et al., 1996), or a polyclonal rabbit antibody against NM-MHC-B (Sjuve et al., 2001). Immunoreactivity was detected using EnhancedChemiLuminescence (ECL; Amersham Biosciences) and visualized with a Fluo-S Max (Bio-Rad Laboratories). For immunohistochemistry, urinary bladders were cut open, pinned to pieces of cork, fixed in 2% formaldehyde containing 0.2% picric acid in 0.1 M phosphate buffer (pH 7.2) over night, rinsed in a Tyrode solution with 10% sucrose, and embedded for cryosectioning (Sjuve et al., 1998). Sections were stained with NM-MHC-A or NM-MHC-B antibodies. Double staining with a monoclonal antibody raised in mouse against smooth muscle α-actin (Cy3 conjugated; C6198, Sigma-Aldrich) was used to determine the colocalization of the proteins in cells/regions.
Electron Microscopy
Strips of urinary bladder tissue were pinned to silica gel and fixed using glutaraldehyde and paraformaldehyde as described previously (Sjuve et al., 1998). The preparations were stored at 4°C in cacodylate buffer (0.125 M) with 0.1% glutaraldehyde. Post fixation was in 1% OsO2, followed by contrasting with uranyl acetate and Reynolds' lead citrate, and embedding in Epon. Sections, 50-nm thick, were examined using a Jeol JEM 1230 microscope equipped with a Gatan Multiscan CCD camera Model 791.
Statistics
Values are given as means ± SEM. Curve fitting was performed using routines implemented in Sigma Plot (SPSS Science). Statistical comparisons were made using Student's t test or repeated measures analysis of variance (using SPSS 10.1.3).
RESULTS
Force and Intracellular Calcium Transients in Intact Preparations
Fig. 1 shows original recordings of force and intracellular [Ca2+] in relaxed and contracted urinary bladder tissue from a SM-MHC–expressing and a SM-MHC–deficient mouse. Activation with high K+ in the SM-MHC–expressing muscle resulted in a contraction with an initial force peak followed by a decay to a stable force level. In the preparation from the SM-MHC–deficient mouse, force increased gradually to a steady force level without an initial peak. In both preparations, a similar Ca2+ transient was observed with a peak reaching between 400–500 nM and a fall to a stable lower level during the sustained phase of contraction. Table I summarizes the results on intracellular [Ca2+] and force. Although force transients were different, no significant differences in calcium levels between the tissues were observed in the relaxed state (with and without external calcium) or during activation with high K+.
Figure 1.

Isometric force (A and B) and intracellular calcium (C and D) in intact urinary bladder preparations from new-born SM-MHC–expressing (A and C) and SM-MHC–deficient (B and D) mice. Preparations were activated with 80 mM K+ (arrows).
TABLE I.
Force and Intracellular Calcium Concentration in Urinary Bladder Preparations from SM-MHC–expressing and SM-MHC–deficient Mice
| SM-MHC expressing | SM-MHC deficient | |
|---|---|---|
| Initial/sustained force | 2.57 ± 0.26 | 0.84 ± 0.04 P < 0.001 |
| Relaxed, calcium-free (nM) | 181 ± 26 | 206 ± 44 |
| Relaxed, 2.5 mM CaCl2 (nM) | 284 ± 35 | 302 ± 47 |
| Peak (nM) | 414 ± 46 | 482 ± 64 |
| Plateau (nM) | 313 ± 31 | 326 ± 41 |
Initial (maximal value measured within 60 s after activation) relative to sustained (plateau after 300 s) force was evaluated. Note that for the SM-MHC–deficient mice, no clear initial peak in force was detected. Calcium was measured in the relaxed state (at 0 and 2.5 mM external CaCl2) and during peak and plateau phase (after 300 s) of high K+–induced contractions. No significant differences in calcium transients were observed between the two muscle groups, although force transients were markedly different. n = 6–12.
Force-velocity Relation and Maximal Isometric Force
Original force-velocity relations of urinary bladder preparations from a new-born and a 3-wk-old SM-MHC–expressing and a new-born SM-MHC–deficient mouse are shown in Fig. 2 A. The SM-MHC–deficient preparation had markedly lower shortening velocity. In Fig. 2 B, mean values of maximal shortening velocity (Vmax) at 3.2 mM MgATP for the respective groups are shown. Vmax of the urinary bladder preparations of the SM-MHC–deficient mice was ∼40% of that in the SM-MHC–expressing tissues. The Vmax values in Fig. 2 B were not corrected for the influence of velocity at zero MgATP (see below). If these corrections were made, the velocity of SM-MHC–deficient tissue would be ∼30% of that of the new-born SM-MHC–expressing tissue at 3.2 mM MgATP. No significant difference in Vmax was observed between the new-born and the 3-wk-old SM-MHC–expressing tissues. The parameters a/P o of Eq. 1 was: 0.426 ± 0.165 for SM-MHC–deficient mice, 0.144 ± 0.017 for new-born SM-MHC–expressing and 0.127 ± 0.024 for 3-wk-old SM-MHC–expressing mice (3.2 mM MgATP). The parameter b was: 0.028 ± 0.009 for SM-MHC–deficient mice, 0.027 ± 0.002 for new-born SM-MHC–expressing mice, and 0.021 ± 0.003 ML/s for 3-wk-old SM-MHC–expressing mice (3.2 mM MgATP). The low Vmax of the SM-MHC–deficient mice was thus not associated with a lower value of parameter b in Eq. 1, instead the a/P o value was increased, indicating a less concave force-velocity relation.
Figure 2.

Shortening velocity (V, muscle lengths/s) and force generation of skinned preparations. A shows force-velocity relations at 3.2 mM MgATP of skinned urinary bladder preparations from a new-born SM-MHC–expressing mouse (open circles), a 3-wk-old SM-MHC–expressing mouse (open squares) and a SM-MHC–deficient mouse (filled circles). B and C show mean values of the maximal shortening velocity (Vmax) and stress (i.e., isometric force per cross-sectional area) of SM-MHC–deficient (filled bars), new-born SM-MHC–expressing mice (open bars), and 3-wk-old SM-MHC–expressing mice (hatched bars). ** and *** indicate statistical differences of P < 0.01 and P < 0.001, respectively, compared with both SM-MHC–expressing groups, Bonferroni method. n = 5–8.
Fig. 2 C shows summarized data regarding force generation of maximally activated (thiophosphorylated), permeabilized preparations contracted at saturating MgATP (3.2 mM). Data are given as stress, i.e., force per cross-sectional area, calculated from maximal isometric force and preparation cross-sectional area (determined from histological sections). The force of the SM-MHC–deficient mice was markedly lower than that of the SM-MHC–expressing muscles.
To ensure that the preparations were fully activated with the ATP-γ-S thiophosphorylation procedure, we performed experiments (n = 2 in each group) where new-born SM-MHC–expressing and SM-MHC–deficient tissues were compared. The preparations were subsequently (a) suboptimally activated with 3 min ATP-γ-S treatment, (b) activated for 15 min with ATP-γ-S according to our standard protocol and (c) activated for 30 min with ATP-γ-S in the presence of 30 nM microcystin to fully inhibit the phosphatase. The force after 3 min thiophosphorylation (SM-MHC–deficient: 0.49 ± 0.08; SM-MHC–expressing: 0.58 ± 0.03) and after 30 min (SM-MHC–deficient: 1.12 ± 0.03; SM-MHC–expressing: 1.15 ± 0.04) relative to that after the 15-min thiophosphorylation period was similar in the two groups. These results suggest that no difference in the rate of Ca2+-induced light chain phosphorylation is present between the two groups. Experiments in which free Ca2+ level was increased (pCa 9–4.5) in the presence of calmodulin at the plateau of contractions after the 15- and 30-min thiophosphorylatyion periods were also performed. No further increase in force was detected, showing that phosphorylation was maximal and sufficient to fully activate the tissues.
Nucleotide Dependence of Maximal Shortening Velocity
To examine the dependence of Vmax on nucleotides, separate experiments in order to construct concentration-response curves of MgATP and of MgADP (at 6 and 10 mM [MgATP]) were performed on the SM-MHC–deficient and SM-MHC–expressing tissues. We observed, in accordance with our previous study on the guinea pig aorta and taenia coli, that a significant shortening velocity could be recorded in ATP-free rigor solution (Löfgren et al., 2001). The mechanism behind this phenomenon is unknown, but most likely it reflects a visco-elastic property of the tissue. To correct for the influence of this process, the apparent Vmax at zero MgATP was determined on each muscle preparation at the end of the experiment. The mean value of this velocity was subtracted from each Vmax value as described previously (Löfgren et al., 2001). These velocities were 26.2 ± 2.4% (new-born SM-MHC expressing, n = 6), 27.0 ± 1.5% (3-wk-old SM-MHC–expressing, n = 6) and 41.7 ± 7.0% (SM-MHC–deficient, n = 6) of the Vmax at saturating MgATP. Fig. 3 shows the influence of varied concentrations of MgATP (Fig. 3 A) and MgADP (Fig. 3 B) on Vmax. The data at different MgATP were fitted to a hyperbolic equation (Eq. 2), giving an apparent binding constant (Km) for MgATP and Vmax.
Figure 3.

Maximal shortening velocity dependence on nucleotides of skinned preparations. A shows the MgATP dependence and B and C show the MgADP dependence of shortening velocity in skinned urinary bladder preparations from new-born SM-MHC–expressing (open circles), 3-wk-old SM-MHC–expressing (open squares), and SM-MHC–deficient mice (filled circles). Velocity data in A are presented relative to the velocity at 3.2 mM MgATP, determined for each muscle preparation. Vmax at 3.2 mM MgATP was for the new-born SM-MHC–expressing mice 0.143 ± 0.012, for the SM-MHC–deficient mice 0.044 ± 0.012, and for the 3-wk-old SM-MHC–expressing mice 0.128 ± 0.018 ML/s. The experiments shown in B are performed in the presence of 6 mM MgATP and those in C with 10 mM MgATP and the values are given relative to velocities at 0 mM MgADP at the respective MgATP. Mean values of the apparent maximal shortening velocity in rigor from the respective groups have been subtracted from the individual values. Lines in A show fits of Eq. 2 to the mean values. Lines in B and C show fits of Eq. 3 to the mean values. Thick lines indicate relations used to estimate Ki values. n = 4–6.
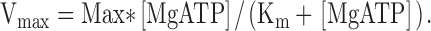 |
(2) |
The parameter “Max” denotes the estimated maximal shortening velocity at saturating [MgATP]. This parameter was in the curve fitting not different from the measured maximal velocities at 3.2 and 10 mM MgATP for each group. The mean Km values for MgATP and Vmax were for the new-born SM-MHC–expressing: 0.013 ± 0.004, for the 3-wk-old SM-MHC–expressing: 0.024 ± 0.009, and for the SM-MHC–deficient mice: 0.023 ± 0.017 mM, n = 5–6. In view of the low velocities, especially in the SM-MHC–deficient tissue, the number of data points below maximal were limited and therefore we cannot exclude a small systematic error in the determination of Km. However, based on the values from the individual experiments presented above and the mean data presented in Fig. 3 A, we conclude that the Km values for the three groups were similar.
Experiments with varied concentrations of MgADP have to be performed in the absence of an ATP-regenerating system. High MgATP concentrations (6 and 10 mM) were used in these experiments, to ensure that the ATP level was saturating even in the absence of ATP-regenerating system. The Vmax at 6 and 10 mM without ATP-regenerating system were for all groups between 98 and 112% of Vmax at 10 mM MgATP with ATP regeneration. As seen in Fig. 3, B and C, MgADP inhibited shortening velocity. The effects of MgADP were dependent on the [MgATP] (compare Fig. 3, B and C), suggesting a competition between ATP and ADP. For the urinary bladders from SM-MHC–expressing mice, minimal inhibition of shortening velocity was observed at 10 mM MgATP (Fig. 3 C) whereas at 6 mM MgATP (Fig. 3 B), velocity was inhibited in a dose-dependent manner by MgADP. In the SM-MHC–deficient tissue the dependence of Vmax on MgADP was very steep at 6 mM MgATP. At 10 mM MgATP for the SM-MHC–deficient tissues and at 6 mM MgATP for the SM-MHC–expressing tissues, a graded inhibition by MgADP was observed. As seen in Fig. 3, the maximal effect of MgADP seemed to level off at an inhibition of Vmax to ∼50% of maximal. This is similar to previous observations in fast and slow smooth muscle from the guinea-pig (Löfgren et al., 2001). Since velocity could not be completely inhibited by MgADP, Michaelis-Menten's kinetics for complete inhibition in its simplest form could not be applied. Since the data suggest an inhibition to ∼50% we used a modified equation (Eq. 3) to describe the inhibition of Vmax by MgADP at constant [MgATP].
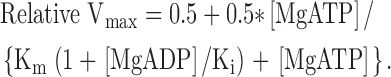 |
(3) |
Using the Km values for MgATP (compare text above and Fig. 3 A) we fitted Eq. 3 to the to the mean data in Fig. 3, B and C, to obtain an estimate of the apparent Ki values for MgADP. Best fits were obtained at 6 mM MgATP for the SM-MHC–expressing groups and at 10 mM MgATP for the SM-MHC–deficient tissue. The mean values for the Ki were for the new-born SM-MHC–expressing: 5.8; for the 3-wk-old SM-MHC–expressing: 8.8 and for the SM-MHC–deficient mice: 0.64 μM (n = 5–6), showing an ∼10-fold lower Ki in the SM-MHC–deficient compared with the SM-MHC–expressing tissues. If the data were analyzed using a standard Michaelis-Menten's equation for competitive inhibition using the lowest concentrations of MgADP only, an ∼10–fold lower Ki for MgADP for the SM-MHC–deficient compared with the SM-MHC–expressing tissue was also observed.
Phosphate Dependence of Force
The effects of varied concentrations of inorganic phosphate (Pi) on active force of skinned preparations are shown in Fig. 4 . Force was inhibited by Pi in a dose-dependent manner in all preparations. The 3-wk-old SM-MHC–expressing group was most sensitive to Pi, with ∼50% inhibition of force at 40 mM Pi. The SM-MHC–deficient group was markedly less sensitive compared with the SM-MHC–expressing groups.
Figure 4.

Measurements of active force at different concentrations of inorganic phosphate. Filled circles show results from SM-MHC–deficient mice, open circles show results from new-born SM-MHC–expressing mice, and open squares from 3-wk-old SM-MHC–expressing mice. Force is normalized to the value at zero [Pi]. n = 6–8. Using repeated measures analysis of variance and correction for multiple comparisons (Bonferroni), all three groups differed (P < 0.001) relative to each other.
Tissue Expression of Actin and Nonmuscle Myosin
Immunohistochemistry showed that the majority of the cells in the muscular layers of the bladder wall were smooth muscle cells as indicated by the smooth muscle α-actin staining. The NM-MHC-B antibody stained α-actin–negative cells located in the interstitium between the muscle bundles and in the submucosa and serosa. These cells comprised <5% of the wall cross-sectional area. NM-MHC-B immunoreactive interstitial cells were small and stellate. They were more numerous and smaller in the new-born mice, both SM-MHC–deficient and SM-MHC–expressing, and less frequent in 3-wk-old SM-MHC–expressing mice (Fig. 5 A). In the new-born mice, the NM-MHC-B antibody gave a very weak signal also from the smooth muscle cells. The NM-MHC-A antibody showed a weak staining of α-actin positive cells in all groups of tissues (unpublished data).
Figure 5.

Immunohistochemistry and gel electrophoresis. A shows a immunohistochemical section from a 3-wk-old mouse bladder stained with the NM-MHC-B antibody. Immunoreactivity is observed in the serosa and submucosa and in small stellate cells in the muscle layer. Bar, 0.1 mm. B shows Coomassie-stained SDS-PAGE and corresponding Western blots with NM-MHC-A and -B antibodies of extracts from the muscle layer of new-born SM-MHC–deficient and SM-MHC–expressing mice and from a 3-wk-old SM-MHC–expressing mouse.
Coomassie-stained gels showed that the intensity of the myosin heavy chain was lower (∼1/5) in the SM-MHC–deficient preparation compared with the SM-MHC–expressing tissue (Fig. 5 B). Extracts of bladders from new-born SM-MHC–expressing and SM-MHC–deficient mice reacted with the NM-MHC-A and NM-MHC-B antibodies on Western blots. For the 3-wk-old SM-MHC–expressing bladders NM-MHC-A was present. Also NM-MHC-B could be detected on the Western blots, but the staining was much weaker than in the new-born bladders (Fig. 5 B).
Ultrastructure
Transmission electron microscopy was performed on transverse sections of the urinary bladder preparations. The main focus of these experiments was to identify myosin filaments in SM-MHC–deficient smooth muscle cells. Structures with sizes corresponding to cross sectioned actin and myosin filaments were observed. The measured diameter of the latter was 14.3 ± 0.41 nm, n = 36. In smooth muscle cells from SM-MHC–expressing mice regions with comparatively abundant thick filaments could easily be identified. Regions with thick filaments (Fig. 6 B) were observed also in smooth muscle cells of the SM-MHC–deficient mice; however, such regions were very sparse and contained fewer thick filaments compared with those in SM-MHC–expressing tissue.
Figure 6.

Electron microscopy of the urinary bladder of a SM-MHC–expressing (A) and a SM-MHC–deficient (B) mouse. The arrows indicate filaments with the size of thick myosin filaments (∼14 nm). Bars, 0.2 μm.
DISCUSSION
We present data on cross-bridge kinetics of filamentous nonmuscle myosin in smooth muscle using a novel transgenic mouse model where the SM-MHC expression is ablated (Morano et al., 2000). To date, two main nonmuscle myosin heavy chain variants have been described in smooth muscle tissue, NM-MHC-A and NM-MHC-B (Kawamoto and Adelstein, 1987, 1991; Katsuragawa et al., 1989; Kuro-o et al., 1991; Simons et al., 1991). Recently, a third nonmuscle myosin heavy chain has been proposed on the basis of genscan predictions (Berg et al. 2001). The structure and role of this putative novel nonmuscle myosin is unknown at present; an ∼75% homology with the motor domain of the nonmuscle myosin B isoform seems to exist. A previous study (Morano et al., 2000) has shown that both nonmuscle myosin A and B are expressed in smooth muscle cells from SM-MHC–deficient transgenic mice. In this study we examined the tissue expression of the NM-MHC-B and found that this type was mainly expressed in smooth muscle α-actin negative cells in the interstitium, similarly to observations from adult rat bladders (Sjuve et al., 2001). In the new-born SM-MHC–deficient tissue, both NM-MHC-A and -B are identified with Western blots. NM-MHC-B seemed to be mainly localized to the interstitial cells, but the smooth muscle cells, identified by the α-actin antibody, also expressed a very weak staining. NM-MHC-A did not appear to be expressed in the interstitial cells, but rather showed a weak distribution in the bladder wall. Our conclusion is thus that the smooth muscle tissue express nonmuscle myosins consistent with the previous report by Morano et al. (2000). The main isoform seems to be NM-MHC-A, but a contribution from NM-MHC-B and possibly also a third, yet uncharacterized, NM-MHC isoform cannot be excluded. The nonmuscle myosins belong to the myosin II family and has previously been shown to form filaments in vitro (Scholey et al., 1980; Smith et al., 1983). Using electron microscopy we demonstrated myosin filaments in the urinary bladder smooth muscle cells of the SM-MHC–deficient mice, showing that the NM-MHC form thick filaments in vivo, at least in tissues lacking SM-MHC. The size of these filaments in cross section appears to be similar to that of smooth muscle myosin filaments, although the detailed structure remains to be clarified.
We interpret the contractions of bladder muscle from SM-MHC–deficient mice to reflect active force generation from a contractile apparatus containing smooth muscle actin and NM-MHC. It should, however, be noted that a very small number of interstitial cells expressing NM-MHC and most likely nonmuscle actin isoforms are present in the tissue. We do not have data suggesting myosin filaments in these cells and cannot at present determine if they contribute to force generation and shortening of the tissue. Different actin isoforms (skeletal and smooth) have been shown to have similar function in the actin–myosin interaction (Mossakowska and Strzelecka-Golaszewska, 1985; Harris and Warshaw, 1993) and cytoplasmic and smooth muscle specific actin isoforms can coexist in the same thin filament (Drew and Murphy, 1997). This suggests that our data, independent of cell type, reflect properties of the nonmuscle myosin.
The nonmuscle myosin IIs are considered to be regulated by myosin light chain phosphorylation (Bresnick, 1999), mainly via myosin light chain kinase (MLCK) activated by Ca2+-calmodulin. We show that the activation by depolarization was associated with a transient in intracellular [Ca2+], exhibiting an initial peak and a plateau during the sustained phase of the contraction. The transients in [Ca2+] after activation were similar in SM-MHC–expressing and SM-MHC–deficient muscles. In the SM-MHC–expressing muscles the force responses to high K+ showed an initial transient and a sustained tonic phase while in the SM-MHC–deficient tissue the initial force transient is absent (compare Morano et al., 2000). This difference in force responses cannot be explained by differences in Ca2+ transients, but rather reflect differences in cellular activation processes or in properties of the contractile myosin.
We examined cross-bridge kinetics of organized nonmuscle myosin in the SM-MHC–deficient tissue. Comparisons were made with the SM-MHC–expressing tissue with low relative contents of nonmuscle myosin. Skinned preparations were used to achieve maximal activation with thiophosphorylation of the regulatory light chains and to control substrate and product levels. Isometric force of the skinned preparations from SM-MHC–deficient tissue was 7–9-fold lower that than of the SM-MHC–expressing tissues. This difference was also present in intact tissue if the maximal forces were compared. The lower force could theoretically reflect shorter contractile filament length (altered parallel coupling), lowered intrinsic force generation of the contractile proteins, or lower content of contractile proteins. Large alterations in filament length seems to be excluded by the shortening velocity data and by reports that unitary cross-bridge force differs little between muscle myosins (Guilford et al., 1997). We found a low total myosin content in the SM-MHC–deficient bladders and the most likely explanation for the lower maximal force compared with the SM-MHC–expressing tissue is therefore a lower total myosin content. The lower myosin content would not, however, primarily affect the maximal shortening velocity, which reflects cross-bridge turnover under unloaded conditions.
The maximal shortening velocity (Vmax) of the SM-MHC–deficient tissue was ∼30% of that in the SM-MHC–expressing tissue. In the in vitro motility assay the NM-MHC-A gives a higher actin filament sliding velocity and ATPase activity compared with the NM-MHC-B isoform (Kelley et al., 1996). In a study by Umemoto and Sellers (1990), cytoplasmic (platelet, nonmuscle A) and smooth (turkey gizzad) muscle myosins were compared using in vitro motility assays at 25°C. They showed that the nonmuscle myosin had a velocity (0.054 μm/s) which is ∼20–25% of that of the smooth muscle myosin (0.237 μm/s). The MgATPase activity of these two myosins is similar, which could reflect that the filament translocation velocity in vitro and ATPase are limited by different cross-bridge reactions (Sellers et al., 1981; Umemoto and Sellers, 1990). The difference in filament velocity is similar to the difference recorded in the present study of the filamentous forms of nonmuscle myosins. If we use the in vitro motility data, in combination with our determinations of Vmax, we can estimate the length of the contractile unit in the smooth muscle (compare half sarcomere length in striated muscle) to ≈1.7 μm in control and 1.2 μm in SM-MHC–deficient tissue. Although these values are very crude estimates, they seem reasonable in view of the estimated thick filament length (2.2 μm) in smooth muscle (Ashton et al., 1975).
In general, differences in shortening velocity between muscles are considered to reflect properties of the myosin molecules. Shortening velocity is considered to be rate-limited by the rate of cross-bridge dissociation (Weiss et al., 2001). In smooth muscle cells, the second-order rate constant of the ATP-induced dissociation of the rigor complex has been reported to be lower than that of skeletal muscle (Nishiye et al., 1993) and lower in slow compared with fast smooth muscles (Somlyo et al., 1988; Khromov et al., 1996). The Km values for ATP and Vmax of the SM-MHC–deficient and SM-MHC–expressing urinary bladders were approximately equal. The ratio between Vmax and Km for ATP are correlated with the binding constant for ATP to the actin–myosin complex (Stienen et al., 1988). Since the Vmax differed by a factor of ∼3 at similar Km, this could suggest that the binding of MgATP to the NM-MHC is weaker compared with that of the SM-MHC. However, the ATP-induced dissociation is fast at saturating MgATP and is thus most likely not limiting the maximal velocity of sliding in smooth muscles.
The ADP-release reaction preceding the cross-bridge dissociation is considered to be important for the velocity of filament sliding (Siemankowski et al., 1985; Weiss et al., 2001). In the slow smooth muscle, the binding of ADP is generally stronger than for the faster skeletal muscle (Arheden and Arner, 1992; Nishiye et al., 1993; Cremo and Geeves, 1998). This relation between velocity and ADP binding is also observed within the smooth muscle group, where the fast and slow muscle types exhibit differences in binding of ADP to both the rigor state (Fuglsang et al., 1993) and to phosphorylated cycling cross-bridges (Löfgren et al., 2001). Here we present data on the effects of ADP on organized nonmuscle myosins. ADP inhibited the Vmax in both SM-MHC–expressing and SM-MHC–deficient tissue. Interestingly, the velocity could not be completely inhibited with ADP within the range studied, but the effect saturated at a relative velocity of ∼50% of that in the absence of ADP, a phenomenon also observed in fast and slow smooth muscles of the guinea-pig (Löfgren et al., 2001). One possible interpretation of this is that the incomplete inhibition is due to differential effects of ADP on the two heads in the myosin molecule during cycling (Löfgren et al., 2001). Due to the complexity of the incomplete inhibition by MgADP, an apparent inhibition constant (Ki) could not be directly derived using Michaelis-Menten's kinetics. Using a modified model assuming 50% maximal inhibition, a 10-fold lower Ki in the SM-MHC–deficient tissues was found. This suggests that the maximally phosphorylated NM-MHC has a stronger ADP binding during cross-bridge cycling.
Fast and slow smooth muscles also differ significantly regarding the inhibitory effect of inorganic phosphate (Pi) on isometric force. Force of smooth muscle is less sensitive to Pi than force of skeletal muscle (Österman and Arner, 1995) and slow smooth muscle is less influenced by Pi than fast smooth muscle (Löfgren et al., 2001). Our data from the SM-MHC–deficient mice show that the force of nonmuscle myosin has a lower sensitivity to Pi compared with the smooth muscle myosin. This shows that the NM-MHC in comparison to the SM-MHC, not only differs in ADP and cross-bridge dissociation reactions, but also in reactions associated with force generation. The Pi effects also provide a measure of cross-bridge kinetics different from the shortening velocity parameter. The results also argue that the difference in Vmax is not only due to structural differences in, for example, filament length.
NM-MHCs have important functions in several nonmuscle contractile systems and experiments on the organized NM-MHC in the smooth muscle tissue can provide a tool for analysis of the general contractile function of this myosin type. In summary, our results show that the NM-MHC can form filaments in smooth muscle cells and that the NM-MHC has a lower shortening velocity, and has different kinetics of reactions involved in cross-bridge dissociation and in force generation compared with the smooth muscle myosin.
Acknowledgments
We thank Rita Wallén for kindly helping with preparations for electron microscopy and Doris Persson for helping with the immunohistochemistry.
The study was supported by grants from Swedish Medical Research Council (A. Arner: 04x-8268, EE: 04x-13406), Medical Faculty Lund University, Crafoord foundation, Swedish Heart Lung Foundation, and Deutsche Forschungsgemeinschaft (I. Morano: Mo. 362/16-2).
Olaf S. Andersen served as editor.
Footnotes
Abbreviations used in this paper: NM-MHC, nonmuscle myosin heavy chain; SM-MHC, smooth muscle myosin heavy chain.
References
- Arheden, H., and A. Arner. 1992. Effects of magnesium pyrophosphate on mechanical properties of skinned smooth muscle from the guinea pig taenia coli. Biophys. J. 61:1480–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arheden, H., A. Arner, and P. Hellstrand. 1988. Cross-bridge behaviour in skinned smooth muscle of the guinea-pig taenia coli at altered ionic strength. J. Physiol. 403:539–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner, A., and P. Hellstrand. 1985. Effects of calcium and substrate on force-velocity relation and energy turnover in skinned smooth muscle of the guinea-pig. J. Physiol. 360:347–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton, F.T., A.V. Somlyo, and A.P. Somlyo. 1975. The contractile apparatus of vascular smooth muscle: intermediate high voltage stereo electron microscopy. J. Mol. Biol. 98:17–29. [DOI] [PubMed] [Google Scholar]
- Berg, J.S., B.C. Powell, and R.E. Cheney. 2001. A millennial myosin census. Mol. Biol. Cell. 12:780–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnick, A.R. 1999. Molecular mechanisms of nonmuscle myosin-II regulation. Curr. Opin. Cell Biol. 11:26–33. [DOI] [PubMed] [Google Scholar]
- Cremo, C.R., and M.A. Geeves. 1998. Interaction of actin and ADP with the head domain of smooth muscle myosin: implications for strain-dependent ADP release in smooth muscle. Biochemistry. 37:1969–1978. [DOI] [PubMed] [Google Scholar]
- Drew, J.S., and R.A. Murphy. 1997. Actin isoform expression, cellular heterogeneity, and contractile function in smooth muscle. Can. J. Physiol. Pharmacol. 75:869–877. [PubMed] [Google Scholar]
- Feldhaus, P., T. Frohlich, R.S. Goody, M. Isakov, and R.H. Schirmer. 1975. Synthetic inhibitors of adenylate kinases in the assays for ATPases and phosphokinases. Eur. J. Biochem. 57:197–204. [DOI] [PubMed] [Google Scholar]
- Fuglsang, A., A. Khromov, K. Torok, A.V. Somlyo, and A.P. Somlyo. 1993. Flash photolysis studies of relaxation and cross-bridge detachment: higher sensitivity of tonic than phasic smooth muscle to MgADP. J. Muscle Res. Cell Motil. 14:666–677. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz, G., M. Poenie, and R.Y. Tsien. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260:3440–3450. [PubMed] [Google Scholar]
- Guilford, W.H., D.E. Dupuis, G. Kennedy, J. Wu, J.B. Patlak, and D.M. Warshaw. 1997. Smooth muscle and skeletal muscle myosins produce similar unitary forces and displacements in the laser trap. Biophys. J. 72:1006–1021 (see comments). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, D.E., and D.M. Warshaw. 1993. Smooth and skeletal muscle actin are mechanically indistinguishable in the in vitro motility assay. Circ. Res. 72:219–224. [DOI] [PubMed] [Google Scholar]
- Heath, K.E., A. Campos-Barros, A. Toren, G. Rozenfeld-Granot, L.E. Carlsson, J. Savige, J.C. Denison, M.C. Gregory, J.G. White, D.F. Barker, et al. 2001. Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal dominant macrothrombocytopenias: May-Hegglin anomaly and Fechtner, Sebastian, Epstein, and Alport-like syndromes. Am. J. Hum. Genet. 69:1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, A.V. 1938. The heat of shortening and the dynamic constants of shortening. Proc. Royal Soc. 126:231–252. [Google Scholar]
- Katsuragawa, Y., M. Yanagisawa, A. Inoue, and T. Masaki. 1989. Two distinct nonmuscle myosin-heavy-chain mRNAs are differentially expressed in various chicken tissues. Identification of a novel gene family of vertebrate non-sarcomeric myosin heavy chains. Eur. J. Biochem. 184:611–616. [DOI] [PubMed] [Google Scholar]
- Kawamoto, S., and R.S. Adelstein. 1987. Characterization of myosin heavy chains in cultured aorta smooth muscle cells. A comparative study. J. Biol. Chem. 262:7282–7288. [PubMed] [Google Scholar]
- Kawamoto, S., and R.S. Adelstein. 1991. Chicken nonmuscle myosin heavy chains: differential expression of two mRNAs and evidence for two different polypeptides. J. Cell Biol. 112:915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, C.A., J.R. Sellers, D.L. Gard, D. Bui, R.S. Adelstein, and I.C. Baines. 1996. Xenopus nonmuscle myosin heavy chain isoforms have different subcellular localizations and enzymatic activities. J. Cell Biol. 134:675–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, M.J., W. Jawien, T.L. Ortel, and J.F. Korczak. 2000. Mutation of MYH9, encoding non-muscle myosin heavy chain A, in May- Hegglin anomaly. Nat. Genet. 26:106–108. [DOI] [PubMed] [Google Scholar]
- Khromov, A., A.V. Somlyo, and A.P. Somlyo. 1998. MgADP promotes a catch-like state developed through force-calcium hysteresis in tonic smooth muscle. Biophys. J. 75:1926–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khromov, A.S., A.V. Somlyo, and A.P. Somlyo. 1996. Nucleotide binding by actomyosin as a determinant of relaxation kinetics of rabbit phasic and tonic smooth muscle. J. Physiol. 492:669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuro-o, M., R. Nagai, K. Nakahara, H. Katoh, R.C. Tsai, H. Tsuchimochi, Y. Yazaki, A. Ohkubo, and F. Takaku. 1991. cDNA cloning of a myosin heavy chain isoform in embryonic smooth muscle and its expression during vascular development and in arteriosclerosis. J. Biol. Chem. 266:3768–3773. [PubMed] [Google Scholar]
- Löfgren, M., U. Malmqvist, and A. Arner. 2001. Substrate and product dependence of force and shortening in fast and slow smooth muscle. J. Gen. Physiol. 117:407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucius, C., A. Arner, A. Steusloff, M. Troschka, F. Hofmann, K. Aktories, and G. Pfitzer. 1998. Clostridium difficile toxin B inhibits carbachol-induced force and myosin light chain phosphorylation in guinea-pig smooth muscle: role of Rho proteins. J. Physiol. 506:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morano, I., G.X. Chai, L.G. Baltas, V. Lamounier-Zepter, G. Lutsch, M. Kott, H. Haase, and M. Bader. 2000. Smooth-muscle contraction without smooth-muscle myosin. Nat. Cell Biol. 2:371–375. [DOI] [PubMed] [Google Scholar]
- Mossakowska, M., and H. Strzelecka-Golaszewska. 1985. Identification of amino acid substitutions differentiating actin isoforms in their interaction with myosin. Eur. J. Biochem. 153:373–381. [DOI] [PubMed] [Google Scholar]
- Nishiye, E., A.V. Somlyo, K. Torok, and A.P. Somlyo. 1993. The effects of MgADP on cross-bridge kinetics: a laser flash photolysis study of guinea-pig smooth muscle. J. Physiol. 460:247–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Österman, A., and A. Arner. 1995. Effects of inorganic phosphate on cross-bridge kinetics at different activation levels in skinned guinea-pig smooth muscle. J. Physiol. 484:369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholey, J.M., K.A. Taylor, and J. Kendrick-Jones. 1980. Regulation of non-muscle myosin assembly by calmodulin-dependent light chain kinase. Nature. 287:233–235. [DOI] [PubMed] [Google Scholar]
- Sellers, J.R., M.D. Pato, and R.S. Adelstein. 1981. Reversible phosphorylation of smooth muscle myosin, heavy meromyosin, and platelet myosin. J. Biol. Chem. 256:13137–13142. [PubMed] [Google Scholar]
- Siemankowski, R.F., M.O. Wiseman, and H.D. White. 1985. ADP dissociation from actomyosin subfragment 1 is sufficiently slow to limit the unloaded shortening velocity in vertebrate muscle. Proc. Natl. Acad. Sci. USA. 82:658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons, M., M. Wang, O.W. McBride, S. Kawamoto, K. Yamakawa, D. Gdula, R.S. Adelstein, and L. Weir. 1991. Human nonmuscle myosin heavy chains are encoded by two genes located on different chromosomes. Circ. Res. 69:530–539. [DOI] [PubMed] [Google Scholar]
- Sjuve, R., A. Arner, Z. Li, B. Mies, D. Paulin, M. Schmittner, and J.V. Small. 1998. Mechanical alterations in smooth muscle from mice lacking desmin. J. Muscle Res. Cell Motil. 19:415–429. [DOI] [PubMed] [Google Scholar]
- Sjuve, R., H. Haase, E. Ekblad, U. Malmqvist, I. Morano, and A. Arner. 2001. Increased expression of non-muscle myosin heavy chain-B in connective tissue cells of hypertrophic rat urinary bladder. Cell Tissue Res. 304:271–278. [DOI] [PubMed] [Google Scholar]
- Smith, R.C., W.Z. Cande, R. Craig, P.J. Tooth, J.M. Scholey, and J. Kendrick-Jones. 1983. Regulation of myosin filament assembly by light-chain phosphorylation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 302:73–82. [DOI] [PubMed] [Google Scholar]
- Somlyo, A.V., Y.E. Goldman, T. Fujimori, M. Bond, D.R. Trentham, and A.P. Somlyo. 1988. Cross-bridge kinetics, cooperativity, and negatively strained cross-bridges in vertebrate smooth muscle. A laser-flash photolysis study. J. Gen. Physiol. 91:165–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich, J.A. 1989. In pursuit of myosin function. Cell Regul. 1:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stienen, G.J., W.J. van der Laarse, and G. Elzinga. 1988. Dependency of the force-velocity relationships on Mg ATP in different types of muscle fibers from Xenopus laevis. Biophys. J. 53:849–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemoto, S., and J.R. Sellers. 1990. Characterization of in vitro motility assays using smooth muscle and cytoplasmic myosins. J. Biol. Chem. 265:14864–14869. [PubMed] [Google Scholar]
- Wede, O.K., M. Löfgren, Z. Li, D. Paulin, and A. Arner. 2002. Mechanical function of intermediate filaments in arteries of different size examined using desmin deficient mice. J. Physiol. 540:941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, S., R. Rossi, M.A. Pellegrino, R. Bottinelli, and M.A. Geeves. 2001. Differing ADP release rates from myosin heavy chain isoforms define the shortening velocity of skeletal muscle fibers. J. Biol. Chem. 276:45902–45908. [DOI] [PubMed] [Google Scholar]