

Abstract
TRPV1 ion channels mediate the response to painful heat, extracellular acidosis, and capsaicin, the pungent extract from plants in the Capsicum family (hot chili peppers) (Szallasi, A., and P.M. Blumberg. 1999. Pharmacol. Rev. 51:159–212; Caterina, M.J., and D. Julius. 2001. Annu. Rev. Neurosci. 24:487–517). The convergence of these stimuli on TRPV1 channels expressed in peripheral sensory nerves underlies the common perceptual experience of pain due to hot temperatures, tissue damage and exposure to capsaicin. TRPV1 channels are nonselective cation channels (Caterina, M.J., M.A. Schumacher, M. Tominaga, T.A. Rosen, J.D. Levine, and D. Julius. 1997. Nature. 389:816–824). When activated, they produce depolarization through the influx of Na+, but their high Ca2+ permeability is also important for mediating the response to pain. In particular, Ca2+ influx is thought to be required for the desensitization to painful sensations over time (Cholewinski, A., G.M. Burgess, and S. Bevan. 1993. Neuroscience. 55:1015–1023; Koplas, P.A., R.L. Rosenberg, and G.S. Oxford. 1997. J. Neurosci. 17:3525–3537). Here we show that in inside-out excised patches from TRPV1 expressed in Xenopus oocytes and HEK 293 cells, Ca2+/calmodulin decreased the capsaicin-activated current. This inhibition was not mimicked by Mg2+, reflected a decrease in open probability, and was slowly reversible. Furthermore, increasing the calmodulin concentration in our patches by coexpression of wild-type calmodulin with TRPV1 produced inhibition by Ca2+ alone. In contrast, patches excised from cells coexpressing TRPV1 with a mutant calmodulin did not respond to Ca2+. Using an in vitro calmodulin-binding assay, we found that TRPV1 in oocyte lysates bound calmodulin, although in a Ca2+-independent manner. Experiments with GST-fusion proteins corresponding to regions of the channel NH2-terminal domain demonstrated that a stretch of ∼30 amino acids adjacent to the first ankyrin repeat bound calmodulin in a Ca2+-dependent manner. The physiological response to pain involves an influx of Ca2+ through TRPV1. Our results indicate that this Ca2+ influx may feed back on the channels, inhibiting their gating. This type of feedback inhibition could play a role in the desensitization produced by capsaicin.
Keywords: desensitization, capsaicin, TRPV1, calcium, calmodulin
INTRODUCTION
The first TRP channel was identified in 1977 as a phototransduction mutant in Drosophila (Minke, 1977). Since that time the TRP channel family has grown to include at least 20 members (Clapham et al., 2001). The importance of TRP channels is demonstrated by the wide variety of their proposed functions: invertebrate phototransduction, responding to painful stimuli, responding to moderate temperature changes; repletion of intracellular calcium stores, receptor–mediated excitation, and modulation of the cell cycle (for reviews see Clapham et al., 2001; Montell, 2001; Montell et al., 2002).
Despite the clear physiological importance of TRP channels, little is known about what regulates their function. One type of TRP channel, TRPV1, is a polymodal receptor that integrates a number of painful stimuli. These stimuli include: noxious heat, with a threshold of ∼42°C; extracellular acidification, with a pKa of ∼5.3 (Caterina et al., 1997; Jordt et al., 2000); anandamide and other arachidonic acid metabolites (Smart et al., 2000); and capsaicin, a pungent extract from plants in the Capsicum family (Szallasi and Blumberg, 1999; Caterina and Julius, 2001). Although all the details of the capsaicin binding site are not known, it has been shown to bind from the intracellular side (Jung et al., 1999), though this result is controversial (Vyklicky et al., 2003).
With intracellular NH2- and COOH-terminal domains, six putative membrane-spanning domains, and sequence similarity in the pore region, TRP channels fall within the superfamily of ion channels that includes voltage-gated Na+, K+, and Ca2+ channels and cyclic nucleotide–gated (CNG) channels (Jan and Jan, 1990, 1992). For those ion channels within this superfamily for which stoichiometry has been directly examined, all have been shown to be composed of four six transmembrane domain subunits or pseudosubunits, with auxiliary subunits sometimes present as well (Clapham et al., 2001).
TRPV1 channels are nonselective cation channels that do not discriminate among monovalent cations and are highly permeable to Ca2+ (Oh et al., 1996). Ca2+ is especially important for TRPV1 function, as extracellular Ca2+ mediates desensitization (Koplas et al., 1997), a process which enables a neuron to adapt to specific stimuli by diminishing its overall response to a sustained chemical or physical signal. Desensitization of TRPV1 ion channels is composed of at least two separate cellular mechanisms. One type of desensitization has been shown to depend on an increase in intracellular Ca2+ (Cholewinski et al., 1993; Docherty et al., 1996a; Liu and Simon, 1996; Koplas et al., 1997) and accounts for most of the acute (seconds) desensitization. There is also a slower (minutes) process of desensitization that can occur in the absence of extracellular Ca2+ (Docherty et al., 1996a; Liu and Simon, 1996; Koplas et al., 1997). There is good evidence that the rate at which acute Ca2+-dependent desensitization develops is increased by dephosphorylation of the ion channels by the Ca2+ and calmodulin-dependent enzyme calcineurin (Docherty et al., 1996b).
Although it is known that desensitization of the TRPV1 channel depends on the presence of intracellular Ca2+, the molecular mechanism of this regulation has remained elusive. It has recently been shown that a 35–amino acid region in the COOH-terminal region of TRPV1 (amino acids 767–801) binds calmodulin (CaM) in an in vitro assay in a Ca2+-dependent manner (Numazaki et al., 2003). Mutant channels in which this region was deleted continued to show desensitization in whole-cell experiments, albeit with altered kinetics. We directly addressed whether CaM might act as a Ca2+ sensor, transducing changes in Ca2+ concentration into changes in channel behavior. We found that application of Ca2+ alone to the intracellular surface of excised patches produced a small decrease in capsaicin-activated current that could not be mimicked by Mg2+. Application of Ca2+ with CaM produced a much larger reduction in current. Overexpression of CaM also potentiated the inhibitory effect of Ca2+, whereas coexpression of a mutant CaM with TRPV1 did not produce a Ca2+ mediated inhibition. Using GST-fusion proteins corresponding to regions of the TRPV1 NH2 terminus, we localized a CaM binding site to a region including amino acids 189–222. CaM binding to this NH2-terminal region was highly Ca2+ dependent. However, in vitro binding assays using full-length channels from cell lysates suggests a second, Ca2+-independent binding site may be present as well. Our data indicate that CaM can mediate Ca2+-dependent inhibition of capsaicin-activated currents, with the NH2-terminal region as a candidate binding site mediating this inhibitory action of Ca2+/CaM.
MATERIALS AND METHODS
Oocyte Electrophysiological Recordings
Rat TRPV1 (a gift from Dr. D. Julius) was subcloned into either pGEMHE, a gift of Dr. E.R. Liman, or pCDNA3 modified to contain the 5′ and 3′ untranslated regions of the Xenopus β-globin genes. A FLAG epitope tag was appended to the COOH-terminal end of the channel and did not affect measured functional properties (unpublished data). Rat wild-type calmodulin and mutant calmodulin1,2,3,4 in pGEMHE were a gift from Dr. J. Adelman. Inside-out patch clamp recordings were made using symmetrical solutions consisting of 130 mM NaCl, 3 mM HEPES (pH 7.2), and 200 μM EDTA for Ca2+-free conditions. For solutions containing Ca2+, EDTA was replaced with the given concentrations of Ca2+, and the free Ca2+ concentrations were confirmed using a Ca2+ electrode (Qwick-Tip; World Precision Instruments). The measured free Ca2+ concentration in our bath (before addition of Ca2+) was 64 nM. We originally used MaxChelator (http://www.stanford.edu/~cpatton/maxc.html) to determine appropriate buffer and Ca2+ concentrations for our experiments. Two lines of evidence suggested that this method did not appropriately control the Ca2+ concentration with either EGTA or HEDTA. First, the efficacy of Ca2+ in modulating channel function varied from one batch of solutions to the next. Second, the free Ca2+ concentration measured with the Ca2+ electrode similarly varied between batches of solution. Surprisingly, using unbuffered solution we observed consistent readings of both the free Ca2+ with the Ca2+ electrode (within a factor of two or less), as well as inhibition of the channels in the presence of calmodulin.
Calmodulin (Calbiochem) stocks were prepared in water and dilutions were made to obtain a final concentration of 500 nM. All solutions were applied to the intracellular surface of the patches with an RSC-200 rapid solution changer (Molecular Kinetics). Capsaicin (Sigma-Aldrich) stocks were prepared in ethanol and diluted to final concentrations of 4 or 20 μM, both saturating concentrations.
Currents were low pass-filtered at 2 kHz with an Axopatch 200B amplifier (Axon Instruments, Inc.) and sampled at 10 kHz. Data were acquired and analyzed with PULSE data acquisition software (HEKA, Inc.) and were plotted and fitted using Igor Pro (Wavemetrics, Inc.). For macroscopic recordings, the following voltage protocol was used. Patches were held at 0 mV and given a prepulse of −120 mV for 30 ms. The voltage was then stepped to voltages ranging from −150 to 110 mV in steps of 20 mV, for 60 ms. Currents in the absence of capsaicin were subtracted. For single-channel experiments, patches were held at 0 mV and stepped to +60 mV for 500 ms. In these experiments open probability (Po) was calculated from fits to the data with two Gaussians. The area under the curve representing open channels relative to the total area was taken as Po. Another way of estimating Po for each experimental condition is to measure the instantaneous tail currents. Tail currents were measured using a protocol in which the patch was stepped to −120 mV for 10 ms, and then from −150 to 110 mV for 60 ms in increments of 20 mV, and then finally to −120 mV for 10 ms again to measure the tail currents. We assumed that I T(V) = Po(V) · N · i(V), where I T(V) is the tail currents at a voltage V, Po(V) is the open probability as a function of voltage, N is the number of channels, and i(V) is the unitary conductance at V. In this case, the values obtained from the tail currents are proportional to Po since N and i(V) did not vary (as seen from our single channel experiments). Student's t test (two-tailed, unpaired) was used as a statistical test in all cases where significance is discussed, with the threshold of significance set as P < 0.05.
Mammalian Cell Culture and Recording
HEK 293 cells expressing large T antigen were transfected with TRPV1-pCDNA3 and pIRES-GFP (BD Biosciences; to visualize successfully transfected cells) with Fugene (Roche Diagnostics) according to manufacturer's instructions. Two days after transfection had been initiated, cells were passaged onto coverslips. Cells were used for recording three days after passaging. For coexpression with wild-type or mutant CaM, HEK 293 cells were transfected as described above using TRPV1 and either cYFP-CaM in pCDNA3 or CaM1,2,3,4 in pCDNA3 (both gifts of Dr. D. Yue) and pIRES-GFP. Recording solutions consisted of 130 mM NaCl and 3 mM HEPES (pH 7.2) either without added Ca2+ or with the addition of the stated concentrations of Ca2+. In every case, the free Ca2+ concentration was confirmed using a Ca2+ electrode as described above.
CaM Pull-down Assay
Oocytes were prepared as described (Rho et al., 2000; Rosenbaum et al., 2002). 2 mM Ca2+ or 2 mM EGTA were added to the lysis buffer as indicated in the text and figures. Cell lysates were incubated at 4°C overnight with 50 μl of a 50% CaM-agarose suspension (Sigma-Aldrich). The material was spun and washed with 1 ml of frog Ringer's solution with either 2 mM Ca2+ or 2 mM EGTA. After 15 washes, protein adhering to the beads was eluted with SDS sample buffer (with β-mercaptoethanol, final concentration of 0.71 M) and run on a Nupage 3–8% Tris-Acetate gel (Invitrogen). Western blots were performed as described previously (Rosenbaum et al., 2002) with anti-FLAG antibody (Sigma-Aldrich). Densitometry was performed using FlourChem software (Alpha Innotech) directly from chemiluminescent blots.
For the CaM pulldown assay with GST-fusion proteins, the fusion proteins purified from bacteria (see below) were incubated with CaM-agarose beads overnight in the presence of either 2 mM Ca2+ or 2 mM EGTA. The beads were then washed five times with 1 ml of the lysis buffer described above (Rho et al., 2000; Rosenbaum et al., 2002), also supplemented with either Ca2+ or EGTA. The proteins were then eluted using SDS sample buffer (with β-mercaptoethanol, final concentration of 0.71 M). Western blots for these experiments were done using either anti-GST or anti-FLAG antibodies (Santa Cruz Biotechnology, Inc.).
GST Fusion Protein Expression and Purification
Fragments of the NH2-terminal region of TRPV1 (accession no. AF029310) were amplified using PCR primers with EcoRI and XhoI restriction sites with the addition of a FLAG epitope to the COOH terminus of some of the constructs, and cloned into the multicloning site of pGEX-6P-1. BL21-competent cells were transformed with the GST-TRPV1 clones. 250 ml of LB with ampicillin (50 mg/L) were inoculated 1:10 with an overnight culture of bacteria and incubated at 37°C until OD600 ∼0.4. The cells were induced with 0.1 mM IPTG for 1.5 h, sonicated in 5 ml of PBS with protease inhibitors for 60 s and Triton X-100 was added to reach a final concentration of 0.1%. This mixture was then agitated at 4°C for 30 min. The proteins were purified using a batch method by incubation with 800 μl of a 50% GSH-agarose slurry for 30 min at room temperature, and then washed extensively with PBS. The proteins were eluted with 50 mM Tris pH 8, 15 mM GSH, 0.1% Triton X-100 for 30 min. For some experiments, a final concentration of 1 mM DTT was used in all steps starting with sonication.
RESULTS
To investigate the molecular basis for Ca2+ regulation of TRPV1 activity, we expressed TRPV1 channels in Xenopus oocytes and studied their activity using the inside-out configuration of the patch-clamp technique. This approach allowed us to control the solution bathing the intracellular surface of the channels, so that the capsaicin concentration activating the channels and the Ca2+concentration at the intracellular surface could be controlled independently. Activation of TRPV1, and the subsequent entry of extracellular Ca2+ through the channels into cells, is believed to initiate a negative feedback loop that results in decreased TRPV1 current. This negative feedback is known as desensitization. We hypothesized that CaM may act as a Ca2+ sensor for TRPV1 channels, modulating channel activity in response to increases in intracellular Ca2+ concentration. To test whether Ca2+/CaM could alter TRPV1 activity in our patches, we applied purified CaM along with 50 μM free Ca2+ to the inside surface of the patches. We found that application of Ca2+/CaM produced a large decrease in the capsaicin-activated current (57.0% ± 9.7%, n = 8; Figs. 1 and 3 A). This decrease did not occur with CaM alone (Fig. 1 A), was rather slow, and reversed over several minutes in the absence of Ca2+ (Fig. 1 C).
Figure 1.

CaM mediates Ca2+ inhibition of TRPV1 channels expressed in Xenopus oocytes. (A) Current families obtained in the presence of 20 μM capsaicin, 20 μM capsaicin + 500 nM CaM + 50 μM free Ca2+, 20 μM capsaicin + 500 nM CaM, or 20 μM capsaicin + 50 μM free Ca2+, as indicated by the labels. (B) IV relation for currents shown in (A). Circles, capsaicin; squares, capsaicin + Ca2+; and triangles, capsaicin + Ca2+/CaM. (C) Time course of inhibition by and recovery from Ca2+/CaM. The intracellular (bath) solution contained 20 μM capsaicin, 50 μM free Ca2+, and/or 500 nM CaM, as indicated by the bars above the graph. The bar labels correspond to the bar immediately beneath each label.
Figure 3.

Coexpression of wild-type and mutant CaM with TRPV1 alters the effects of Ca2+ on channel function in oocytes and HEK cells. (A) Box-plot of the percent decrease of capsaicin-activated (20 μM) current in Xenopus oocytes produced by 50 μM free Ca2+ and no added CaM (left, n = 8), 500 nM added CaM, (center, n = 8), or CaMwt coexpressed with TRPV1 and no added CaM (right, n = 4). For the boxes, the line indicates the median of the data, the box surrounds the 25th through 75th percentile of the data, and the whiskers reach the 10th and 90th percentiles. As indicated by the asterisks, both the center and right groups had statistically significant differences compared with Ca2+ and no added CaM. (B) Box-plot of the percent decrease of capsaicin-activated (20 μM) current in HEK cells produced by 50 μM free Ca2+ and either no added CaM (first box, n = 7), 500 nM added CaM (second box, n = 3), CaMwt coexpressed with TRPV1 and no added CaM (third box, n = 6), or CaM1,2,3,4v. coexpressed with TRPV1 and no added CaM (fourth box, n = 6). As indicated by the asterisks, both the center groups had statistically significant differences compared with Ca2+ and no added CaM. For the fourth condition, p exactly equalled 0.05, and therefore did not meet our criterion for significance. (C) Effects of CaMwt and CaM1,2,3,4 overexpression in HEK cells on the response to Ca2+ (50 μM). Open circles represent the data from coexpression of TRPV1 and CaMwt and filled circles represent data from coexpression of TRPV1 and CaM1,2,3,4. Data were obtained from pulses to +60 mV every 3 s. Ca2+ was introduced at time t = 0. (D) Dose–response to different Ca2+ concentrations in the presence of exogenous CaM. Data were obtained at a voltage of +60 mV and are normalized to the current obtained in the presence of each Ca2+ concentration with CaM. Data are the mean values from three patches. The smooth curve is a fit with the Hill equation, giving an IC50 for inhibition by Ca2+ in the presence of CaM of 60 μM and a Hill slope of 1.25.
The application of 50 μM free Ca2+ without CaM to the intracellular surface of patches produced a small decrease in the capsaicin-activated current (Figs. 1 and 3 A). The mean decrease with 50 μM free Ca2+ was 7.4% (±1.7%, SEM, n = 8). Could this small decrease in current have been due to endogenous CaM? For this to be the case, CaM must remain associated with the patches even after excision of the membrane from the cell, so that the addition of Ca2+ would produce a Ca2+/CaM complex that could regulate channel function. This might prove to be a very inefficient process, giving a relatively low CaM concentration in our patches and producing only the small reduction of current observed
It has recently been reported that, in whole-cell experiments, Ca2+/CaM did not appear to modulate TRPV1 channels expressed in HEK 293 cells (Numazaki et al., 2003). It was found that including CaM inhibitors in the pipette and coexpression of mutant CaM did not alter desensitization. We wondered if Ca2+/CaM regulation of TRPV1 might have some cell specificity that would indicate the requirement for the presence of an ancillary factor. If this were the case, then the Ca2+/CaM effect we observe might be an artifact of the oocyte expression system. To examine this question, we expressed TRPV1 in HEK 293 cells and tested whether Ca2+/CaM would inhibit capsaicin-activated currents. As shown in Figs. 2 and 3 B, we found that Ca2+/CaM produced inhibition of capsaicin-activated currents that was comparable in magnitude (Fig. 3 B) and kinetics (Fig. 2 C) to that observed with channels expressed in Xenopus oocytes. The current decrease produced by Ca2+ and Ca2+/CaM, respectively, were 5% (± 2%, n = 3) and 43% (± 3%, n = 3). Thus, it does not seem likely that a difference in the complement of cellular proteins/factors underlies the difference between our data and that previously reported (Numazaki et al., 2003).
Figure 2.

Ca2+/CaM inhibits TRPV1 channels expressed in HEK 293 cells. (A) Current families activated by 20 μM capsaicin, capsaicin + 50 μM free Ca2+, or capsaicin + Ca2+ + 500 nM CaM, as indicated. The current family labeled “wash” was recorded in capsaicin after Ca2+/CaM had been extensively washed from the patch. (B) IV relations from the current families shown in A. Circles, capsaicin; squares, capsaicin + Ca2+; and triangles, capsaicin + Ca2+/CaM. (C) Kinetics of onset and recovery from Ca2+/CaM regulation. The presence of capsaicin, Ca2+, and CaM were as indicated by the labeled bars.
The ability of purified CaM to modulate TRPV1 function does not directly address whether the small reduction in current observed with Ca2+ alone was mediated by CaM. As discussed above, for this to be the case, CaM would have to remain associated with the patches after their excision from the cell. To determine whether we could manipulate the response to Ca2+ in patches by manipulating expression levels of CaM, we coexpressed CaMwt with TRPV1 in both Xenopus oocytes and HEK 293 cells. If CaM can be retained in cell-free excised patches, then increasing CaM levels through overexpression should lead to an increased response to Ca2+. We found that in cells overexpressing CaMwt and TRPV1 application of Ca2+ alone produced a large reduction in current in both oocytes (Fig. 3 A; 43.5% ± 11.8%, n = 4) and HEK cells (Fig. 3, B and C; 61.9% ± 6.6%, n = 6). This current reduction was statistically significant compared with application of Ca2+ onto patches from cells expressing only TRPV1. Thus, increasing the expression level of CaM facilitated the Ca2+-induced inhibition of TRPV1 channels. The simplest explanation of this phenomenon is that CaM remains associated with the patches even after excision, and that it can then modulate TRPV1 function when Ca2+ is added.
From these experiments we can conclude that CaM remains associated with the patches upon excision, but not whether CaM is associated with the channels themselves. If we could potentiate the effect of Ca2+ by overexpressing wild-type CaM, then we should be able to attenuate or eliminate the effect of Ca2+ by overexpressing a nonfunctional CaM mutant. The idea here is that the mutant CaM ought to compete with wild-type CaM for association with the channels, but that it would not be able to transduce the changes in Ca2+ concentration into changes in channel function. To test this prediction, we cotransfected HEK 293 cells with TRPV1 and CaM1,2,3,4, a mutant in which all four Ca2+-binding sites have been crippled. When Ca2+ was applied to patches from these cells, the current was not reduced (Fig. 3, B and C). In fact, we observed a slight increase in current upon application of Ca2+ to these channels (Fig. 3, B and C). The simplest interpretation of these results is that CaM1,2,3,4 competes with endogenous CaM for association with the channels. These data, together with the biochemical data discussed below, suggest that CaM can remain associated with the channels in the absence of Ca2+.
The intracellular concentration of Ca2+ varies not only with the activation state of the cell, but also among compartments within the same cell. Furthermore, the apparent affinity of CaM for Ca2+ depends on the target protein with which Ca2+/CaM interacts. To determine the Ca2+ sensitivity of CaM inhibition of the TRPV1, we measured a dose–response relation for inhibition of the channels by different Ca2+ concentrations, in the presence of a fixed CaM concentration. As shown in Fig. 3 D, we found that the IC50 for inhibition by Ca2+ in the presence of 500 nM CaM was 60 μM. This is well within the range of Ca2+ concentrations expected near these Ca2+-permeable channels.
The outward rectification of TRPV1 channels arises from voltage dependence of both gating and unitary conductance (Caterina et al., 1997; but see also Hui et al., 2003). To determine which of these properties is affected by Ca2+/CaM, we examined the effect of Ca2+ on patches containing only a single TRPV1 channel. As shown in Fig. 4 A, Ca2+ decreased the channel open probability, but produced little or no change in unitary conductance. These data indicate that Ca2+ altered TRPV1 function by biasing gating toward the closed state. We next examined this question in a second way, looking at tail currents following steps to a range of voltage from −150 to +110 in steps of 20 mV. By examining instantaneous tail currents we could thus get a measure of the relative open probability elicited by the voltage immediately preceding the tail. Fig. 4 B shows an example of a typical current family measured using our tail current protocol, along with normalized tail currents (Fig. 4 C) under the following conditions: capsaicin only (circles), capsaicin + Ca2+ (squares), and capsaicin + Ca2+/CaM (triangles). The solid curves shown are fits to the data with the Boltzmann equation. Together with the absence of an effect of Ca2+/CaM on unitary conductance and channel number in single-channel experiments (Fig. 4 A), these data indicate that the primary effect of Ca2+/CaM on channel function is to decrease open probability.
Figure 4.

Ca2+/CaM inhibition of TRPV1 reflects a decrease in channel open probability. (A) Currents from a patch containing a single TRPV1 channel in the absence of both capsaicin and Ca2+ (top), in the presence of 4 μM capsaicin but no Ca2+ (middle), and in the presence of 4 μM capsaicin and 300 nM free Ca2+ (bottom). Consecutive traces at +60 mV are shown. All-points histograms were made from all data collected under each condition (2 s without capsaicin, 12 s with capsaicin in the absence of Ca2+, and 6.1 s for capsaicin plus Ca2+). Open probabilities were taken from the area under the Gaussian representing open channels relative to the sum of the area under the Gaussians representing the closed and open channels, and were as follows: capsaicin alone = 0.99, capsaicin + Ca2+ = 0.72. For this experiment, the channels were expressed in Xenopus oocytes. (B) Currents in response to steps from a holding potential of −120 mV to between −150 mV and +110 mV, in steps of 20 mV. After the voltage steps the potential was returned to −120 mV. Currents activated by 20 μM capsaicin (top), capsaicin + 50 μM free Ca2+ (middle), or capsaicin + 50 μM free Ca2+ and 500 nM CaM (bottom). (C) Instantaneous currents measured upon the return of the potential to −120 mV plotted versus the step potential preceding this repolarization. Currents activated by 20 μM capsaicin (circles), capsaicin + 50 μM free Ca2+ (squares), or capsaicin + 50 μM free Ca2+ and 500 nM CaM (triangles). The currents were normalized to the maximum current activated by capsaicin (without Ca2+ or CaM). Solid curves are fits with the Boltzmann equation with the following parameters: capsaicin: z = 0.41e0 (elemental charge), V1/2 = −122 mV; Ca2+: z = 0.31e0, V1/2 = −109 mV; Ca2+/CaM: z = 0.2e0, V1/2 = +40 mV.
Fast (but not slow) Ca2+ chelators eliminate desensitization in whole-cell experiments (Piper et al., 1999), suggesting that the Ca2+ sensor is either part of, or is tightly associated with, TRPV1. To further investigate whether CaM fits this criterion, we examined whether CaM immobilized on agarose beads could bind TRPV1. Cell lysates from TRPV1-expressing oocytes were incubated with CaM-agarose beads and then extensively washed. As shown in Fig. 5 A, the channels remained on the CaM beads and could be subsequently eluted with denaturing sample buffer. The channels did not interact with either naked sepharose beads or with GSH immobilized on agarose beads (Fig. 5 B). We next asked whether the interaction between TRPV1 and CaM in the pull-down assay was Ca2+ dependent. We found that the fraction of TRPV1 that bound CaM was the same in the presence of 2 mM Ca2+ and 2 mM EGTA with no added Ca2+ (Fig. 5 C). The Ca2+-independent association between CaM and TRPV1 is consistent with the association between CaM and TRPV1 we observed in cell-free excised patches, and also consistent with the ability of CaM1,2,3,4 to compete with endogenous wild-type CaM for association with the channels.
Figure 5.
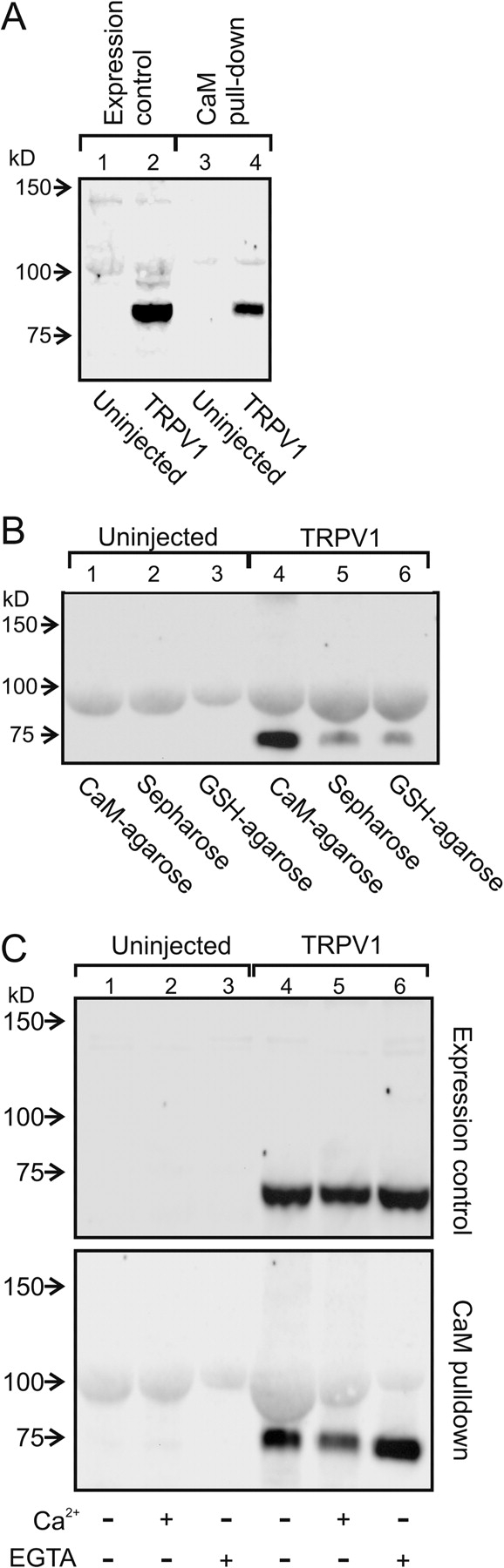
TRPV1 channels interact directly with CaM. (A) TRPV1 binds to CaM immobilized on agarose beads. Lanes 1 and 2 represent the input and lanes 3 and 4 represent the protein eluted from the CaM-agarose beads. Lanes 1 and 3 were from uninjected oocytes and lanes 2 and 4 were from oocytes injected with TRPV1 cRNA. (B) TRPV1 does not interact with beads alone. This control demonstrates that the interaction of TRPV1 and CaM is specific. Lanes 1–3 are from uninjected oocytes and lanes 4–6 are from oocytes injected with TRPV1 cRNA. The beads used in each case were as indicated in the figure. (C) The top panel represents inputs and the lower panel represents protein eluted from the CaM beads. Lanes 1–3 are from uninjected oocytes and lanes 4–6 are from oocytes injected with TRPV1 cRNA. Added Ca2+ and EGTA were as indicated.
TRPV1 channels span the membrane six times and have extensive intracellular NH2- and COOH-terminal regions. We hypothesized that (a) the interaction between CaM and TRPV1 was direct, i.e., not mediated by another protein, and (b) the NH2-terminal region might be involved in CaM binding. We therefore made seven constructs corresponding to overlapping segments of the TRPV1 NH2-terminal domain (Fig. 6 A) and expressed them in bacteria as GST-fusion proteins. We purified the GST-fusion proteins, and examined whether (a) they bound to CaM, and (b) binding to CaM was Ca2+ dependent. The input (total protein added to each experiment), fusion protein bound in the presence of Ca2+, and fusion protein bound in the presence of EGTA are shown for each of these constructs in Fig. 6 B. For two of these fusion proteins, encoding amino acids 149–222 and 189–250, more than half the input was retained by binding CaM. A third protein, consisting of amino acids 247–334, bound more weakly. For all three of these constructs, binding was strongly Ca2+ dependent, with 50–100 times as much protein binding in the presence of 2 mM Ca2+ compared with 2 mM EGTA and no added Ca2+.
Figure 6.
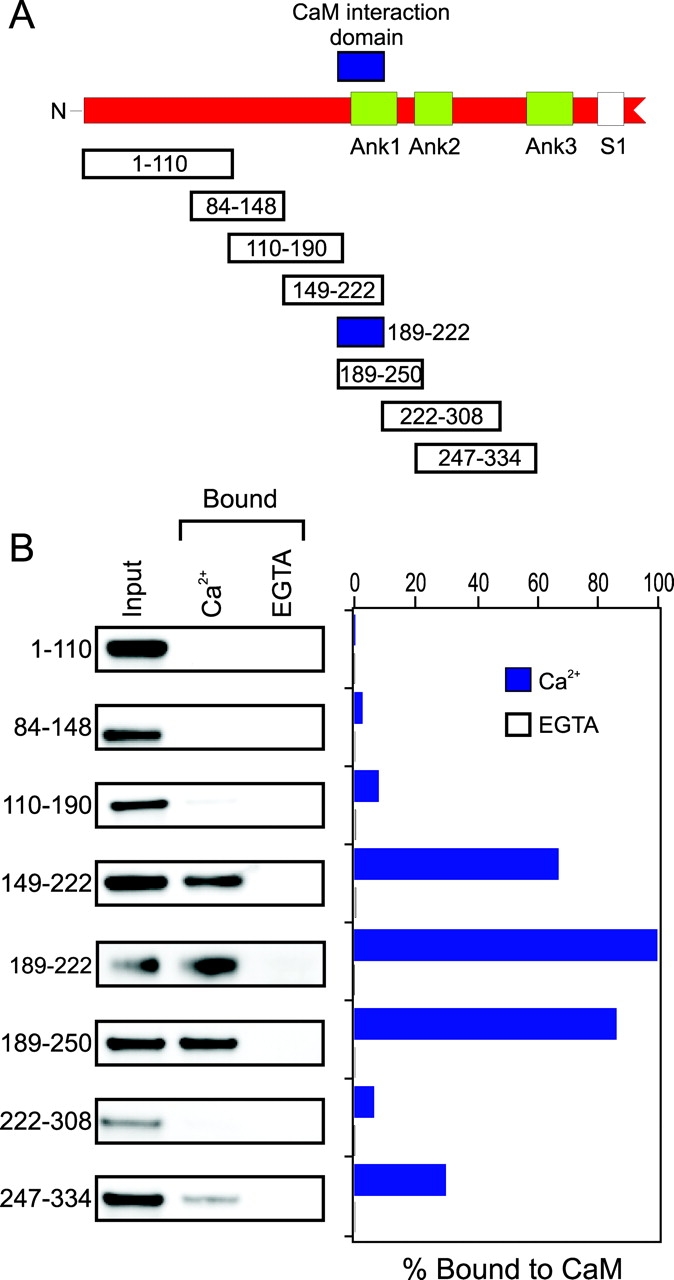
The TRPV1 NH2-terminal region interacts directly with CaM in a Ca2+-dependent manner. (A) Cartoon diagram of TRPV1 NH2-terminal region (not to scale). The three Ankyrin repeats are shown in green (Ank1–3), the region required for interaction with CaM is shown in blue, and the first transmembrane domain (S1) is shown in white. GST-TRPV1 fusion proteins designated below, aligned in-scale to their corresponding sequence in the NH2 terminus. The numbers within each box refer to the amino acid numbers from TRPV1 encoded in each GST-fusion protein. (B) In vitro CaM binding assay shows that two overlapping GST-fusion proteins can bind CaM in a Ca2+-dependent manner. (Left) Western blot of proteins shown in A under three conditions: (1) Input. This represents the amount of each protein that was loaded on the CaM-agarose beads. For the subsequent two lanes, this amount of protein represents that maximum possible amount that can bind. (2) Proteins that bound to CaM agarose beads in the presence of 2 mM Ca2+. (3) Proteins that bound to CaM agarose beads in the presence of 2 mM EGTA with no added Ca2+. (Right) Bar graph showing the percent of each GST-fusion protein that bound CaM in the presence of Ca2+ (blue) or EGTA (white), as determine by densitometry of the experiment on the left. For each experimental condition (Ca2+ and EGTA), the values were normalized to that of the input lane.
The two GST fusion proteins that interacted most strongly with Ca2+/CaM overlapped in the region corresponding to amino acids 189–222. These amino acids partly overlap with the first of three Ankyrin repeats in the NH2-terminal region. Although there is no precedent for Ankyrin repeats binding CaM, these data suggest that Ca2+/CaM may modulate channel function by binding directly to this region. We next produced a GST-fusion protein corresponding to amino acids 189–222 to determine if this region is sufficient to allow CaM binding. As shown in Fig. 6 B, this region was indeed sufficient to allow Ca2+-dependent CaM binding. To examine whether CaM binding to amino acids 189–222 accounted for the inhibition of functional TRPV1 channels by Ca2+/CaM, we made three mutant channels containing deletions in the region of the CaM-binding site: TRPV1Δ189–222, TRPV1Δ200–232, and TRPV1Δ205–222. When expressed in Xenopus oocytes, none of these deletion mutants formed capsaicin-activated channels in the plasma membrane.
The lack of expression of functional channels in which the CaM-binding domain has been removed is not unprecedented. For example, KCNQ potassium channels in which the CaM-binding domain is deleted also do not produce detectable currents (Wen and Levitan, 2002). We therefore examined whether lack of protein might account for the lack of capsaicin-activated current we observed. When oocyte lysates were examined with SDS/PAGE and Western blot analysis for expression of these mutants, TRPV1Δ189–222 produced no detectable protein. Expression of TRPV1Δ200–232 and TRPV1Δ205–222 protein was detectable, but reduced compared with wild-type channels (Fig. 7) .
Figure 7.

NH2-terminal deletions reduce proteins levels as well as the fraction of the protein that binds to CaM. Lanes 1–4 represent the input and lanes 5–8 represent the protein eluted from the CaM beads. Each channel type is as indicated. The experiment was performed in the presence of 2 mM Ca2+.
Although the TRPV1Δ200–232 and TRPV1Δ205–222 proteins did not produce capsaicin-activated currents, we asked whether they would bind to CaM in the pull-down assay. We found that the binding of TRPV1Δ200–232 and TRPV1Δ205–222 mutant channels to CaM-agarose beads was decreased by approximately a factor of 10 compared with wild-type channels (Fig. 7). The residual binding of the deletion mutants could have been due to binding of CaM to amino acids 247–334 in the NH2-terminal region. Alternatively, it could have been due to another binding site within the channel. Although wild-type TRPV1 bound CaM in a Ca2+-independent manner in the assay shown in Fig. 5 C, the protein examined in Fig. 7 was not functional, and we have no evidence it was properly folded. These data, then, hint at a second CaM binding region, but their CaM binding cannot be quantitatively compared with functional channels. Nevertheless, the CaM binding ability of the deletion mutants together with the Ca2+-independent binding of full-length TRPV1 suggests that multiple regions of channel sequence may interact with CaM in a synergistic manner.
DISCUSSION
Our data show that Ca2+/CaM binds to the NH2-terminal region of TRPV1 channels. The interaction of Ca2+/CaM with the channels inhibits gating to reduce open probability. In the peripheral sensory neurons that typically express TRPV1, this would have the effect of interfering with depolarization and decreasing Ca2+ influx into the cells. In this way the entry of Ca2+ through open channels could feed back on the channels, via their interaction with CaM, to cause channel closure and, perhaps, desensitization to noxious sensory stimuli.
Ion channels in the TRPV and TRPC families contain ankyrin repeats in their intracellular NH2-terminal regions (Montell et al., 2002). Ankyrin repeats consist of an ∼33-residue repeating motif named after the cytoskeletal protein ankyrin, which contains 24 copies of these repeats (Sedgwick and Smerdon, 1999). Present in over 400 proteins, they appear to have diverse functions, many of which involve protein–protein interactions. The function of the ankyrin repeats in TRP channels is not known; speculation has included roles in mediating subunit interactions, interactions with cytoskeletal elements, and interactions with elements of signal transduction cascades. Ankyrin repeats have not been previously shown to interact with calmodulin or to regulate dynamic response properties in ion channels.
It has previously been shown that phosphorylation of an amino acid in the NH2-terminal region of TRPV1 (S116) by PKA interferes with desensitization to capsaicin in whole-cell experiments (Bhave et al., 2002). Furthermore, S116 and three other residues in the TRPV1 NH2-terminal region were phosphorylated by PKA in an in vitro PKA phosphorylation assay. None of these four residues fall within the overlapping region of the NH2-terminal GST fusion proteins that bound CaM in our experiments. However, the localization within the NH2-terminal region of two apparently disparate mechanisms involved in desensitization raises the question of whether, and to what extent, phosphorylation by PKA and binding of Ca2+/CaM interact to regulate TRPV1 open probability.
Several features of the TRPV1 interaction with Ca2+/CaM resemble the physiological adaptation to odorants by olfactory receptors (Trudeau and Zagotta, 2003). In that case, Ca2+ entering through CNG channels complexes with CaM to inhibit CNG channel activation. CNG channels and TRPV1 channels are both members of the six-membrane spanning domain super-family of ion channels that includes the voltage gated Na+, K+, and Ca2+ channels (Jan and Jan, 1990, 1992), but show no appreciable sequence similarity outside the core transmembrane regions. Interestingly, in olfactory CNG channels CaM also binds to the NH2-terminal region.
Recently, a CaM-binding site in the COOH-terminal domain of TRPV1 has been identified (Numazaki et al., 2003). This 35–amino acid region bound CaM in GST pull-down assays in a Ca2+-dependent manner. Desensitization persisted in functional channels lacking these 35 amino acids, indicating that CaM binding to this region is not required for desensitization. Nevertheless, these data, in combination with the Ca2+-independent binding of CaM and full-length TRPV1, indicate that multiple regions of the channels may bind CaM.
Our experiments localized a Ca2+-dependent CaM binding site in the TRPV1 NH2-terminal region. The Ca2+ independence of the binding of full-length TRPV1 to CaM suggests that the channels may contain a second, Ca2+-independent CaM binding site. There is precedent for the coexistence of Ca2+-dependent and Ca2+-independent binding sites for CaM within a given protein (Saimi and Kung, 2002). In SK channels for example, the C-lobe of CaM appears to anchor it to the channels in a Ca2+-independent manner, whereas the N-lobe influences gating in a Ca2+-dependent manner (Keen et al., 1999; Peterson et al., 1999). In L-type Ca2+ channels, it is also the N-lobe that is involved in gating and the C-lobe that is involved in anchoring CaM to the channels (Peterson et al., 1999; Zuhlke et al., 1999; Pitt et al., 2001). Thus, the Ca2+-independent interaction of full-length TRPV1 with CaM provides a tantalizing suggestion that TRPV1 may indeed participate in such an intricately regulated CaM system.
Acknowledgments
We would like to thank Ms. Emilie Wyrick for profoundly excellent technical assistance. We also thank Drs. Anita L. Zimmerman, William N. Zagotta, León D. Islas, and Matthew Trudeau for helpful discussions.
Olaf S. Andersen served as editor.
Tamara Rosenbaum and Ariela Gordon-Shaag contributed equally to this work.
Abbreviations used in this paper: CaM, calmodulin; CNG, cyclic nucleotide–gated.
References
- Bhave, G., W. Zhu, H. Wang, D.J. Brasier, G.S. Oxford, and R.W.T. Gereau. 2002. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 35:721–731. [DOI] [PubMed] [Google Scholar]
- Caterina, M.J., and D. Julius. 2001. The vanilloid receptor: a molecular gateway to the pain pathway. Annu. Rev. Neurosci. 24:487–517. [DOI] [PubMed] [Google Scholar]
- Caterina, M.J., M.A. Schumacher, M. Tominaga, T.A. Rosen, J.D. Levine, and D. Julius. 1997. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 389:816–824. [DOI] [PubMed] [Google Scholar]
- Cholewinski, A., G.M. Burgess, and S. Bevan. 1993. The role of calcium in capsaicin-induced desensitization in rat cultured dorsal root ganglion neurons. Neuroscience. 55:1015–1023. [DOI] [PubMed] [Google Scholar]
- Clapham, D.E., L.W. Runnels, and C. Strubing. 2001. The TRP ion channel family. Nat. Rev. Neurosci. 2:387–396. [DOI] [PubMed] [Google Scholar]
- Docherty, R.J., J.C. Yeats, S. Bevan, and H.W. Boddeke. 1996. a. Inhibition of calcineurin inhibits the desensitization of capsaicin- evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflugers Arch. 431:828–837. [DOI] [PubMed] [Google Scholar]
- Docherty, R.J., J.C. Yeats, S. Bevan, and H.W. Boddeke. 1996. b. Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflugers Arch. 431:828–837. [DOI] [PubMed] [Google Scholar]
- Hui, K., B. Liu, and F. Qin. 2003. Capsaicin activation of the pain receptor, VR1: multiple open states from both partial and full binding. Biophys. J. 84:2957–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan, L.Y., and Y.N. Jan. 1990. A superfamily of ion channels. Nature. 345:672. [DOI] [PubMed] [Google Scholar]
- Jan, L.Y., and Y.N. Jan. 1992. Tracing the roots of ion channels. Cell. 69:715–718. [DOI] [PubMed] [Google Scholar]
- Jordt, S.E., M. Tominaga, and D. Julius. 2000. Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc. Natl. Acad. Sci. USA. 97:8134–8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, J., S.W. Hwang, J. Kwak, S.Y. Lee, C.J. Kang, W.B. Kim, D. Kim, and U. Oh. 1999. Capsaicin binds to the intracellular domain of the capsaicin-activated ion channel. J. Neurosci. 19:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen, J.E., R. Khawaled, D.L. Farrens, T. Neelands, A. Rivard, C.T. Bond, A. Janowsky, B. Fakler, J.P. Adelman, and J. Maylie. 1999. Domains responsible for constitutive and Ca2+-dependent interactions between calmodulin and small conductance Ca2+-activated potassium channels. J. Neurosci. 19:8830–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koplas, P.A., R.L. Rosenberg, and G.S. Oxford. 1997. The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J. Neurosci. 17:3525–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L., and S.A. Simon. 1996. Capsaicin-induced currents with distinct desensitization and Ca2+ dependence in rat trigeminal ganglion cells. J. Neurophysiol. 75:1503–1514. [DOI] [PubMed] [Google Scholar]
- Minke, B. 1977. Drosophila mutant with a transducer defect. Biophys. Struct. Mech. 3:59–64. [DOI] [PubMed] [Google Scholar]
- Montell, C. 2001. Physiology, phylogeny, and functions of the TRP superfamily of cation channels. Sci STKE. 2001:RE1. [DOI] [PubMed]
- Montell, C., L. Birnbaumer, and V. Flockerzi. 2002. The TRP channels, a remarkably functional family. Cell. 108:595–598. [DOI] [PubMed] [Google Scholar]
- Numazaki, M., T. Tominaga, K. Takeuchi, N. Murayama, H. Toyooka, and M. Tominaga. 2003. Structural determinant of TRPV1 desensitization interacts with calmodulin. Proc. Natl. Acad. Sci. USA. 100:8002–8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, U., S.W. Hwang, and D. Kim. 1996. Capsaicin activates a nonselective cation channel in cultured neonatal rat dorsal root ganglion neurons. J. Neurosci. 16:1659–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, B.Z., C.D. DeMaria, J.P. Adelman, and D.T. Yue. 1999. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 22:549–558. [DOI] [PubMed] [Google Scholar]
- Piper, A.S., J.C. Yeats, S. Bevan, and R.J. Docherty. 1999. A study of the voltage dependence of capsaicin-activated membrane currents in rat sensory neurones before and after acute desensitization. J. Physiol. 518:721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt, G.S., R.D. Zuhlke, A. Hudmon, H. Schulman, H. Reuter, and R.W. Tsien. 2001. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J. Biol. Chem. 276:30794–30802. [DOI] [PubMed] [Google Scholar]
- Rho, S., H.M. Lee, K. Lee, and C. Park. 2000. Effects of mutation at a conserved N-glycosylation site in the bovine retinal cyclic nucleotide-gated ion channel. FEBS Lett. 478:246–252. [DOI] [PubMed] [Google Scholar]
- Rosenbaum, T., M. Awaya, and S.E. Gordon. 2002. Subunit modification and association in VR1 ion channels. BMC Neurosci. 3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saimi, Y., and C. Kung. 2002. Calmodulin as an ion channel subunit. Annu. Rev. Physiol. 64:289–311. [DOI] [PubMed] [Google Scholar]
- Sedgwick, S.G., and S.J. Smerdon. 1999. The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem. Sci. 24:311–316. [DOI] [PubMed] [Google Scholar]
- Smart, D., M.J. Gunthorpe, J.C. Jerman, S. Nasir, J. Gray, A.I. Muir, J.K. Chambers, A.D. Randall, and J.B. Davis. 2000. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br. J. Pharmacol. 129:227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szallasi, A., and P.M. Blumberg. 1999. Vanilloid (Capsaicin) receptors and mechanisms. Pharmacol. Rev. 51:159–212. [PubMed] [Google Scholar]
- Trudeau, M.C., and W.N. Zagotta. 2003. Calcium/calmodulin modulation of olfactory and rod cyclic nucleotide-gated ion channels. J. Biol. Chem. 278:18705–18708. [DOI] [PubMed] [Google Scholar]
- Vyklicky, L., A. Lyfenko, D.P. Kuffler, and V. Vlachova. 2003. Vanilloid receptor TRPV1 is not activated by vanilloids applied intracellularly. Neuroreport. 14:1061–1065. [DOI] [PubMed] [Google Scholar]
- Wen, H., and I.B. Levitan. 2002. Calmodulin is an auxiliary subunit of KCNQ2/3 potassium channels. J. Neurosci. 22:7991–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhlke, R.D., G.S. Pitt, K. Deisseroth, R.W. Tsien, and H. Reuter. 1999. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 399:159–162. [DOI] [PubMed] [Google Scholar]