

Abstract
The putative voltage-sensitive release mechanism (VSRM) was investigated in rabbit cardiac myocytes at 37°C with high resistance microelectrodes to minimize intracellular dialysis. When the holding potential was adjusted from −40 to −60 mV, the putative VSRM was expected to operate alongside CICR. Under these conditions however, we did not observe a plateau at positive potentials of the cell shortening versus voltage relationship. The threshold for cell shortening changed by −10 mV, but this resulted from a similar change of the threshold for activation of inward current. Cell shortening under conditions where the putative VSRM was expected to operate was blocked in a dose dependent way by nifedipine and CdCl2 and blocked completely by NiCl2. “Tail contractions” persisted in the presence of nifedipine and CdCl2 but were blocked completely by NiCl2. Block of early outward current by 4-aminopyridine and 4-acetoamido-4′-isothiocyanato-stilbene-2,2′-disulfonic acid (SITS) demonstrated persisting inward current during test depolarizations despite the presence of nifedipine and CdCl2. Inward current did not persist in the presence of NiCl2. A tonic component of cell shortening that was prominent during depolarizations to positive potentials under conditions selective for the putative VSRM was sensitive to rapidly applied changes in superfusate [Na+] and to the outward Na+/Ca2+ exchange current blocking drug KB-R7943. This component of cell shortening was thought to be the result of Na+/Ca2+ exchange–mediated excitation contraction coupling. Cell shortening recorded under conditions selective for the putative VSRM was increased by the enhanced state of phosphorylation induced by isoprenaline (1 μM) and by enhancing sarcoplasmic reticulum Ca2+ content by manipulation of the conditioning steps. Under these conditions, cell shortening at positive test depolarizations was converted from tonic to phasic. We conclude that the putative VSRM is explained by CICR with the Ca2+ “trigger” supplied by unblocked L-type Ca2+ channels and Na+/Ca2+ exchange.
Keywords: EC coupling, L-type Ca2+ channels, VSRM, CICR
INTRODUCTION
The voltage-sensitive release mechanism (VSRM)* is a recently described and controversial (Piacentino et al., 2000; Trafford and Eisner, 1998; Wier and Balke, 1999) mechanism proposed for excitation-contraction (EC) coupling in cardiac muscle (Ferrier and Howlett, 1995; Hobai et al., 1997b). Under certain experimental conditions, the putative VSRM is described as capable of mediating SR Ca2+ release and cell shortening in isolated myocytes in response to membrane depolarization per se. These conditions include a physiological experimental temperature of 37°C, a fully loaded SR and a holding potential negative to −40 mV as the putative VSRM is said to undergo complete steady-state inactivation at this potential (Ferrier et al., 1998; Howlett et al., 1998). If intracellular dialysis with patch pipettes occurs, the putative VSRM is supported only when the phosphorylation state of the cell is enhanced by inclusion of cAMP in the pipette filling solution (Hobai et al., 1997b; Ferrier et al., 1998). When these conditions are fulfilled, properties of the EC coupling observed include a voltage threshold for activation negative to that for L-type Ca2+ current (ICa(L)), persisting cell shortening or SR Ca2+ release at positive potentials in the region of ECa resulting in sigmoid shaped voltage dependence, and persisting cell shortening or SR Ca2+ release during exposure to L-type Ca2+ channel blocking agents. These have been proposed as defining features of the putative VSRM that has now been described in guinea-pig (Ferrier and Howlett, 1995; Ferrier et al., 1998, 2000; Howlett et al., 1998; Mason and Ferrier, 1999), rabbit (Hobai et al., 1997b), rat (Hobai et al., 1997b; Howlett et al., 1998), hamster (Howlett et al., 1999), human atrial myocytes (Van Wagoner et al., 1999), and also in heart failure models in hamster (Howlett et al., 1999) and rat (Sjaastad et al., 2000).
The experimental conditions necessary for demonstration of the putative VSRM have significant effects on the properties of cardiac myocyte calcium handling. This raises the possibility that modification of CICR by these conditions may produce EC coupling with features of the putative VSRM. There are a number of examples of this effect.
Small amplitude depolarizations in rat myocytes to negative test potentials from a holding potential of −50 mV have been shown to elicit ICa(L), Ca2+ transients, and cell shortening (Talo et al., 1990). In another study, Ca2+ sparks were recorded in rat myocytes during voltage ramps over a negative voltage range between −80 and −40 mV (Cannell et al., 1995). The spatial and temporal distribution as well as the stochastic nature of the sparks led to the conclusion that they were under the control of single L-type Ca2+ channel openings. These studies illustrate the possibility that negative holding potentials permit contractions mediated by CICR at negative test potentials.
Phosphorylation of the L-type Ca2+ channel, e.g., by intracellular cAMP results in an increased open probability of the channel and whole cell ICa(L) (McDonald et al., 1994). The voltage threshold for activation of ICa(L) has been shown in rat and canine myocytes to shift to more negative potentials when intracellular [cAMP] was increased after application of isoprenaline (1–3 μM). In these experiments, fractional potentiation of ICa(L) was greatest during depolarizations to negative potentials at the foot of its activation curve (Tiaho et al., 1991) and was shown to be capable of eliciting SR Ca2+ release (Sipido et al., 2000). These results also indicate the possibility that increased intracellular [cAMP] will allow contractions mediated by CICR at negative test potentials.
In guinea-pig ventricular myocytes at 37°C, the voltage threshold for cell shortening shifted from −35 to −40 mV and the cell shortening versus voltage curve became sigmoidal when measures were taken to preserve SR Ca2+ stores (Isenberg and Wendt Gallitelli, 1989). In these experiments, the holding potential of −45 mV would have inactivated the putative VSRM and SR Ca2+ overload may have permitted CICR to mimic its described features.
The response to L-type Ca2+ channel blockers is also influenced by the conditions necessary for demonstration of the putative VSRM. Block by dihydropyridines or multivalent heavy metal cations is voltage dependent. Block is therefore less complete when depolarizations are applied from more negative holding potentials or when depolarizations are applied to negative activating potentials (Kokubun et al., 1986; Lansman et al., 1986). ICa(L) amplitude is steeply temperature dependent and experimental temperature also affects the response to L-type Ca2+ channel blockers. In one study, the Q10 for the maximum amplitude of ICa(L) increased from 2.4 to 4.2 during inhibition by nifedipine (Allen and Chapman, 1992). Phosphorylation of the L-type Ca2+ channel also results in reduced sensitivity to channel blockers (Hobai et al., 2000). These phenomena illustrate the potential for ICa(L) to persist under conditions believed to be selective for the putative VSRM when ICa(L) is presumed to be blocked completely.
A further effect of negative holding potential is that on steady-state inactivation of the transient outward current (Ito). Ito starts to inactivate at −70 mV and the half maximal inactivation point lies at −40 mV. The current is manifest as a depolarization-activated immediate outward current with an activation voltage threshold of −20 mV and maximum amplitude increasing with depolarization to more positive potentials (Giles and Imaizumi, 1988; Hiraoka and Kawano, 1989). It follows, therefore, that Ito amplitude will always be greater under conditions necessary for demonstration of the putative VSRM. The presence of Ito as the dominant early membrane current during conditions supporting the putative VSRM may mask coexisting ICa(L).
The effects of SR Ca2+ “overload” have been well studied. In Ca2+ overloaded cells, the probability of spontaneous Ca2+ sparks is increased (Cheng et al., 1993), spontaneous propagating waves of contraction (Berlin et al., 1987), or intracellular Ca2+ (Orchard et al., 1983; Wier et al., 1987; Trafford et al., 1993) may be observed and fractional SR Ca2+ release activated by CICR is increased (Han et al., 1994; Janczewski et al., 1995; Levi et al., 1996; Shannon et al., 2000). It has also been pointed out that after SR Ca2+ loading, a Ca2+ transient could be released by a Ca2+ trigger that would usually be too small to initiate release under control levels of SR Ca2+ content (Fabiato, 1983; Trafford et al., 1993; Han et al., 1994; Bassani et al., 1995; Spencer and Berlin, 1995; Dupont et al., 1996; Shannon et al., 2000). Phosphorylation of phospholamban, e.g., by intracellular cAMP, increases the rate of SR Ca2+ pumping and therefore SR Ca2+ content (Sham et al., 1991; Bers, 2001). Phosphorylation of the SR Ca2+ release channel increases its sensitivity to activating Ca2+ although varying effects may be produced by different phosphorylation systems (Hain et al., 1995; Lokuta et al., 1995; duBell et al., 1996). Elevated intracellular cAMP has been a mandatory condition for the putative VSRM in a number of reports (Hobai et al., 1997b; Ferrier et al., 1998; Zhu and Ferrier, 2000). These further effects on CICR of elevated intracellular cAMP may have contributed to EC coupling mimicking the putative VSRM.
Na+/Ca2+ exchange is able to mediate cell shortening in the absence of L-type Ca2+ channel activation (Eisner et al., 1983; Barcenas Ruiz et al., 1987; Bers et al., 1988; Beuckelmann and Wier, 1989). Steps have rarely been taken to block Na+/Ca2+ exchange during routine demonstration of the putative VSRM. It has been shown in guinea-pig myocytes (Vornanen et al., 1994) and in rat myocytes (Wasserstrom and Vites, 1996) that the effect of increased experimental temperature on Na+/Ca2+ exchange resulted in conversion of the phasic tension versus voltage curve from bell shaped to sigmoid. In the study of Wasserstrom and Vites (1996), the amplitude of cell shortening was also increased when the holding potential was adjusted to a more negative value. Under conditions where EC coupling was mediated by Na+/Ca2+ exchange in rabbit myocytes, the cell shortening versus voltage curve changed from bell shaped to sigmoid with regenerative, oscillatory Ca2+ release when SR Ca2+ content was increased by intracellular dialysis with high [Na+] (Levi et al., 1996). These studies demonstrate that Na+/Ca2+ exchange is capable of reproducing one of the defining features of the putative VSRM. It has also been shown that Na+/Ca2+ exchange is enhanced by dialysis with intracellular cAMP (Perchenet et al., 2000).
In many studies, failure of EC coupling has been observed despite experimental conditions compatible with operation of the putative VSRM. Thus, cell shortening in guinea-pig was a bell-shaped function of voltage during depolarization from a holding potential of −68 mV (Beuckelmann and Wier, 1988) and was blocked by NiCl2 (5 mM) in myocytes at their resting membrane potential at 34–37°C (Levesque et al., 1994; Levi et al., 1994). The Ca2+ transient elicited in rat by depolarization from a holding potential between −70 and −80 mV at 34–37°C was blocked by NiCl2 (5 mM) ± nifedipine (20 μM) (Hancox and Levi, 1995; Wasserstrom and Vites, 1996).
The most notable failure to elicit the putative VSRM occurred when all necessary experimental conditions were present together in field stimulated rabbit myocytes at their resting membrane potential at 36°C. In this experiment, the Ca2+ transient and cell shortening were blocked by nifedipine (20 μM) and NiCl2 (5 mM) (Levi and Issberner, 1996). The putative VSRM does not, therefore, appear to be a robust mechanism of EC coupling.
A large body of evidence suggests therefore that under appropriate conditions, CICR is able to generate EC coupling with properties resembling the hypothesized VSRM. The present series of experiments have been devised to determine the contribution of CICR to the hypothesized VSRM. We have used a physiological superfusing solution in preference to the low [Na+] solution used by Ferrier and Howlett and intracellular dialysis was minimized with the use of sharp microelectrodes. We find that under the conditions recommended for demonstration of the putative VSRM, either the properties of CICR are modified to the point where VSRM-like behavior is produced or EC coupling is blocked completely. In this respect our results are in broad agreement with those of Piacentino et al. (2000).
MATERIALS AND METHODS
Myocyte Isolation
Isolated cardiac myocytes were prepared from adult male New Zealand white rabbits using a modification of methods which have been described in detail elsewhere (MacLeod and Harding, 1991). Rabbits were anticoagulated and killed by intravenous injection of 2,500 i.u. heparin (Leo Laboratories Ltd.) and 300 mg pentobarbitone (Rhone Merieux), respectively. Left ventricular myocytes were isolated using 0.5 mg ml−1 collagenase (204 i.u. mg−1, Worthington Biochemical Corporation) and 0.5 mg ml−1 hyaluronidase (330 i.u. mg−1, Sigma-Aldrich). After isolation, the cells were stored in Dulbecco's modified Eagle's medium (GIBCO BRL) at room temperature and used within 10 h.
Experimental Setup
A cell superfusion system was designed to maximize switching speed between solutions while minimizing disturbance of experimental temperature. The setup was similar to that described previously (Terracciano et al., 2001). Briefly, a superfusion chamber with a volume of 20 μl was mounted on the stage of an inverted microscope. The floor of the superfusion chamber was formed by a glass microscope coverslip. Cells were allowed to settle for up to 5 min on the glass coverslip forming the base of the chamber. Adhesion was facilitated by prior application of mouse laminin. Superfusing solutions were conducted to the chamber by means of polyethylene tubing enclosed within a 1.5-m long silicon rubber insulating jacket extending to within 15 mm of the chamber inlet. A “zero dead space” 4:1 teflon manifold was incorporated into the wall of the chamber and solution switching was controlled by means of four solenoid valves. Flow rate was maintained at ≈3 ml min−1 which permitted solution switching to be 95% complete within 1 s and was increased to ≈8 ml min−1 before application of caffeine. All experiments were performed at 37°C and the temperature profile during a solution switch demonstrated a maximum perturbation of −0.5°C with recovery to baseline within 4 s.
Electrophysiological Recordings
Micropipettes were pulled from 1.5-mm filamented borosilicate glass capillaries (Harvard Apparatus Ltd.) with a mechanical pipette puller (P-87; Sutter Instrument Co.) adjusted to obtain microelectrode resistances of 12–30 MΩ when filled with a 2-M KCl–based solution. Microelectrodes were connected to an Axoclamp-2A amplifier (Axon Instruments, Inc.). The amplifier was coupled via a Digidata 1200 interface (Axon Instruments, Inc.) to a computer that generated electrophysiological protocols, controlled the solenoid valves, and stored experimental data using pCLAMP 6.0.3 software (Axon Instruments, Inc.). Membrane current was recorded in discontinuous single electrode voltage clamp (dSEVC) mode enabling voltage clamp with a single high resistance microelectrode. Continuous monitoring on a dedicated oscilloscope of the “monitor output” triggered at the switching frequency enabled optimal micropipette capacitance compensation. With optimal capacitance compensation, current flow settled rapidly to baseline without overshoot during the latter part of the duty cycle, ensuring faithful voltage recording. Gain (0.3–0.8 nA mV−1) and sampling rate (7–12 kHz) were advanced to maximum values allowing complete settling of current without overshoot. These settings allowed maximally square voltage steps to be obtained. With minimization of sources of external noise and optimal setup of the voltage clamp, dSEVC resulted in ≤2 mV overshoot beyond the command potential during activation of ICa(L) with peak amplitudes up to −2 nA at 37°C as encountered in these experiments (Fig. 1) . dSEVC has previously been used successfully in the investigation of ICa(L) in isolated mammalian myocytes (e.g., Mitchell et al., 1987; Egan et al., 1989; Levi et al., 1993). Cell capacitance was determined by integration of the capacitance current recorded during 5 mV hyperpolarizing and depolarizing steps under voltage clamp between −70 and −75 mV. Changes in cell length were measured by video edge detection as described previously (Steadman et al., 1988).
Figure 1.

Membrane current and recorded membrane potential during a command voltage step from −40 to 0 mV under dSEVC (Cm=165 pF). The capacitance transient in this and all subsequent current records is blanked.
Solutions
The following solutions were made up in ultra-pure water produced by running distilled water through a Milli-Q water purification system (Millipore). Concentrations are given in mM unless indicated otherwise. Chemicals were obtained from B.D.H., Merck Ltd unless indicated otherwise.
Normal tyrode: NaCl 140, KCl 6, CaCl2 2, MgCl2 1, glucose 10, HEPES 10, pH titrated to 7.4 at 37°C with NaOH ≈6.
Electrode-filling solution: KCl 2M, HEPES 10, EGTA 100 μM, pH titrated to 7.2 with KOH.
Low sodium/calcium solution: NaCl 50, LiCl 90, KCl 6, CaCl2 0.7, MgCl2 2, glucose 10, HEPES 10, pH titrated to 7.4 at 37°C with LiOH ≈6.
Inward Na+/Ca2+ exchange current activating solution: NaCl 140, KCl 6, CaCl2 0.7, MgCl2 2, glucose 10, HEPES 10, pH titrated to 7.4 at 37°C with NaOH ≈6.
Outward Na+/Ca2+ exchange current activating solution: LiCl 140, KCl 6, CaCl2 4, MgCl2 1, glucose 10, HEPES 10, pH titrated to 7.4 at 37°C with NaOH ≈6.
The following stock solutions were made up in Milli-Q water and refrigerated unless indicated otherwise. They were added to experimental solutions as required.
Lignocaine 250 (Sigma-Aldrich), TTX 3 (citrate buffered, lyophilized solid form, solution frozen at −20°C, obtained from Calbiochem and Alomone Labs), CdCl2 100, NiCl2 500, nifedipine 30 (Sigma-Aldrich) (in ethanol, stored at −20°C, final solution protected from light), L-isoprenaline 0.1 (Sigma-Aldrich) (in ascorbic acid 0.1, frozen and stored for <8 h), 4 aminopyridine (4AP) 500 (Sigma-Aldrich), KB-R7943 10 (in dimethyl sulphoxide (DMSO), stored at −20°C, gift from Kanebo Co. Ltd., Osaka 534, Japan). Solvents in appropriate dilutions were added to control solutions. The drugs 4-acetoamido-4′-isothiocyanato-stilbene-2,2′-disulfonic acid (SITS, final solution protected from light) (Sigma-Aldrich) and caffeine were added directly to experimental solutions in powder form.
Analysis
Averaging and integration of membrane currents was achieved using the “Analyze/Operators” function of CLAMPFIT 6.0.3. Data are expressed as mean ± SE. Statistical tests of differences between two groups were performed with the paired, two-tailed Student's t test.
RESULTS
The Effect of Holding Potential on Cell Shortening
The first series of experiments was designed to establish the effect on cell shortening of adjusting the holding potential to a value supporting the putative VSRM. The experimental protocol is shown in Fig. 2 A. SR Ca2+ content was maintained and kept constant by the application under voltage clamp of a conditioning train consisting of 10 steps from −80 to 60 mV for 300 ms repeated at 1 Hz. The membrane was then depolarized to a holding potential of −45 or −40 mV to establish CICR-selective recording conditions or to a holding potential of −65 or −60 mV to establish conditions where the putative VSRM could operate alongside CICR. This holding period was imposed for 4 s and when the potential was −65 or −60 mV, lignocaine (500 μM) or TTX (30 μM) was applied in order to block INa. In a proportion of cells, lignocaine or TTX was also applied when the holding potential was −45 or −40 mV. After the holding period, the membrane was depolarized to test potentials between −65 and −30 mV in 5-mV increments and between −30 and 80 mV in 10-mV increments.
Figure 2.

(A) CICR protocol and protocol for CICR + VSRM combined. (B) Membrane current and cell shortening during depolarizations from a holding potential of −45 mV in the presence of TTX (30 μM) (Cm = 254 pF) and in a different cell from a holding potential of −60 mV in the presence of lignocaine (500 μM) (Cm = 180 pF). (C) I-V and cell shortening versus voltage curves for depolarizations from a holding potential of −65 or −60 mV (♦, solid line, n = 8) and a holding potential of −45 or −40 mV (▪, dashed line, n = 32). (D) Selected records from (B) redrawn to larger scale. (E) Peak inward current was scaled to show the more negative threshold for inward current with a holding potential of −65 or −60 mV.
Under CICR-selective conditions, the voltage threshold for cell shortening occurred at −40 mV (Fig. 2 B). With depolarization from the holding potential of −65 or −60 mV, the threshold for cell shortening became more negative and occurred at −50 mV. This negative movement of the voltage threshold for cell shortening was, however, accompanied by a negative movement of the threshold for activation of phasic inward current, from −35 to −40 mV. This is shown more clearly in Fig. 2 D, which demonstrates selected records from Fig. 2 B drawn to a larger scale. The threshold for cell shortening under either CICR-selective conditions or conditions where the putative VSRM was supported occurred at a potential negative to that for phasic inward current. Cell shortening at these potentials was tonic and maintained. This demonstrated that the appearance of cell shortening at a voltage negative to the voltage threshold for phasic inward current was not a defining property of the putative VSRM. Although cell shortening with small depolarizations was not accompanied by phasic inward current, it is seen clearly in Fig. 2 B that noninactivating inward current occurred.
Under CICR-selective conditions, peak shortening occurred between 0 and 20 mV and its voltage dependence corresponded closely with that of peak inward current (Fig. 2 C). Inward current amplitude was measured as peak inward current subtracted from steady-state current at the end of the depolarizing step and was presumed to represent ICa(L). Cell shortening at positive potentials was not eliminated, despite depolarizations to 80 mV. When the holding potential was adjusted to −65 or −60 mV, peak cell shortening continued to occur around 0 mV and decreased with more positive test depolarizations. Cell shortening under these conditions therefore continued to reflect the voltage dependence of inward current as it did under CICR-selective conditions. Thus, although conditions permitting operation of the putative VSRM were established, the voltage dependence of cell shortening failed to fulfil the predictions of the VSRM hypothesis and did not reach a plateau at positive potentials (Ferrier and Howlett, 1995; Hobai et al., 1997b).
The Effect of Holding Potential on L-type Ca2+ Current
An experiment to confirm the dependence of the voltage threshold for ICa(L) on holding potential is illustrated in Fig. 3 A. Inward currents were elicited from a holding potential of −40 mV by voltage steps of 100 ms. Depolarizations were applied to test potentials between −40 and 0 mV in 5-mV increments and repeated at a frequency of 2 Hz. TTX (30 μM) was applied 4 s before the first step. Nifedipine (60 μM) was then added during steps to 0 mV at 0.5 Hz for 30 s while its use-dependent effect was becoming established and the protocol was repeated. L-type calcium current was measured as the nifedipine-sensitive component by subtraction of corresponding traces. The experimental protocol was repeated with measurement of L-type Ca2+ current elicited by depolarizations from a holding potential of −60 mV. These experiments were conducted in a separate population of cells as the effects of nifedipine were only partially reversible. Conditioning steps were not used in order to minimize time between obtaining corresponding traces in the presence and absence of nifedipine.
Figure 3.

(A) Voltage protocol for nifedipine-sensitive ICa(L). Inward current was elicited in the presence of TTX (30 μM) by depolarization from a holding potential of −40 mV or, in a separate population of cells, −60 mV. The protocol was repeated in the presence of nifedipine (60 μM) and L-type Ca2+ current records were obtained by subtraction. (B) Nifedipine-sensitive ICa(L), during depolarizations from a holding potential of −60 mV (Cm = 118 pF). (C) I-V curves for the phasic nifedipine sensitive current density obtained with a holding potential of −60 mV (♦, n = 19) and −40 mV (▪, n = 13). (D) Nifedipine-sensitive ICa(L), during depolarizations from a holding potential of −60 mV (Cm = 117 pF). (E) I-V curve for the steady-state nifedipine sensitive current density measured at the end of the 100-ms depolarizing step (n = 19).
When the holding potential was −60 mV, phasic ICa(L) was detectable during depolarization to −50 mV in 4/19 cells, to −45 mV in 5/19 cells, and to −40 mV in 9/19 cells. An example of a cell with a voltage threshold for ICa(L) of −50 mV is shown in Fig. 3 B. The rapid initial currents seen during the steps to −50 and −45 mV represent an unsubtracted component of capacitance current. As the peak amplitude of capacitance current flowing during these experiments was of the order of 3 nA, these residuals of ≈100 pA amplitude and <3 ms duration were felt to represent satisfactory subtraction. The I-V curve obtained from these data demonstrated the voltage threshold for ICa(L) to be −50 mV when measured from a holding potential of −60 mV (Fig. 3 C). This corresponded closely with the voltage threshold for activation of cell shortening shown in Fig. 2 B. Mean data from identical experiments performed from a holding potential of −40 mV were plotted on the same axes for comparison (Fig. 3 C). This demonstrated that when holding potential was adjusted from −40 to −60 mV, there was a corresponding −15-mV shift in the voltage threshold for ICa(L) activation from −35 to −50 mV.
In other cells, small depolarizations from a holding potential of −60 mV resulted in activation of maintained ICa(L) in the absence of a phasic component. An example of this phenomenon is shown in Fig. 3 D, where inward current activated with depolarization to −50, −40, and −30 mV before phasic ICa(L) appeared with depolarization to −20 mV. The maintained component of the nifedipine-sensitive current is plotted as a function of voltage (Fig. 3 E). The voltage threshold for these maintained currents lay also at −50 mV. Their voltage dependence was compatible with their identity as ICa(L) window current resulting from incomplete steady-state inactivation of L-type Ca2+ channels within the voltage range shown. Thus, the effect of holding potential on the voltage threshold of cell shortening was shown to operate via an effect of holding potential on the voltage threshold for ICa(L). It seems likely that the tonic cell shortening recorded with small depolarizations corresponds with the maintained component of ICa(L).
Cell Shortening under Recording Conditions Selective for the Putative VSRM
An experiment to determine whether the putative VSRM could mediate cell shortening in the presence of L-type Ca2+ channel blockers is shown in Fig. 4 . Voltage protocols were based on those shown in Fig. 2 A, but the conditioning train under conditions selective for the putative VSRM was extended to 30 steps. When CdCl2 (120–200 μM) was used, it was applied transiently during the 4-s holding period and removed after completion of the test depolarizing step. The use dependence of nifedipine (30–60 μM) mandated it to be present continuously throughout the experiment. In later experiments, NiCl2 (12 mM) was used. Experiments under CICR-selective control conditions were completed before application of L-type Ca2+ channel blockers.
Figure 4.
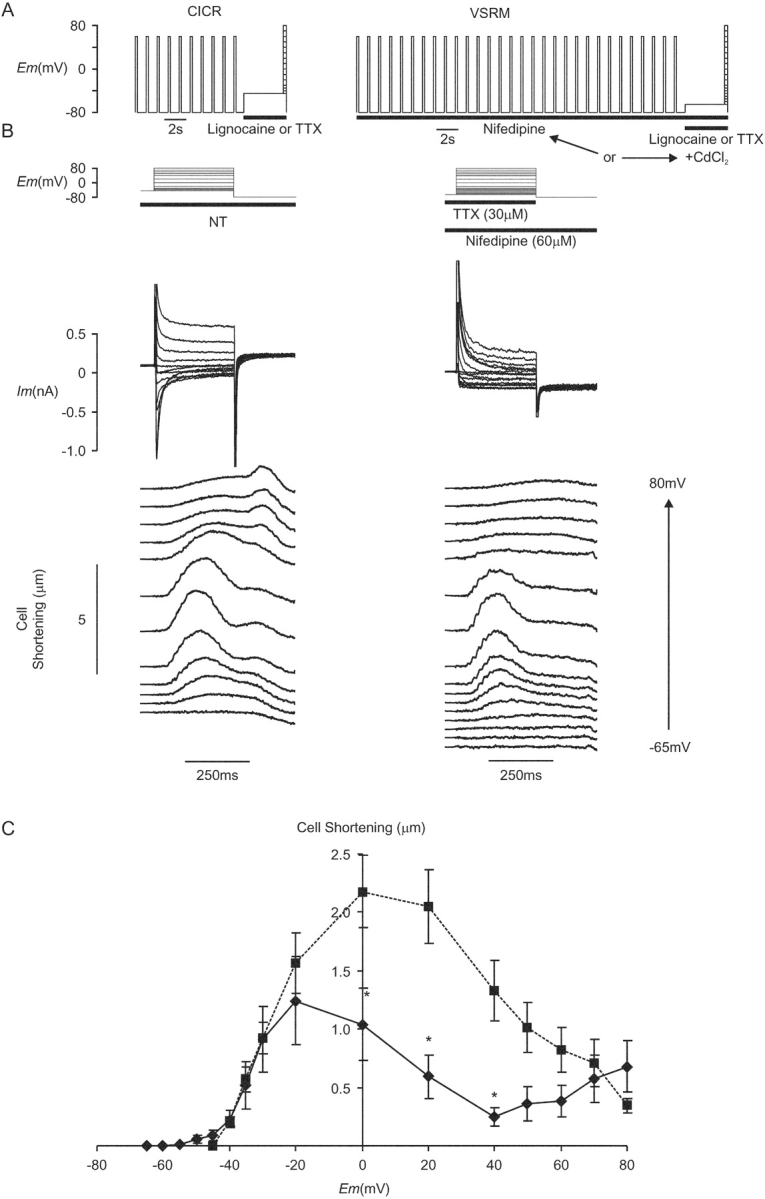
(A) Voltage protocols for experiments under conditions selective for CICR and the putative VSRM. (B) Membrane current and cell shortening under CICR-selective conditions during superfusion with normal Tyrode solution (NT) and under conditions selective for the putative VSRM in the presence of TTX (30 μM) and nifedipine (60 μΜ) (Cm=134 pF). Each test depolarization was preceded by a conditioning protocol. For clarity, cell shortening records have been offset vertically in proportion to the test potential. (C) Maximum cell shortening versus voltage curves under paired conditions selective for CICR (▪, dashed line) and the putative VSRM (♦, solid line, n = 14, asterisk indicates significant difference at the 95% level by Student's paired t test).
In some cells, small phasic inward currents were activated by depolarization despite the presence of L-type Ca2+ channel blockers. Data from these cells were not included in the calculation of the cell shortening curve seen in Fig. 4 C. With depolarizations to test potentials negative to 0 mV, membrane current in the remainder of cells was inward and maintained (Fig. 4 B). With test depolarizations to potentials of −20 mV and more positive, inactivating early outward current typical of Ito was activated.
Application of the L-type Ca2+ channel blockers nifedipine or CdCl2 did not abolish cell shortening but maximum amplitude was reduced to 60% of that under CICR-selective conditions in paired controls (Fig. 4 C). The voltage threshold for cell shortening remained at −50 mV and cell shortening reached a peak at −20 mV. With depolarization to more positive potentials cell shortening did not reach a plateau but decreased, reaching a nadir at 40 mV. The shape of the cell shortening versus voltage curve was therefore analogous to that shown in Fig. 2 C when ICa(L) was present and reflected to some extent the voltage dependence of ICa(L). With depolarization to yet further positive potentials, the shape of the curve deviated from that recorded with ICa(L) present and described a second rise.
The effect on maximum cell shortening amplitude of different L-type Ca2+ channel blockers in varying concentration is shown in Fig. 5 . With small numbers of cells in each group, increasing potency of L-type Ca2+ channel blockade seemed to produce increasing suppression of cell shortening amplitude.
Figure 5.

Peak maximum cell shortening as a function of L-type Ca2+ channel blocker under conditions selective for the putative VSRM. Number of cells indicated above individual bars.
Tail contractions occurred on repolarization to −80 mV from test potentials positive to 40 mV in a proportion of cells (18/29) under CICR-selective conditions (Fig. 4 B). This phenomenon occurred also in a proportion of cells (9/26) under conditions selective for the putative VSRM (Fig. 6 A). This suggested that L-type Ca2+ channels were open during test depolarizations under conditions selective for the putative VSRM. The effect of different L-type Ca2+ channel blockers in varying concentration on repolarization-activated tail contractions is shown in Fig. 6 B. Once again, in groups with small numbers of cells, repolarization-activated tail contractions appeared less frequently as potency of L-type Ca2+ channel blockade increased.
Figure 6.
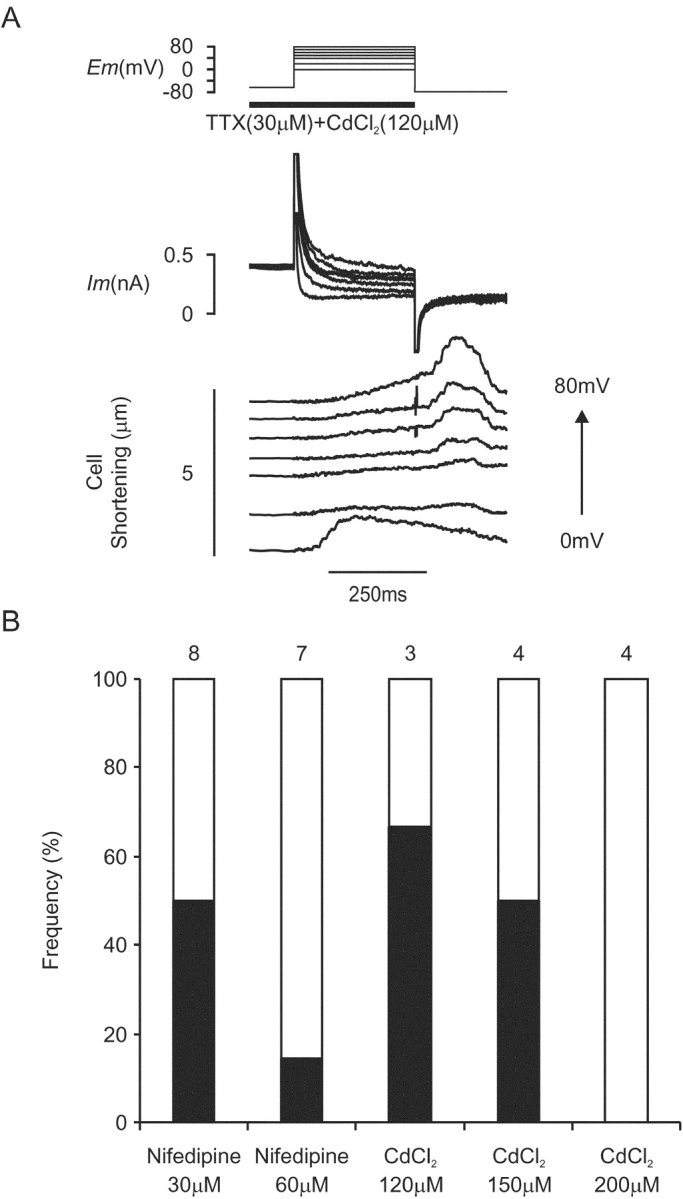
(A) Membrane current and cell shortening during depolarizations applied from a holding potential of −65 mV under conditions selective for the putative VSRM in the presence of TTX (30 μM) and CdCl2 (120 μM) (Cm=131 pF). Each test depolarization was preceded by a conditioning protocol. (B) Frequency histogram of repolarization induced tail contraction as a function of L-type Ca2+ channel blocker under conditions selective for the putative VSRM. Numbers above bars indicate numbers of cells exposed to each blocker.
In a separate series of experiments, the effect was established of NiCl2 (12 mM) on cell shortening under conditions selective for the putative VSRM (Fig. 7 A). Voltage protocols were based on those shown in Fig. 4 A but the conditioning train consisted of action potentials at 0.5 Hz for 30 s under current clamp before depolarization to the holding potential of −60 mV under voltage clamp. Lignocaine (400 μM) and NiCl2 (12 mM) were applied transiently during the 4-s holding period and removed after completion of the test depolarizing step. Under CICR-selective conditions in paired controls, the conditioning period under current clamp was reduced to 10 s, the holding potential was −40 mV, and lignocaine and NiCl2 were not applied. The effect of NiCl2 was unequivocal. Maintained inward current during small depolarizations was blocked. There were no occurrences of phasic inward current. Cell shortening and repolarization tail contractions were blocked completely at all potentials (Fig. 7, B and C).
Figure 7.

(A) Voltage protocol for experiments under conditions selective for CICR and the putative VSRM in the presence of lignocaine (400 μM) and NiCl2 (12 mM). (B) Membrane current and cell shortening during paired depolarization under the selective conditions illustrated (Cm = 159 pF). (C) I-V curve under CICR-selective conditions (n = 11). Corresponding cell shortening versus voltage curves under conditions selective for CICR (▪, dashed line) and the putative VSRM (♦, solid line, n = 11).
The Persistence of L-type Ca2+ Current under Conditions Presumed to be Selective for the Putative VSRM
Together, the results described above suggested strongly the presence of unblocked ICa(L) under conditions selective for the putative VSRM in the presence of nifedipine (30–60 μM) or CdCl2 (120–200 μM). This may have been undetectable in the presence of overlying Ito. An experiment was designed to test directly this hypothesis (Fig. 8 A). Cells were held under voltage clamp at a holding potential of −60 mV and depolarizing steps were applied to potentials between −50 mV and 80 mV at a repeat frequency of 2 Hz. Na+ channels were blocked by application of lignocaine (500 μM) 3 s before the first step and L-type Ca2+ channel blockers were applied. Either CdCl2 (120 μM) or NiCl2 (12 mM) were applied 3 s before the start of the voltage protocol and simultaneously with lignocaine. When nifedipine (30 μM) was used, it was added during voltage clamp steps from −45 to 0 mV at 0.5 Hz while its use-dependent effect was becoming established and the voltage protocol was started 30 s later. Conditioning voltage clamp steps were otherwise not used. The Ito blocker, 4AP (5 mM), and the ICl(Ca) blocker SITS (2 mM) were then added and the cell was superfused for 5 min before the protocol was repeated. Inward current records during depolarization to −10 mV are shown before (control) and after block of Ito and ICl(Ca) (Fig. 8 B). A component of unblocked inward current was detectable and was maximal at −10 mV but was blocked completely by NiCl2 (12 mM) (Fig. 8 C).
Figure 8.
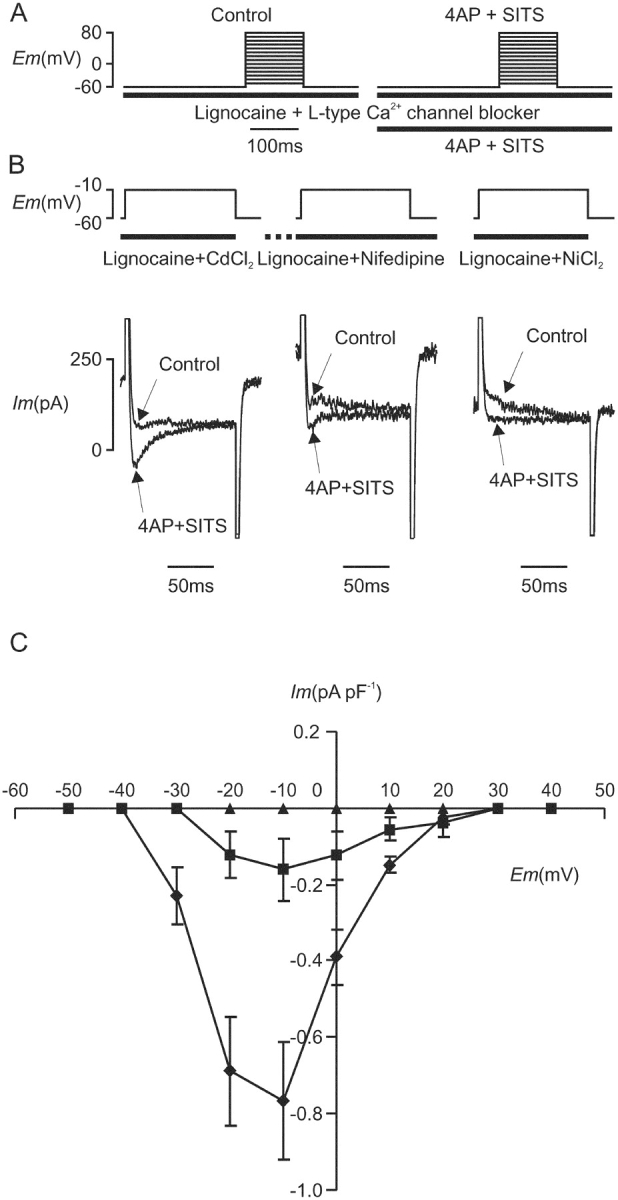
(A) Voltage protocol for demonstration of unblocked inward current in the presence of L-type Ca2+ channel blockers under “Control” conditions and after treatment with “4AP + SITS” for 5 min. (B) Inward current during depolarization from −60 to −10 mV in the presence of lignocaine (500 μM) and either CdCl2 (120 μM), nifedipine (30 μM), or NiCl2 (12 mM). Records are shown before and after exposure to 4AP (5 mM) and SITS (2 mM) as indicated. (C) Corresponding I-V curves for the unblocked inward current during exposure to 4AP and SITS in the presence of CdCl2 (120 μM, ♦, n = 4), nifedipine (30 μM, ▪, n = 3), and NiCl2 (12 mM, ▴, n = 5).
The Contribution of Na+/Ca2+ Exchange to the Putative VSRM
Typically, cell shortening under conditions selective for the putative VSRM was tonic and maintained at test potentials between −50 and −40 mV (Fig. 4 B). At test potentials between −40 and 20 mV, the amplitude of cell shortening was dependent on the potency of L-type Ca2+ channel blockade (ibid.). It was noted also that the potency of L-type Ca2+ channel blockade affected the form of cell shortening. Cell shortening under conditions selective for the putative VSRM could be phasic in the presence of nifedipine (30–60 μM) or CdCl2 (120 μM) (e.g., Figs. 4 B, 6 A, 9 A, and 14 A). Cell shortening in the presence of CdCl2 (≥150 μM) or NiCl2 (12 mM) was never phasic (e.g., Figs. 7 B and 11 A).
With depolarizations to strongly positive potentials, cell shortening was tonic and progressive throughout the duration of the test step. The form of cell shortening under conditions selective for CICR and the putative VSRM was examined quantitatively (Fig. 9 A). The times to onset and peak of cell shortening are shown as functions of test potential in Fig. 9, B and C, respectively. Under CICR-selective conditions, these time intervals shortened as test depolarizations were made to potentials approaching 0 mV where they were at a minimum. This corresponded with the well known effect of shortening of the time to peak of ICa(L) over this voltage range. As depolarizations were made to more positive potentials, the time intervals became progressively longer and time to peak shortening tended asymptotically toward the depolarizing step duration of 310 ms. This reflected the tonic progressive nature of cell shortening at positive potentials whereby cells continued to shorten throughout the period of depolarization. The curves defining the form of cell shortening under conditions selective for the putative VSRM were comparable in shape to those under CICR-selective conditions.
Figure 9.
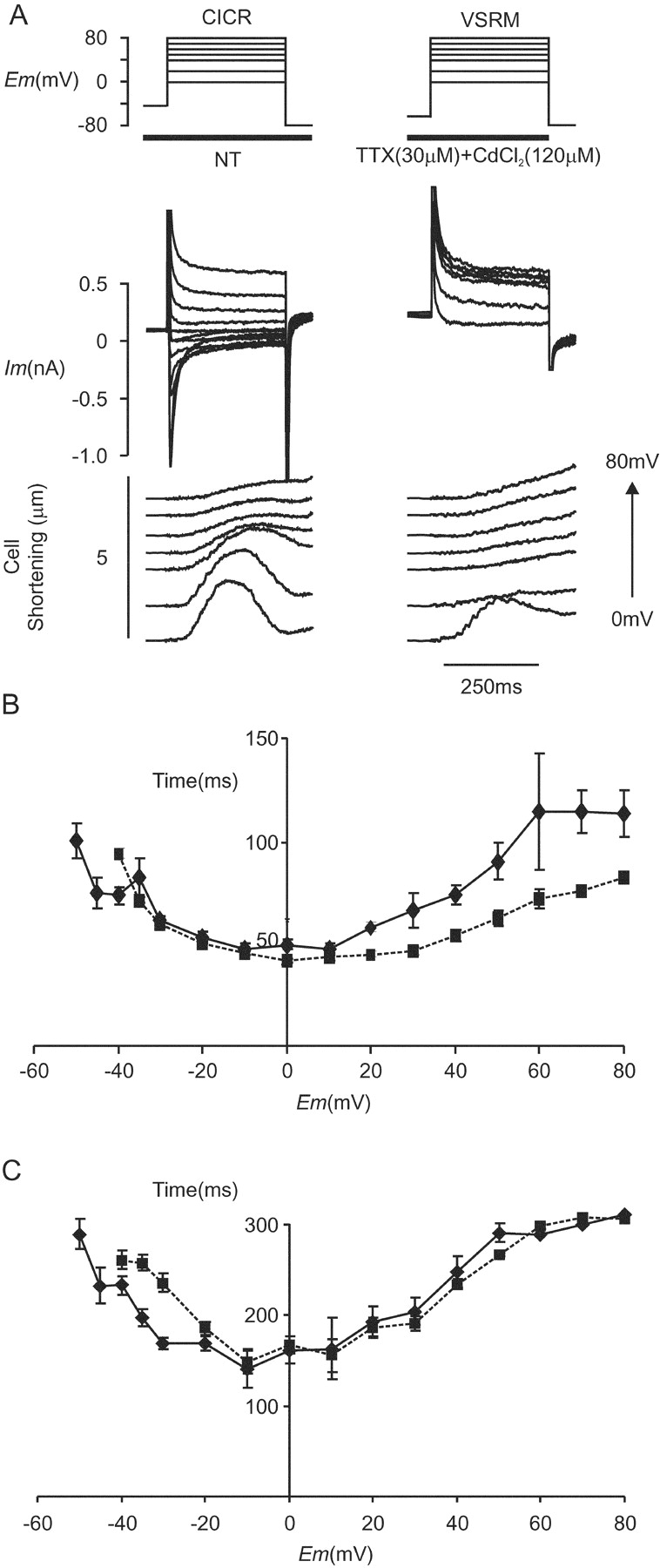
(A) Membrane current and cell shortening during depolarization to positive potentials under conditions selective for CICR (Cm = 134 pF) and the putative VSRM (Cm = 105 pF). The record shown for CICR-selective conditions is shown for comparison and taken from Fig. 4 B. (B) Time from depolarization to onset of cell shortening for CICR (▪, dashed line, n = 31) and the putative VSRM (♦, solid line, n = 22). (C) Time from depolarization to peak cell shortening for CICR (▪, dashed line, n = 31) and the putative VSRM (♦, solid line, n = 22).
These features of late-peaking progressive cell shortening or an increase in intracellular [Ca2+] occurring with a significant delay after depolarization are typical of those described for EC coupling mediated by Na+/Ca2+ exchange (Barcenas Ruiz et al., 1987; Barcenas Ruiz and Wier, 1987; Beuckelmann and Wier, 1989; Nuss and Houser, 1992; Sham et al., 1992; Bouchard et al., 1993). This mode of EC coupling is expected to operate during depolarizations to strongly positive potentials under our CICR-selective conditions. The similarity between the shapes of the curves obtained under the two sets of conditions illustrated in Fig. 9, B and C, raised the possibility that EC coupling mediated by Na+/Ca2+ exchange might also have operated under conditions selective for the putative VSRM. In turn, EC coupling mediated by Na+/Ca2+ exchange may have contributed to the upturn at positive potentials of the cell shortening versus voltage curve. A number of experiments were performed to test this hypothesis.
The driving force for Na+/Ca2+ exchange is given by Em–ENCX (the equilibrium potential for Na+/Ca2+ exchange) so an experiment was devised to produce an abrupt increase in ENCX by increasing rapidly extracellular [Na+] (Fig. 10 A). This produced selective inhibition of outward Na+/Ca2+ exchange current. For this series of experiments, cells were stored in a low sodium/calcium solution until use and this solution was also used as the control superfusing solution. Cells underwent 10 conditioning steps of 300 ms duration under voltage clamp before each test step. The conditioning steps were applied to 0 mV rather than 60 mV in order to produce calcium influx via L-type Ca2+ channels. A holding potential of −65 mV was set for conditions selective for the putative VSRM and −45 mV for CICR. Na+ and L-type Ca2+ current were blocked under conditions selective for the putative VSRM by rapid application of lignocaine (500 μM) and CdCl2 (150 μM) at the onset of the 4-s holding period. Cell shortening was elicited by voltage steps between −40 and 80 mV in 40-mV increments. The rapid increase in extracellular [Na+] was achieved by switching the superfusate to a Tyrode solution containing 146 mM Na+ 2 s before test depolarizations. The experiments were performed in paired fashion with CICR experiments completed before application of lignocaine and CdCl2 under conditions selective for the putative VSRM. Assuming no change in intracellular [Na+] or [Ca2+] during the time course of the switch, we calculated that ENCX would change by 86 mV. However, subsarcolemmal Na+ accumulation during exposure to the higher [Na+]-containing solution must have attenuated the ΔENCX achieved in practice.
Figure 10.

(A) Voltage protocol for the assessment of the effect of a rapid switch in superfusate [Na+] on cell shortening under conditions selective for CICR and the putative VSRM. Conditioning steps to 0 mV were applied before each test depolarization. The “Control” run was performed in a solution containing 50 mM Na+ before the run incorporating a rapid solution “Switch” to 146 mM Na+. This sequence was repeated under conditions selective for the putative VSRM with exposure to lignocaine (500 μM) and CdCl2 (150 μM). (B) Membrane current and cell shortening during depolarizations under the conditions described in A. Brief periods of loss of edge detection are blanked for clarity (Cm = 161 pF). (C) Paired cell shortening versus voltage relationships under conditions selective for CICR and the putative VSRM (n = 12). Open bars indicate control conditions and black bars indicate the switch to 146 mM Na+. (D, i) Phasic and tonic components of cell shortening under CICR selective conditions. Open bars indicate control conditions and black bars indicate the switch to 146 mM Na+ (asterisk indicates significant difference at the 95% level by Student's paired t test). (ii) Cell shortening under conditions selective for the VSRM consisted of a tonic component only.
After the solution switch, holding current under voltage clamp deviated over the following 2 s, by −0.29 ± 0.04 pA pF−1 at −45 mV under CICR-selective conditions and by −0.24 ± 0.04 pA pF−1 at −65 mV under conditions selective for the putative VSRM (n = 13). This deviation was not significantly different between the two sets of conditions (P = 0.55). Peak ICa(L) amplitude was −3.2 ± 0.18 pA pF−1 at 0 mV in low sodium/calcium solution and −3.1 ± 0.21 pA pF−1 after the switch to Tyrode solution containing 146 mM Na+ (n = 13, P = 0.57, Fig. 10 B). Within this population of cells, the form of cell shortening under conditions selective for the putative VSRM in the presence of CdCl2 (150 μM) was never phasic. Application of 146 mM Na+ had a prominent inhibitory effect on cell shortening under conditions selective either for CICR or the putative VSRM (Fig. 10 C). The tonic cell shortening seen under conditions selective for the putative VSRM was inhibited substantially (Fig. 10 D). Phasic cell shortening under CICR-selective conditions was also inhibited.
KB-R7943 is an Na+/Ca2+ exchange–blocking drug with preferential effects on outward over inward Na+/Ca2+ exchange current (Watano et al., 1996). Its effects on cell shortening were examined. Voltage protocols were based on those shown in Fig. 10 A, but the basic superfusate was normal Tyrode, lignocaine (500 μM) was present during the holding period under CICR-selective conditions, and all solutions contained 0.02% DMSO (Fig. 11 A). The onset of action of KB-R7943 took up to 3 min and was only partly reversible. For these reasons, control measurements were made under conditions selective for first CICR and then the putative VSRM before adding KB-R7943 (2 μM) and remeasuring.
Figure 11.

(A) The effect of KB-R7943 (2 μM) on membrane current and cell shortening under paired conditions selective for CICR and the putative VSRM (Cm = 93 pF). Conditioning steps to 0 mV were applied before each test depolarization. Control runs under conditions selective for CICR and the putative VSRM were performed first and repeated after exposure to KB-R7943 for 3 min. (B) Paired cell shortening versus voltage relationships under conditions selective for CICR and the putative VSRM (n = 10). Open bars indicate control conditions and black bars indicate exposure to KB-R7943. (C) Phasic and tonic components of cell shortening under CICR-selective conditions. Open bars indicate control conditions and black bars indicate exposure to KB-R7943 (asterisk indicates significant difference at the 95% level by Student's paired t test). Cell shortening under conditions selective for the VSRM consisted of a tonic component only.
KB-R7943 (2 μM) produced a −0.30 ± 0.11 pA pF−1 change in holding current under voltage clamp at −45 mV and a −0.36 ± 0.13 pA pF−1 change at −65 mV (n = 10, P = 0.36) compatible with its IK1 blocking effect. L-type Ca2+ current at 0 mV under CICR-selective conditions was reduced from −6.4 ± 0.7 pA pF−1 to −3.5 ± 0.6 pA pF−1 in KB-R7943 (53 ± 7% of control, n = 10, P < 0.001). In this population of cells in which conditions selective for the putative VSRM were established in the presence of CdCl2 (150 μM), the form of cell shortening was never phasic. Under these conditions, cell shortening was reduced by KB-R7943 (2 μM) at all potentials, and to 29 ± 7% of control at 80 mV (P < 0.001; Fig. 11, B and C). KB-R7943 inhibited phasic as well as tonic cell shortening under CICR-selective conditions compatible with the demonstrated inhibitory effect on ICa(L).
NiCl2 (12 mM) produced complete block of cell shortening at all potentials (Fig. 7). NiCl2 is known to be a potent blocker of Na+/Ca2+ exchange as well as L-type Ca2+ current and its effects may have been mediated partly by this effect. An experiment was performed to establish the completeness of block of Na+/Ca2+ exchange by NiCl2. Inward Na+/Ca2+ exchange was activated by rapid application of caffeine to myocytes held under voltage clamp at −70 mV (Fig. 12 A(i)). This produced inward Na+/Ca2+ exchange current and cell shortening. NiCl2 (12 mM) was then applied 4 s before reapplication of caffeine. The conditioning protocol was identical to that used in experiments investigating the effect of NiCl2 on the putative VSRM (Fig. 7 A) and consisted of action potentials under current clamp at 0.5 Hz for 30 s. NiCl2 produced an outward change in the holding current, often with the development of cell shortening, compatible with block of background inward Na+/Ca2+ exchange current. Cell shortening on reapplication of caffeine was exaggerated by NiCl2. This was compatible with inhibition of extrusion by Na+/Ca2+ exchange of released Ca2+. Peak inward current was measured with respect to steady-state current in the presence of caffeine and was reduced by NiCl2 to 3 ± 3% of control (P < 0.01; Fig. 12 B(i)). In 6/7 cells, block was complete. In an analogous experiment, outward Na+/Ca2+ exchange was activated by rapid application of outward Na+/Ca2+ exchange–activating solution to cells held under voltage clamp at 0 mV (Fig. 12 A(ii)). Cell shortening under these conditions was attenuated by NiCl2. This was compatible with inhibition of Na+/Ca2+ exchange–mediated inward flux of Ca2+. Peak outward current was reduced by NiCl2 (12 mM) to 20 ± 6% of control although part of the residual current may have represented ICl(Ca) (P < 0.01; Fig. 12 B(ii)).
Figure 12.

(A, i) The effect of NiCl2 (12 mM) on inward Na+/Ca2+ exchange current activated under voltage clamp at −70 mV by rapid application of caffeine (10 mM) (Cm = 164 pF). (ii) The effect of NiCl2 (12 mM) on outward Na+/Ca2+ exchange current activated under voltage clamp at 0 mV by a rapid switch to outward Na+/Ca2+ exchange activating solution (RMP = resting membrane potential). Cell shortening is shown in the bottom panel. (B, i). Peak inward Na+/Ca2+ exchange current density following application of 12 mM NiCl2 (n = 7). (ii) Peak outward Na+/Ca2+ exchange current after application of 12 mM NiCl2 (n = 7, asterisk indicates significant difference at the 95% level by Student's paired t test).
The Effect of Isoprenaline and Increased SR Ca2+ Content on the Putative VSRM
Published work on the putative VSRM has demonstrated that dialysis with a high concentration of cAMP is necessary for its demonstration when patch pipettes are used (Hobai et al., 1997b; Ferrier et al., 1998). A prominent affect of elevated cAMP is to increase SR Ca2+ content and this is known to be a major determinant of the gain of CICR. This activation within the β-adrenergic pathway is known also to increase ICa(L) amplitude and increase resistance to L-type Ca2+ channel blockers (McDonald et al., 1994; Hobai et al., 2000). All of these effects are important under conditions where ICa(L) is blocked incompletely. Preliminary experiments in our lab using the twostep voltage protocol proposed by Ferrier and Howlett (1995) demonstrated that putative VSRM contractions at −40 mV were present only in cells exhibiting signs of SR Ca2+ overload. The hypothesis was developed, therefore, that under the influence of elevated intracellular cAMP, CICR could mediate EC coupling with features resembling the putative VSRM. To test this hypothesis, intracellular levels of cAMP were increased by inclusion of isoprenaline in the superfusate (Callewaert et al., 1988; Hussain and Orchard, 1997).
Voltage protocols were based on those shown in Fig. 11 A, but solutions did not contain DMSO. 20 conditioning steps to 60 mV were applied and the membrane was repolarized from the final conditioning step to the holding potential (Fig. 13 A). L-type Ca2+ channels were blocked by CdCl2 (120 μM). Experiments under control conditions were always performed before application of isoprenaline (1 μM) at the onset of the conditioning period.
Figure 13.

(A) The effect of isoprenaline (1 μM) on membrane current and cell shortening under conditions selective for CICR (Cm = 230 pF) and the putative VSRM (Cm = 158 pF). 20 conditioning steps were applied to 60 mV before the membrane was repolarized to the conditioning potential. (B) Cell shortening versus voltage relationships under conditions selective for CICR (n = 11) and the putative VSRM (n = 12). Open bars indicate control conditions and black bars indicate exposure to isoprenaline.
SR Ca2+ content was assessed by integration of the inward current activated by rapid application of caffeine (10 mM). This represented transsarcolemmal extrusion by Na+/Ca2+ exchange of Ca2+ released from the SR. Released Ca2+ distributes chiefly within the cytoplasmic intracellular compartment and a scaling factor of 0.65 was applied to account for the proportion of intracellular volume occupied by mitochondria. SR Ca2+ content was therefore expressed in units of μmol l−1 of accessible cell volume (a.c.v.). Isoprenaline (1 μM) produced an increase in SR Ca2+ content from 25.9 ± 4.3 μmol l−1 a.c.v. to 59.0 ± 8.6 μmol l−1 a.c.v. (n = 7, P < 0.001). Spontaneous cell shortening did not occur. Under CICR-selective conditions, ICa(L) increased from −5.5 ± 0.7 pA pF−1 to −8.3 ± 1.1 pA pF−1 (n = 11, P < 0.05). Isoprenaline (1 μM) had little effect on the amplitude of cell shortening under these conditions. A reduction in cell shortening during the voltage step to 40 mV was observed with no significant change at other potentials (Fig. 13, A and B). Under conditions selective for the putative VSRM, marked effects of isoprenaline were observed on the form of cell shortening in individual cells. In two cells, shortening at test depolarizations to ≥40 mV became rapid and phasic (Fig. 13 A, right column). At these potentials, cell shortening was always of late onset, progressive and with a late peak in the absence of isoprenaline. In addition, maximum cell shortening amplitude at test depolarizations of −40 and 0 mV was increased in the presence of isoprenaline.
The effect on cell shortening of adjustment of the conditioning step duration from 300 to 600 ms was assessed (Fig. 14) . This experiment was devised as a more selective test of the effect of increased SR Ca2+ content on cell shortening. Voltage protocols were based on those shown in Fig. 13 A, but Na+ channels were blocked by TTX (30–40 μM) or lignocaine (500 μM). Experiments under CICR-selective conditions were completed before conditions selective for the putative VSRM were established in the presence of CdCl2 (120 μM).
Figure 14.

(A) The effect of prolonging conditioning steps from 300 to 600 ms on membrane current and cell shortening under conditions selective for CICR (Cm = 174 pF) and the putative VSRM (Cm=170 pF). 20 conditioning steps were applied to 60 mV before the membrane was repolarized to the conditioning potential. Brief periods of loss of edge detection are blanked for clarity. (B) Cell shortening versus voltage plots under conditions selective for CICR (n = 19) and the putative VSRM (n = 22). Open bars indicate conditioning steps of 300 ms duration and black bars indicate conditioning steps of 600 ms duration (asterisk indicates significant difference at the 95% level by Student's paired t test).
Under these conditions SR Ca2+ content was increased from 35.8 ± 5.6 μmol l−1 a.c.v. to 49.4 ± 5.0 μmol l−1 a.c.v. (n = 11, P < 0.01). Spontaneous cell shortening was not induced and ICa(L) amplitude was reduced by 600 ms conditioning steps from −5.3 ± 0.6 pA pF−1 to −4.6 ± 0.5 pA pF−1 (n = 19, P < 0.05). Under CICR-selective conditions, cell shortening at 0 mV was increased, but remained unchanged at other test depolarizations (Fig. 14 B). Under conditions selective for the putative VSRM marked effects were seen in individual cells. In one cell, shortening at test depolarizations of ≥40 mV became rapid and phasic (Fig. 14 A, right column). At these potentials, cell shortening was always of late onset, progressive, and with a late peak during conditioning with 300-ms steps. Maximum cell shortening amplitude during conditioning with 600-ms steps was increased at −40, 0, and 40 mV, but remained unchanged at 80 mV (Fig. 14 B).
DISCUSSION
We have demonstrated that when holding potential was adjusted in order to allow the hypothesized VSRM to operate alongside CICR, there was a change in the voltage threshold for cell shortening to a more negative value. This was in qualitative agreement with the result of Howlett et al. (1998), but we recorded a change of −10 mV in contrast to the −20 mV recorded in that study. This was accompanied by a change of the same magnitude in the voltage threshold for ICa(L). Indeed, this phenomenon is also evident in the work of Howlett et al. (1998) (see their Fig. 2 D). Inspection of membrane current records, however, suggested that cell shortening at threshold potentials was occurring in the absence of inward current. As this was the case when holding potential was set in a range for selective demonstration of CICR as well as after adjustment to more negative values, we reasoned that ICa(L) might be present but undetected. The nifedipine-sensitive current obtained by subtraction demonstrated that cell shortening at these negative test potentials was always accompanied by ICa(L) and confirmed that ICa(L) could be detected at a test potential of −50 mV when depolarizations were applied from a holding potential of −60 mV. This finding suggested strongly that under these conditions, cell shortening was dependent on CICR triggered by ICa(L) and not on a combination of CICR and putative VSRM. These findings in relation to the putative VSRM are in accordance with the earlier study of “small depolarizations” by Talo et al. (1990).
Under the terms of the local control theory for CICR (Stern, 1992), gain is variable and greatest during test depolarizations to negative potentials. It is at these potentials that the electrochemical gradient for Ca2+ and therefore unitary L-type channel Ca2+ flux is greatest (Wier et al., 1994). At these potentials therefore, the low amplitude ICa(L) recorded by nifedipine subtraction is expected to be significant with respect to its ability to trigger CICR and cell shortening. This argument emphasizes the importance of complete exclusion of ICa(L) before ascribing cell shortening to the hypothesized VSRM. The approach taken by Ferrier and Howlett (1995) and also in their later work relied heavily on the demonstration of cell shortening at −40 mV. They reasoned, but did not demonstrate convincingly, that ICa(L) was not activated at this potential. It is now clear that this is not the case.
The persistence of repolarization-activated tail contractions (London and Krueger, 1986; Barcenas Ruiz and Wier, 1987; Beuckelmann and Wier, 1988) in a proportion of cells exposed to L-type Ca2+ channel blockers suggested strongly that ICa(L) was blocked incompletely. This occurred despite the use of nifedipine in concentrations an order of magnitude greater and CdCl2 in concentrations up to twice those in earlier studies. It was clear from our preliminary experiments that the Ito became prominent when the holding potential was −60 mV. This current is known to have a comparable time course but an opposite direction to ICa(L) and was likely to have played a part in masking underlying ICa(L). In the presence of verapamil (2 μM), persisting inward current may be observed even in the raw membrane currents in the “control experiment” of Howlett et al. (1998) (their Fig. 1 B). We too observed this occasionally during the experiments illustrated in Fig. 4. We proceeded therefore, by blocking Ito, to establish to what extent ICa(L) was present under experimental conditions designed to demonstrate the putative VSRM. It was shown that even at high concentration, nifedipine and CdCl2 were unable to block completely ICa(L). Dependency of cell shortening on CICR was confirmed by the correlation between cell shortening and the concentration of L-type Ca2+ channel blocker present (Fig. 5). To emphasize this point, both ICa(L) and cell shortening were blocked completely by NiCl2. It is further emphasized that L-type Ca2+ channel blockers are less effective as holding potential becomes more negative (Kokubun et al., 1986; Lansman et al., 1986) with phosphorylation of the channel (Hobai et al., 2000) or as experimental temperature increases (Allen and Chapman, 1992). NiCl2 in the lower concentration of 200 μM had a significant inhibitory effect on cell shortening in guinea-pig (Ferrier and Howlett, 1995). At this concentration, T-type Ca2+ current is blocked selectively over L-type Ca2+ current and this current is expressed in guinea-pig but not rabbit myocytes (Vassort and Alvarez, 1994; Bers, 2001). This result suggested strongly a role for T-type Ca2+ current triggered CICR (Sipido et al., 1998).
When holding potential was adjusted in order to allow the hypothesized VSRM to operate alongside CICR, the overall shape of the cell shortening versus voltage curve remained bell shaped without a plateau at positive potentials. In this respect we failed to reproduce the findings of Ferrier and Howlett (1995). A broad maximum of cell shortening or the Ca2+ transient as a function of voltage has often been observed in previous work on CICR however. Thus, large amplitude responses were recorded at negative and positive potentials despite low amplitude ICa(L) (Cannell et al., 1987; Callewaert et al., 1988). This can be ascribed to high gain at negative potentials (Wier et al., 1994) and can be explained by the many nonlinearities in the system at positive potentials (Callewaert, 1992). In many of their experiments, Ferrier and Howlett (1995) did not extend their observations to very positive potentials and may have interpreted a broad peak in their curves mistakenly as a plateau. In one experiment, a sigmoid curve was obtained with observations to potentials of 80 mV (Ferrier and Howlett, 1995). In that experiment the holding potential was −55 mV and Na+ channel blockers were not used. It is relevant, therefore, that INa has been shown to contribute to a sigmoid cell shortening versus voltage curve under just these conditions (Piacentino et al., 2000). In the current report, lignocaine in a concentration of at least 400 μM or TTX in a concentration of 30 μM were used and there was never any reason to suspect that INa was present. Furthermore, in the experiment of Ferrier and Howlett (1995) demonstrating a sigmoid curve to potentials of 80 mV, L-type Ca2+ channel blockers were not present and CICR therefore operated alongside the putative VSRM. A further factor in the experiment of Ferrier and Howlett (1995) was the use of reduced [Na+] in the superfusate and this may have contributed to increased SR Ca2+ content. SR Ca2+ content is an important determinant of CICR gain and instances have already been quoted of increased SR Ca2+ content or increased experimental temperature generating a sigmoid cell shortening vs. voltage curve (Vornanen et al., 1994; Levi et al., 1996; Wasserstrom and Vites, 1996). In our experiments, increased SR Ca2+ content did not generate a sigmoid curve but responses in individual cells were fundamentally changed in that high amplitude phasic cell shortening occurred at positive potentials. That isoprenaline did not increase maximum cell shortening may be explained by phosphorylation of troponin I and reduced myofilament sensitivity to Ca2+ (Okazaki et al., 1990; Bers, 2001).
Our approach to assessment of the contribution of Na+/Ca2+ exchange to the hypothesized VSRM differed from that of Ferrier et al. (2000). They measured the amplitude and voltage dependence of the sustained Ca2+ transient in cells under steady-state superfusion with 50 or 100 mM Na+ and found it to be independent of [Na+]. The sustained Ca2+ transient persisted under these conditions. It was concluded that the sustained component was not dependent on Na+/Ca2+ exchange. Consideration of the mode of operation of Na+/Ca2+ exchange calls this conclusion into doubt. In the steady-state, intracellular [Na+] would rise with or without a significant drop in subsarcolemmal [Ca2+] in response to increased superfusate [Na+]. One or both of these effects would attenuate any possible effect on ENCX. For this reason, we investigated the effects of an abrupt increment in superfusate [Na+] that produced an 86 mV change in ENCX during the time course of the test depolarization. Under the conditions of this experiment, a significant reduction in tonic cell shortening was observed. Superfusate [Ca2+] remained unchanged during the solution switches and no effects on CICR were expected. Phasic shortening under CICR-selective conditions, however, was reduced and this finding is difficult to explain. It could be argued that the reduced [Na+] superfusate used in these experiments might have contributed to SR Ca2+ overload, but no signs of this were observed. Even if SR Ca2+ overload had been present, individual cell shortening records and the cell shortening versus voltage curves were of conventional form. In a separate experiment, they found that sustained cell shortening persisted when NiCl2 (2 mM) was applied rapidly some seconds after its initiation (Ferrier et al., 2000). It is likely, however, that the maintaining factors for the sustained component of cell shortening are different from those governing its initiation. As NiCl2 was applied during a steady-state period, then net sarcolemmal flux was by definition zero. It is conceivable that the capacity of the SR to take up Ca2+ was exhausted by inward Ca2+ flux during a prolonged period at a positive membrane potential. Application of NiCl2 and block of the Na+/Ca2+ exchange at this time might have unpredictable effects. The traces seen in Fig. 12 A show that NiCl2 caused membrane current to become more outward and provoked low amplitude cell-shortening oscillation. This implies that under these conditions, NiCl2 caused intracellular [Ca2+] to rise and, therefore, by inference, blocked background inward Na+/Ca2+ exchange current. In our experiments with NiCl2 it was applied 4 s before test depolarizations thus allowing its effects to develop. Used in this way, cell shortening was abolished.
When ICa(L) was blocked partially by nifedipine or CdCl2, cell shortening was suppressed partially and the curve retained its basic bell shape, but a secondary rise was present at positive potentials. This curve resembled superficially the sigmoid relationship claimed for the hypothesized VSRM. Under the influence of a hypothesized mechanism for EC coupling dependent only on membrane voltage, time to the onset and peak of cell shortening would be expected to shorten with more positive test depolarizations as the voltage sensor moved more rapidly through a stronger electric field. However, this feature was not observed and the form of cell shortening at these potentials under conditions selective for the putative VSRM resembled that under CICR-selective conditions.
A possible explanation for the appearance of a secondary rise in the shape of the curve relates to the interrelationship between L-type Ca2+ channel flux and operation of Na+/Ca2+ exchange. Depolarization-activated ICa(L) causes Ca2+ to accumulate within the subsarcolemmal space and therefore at the cytoplasmic face of the Na+/Ca2+ exchange protein. This accumulation results in a positive change of ENCX, thus reducing the electrochemical gradient for inward Ca2+ flux by Na+/Ca2+ exchange. When ICa(L) is blocked, this inhibitory effect on Na+/Ca2+ exchange is reduced, thus allowing unhindered Na+/Ca2+ exchange–mediated Ca2+ entry at positive potentials. It was seen that while CdCl2 (120 μM) produced less complete block of ICa(L) than nifedipine (30 μM), the block of cell shortening was greater with CdCl2. This apparent anomaly may be explained by the inhibitory effect of CdCl2 on Na+/Ca2+ exchange (Hobai et al., 1997a).
In conclusion, a fraction of ICa(L) remains unblocked in the presence of L-type Ca2+ channel blockers under conditions necessary for demonstration of the hypothesized VSRM. The currents are masked by Ito, which becomes less inactivated at negative holding potentials. When the sensitive method of current subtraction is employed, it is clear that cell shortening never occurs in the absence of ICa(L). When complete block of ICa(L) is achieved, cell shortening is abolished. Our results are also compatible with a contribution from Na+/Ca2+ exchange to the hypothesized VSRM with particular reference to the tonic component of shortening at positive potentials. We are in agreement with the conclusions of Piacentino et al. (2000). Our results show no significant VSRM and thus any possible contribution of a VSRM to cardiac EC coupling appears to be negligible. A quantitative analysis of the contribution of ICa(L) to cell shortening under these experimental conditions will follow. It has been asserted that it is not possible to draw conclusions about the putative VSRM when its properties have not been reproduced exactly (Ferrier and Howlett, 2001). We reinforce the argument that cell shortening or a calcium transient cannot be ascribed to the hypothesized VSRM unless exclusion of trans sarcolemmal Ca2+ entry is demonstrated.
Acknowledgments
We are grateful to the British Heart Foundation who provided funding by means of a junior research fellowship grant to Dr. Huw Griffiths.
Olaf S. Andersen served as editor.
Footnotes
Abbreviations used in this paper: a.c.v., accessible cell volume; dSEVC, discontinuous single electrode voltage clamp; EC, excitation-contraction; ENCX, equilibrium potential for Na+/Ca2+ exchange; 4AP, 4-aminopyridine; SITS, 4-acetoamido-4′-isothiocyanato-stilbene-2,2′-disulfonic acid; TTX, tetrodotoxin; VSRM, voltage-sensitive release mechanism.
References
- Allen, T.J.A., and R.A. Chapman. 1992. Temperature dependence of L-type calcium currents in single isolated guinea-pig ventricular myocytes. J. Physiol. 446:554P. [Google Scholar]
- Barcenas Ruiz, L., D.J. Beuckelmann, and W.G. Wier. 1987. Sodium-calcium exchange in heart: membrane currents and changes in [Ca2+]i. Science. 238:1720–1722. [DOI] [PubMed] [Google Scholar]
- Barcenas Ruiz, L., and W.G. Wier. 1987. Voltage dependence of intracellular [Ca2+]i transients in guinea pig ventricular myocytes. Circ. Res. 61:148–154. [DOI] [PubMed] [Google Scholar]
- Bassani, J.W., W. Yuan, and D.M. Bers. 1995. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am. J. Physiol. 268:C1313–C1319. [DOI] [PubMed] [Google Scholar]
- Berlin, J.R., M.B. Cannell, and W.J. Lederer. 1987. Regulation of twitch tension in sheep cardiac Purkinje fibers during calcium overload. Am. J. Physiol. 253:H1540–H1547. [DOI] [PubMed] [Google Scholar]
- Bers, D.M. 2001. Excitation-Contraction Coupling and Cardiac Contractile Force. Kluwer Academic Publishers.
- Bers, D.M., D.M. Christensen, and T.X. Nguyen. 1988. Can Ca entry via Na-Ca exchange directly activate cardiac muscle contraction? J. Mol. Cell. Cardiol. 20:405–414. [DOI] [PubMed] [Google Scholar]
- Beuckelmann, D.J., and W.G. Wier. 1988. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. J. Physiol. 405:233–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuckelmann, D.J., and W.G. Wier. 1989. Sodium-calcium exchange in guinea-pig cardiac cells: exchange current and changes in intracellular Ca2+. J. Physiol. 414:499–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard, R.A., R.B. Clark, and W.R. Giles. 1993. Role of sodium-calcium exchange in activation of contraction in rat ventricle. J. Physiol. 472:391–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callewaert, G. 1992. Excitation-contraction coupling in mammalian cardiac cells. Cardiovasc. Res. 26:923–932. [DOI] [PubMed] [Google Scholar]
- Callewaert, G., L. Cleemann, and M. Morad. 1988. Epinephrine enhances Ca2+ current-regulated Ca2+ release and Ca2+ reuptake in rat ventricular myocytes. Proc. Natl. Acad. Sci. USA. 85:2009–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell, M.B., J.R. Berlin, and W.J. Lederer. 1987. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science. 238:1419–1423. [DOI] [PubMed] [Google Scholar]
- Cannell, M.B., H. Cheng, and W.J. Lederer. 1995. The control of calcium release in heart muscle. Science. 268:1045–1049. [DOI] [PubMed] [Google Scholar]
- Cheng, H., W.J. Lederer, and M.B. Cannell. 1993. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 262:740–744. [DOI] [PubMed] [Google Scholar]
- duBell, W.H., W.J. Lederer, and T.B. Rogers. 1996. Dynamic modulation of excitation-contraction coupling by protein phosphatases in rat ventricular myocytes. J. Physiol. 493:793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont, G., J. Pontes, and A. Goldbeter. 1996. Modeling spiral Ca2+ waves in single cardiac cells: role of the spatial heterogeneity created by the nucleus. Am. J. Physiol. 271:C1390–C1399. [DOI] [PubMed] [Google Scholar]
- Egan, T.M., D. Noble, S.J. Noble, T. Powell, A.J. Spindler, and V.W. Twist. 1989. Sodium-calcium exchange during the action potential in guinea- pig ventricular cells. J. Physiol. 411:639–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner, D.A., W.J. Lederer, and R.D. Vaughan Jones. 1983. The control of tonic tension by membrane potential and intracellular sodium activity in the sheep cardiac Purkinje fibre. J. Physiol. 335:723–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato, A. 1983. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245:C1–C14. [DOI] [PubMed] [Google Scholar]
- Ferrier, G.R., and S.E. Howlett. 1995. Contractions in guinea-pig ventricular myocytes triggered by a calcium-release mechanism separate from Na+ and L-currents. J. Physiol. 484:107–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrier, G.R., and S.E. Howlett. 2001. Cardiac excitation-contraction coupling: role of membrane potential in regulation of contraction. Am. J. Physiol. Heart Circ. Physiol. 280:H1928–H1944. [DOI] [PubMed] [Google Scholar]
- Ferrier, G.R., I.M. Redondo, C.A. Mason, C. Mapplebeck, and S.E. Howlett. 2000. Regulation of contraction and relaxation by membrane potential in cardiac ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 278:H1618–H1626. [DOI] [PubMed] [Google Scholar]
- Ferrier, G.R., J.Q. Zhu, I.M. Redondo, and S.E. Howlett. 1998. Role of cAMP-dependent protein kinase A in activation of a voltage-sensitive release mechanism for cardiac contraction in guinea-pig myocytes. J. Physiol. 513:185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles, W.R., and Y. Imaizumi. 1988. Comparison of potassium currents in rabbit atrial and ventricular cells. J. Physiol. 405:123–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hain, J., H. Onoue, M. Mayrleitner, S. Fleischer, and H. Schindler. 1995. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J. Biol. Chem. 270:2074–2081. [DOI] [PubMed] [Google Scholar]
- Han, S., A. Schiefer, and G. Isenberg. 1994. Ca2+ load of guinea-pig ventricular myocytes determines efficacy of brief Ca2+ currents as trigger for Ca2+ release. J. Physiol. 480:411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancox, J.C., and A.J. Levi. 1995. Calcium transients which accompany the activation of sodium current in rat ventricular myocytes at 37°C: a trigger role for reverse Na-Ca exchange activated by membrane potential? Pflugers Arch. 430:887–893. [DOI] [PubMed] [Google Scholar]
- Hiraoka, M., and S. Kawano. 1989. Calcium-sensitive and insensitive transient outward current in rabbit ventricular myocytes. J. Physiol. 410:187–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobai, I.A., J.A. Bates, F.C. Howarth, and A.J. Levi. 1997. a. Inhibition by external Cd2+ of Na/Ca exchange and L-type Ca channel in rabbit ventricular myocytes. Am. J. Physiol. 272:H2164–H2172. [DOI] [PubMed] [Google Scholar]
- Hobai, I.A., J.C. Hancox, and A.J. Levi. 2000. Inhibition by nickel of the L-type Ca channel in guinea-pig ventricular myocytes and effects of internal cAMP. Am. J. Physiol. Heart Circ. Physiol. 279:H692–H701. [DOI] [PubMed] [Google Scholar]
- Hobai, I.A., F.C. Howarth, V.K. Pabbathi, G.R. Dalton, J.C. Hancox, J.Q. Zhu, S.E. Howlett, G.R. Ferrier, and A.J. Levi. 1997. b. “Voltage-activated Ca release” in rabbit, rat and guinea-pig cardiac myocytes, and modulation by internal cAMP. Pflugers Arch. 435:164–173. [DOI] [PubMed] [Google Scholar]
- Howlett, S.E., W. Xiong, C.L. Mapplebeck, and G.R. Ferrier. 1999. Role of voltage-sensitive release mechanism in depression of cardiac contraction in myopathic hamsters. Am. J. Physiol. 277:H1690–H1700. [DOI] [PubMed] [Google Scholar]
- Howlett, S.E., J.Q. Zhu, and G.R. Ferrier. 1998. Contribution of a voltage-sensitive calcium release mechanism to contraction in cardiac ventricular myocytes. Am. J. Physiol. 274:H155–H170. [DOI] [PubMed] [Google Scholar]
- Hussain, M., and C.H. Orchard. 1997. Sarcoplasmic reticulum Ca2+ content, L-type Ca2+ current and the Ca2+ transient in rat myocytes during beta-adrenergic stimulation. J. Physiol. 505:385–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenberg, G., and M.F. Wendt Gallitelli. 1989. Cellular mechanisms of excitation contraction coupling. Isolated Adult Cardiomyocytes. H.M. Piper and G. Isenberg, editors. CRC Press Inc., Boca Raton, Fla. 213–248.
- Janczewski, A.M., H.A. Spurgeon, M.D. Stern, and E.G. Lakatta. 1995. Effects of sarcoplasmic reticulum Ca2+ load on the gain function of Ca2+ release by Ca2+ current in cardiac cells. Am. J. Physiol. 268:H916–H920. [DOI] [PubMed] [Google Scholar]
- Kokubun, S., B. Prod'hom, C. Becker, H. Porzig, and H. Reuter. 1986. Studies on Ca channels in intact cardiac cells: voltage-dependent effects and cooperative interactions of dihydropyridine enantiomers. Mol. Pharmacol. 30:571–584. [PubMed] [Google Scholar]
- Lansman, J.B., P. Hess, and R.W. Tsien. 1986. Blockade of current through single calcium channels by Cd2+, Mg2+, and Ca2+. Voltage and concentration dependence of calcium entry into the pore. J. Gen. Physiol. 88:321–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque, P.C., N. Leblanc, and J.R. Hume. 1994. Release of calcium from guinea pig cardiac sarcoplasmic reticulum induced by sodium-calcium exchange. Cardiovasc. Res. 28:370–378. [DOI] [PubMed] [Google Scholar]
- Levi, A.J., P. Brooksby, and J.C. Hancox. 1993. A role for depolarization induced calcium entry on the Na-Ca exchange in triggering intracellular calcium release and contraction in rat ventricular myocytes. Cardiovasc. Res. 27:1677–1690. [DOI] [PubMed] [Google Scholar]
- Levi, A.J., and J. Issberner. 1996. Effect on the fura-2 transient of rapidly blocking the Ca2+ channel in electrically stimulated rabbit heart cells. J. Physiol. 493:19–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi, A.J., J.S. Mitcheson, and J.C. Hancox. 1996. The effect of internal sodium and caesium on phasic contraction of patch-clamped rabbit ventricular myocytes. J. Physiol. 492:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi, A.J., K.W. Spitzer, O. Kohmoto, and J.H. Bridge. 1994. Depolarization-induced Ca entry via Na-Ca exchange triggers SR release in guinea pig cardiac myocytes. Am. J. Physiol. 266:H1422–H1433. [DOI] [PubMed] [Google Scholar]
- Lokuta, A.-J., T.-B. Rogers, W.-J. Lederer, and H.-H. Valdivia. 1995. Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation-dephosphorylation mechanism. J. Physiol. 487:609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London, B., and J.W. Krueger. 1986. Contraction in voltage-clamped, internally perfused single heart cells. J. Gen. Physiol. 88:475–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod, K.T., and S.E. Harding. 1991. Effects of phorbol ester on contraction, intracellular pH and intracellular Ca2+ in isolated mammalian ventricular myocytes. J. Physiol. 444:481–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason, C.A., and G.R. Ferrier. 1999. Tetracaine can inhibit contractions initiated by a voltage-sensitive release mechanism in guinea-pig ventricular myocytes. J. Physiol. 519:851–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, T.F., S. Pelzer, W. Trautwein, and D.J. Pelzer. 1994. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol. Rev. 74:365–507. [DOI] [PubMed] [Google Scholar]
- Mitchell, M.R., T. Powell, D.A. Terrar, and V.W. Twist. 1987. Electrical activity and contraction in cells isolated from rat and guinea-pig ventricular muscle: a comparative study. J. Physiol. 391:527–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss, H.B., and S.R. Houser. 1992. Sodium-calcium exchange-mediated contractions in feline ventricular myocytes. Am. J. Physiol. 263:H1161–H1169. [DOI] [PubMed] [Google Scholar]
- Okazaki, O., N. Suda, K. Hongo, M. Konishi, and S. Kurihara. 1990. Modulation of Ca2+ transients and contractile properties by beta- adrenoceptor stimulation in ferret ventricular muscles. J. Physiol. 423:221–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard, C.H., D.A. Eisner, and D.G. Allen. 1983. Oscillations of intracellular Ca2+ in mammalian cardiac muscle. Nature. 304:735–738. [DOI] [PubMed] [Google Scholar]
- Perchenet, L., A.K. Hinde, K.C. Patel, J.C. Hancox, and A.J. Levi. 2000. Stimulation of Na/Ca exchange by the beta-adrenergic/protein kinase A pathway in guinea-pig ventricular myocytes at 37 degrees. Pflugers Arch. 439:822–828. [DOI] [PubMed] [Google Scholar]
- Piacentino, V., III, K. Dipla, J.P. Gaughan, and S.R. Houser. 2000. Voltage-dependent Ca release from the SR of feline ventricular myocytes is explained by Ca-induced Ca release. J. Physiol. 523:533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham, J.S., L.R. Jones, and M. Morad. 1991. Phospholamban mediates the beta-adrenergic-enhanced Ca2+ uptake in mammalian ventricular myocytes. Am. J. Physiol. 261:H1344–H1349. [DOI] [PubMed] [Google Scholar]
- Sham, J.S., L. Cleemann, and M. Morad. 1992. Gating of the cardiac Ca2+ release channel: the role of Na+ current and Na+-Ca2+ exchange. Science. 255:850–853. [DOI] [PubMed] [Google Scholar]
- Shannon, T.R., K.S. Ginsburg, and D.M. Bers. 2000. Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophys. J. 78:334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido, K.R., E. Carmeliet, and F. Van de Werf. 1998. T-type Ca2+ current as a trigger for Ca2+ release from the sarcoplasmic reticulum in guinea-pig ventricular myocytes. J. Physiol. 508:439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido, K.R., P.G.A. Volders, G. Antoons, and M. Vos. 2000. L-type Ca2+ currents at negative potentials during β-adrenergic stimulation. Biophys. J. 78:371A. [Google Scholar]
- Sjaastad, I., J.A. Birkeland, G.R. Ferrier, S.E. Howlett, J.A. Wasserstrom, and O.M. Sejersted. 2000. Rats with congestive heart failure exhibit a defect in excitation-contraction coupling caused by suppression of the voltage sensitive release mechanism. Circulation. 102:297. [Google Scholar]
- Spencer, C.I., and J.R. Berlin. 1995. Control of sarcoplasmic reticulum calcium release during calcium loading in isolated rat ventricular myocytes. J. Physiol. 488:267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steadman, B.W., K.B. Moore, K.W. Spitzer, and J.H. Bridge. 1988. A video system for measuring motion in contracting heart cells. IEEE Trans. Biomed. Eng. 35:264–272. [DOI] [PubMed] [Google Scholar]
- Stern, M.D. 1992. Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 63:497–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talo, A., M.D. Stern, H.A. Spurgeon, G. Isenberg, and E.G. Lakatta. 1990. Sustained subthreshold-for-twitch depolarization in rat single ventricular myocytes causes sustained calcium channel activation and sarcoplasmic reticulum calcium release. J. Gen. Physiol. 96:1085–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terracciano, C.M., K.D. Philipson, and K.T. MacLeod. 2001. Overexpression of the Na+/Ca2+ exchanger and inhibition of the sarcoplasmic reticulum Ca2+-ATPase in ventricular myocytes from transgenic mice. Cardiovasc. Res. 49:38–47. [DOI] [PubMed] [Google Scholar]
- Tiaho, F., J. Nargeot, and S. Richard. 1991. Voltage-dependent regulation of L-type cardiac Ca channels by isoproterenol. Pflugers Arch. 419:596–602. [DOI] [PubMed] [Google Scholar]
- Trafford, A.W., and D.A. Eisner. 1998. Another trigger for the heartbeat. J. Physiol. 513:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trafford, A.W., S.C. O'Neill, and D.A. Eisner. 1993. Factors affecting the propagation of locally activated systolic Ca transients in rat ventricular myocytes. Pflugers Arch. 425:181–183. [DOI] [PubMed] [Google Scholar]
- Van Wagoner, D.R., M. Kirian, B. Arakaki, and M. Lamorgese. 1999. Voltage sensitive calcium release in human atrial myocytes. Biophys. J. 76:A459. [Google Scholar]
- Vassort, G., and J. Alvarez. 1994. Cardiac T-type calcium current: pharmacology and roles in cardiac tissues. J. Cardiovasc. Electrophysiol. 5:376–393. [DOI] [PubMed] [Google Scholar]
- Vornanen, M., N. Shepherd, and G. Isenberg. 1994. Tension-voltage relations of single myocytes reflect Ca release triggered by Na/Ca exchange at 35°C but not 23°C. Am. J. Physiol. 267:C623–C632. [DOI] [PubMed] [Google Scholar]
- Wasserstrom, J.A., and A.M. Vites. 1996. The role of Na+-Ca2+ exchange in activation of excitation- contraction coupling in rat ventricular myocytes. J. Physiol. 493:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watano, T., J. Kimura, T. Morita, and H. Nakanishi. 1996. A novel antagonist, No. 7943, of the Na+/Ca2+ exchange current in guinea-pig cardiac ventricular cells. Br. J. Pharmacol. 119:555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wier, W.G., and C.W. Balke. 1999. Ca2+ release mechanisms, Ca2+ sparks, and local control of excitation-contraction coupling in normal heart muscle. Circ. Res. 85:770–776. [DOI] [PubMed] [Google Scholar]
- Wier, W.G., M.B. Cannell, J.R. Berlin, E. Marban, and W.J. Lederer. 1987. Cellular and subcellular heterogeneity of [Ca2+]i in single heart cells revealed by fura-2. Science. 235:325–328. [DOI] [PubMed] [Google Scholar]
- Wier, W.G., T.M. Egan, J.R. Lopez Lopez, and C.W. Balke. 1994. Local control of excitation-contraction coupling in rat heart cells. J. Physiol. 474:463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, J.Q., and G.R. Ferrier. 2000. Regulation of a voltage-sensitive release mechanism by Ca2+-calmodulin dependent kinase in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 279:H2104–H2115. [DOI] [PubMed] [Google Scholar]