

Abstract
CFTR (cystic fibrosis transmembrane conductance regulator), the protein whose dysfunction causes cystic fibrosis, is a chloride ion channel whose gating is controlled by interactions of MgATP with CFTR's two cytoplasmic nucleotide binding domains, but only after several serines in CFTR's regulatory (R) domain have been phosphorylated by cAMP-dependent protein kinase (PKA). Whereas eight R-domain serines have previously been shown to be phosphorylated in purified CFTR, it is not known how individual phosphoserines regulate channel gating, although two of them, at positions 737 and 768, have been suggested to be inhibitory. Here we show, using mass spectrometric analysis, that Ser 768 is the first site phosphorylated in purified R-domain protein, and that it and five other R-domain sites are already phosphorylated in resting Xenopus oocytes expressing wild-type (WT) human epithelial CFTR. The WT channels have lower activity than S768A channels (with Ser 768 mutated to Ala) in resting oocytes, confirming the inhibitory influence of phosphoserine 768. In excised patches exposed to a range of PKA concentrations, the open probability (Po) of mutant S768A channels exceeded that of WT CFTR channels at all [PKA], and the half-maximally activating [PKA] for WT channels was twice that for S768A channels. As the open burst duration of S768A CFTR channels was almost double that of WT channels, at both low (55 nM) and high (550 nM) [PKA], we conclude that the principal mechanism by which phosphoserine 768 inhibits WT CFTR is by hastening the termination of open channel bursts. The right-shifted Po-[PKA] curve of WT channels might explain their slower activation, compared with S768A channels, at low [PKA]. The finding that phosphorylation kinetics of WT or S768A R-domain peptides were similar provides no support for an alternative explanation, that early phosphorylation of Ser 768 in WT CFTR might also impair subsequent phosphorylation of stimulatory R-domain serines. The observed reduced sensitivity to activation by [PKA] imparted by Ser 768 might serve to ensure activation of WT CFTR by strong stimuli while dampening responses to weak signals.
Keywords: ABC transporters, chloride ion-channel gating, multiple phosphorylation sites, mass spectrometry, autoradiography
INTRODUCTION
CFTR (cystic fibrosis transmembrane conductance regulator) is the product of the gene mutated in cystic fibrosis patients (Riordan et al., 1989), and a member of the family of ATP-binding casette (ABC) transporters. Like all family members, CFTR incorporates two cytoplasmic nucleotide binding domains (NBDs) that interact with MgATP. But, unique among ABC proteins, CFTR functions as an ion channel. Also unlike other ABC family members, the CFTR sequence includes a central ∼200-residue regulatory (R) domain that contains multiple serines in consensus sequences for phosphorylation by PKA and PKC (Riordan et al., 1989). Indeed, phosphorylation of CFTR by PKA is a necessary, but not sufficient, condition for CFTR channels to gate; once phosphorylated, CFTR channels require exposure to MgATP to open and close (for reviews see Gadsby and Nairn, 1999; Sheppard and Welsh, 1999).
Recent detailed analyses of the gating of wild-type (WT) and NBD mutant CFTR channels and of their interactions with photolabile nucleotides (Aleksandrov et al., 2001, 2002; Basso et al., 2003; Vergani et al., 2003), together with structural information from crystals of NBD dimers from other ABC proteins (e.g., Hopfner et al., 2000; Locher et al., 2002; Smith et al., 2002; Chen et al., 2003), have begun to clarify the major conformational changes associated with opening and closing of CFTR channels: formation of an NBD1–NBD2 dimer with two MgATPs bound at the two catalytic sites in the dimer interface is suggested to initiate a channel open burst, and hydrolysis of the MgATP at the NBD2 catalytic site to terminate the burst (e.g., Basso et al., 2003; Vergani et al., 2003). In contrast, the mechanisms by which phosphorylation regulates CFTR channel gating remain poorly understood (Gadsby and Nairn, 1999; Seibert et al., 1999; Ostedgaard et al., 2001; Chappe et al., 2004). And yet phosphorylation is probably the principal mechanism of regulation of CFTR function in vivo, because cellular [MgATP] levels are in the millimolar range, concentrations at least an order of magnitude higher than those needed to half-maximally activate CFTR. A major barrier to understanding the regulation by phosphorylation is the large number of phosphorylation sites in each CFTR channel. Of CFTR's many consensus sites for phosphorylation by PKA, to date five serines (R-domain serines 660, 700, 737, 795, and 813) have been shown to be phosphorylated in vivo (Cheng et al., 1991; Picciotto et al., 1992) and eight (those five plus serines 712, 753, and 768) in vitro (Neville et al., 1997).
Because deletion of much (residues 708–835; Rich et al., 1991) or all of the R domain (residues 634–836; Csanády et al., 2000) results in constitutively active CFTR channels that no longer require phosphorylation to open and close in the presence of MgATP, it is clear that the dephosphorylated R domain inhibits channel gating and that this inhibition is relieved by phosphorylating or removing the R domain (Cheng et al., 1991; Rich et al., 1991; Csanády et al., 2000). But the complexity associated with so many phosphorylation sites has made it hard to decipher details of the regulatory mechanisms. Early mutagenesis studies concluded that the multiple phosphorylation sites conferred redundancy, as overall CFTR function during strong stimulation through the cAMP/PKA pathway seemed little affected by mutation of single serines (or threonine) in any of CFTR's 10 dibasic (R/K-R/K-X-S/T) consensus sites (Cheng et al., 1991). Average channel current in excised patches in the presence of PKA was less than halved after combined mutation of four serines (660, 737, 795, and 813) out of the five phosphorylated in vivo, and combined mutation of 8, or all 10, of the dibasic sites appeared to only slightly reduce that remaining current (Chang et al., 1993; Rich et al., 1993; Winter and Welsh, 1997; Mathews et al., 1998). The initial notion of redundancy gained support from the demonstration that simultaneous replacement of several (≥6) of the consensus serines by aspartates (Rich et al., 1993) or glutamates (Aleksandrov et al., 2000), thought to mimic the negative charges of phosphoserines, resulted in CFTR channels that were at least partially active (though with low open probability) in the presence of MgATP without phosphorylation. This led to the suggestion that activation of CFTR by phosphorylation might reflect an electrostatic consequence of accumulation of negative charges in the R domain.
Such a simple mechanism of CFTR activation by phosphorylation based on accumulation of redundant nonspecific events (e.g., Cheng et al., 1991; Rich et al., 1993; Seibert et al., 1999; Ostedgaard et al., 2000) seems unlikely on at least two grounds. First, structural studies of the entire isolated R domain have shown that it undergoes major structural rearrangement upon phosphorylation (Dulhanty et al., 1995; but also see Ostedgaard et al., 2000), consistent with the idea that conformational changes in the R domain upon phosphorylation, rather than accumulation of charged phosphoserines per se, are responsible for its regulatory function. Second, a study of CFTR channels bearing single serine-to-alanine mutations in the R domain (Wilkinson et al., 1997) suggested that, in contrast to most other consensus serines in the R domain, phosphorylation of Ser 737 or Ser 768 exerts an inhibitory effect on CFTR channel function, despite undoubted introduction of negative charge. These results support regulatory mechanisms that involve specific actions of individual phosphoserines in a structured R domain, rather than a nonspecific buildup of electric charge.
However, little information is available about such distinct mechanistic roles of individual serines, or whether individual serines are phosphorylated in a particular sequence. Here, we took a kinetic approach and found that Ser 768 is the most readily phosphorylated serine in the isolated R domain in vitro. Even in vivo, in full-length CFTR expressed in Xenopus oocytes, we found that Ser 768 is already phosphorylated by basal PKA activity in resting, nonstimulated oocytes. We confirmed that this phosphorylation of Ser 768 inhibits activity of WT CFTR channels both at low concentrations of PKA and at high [PKA], and we found that it does so principally by making CFTR channels close sooner after opening than they otherwise would. One consequence of this effect of phosphoserine 768 is that the dose–response curve for activation of CFTR chloride current by PKA is shifted to higher [PKA] for WT channels than for mutant S768A CFTR channels. We suggest that this would help ensure robust responses of CFTR channels to strong activating signals while dampening their responses to weak signals.
MATERIALS AND METHODS
Molecular Biology
pGEMHE-WT CFTR and pGEMHE-Flag-WT CFTR were constructed as previously described (Chan et al., 2000). S768A CFTR cDNA, provided by D. Dawson (OHSU, Portland, OR), was subcloned into the SmaI and XhoI sites of pGEMHE. All DNA sequences were confirmed by automated DNA sequencing. cRNA was prepared by in vitro transcription and quantitated as previously described (Chan et al., 2000).
Isolation and Injection of Xenopus Oocytes, and Two-Microelectrode Voltage Clamp Recordings
Oocytes were isolated, and injected with 1–5 ng cRNA for each CFTR construct (in a constant 50-nl volume), as previously described (Chan et al., 2000). Recordings were made in oocytes continuously superfused with Ca2+-free Ringer's solution at room temperature (∼24°C) and held near their initial resting potential (−40 to −20 mV). Basal, or activated (by 50 μM forskolin plus 1 mM IBMX), conductance was measured as the slope between −60 and −20 mV of the steady-state current/voltage plot obtained with 75- to 250-ms voltage steps to potentials between −100 and +80 mV. To abolish basal PKA activity, oocytes were injected, usually 15–30 min before recording, with 50 nl of 20 mM RpcAMPS (BIOLOG) solution; estimated final concentration, ∼2 mM.
Excised Patch Recording
Single-channel or macroscopic currents were recorded at room temperature in inside-out patches excised from oocytes injected with 0.1–5 ng of cRNA as previously described (Chan et al., 2000; Csanády et al., 2000). In brief, outward unitary currents were recorded at a pipette potential of −40 mV (Vm = +40 mV), with pipette solution: 138 mM NMG, 2 mM MgCl2, 5 mM HEPES, 136 mM HCl (pH 7.4 with HCl). Pipette resistances were ∼1 MΩ or 4–7 MΩ for macropatch or single-channel recordings, respectively, and seal resistances were 100–300 GΩ. The continuously flowing bath solution contained 138 mM NMG, 2 mM Mg-sulfamate, 5 mM HEPES, 0.5 mM EGTA, 134 mM sulfamic acid, pH 7.1 with sulfamic acid. Solution exchange (measured from the decay of endogenous Ca2+-activated Cl− channel current, after brief application and removal of 2 mM Ca-sulfamate; e.g., see Fig. 5) had a time constant of 200–600 ms, and was essentially complete within 1–3 s. Nucleotides were added in the form of MgATP (pH 7.2 with NMG), and NMG-pyrophosphate (pH 7.2 with NMG, supplemented with equimolar Mg-sulfamate). CFTR channels were activated with 30–550 nM PKA catalytic subunit purified from bovine heart (Kaczmarek et al., 1980). Records were filtered on-line at 100 Hz using an 8-pole Bessel filter, and digitized at 1 kHz.
Figure 5.

Activation in excised patches of macroscopic WT and S768A CFTR currents by low and high concentrations of PKA catalytic subunit. (A and B) Currents recorded in patches containing hundreds of WT (A) or S768A (B) CFTR channels. No substantial current is activated in either case by 2 mM MgATP applied ∼2 min after patch excision, but 55 nM, and 550 nM, PKA catalytic subunit activate increasing amounts of current in both cases. Solid green and blue lines show single-exponential fits to the current time courses, with indicated time constants; activation/inactivation time course of endogenous Ca2+-activated Cl− channel current elicited by a brief pulse of 2 mM Ca2+ was used to estimate the speed of solution exchange. (C) Fractional current activated by 55 nM PKA was significantly smaller for WT (gray bar) than S768A (black bar; *, P = 0.0024) CFTR channels; mean steady current in 55 nM PKA was divided by mean steady current in the same patch at 550 nM PKA. (D) Time constants of macroscopic current relaxations upon addition of 55 nM (top) or 550 nM (bottom) PKA, for WT (gray bars) and S768A (black bars) CFTR; activation was faster for S768A at low (*, P = 0.036), but not at high [PKA].
Kinetic Analysis
Records containing one to seven channels were analyzed as previously described (Chan et al., 2000; Csanády et al., 2000). Baseline-subtracted currents (e.g., see Fig. 6; to remove slow drifts and the ≤0.5 pA shift on adding PKA, due to its buffer) were idealized by conventional half-amplitude threshold crossing. Events lists were fitted with a simple three-state closed–open–blocked (C-O-B) scheme in which all principal gating transitions were pooled into a closed–open scheme, and rapid flickery closures were modeled as pore blockage events (Ishihara and Welsh, 1997). Rate constants rCO, rOC, rOB, and rBO were extracted by a simultaneous fit to the dwell-time histograms of all conductance levels, as previously described (Csanády, 2000), and mean closed interburst and open burst durations then calculated as τib = 1/rCO and τb = (1/rOC)(1+ rOB/rBO), respectively. Dead time was 4 ms, and typical rates were rOB = ∼3 s−1, rBO = ∼100 s−1, and rCO and rOC on the order of 1 s−1 (depending on [PKA]). Open probabilities were calculated from the events lists as the time-average of the fraction of open channels. Channels were counted at the end of each record by locking them in the open state with 2 mM PPi (or 1 mM AMPPNP) in the presence of 0.1 mM MgATP. Statistical tests (Csanády et al., 2000) were then applied to evaluate whether the number of active channels in the patch (N) was likely equal to the maximum number of simultaneously open channels seen under the above locked-open conditions (N'). Extracted τib and Po values were included in Fig. 6 (C and D) only if N > N' could be excluded with >90% confidence (N was between 1 and 7 for these patches). Because the extracted value of τb is less sensitive to N, Fig. 6 E also includes τb estimates from two WT patches with N' = 8 in which transitions were well resolved but N > N' could not be excluded with >90% confidence.
Figure 6.

Kinetic behavior of WT and S768A CFTR channels in excised patches exposed to low and high [PKA]. Representative baseline-subtracted current traces of (A) four WT and (B) five S768A channels, recorded from excised patches in the presence of 2 mM MgATP + 55 nM or 550 nM PKA; 20-s segments (indicated by bars) under each condition are shown with 10-fold expanded time scale, below. Channels were counted by locking them in the open-burst state with 0.1 mM MgATP + 2 mM pyrophosphate (PPi) + 300 nM PKA. (C–E) Open probabilities (C), mean interburst (D) and open burst (E) durations at 55 nM (top) or 550 nM PKA (bottom), for WT (gray bars) and S768A (black bars) CFTR channels; asterisks indicate significant differences between S768A and WT (0.001 < P < 0.06).
Calculation of Fractional Activities and Fitting of Macroscopic Current Relaxations
Macroscopic currents, typically originating from hundreds or thousands of channels, were refiltered at 10 Hz, and sampled at 50 Hz. Fractional currents at 55 nM PKA were obtained by normalizing the mean of the steady current at 55 nM PKA to the steady current at 550 nM PKA measured subsequently in the same patch. To allow comparison of activation rates of WT and S768A CFTR channels, current relaxation time courses were fitted with single exponentials by nonlinear least squares (SigmaPlot 7.0). All results are presented as mean ± SEM.
Preparation of R-domain Peptide
The region of the R domain encompassing amino acids 645–835 of CFTR (termed CF-2) was expressed from a pET-8c expression vector in Escherichia coli essentially as previously described (Picciotto et al., 1992). Cells were lysed in a buffer that did not contain detergent or denaturing agents, and the untagged protein was purified using anion exchange chromatography, hydroxylapatite chromatography, and gel filtration as previously described (Picciotto et al., 1992).
Expression, Purification, and Renaturation of Hexa-His–Tagged R Domain
E. coli containing the plasmid pQE60, encoding CFTR amino acids 645–835 with COOH-terminal 6xHis tag, were grown under standard conditions, and protein expression was induced with 1 mM IPTG. Cells were harvested by centrifugation, frozen at −20°C, and lysed at room temperature by stirring with 6 M guanidine-HCl, 0.1 M NaH2PO4, 0.01 M Tris-HCl, 3 mM imidazole, pH 8.0. The lysate was centrifuged for 30 min at 10,000 rpm, the supernatant collected and applied to Ni-NTA resin using the batch method, and the resin mixed end-over-end at 4°C for 2–12 h. The Ni-NTA beads were collected by centrifugation, washed with lysis buffer, and protein was eluted with lysis buffer containing 250 mM imidazole. Protease inhibitors were added to the eluate, and the sample renatured by steps of dialysis into buffers with decreasing concentrations of guanidine-HCl. The first dialysis (2 L, 1 h, room temperature) contained 3 M guanidine-HCl, 50 mM Tris pH 8.0, 150 mM NaCl, 1 mM 2-mercaptoethanol, 6% glycerol; the second dialysis (2 L, 4 h, 4°C) contained 1.5 M guanidine-HCl, 50 mM Tris pH 8.0, 150 mM NaCl, 1 mM 2-mercaptoethanol, 6% glycerol; the third dialysis (2 L, 2 h, 4°C) contained 0.75 M guanidine-HCl, 50 mM Tris pH 8.0, 150 mM NaCl, 1 mM 2-mercaptoethanol, 10% glycerol; the fourth dialysis (4 L, 12 h, 4°C) contained 0.125 M guanidine-HCl, 50 mM Tris pH 8.0, 10 mM NaCl, no mercaptoethanol, 10% glycerol.
Phosphorylation of R-domain Proteins
Phosphorylation was assayed after various times at 30°C in a standard reaction mixture that contained 0.5–5 μg R-domain protein, 50 mM Hepes, pH 7.5, 10 mM Mg-acetate, 1 mM EGTA, 1–50 μM γ32P-MgATP. Typically, the reaction was started by addition of ATP to a large reaction mixture (500–1000 μl) and aliquots were removed at different time points into SDS-containing stop buffer (1% SDS, 60 mM Tris-HCl, pH 6.8, 5% glycerol, and 0.2 M mercaptoethanol; pyronin Y was added as a visible dye front marker). Samples were analyzed by SDS-PAGE (11 or 12% acrylamide) using the method of Laemmli, and gels were stained, dried, and subjected to autoradiography.
Gel pieces containing 32P-labeled R-domain protein were excised from the dried SDS-polyacrylamide gel, and subjected to digestion with TPCK-treated trypsin (50 μg/ml; Worthington) as previously described (Picciotto et al., 1992). Dried samples were resuspended in electrophoresis buffer (10% acetic acid, 1% pyridine, pH 3.5), and spotted on 20 × 20-cm thin layer cellulose plates (Eastman Kodak). Phosphopeptides were separated by electrophoresis at 400 V for ∼90 min in the first dimension, plates were dried, and then phosphopeptides were separated by ascending chromatography in the second dimension in a buffer containing pyridine:1-butanol:water:acetic acid (10:15:12:3, vol/vol). Dried plates were subjected to autoradiography.
Preparation of Oocyte Membranes and Immunoprecipitation of CFTR
For each in vivo assay of CFTR phosphorylation, 1,000 selected oocytes were each injected with 20 ng of Flag-WT CFTR cRNA, and after 2 d of incubation in Ca2+-containing Ringer's solution, they were washed twice with Ca2+-free Ringer's and then lysed with 6–8 ml of oocyte lysis buffer, comprising 10 mM Hepes, 5 mg/ml BSA, 1 mM PMSF, pH 7.5; the lysis buffer also included 5 mM EDTA, 10 mM NaF, 5 mM NaVO4, and 2 μM microcystin-LR to inhibit endogenous phosphatases, and was supplemented with a cocktail of protease inhibitors (Calbiochem; 1 mM AEBSF-HCl, 300 nM aprotinin, 1 mM E-64, 2 μM leupeptin hemisulfate, 1 mM EDTA). The lysate suspension was spun at 3,000 g for 10 min, and the supernatant kept on ice while the pellet was resuspended in a further 6–8 ml lysis buffer and then centrifuged again at 3,000 g for 10 min. The two supernatants were combined and centrifuged at 160,000 g for 1 h to yield a total membrane pellet, which was washed with 10 ml modified lysis buffer (BSA was replaced with 10% glycerol), spun down, and then resuspended in 0.5 ml of the modified buffer.
For immunoprecipitation, membranes (containing ∼2 mg membrane proteins) were dissolved in 2 ml solubilization buffer, containing 1% NP-40, 0.1% SDS, 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 2 μM microcystin, for 1.5–2.5 h at 4°C and then centrifuged at 91,000 g for 1 h. The supernatant was mixed overnight at 4°C with anti-Flag M2 affinity gel, prewashed with solubilization buffer lacking SDS, and the beads collected by centrifugation at 26,000 g for 5 min. The beads were washed twice with solubilization buffer without SDS, twice with high-salt buffer (50 mM Tris/HCl, pH 7.5, 500 mM NaCl, and 0.05% Tween 20), twice with low-salt buffer (50 mM Tris/HCl, pH 7.5, 150 mM NaCl) and then twice with no-salt buffer (50 mM Tris/HCl, pH 7.5); all these buffers included 5 mM EDTA and 2 μM microcystin. Bound proteins were eluted with 150 μl of Laemmli sample buffer (plus 1 mM DTT) at room temperature for 1 h, and then separated by SDS-PAGE overnight (30–40 V) on thin (1 mm) 5–15% gradient gels. The gels were then cut in half: one half contained 10–15% of the samples and was used for immunoblots with anti R-domain Ab, as previously described (Chan et al., 2000); the other half contained the rest of the protein and was stained with zinc sulfate and imidazole for mass spectrometry. 1,000 uninjected oocytes were treated identically to confirm the absence from immunoblot and Zn-stained gel of bands that, in the lanes from CFTR-injected oocytes, could be identified as fully glycosylated CFTR. Protein concentrations were measured with bicinchonic acid (Pierce Chemical Co.).
Mass Spectrometric Analysis of Intact CFTR Protein
The band of mature CFTR was excised from the gel, destained, and subjected to in-gel digestion with bovine trypsin (Roche). The resulting peptides were extracted from the gel and bound to a ZipTip pipette tip (Millipore). The ZipTip resin was washed and the peptides were eluted. The peptide mixture was analyzed using a MALDI-TOF mass spectrometer (model Voyager STR-DE, PerSeptive; Perkin-Elmer) and a MALDI-QqTOF mass spectrometer (Centaur prototype, Sciex, modified in-house; Krutchinsky et al., 2000). Putative phosphopeptides identified in these spectra were further analyzed by MALDI-QqTOF-MS/MS and/or ESI-QqTOF-MS/MS.
Mass Spectrometric Analysis of R-domain Protein
Analysis of the SDS-PAGE gel bands representing the different phosphoforms of the R domain was performed as follows. The individual Coomassie-stained bands were excised, destained, subjected to enzymatic digestion with trypsin, and the resulting peptides extracted from the gel pieces and analyzed by MALDI ion trap mass spectrometry as previously described (Qin and Chait, 1997).
RESULTS
Electrophoretic Mobility Shifts Signal Incremental Phosphorylation of R-domain in Vitro
To examine the order in which R-domain serines are phosphorylated by PKA, purified R domain peptide was incubated with PKA and either relatively high (50 μM) or low (1 μM) γ32P-MgATP, and sampled at various times. As phosphorylation proceeded, the mobility of the R domain on SDS-PAGE declined in discrete steps, implying the existence of as many as six distinct phosphoforms (Fig. 1 A), all of which could also be discerned in the autoradiogram of the same gel (Fig. 1 B). MALDI ion trap mass spectrometry analysis of analogous bands excised from the gel shown in Fig. 2 A yielded the mass spectra displayed in Fig. 2 B, in which several pairs of peaks separated by 98 D could be identified. In this phosphopeptide signature, the higher-mass peak corresponds to the intact phosphopeptide and the lower peak arises from elimination of the elements of phosphoric acid, confirming phosphorylation of a single residue which, in each case, could be inferred to be the unique serine present in a PKA consensus sequence. As Ser 768 was the sole residue found phosphorylated in the lowest band, Band 1 (Fig. 2 B), at low [MgATP], this is the most readily phosphorylated site in the R domain. In the small amount of R domain protein that underwent the major mobility shift, to Band 3, at low [MgATP] (Fig. 2 A, right), Ser 737 was the only additional serine phosphorylated (Fig. 2 B), so linking phosphorylation of Ser 737 to the largest individual mobility shift. The minor mobility shift, from Band 1 to Band 2, seen after prolonged incubation with high [MgATP], was associated with phosphorylation of Ser 700 and Ser 795, in addition to Ser 768. Band 4, obtained at late times, after undergoing the large mobility shift, contained not only phosphorylated Ser 700, Ser 737, Ser 768, and Ser 795, as expected, but also phosphorylated Ser 712. Analysis of the uppermost band, Band 6, that had undergone further incremental mobility shifts after long incubation, revealed additional phosphorylation of Ser 660 and, by inference, of Ser 813 (see Fig. 2 legend), and also of Ser 670 and Ser 753 (Fig. 2 B). Because Ser 768 thus stood out both biochemically, as being the most readily phosphorylated residue (Fig. 2 B; Picciotto et al., 1992), and functionally, as being inhibitory (Wilkinson et al., 1997), we targeted it for further detailed investigation.
Figure 1.

Time course of in vitro phosphorylation of R-domain peptide by PKA revealed by SDS-PAGE. Incubations were with 50 μM γ32P-MgATP for various times as indicated (0–45 min), except for the sample shown in the lower right panel, which was incubated with 1 μM γ32P-MgATP for 2 min. Samples were separated by SDS-PAGE, gels stained with Coomassie blue (A, STAIN), dried, and exposed to X-ray film (B, AUTORAD). Numbers label bands of varying mobility (stain band labeled * is an impurity). The unphosphorylated R domain (sample at 0 min) migrates as a single species of ∼28 kD (band 0, upper panel, extreme right).
Figure 2.

Order of in vitro PKA phosphorylation of R-domain peptide determined by SDS-PAGE and mass spectrometry. (A) R-domain protein was incubated with 50 μM or 1 μM γ32P-MgATP. Samples were separated by SDS-PAGE and exposed to X-ray film. The gel bands indicated (numbered arrows) were excised (Band 6 was excised from the upper part of the top-most band in the 10-min lane at 50 μM MgATP, as indicated by the arrow), digested by trypsin, and the resulting proteolytic products subjected to mass spectrometric analysis. (B) MALDI-ion trap mass spectrometric analysis of the five bands indicated in A showing that serine 768 is the most readily phosphorylated residue. Pairs of peaks separated by 98 D (joined by the red and blue connecting lines) provide signatures for phosphorylated peptides (see text). The blue lines designate peptide that is singly oxidized at methionine 773 (or 721, Band 4); the red lines indicate the corresponding unoxidized peptides. The amino acid residues of these phosphopeptides are indicated above each pair of peaks. For example, 766–785 and 765–785 correspond to two alternative cleavage products of the trypsin digestion. Because the masses of phosphorylated peptides 811–830 and 766–785 are respectively 2411.110 and 2411.164 (Table I), these two species were not resolved in this experiment, but the presence of 811–830 was inferred from the change in ratio of the pair of peaks associated with this mass in Band 6 (bottom panel) compared with other bands, which contain only 766–785. The peptide 766–785 contains a single methionine, which was partially oxidized to give the two pairs of peaks seen in all five panels. As peptide 811–830 contains no methionine residue, the presence of peptide 811–830 together with peptide 766–785 is signaled by a change in the ratio of the pairs of peaks 16 D apart.
Large Resting Conductance of Xenopus Oocytes Expressing Mutant S768A CFTR
We compared the activity of WT and Ser-768-to-Ala mutant CFTR channels after expressing them in oocytes. Two-microelectrode voltage-clamp recordings revealed measurable membrane conductance in resting oocytes expressing WT or S768A CFTR (Fig. 3, B–D, plot c and e). This resting conductance likely reflected basal CFTR channel activity, as it was not present unless CFTR cRNA was injected (Fig. 3, A and D, plot a), it had a reversal potential (−25 mV) appropriate for a chloride current (Fig. 3 D), and it was diminished by injection of the PKA inhibitor RpcAMPS (Fig. 3 E). RpcAMPS injection reduced basal WT CFTR conductance from 12 ± 2 to 4 ± 1 μS (n = 8), and similarly lowered the much larger basal S768A CFTR conductance from 146 ± 9 (n = 9) to 7 ± 2 μS (n = 5; e.g., Fig. 3 E). Stimulation of the endogenous cAMP–PKA pathway by superfusion with 1 mM IBMX + 50 μM forskolin robustly increased membrane conductance in WT CFTR-injected oocytes (to 151 ± 5 μS, n = 10; Fig. 3, B and D, plot d), but only slightly increased membrane conductance in oocytes expressing S768A CFTR (to 178 ± 4 μS, n = 9; Fig. 3, C and D, plot f). Thus, although the maximally activated conductances of oocytes expressing S768A or WT CFTR were comparable, the ratios of their resting conductance to maximally activated conductance were very different (Fig. 3 F), averaging 0.16 ± 0.02, n = 10, for WT but 0.82 ± 0.04, n = 9, for S768A. Because the maximum conductance (∼180 μS) activated by forskolin + IBMX in oocytes injected with ≥2.5 ng CFTR cRNA possibly reflects saturation of some component in the oocyte cAMP–PKA pathway (Csanády et al., 2000), further comparison of S768A and WT channel activity was limited to excised patches (see Figs. 5–7, below).
Figure 3.

Membrane conductance of resting and activated oocytes expressing WT or mutant S768A CFTR. Current time courses, recorded under two-microelectrode voltage clamp, of oocytes injected with water (A), or with cRNA encoding (B) WT or (C) S768A CFTR. Membrane potential was held near the resting potential (between −40 and −20 mV). Membrane conductance was monitored at regular intervals using brief voltage steps (vertical lines); complete current/voltage (I/V) relationships were determined under resting conditions (time points a, c, e), and after activation of cellular PKA with 50 μM forskolin + 1 mM IBMX (time points b, d, f). (D) Representative I/V plots under conditions as indicated in A–C, showing steady currents measured by averaging current samples toward the ends of 75-ms steps to voltages between −100 and +80 mV. (E) Rapid reduction of resting conductance (from 145 to 8 μS) upon injection of RpcAMPS into an oocyte expressing S768A CFTR. (F) Ratios, for WT- and S768A-expressing oocytes, of membrane conductances at rest (Grest), and after maximal activation of CFTR (Gmax); conductance values were similar for oocytes injected with 2.5 or 5 ng of each cRNA, so pooled results are shown.
Figure 7.

Dependence on [PKA] of open probability, Po, of WT (•) and S768A (○) CFTR channels. Steady-state mean Po measured in (n) excised patches, containing few channels, during exposure to 6–550 nM PKA plus near-saturating [MgATP] (1–2 mM), is plotted against [PKA]. Lines show nonlinear least-squares fits to the Hill equation, yielding Po,max = 0.34 ± 0.06, K 0.5 = 149 ± 46 nM, nH = 1.5 ± 0.5 for WT, and Po,max = 0.51 ± 0.05, K 0.5 = 71 ± 12 nM, nH = 1.8 ± 0.5 for S768A.
Insofar as the Ser to Ala mutation at position 768 may be expected to little alter the structure, and hence the function, of CFTR channels in the absence of phosphorylation, the large difference between the activation levels of S768A and WT CFTR channels in resting oocytes suggests that the basal PKA activity in those oocytes was sufficient to phosphorylate Ser 768 in WT CFTR, which then exerted its inhibitory influence on CFTR conductance. Moreover, that limited conductance of WT CFTR, as well as the substantial conductance of S768A CFTR, in resting oocytes suggests that the basal activity of PKA was also able to sustain steady-state phosphorylation of at least one stimulatory site in CFTR.
Serine 768 of WT CFTR Is Phosphorylated in Resting Oocytes
To investigate whether Ser 768, together with some other site, is in fact phosphorylated in resting oocytes, we turned to mass spectrometry. 2 d after injection with 20 ng cRNA encoding WT CFTR containing an NH2-terminal Flag epitope, oocytes were incubated for 10 min in calcium-free frog Ringer's at room temperature and then frozen in liquid nitrogen, and membranes were harvested in a cocktail of phosphatase and protease inhibitors. Flag-CFTR protein was immunoprecipitated with anti-Flag beads, separated by SDS-PAGE, and the ∼180-kD band corresponding to CFTR protein was excised, digested with trypsin, and subjected to MALDI-TOF and MALDI-QqTOF mass spectrometry. Both methods identified several putative phosphopeptides (Fig. 4) that exhibited a mass 80 D greater than expected for unmodified tryptic peptides from CFTR (Table I; HPO3 = 80 D). MALDI-QqTOF-MS/MS spectra of these peptides demonstrated loss of 98 D from the parent peak, providing additional confirmation of the presence of a phosphate group (H3PO4 = 98 D); this is analogous to the loss of phosphate from a phosphopeptide catalyzed by a protein phosphatase. MS/MS data (unpublished data) obtained from the peptides unequivocally localized sites of phosphorylation to serine residues 700, 737, and 795. The MALDI-TOF spectrum also identified phosphopeptide 710–716, which contains Ser 712 (Fig. 4), and MALDI-QqTOF-MS/MS of that ion (at m/z = 928.478) yielded an intense fragment ion peak at the parent ion mass minus 98 D, confirming that this peptide was indeed phosphorylated. Because the 710–716 peptide sequence, KFSIVQK, contains only a single potential phosphorylation site (Ser 712), the site of phosphorylation is unambiguous. Although the precise locations of the phosphates in phosphorylated peptides 658–668 (containing Ser 660) and 765–785 and 766–785 (containing Ser 768) were not ascertained, other Ser/Thr residues in these peptides do not lie in PKA consensus sequences, and none has ever been shown to be phosphorylated either in vivo or in vitro. We therefore infer that both Ser 660 and Ser 768 were phosphorylated.
Figure 4.

Serine 768 of WT CFTR is significantly phosphorylated in vivo in resting oocytes. MALDI-TOF (top) and MALDI-QqTOF (bottom) mass spectra of peptides obtained by trypsin digestion of WT CFTR immunoprecipitated from the membranes of resting oocytes that were lysed in the presence of phosphatase inhibitors to preserve phosphoserines present at that instant. Arrows identify phosphorylated (+P) peptides (molecular masses are listed in Table I).
TABLE I.
Molecular Mass Determination of Phosphorylated Peptides from Tryptic Digests of CFTR Phosphorylated In Vivo by Basally Active PKA in Resting Oocytes (Fig. 4)
| Phosphorylated Residue | Phosphorylated Peptidea | Measured Mass | Theoretical Mass | ΔM |
|---|---|---|---|---|
| D | D | D b | ||
| 660c | 658–668 | 1418.703 | 1418.703 | 0.000 |
| 700 | 698–709 | 1447.752 | 1447.755 | −0.003 |
| 737 | 735–751 | 1958.990 | 1958.982 | 0.008 |
| 768c | 766–785 | 2411.168 | 2411.164 | 0.004 |
| 768c | 765–785 | 2567.269 | 2567.265 | 0.004 |
| 795 | 793–810 | 2097.041 | 2097.051 | −0.010 |
A signal corresponding to phosphorylated peptide 710–716 (containing Ser 712) was also observed in the MALDI-TOF spectrum; MS/MS of this species exhibited loss of 98 D, confirming that it was phosphorylated.
ΔM = measured mass − theoretical mass.
Not confirmed by MS/MS, but inferred as the sole PKA consensus site within the phosphopeptide identified in the second column.
Because Ser768 thus appears to be phosphorylated in WT CFTR (with Flag) in resting oocytes, the relatively high basal activity of S768A CFTR channels can reasonably be attributed to absence of that phosphorylation and, hence, lack of its inhibitory effect on CFTR current. By the same token, our finding that serines 660, 700, 712, 737, and 795 were also phosphorylated means that these are candidates for the stimulatory site (or sites) underlying the observed activity of WT and S768A CFTR channels in resting oocytes.
Fractional Activation by Low [PKA] is Enhanced for S768A Mutant Channels
We examined the inhibitory influence of phosphoserine 768 by comparing channel function in inside-out patches excised from oocytes expressing WT (Fig. 5 A) or S768A mutant (Fig. 5 B) CFTR. Though the patches contained hundreds of channels, no appreciable currents flowed in either WT or S768A channels when their cytoplasmic surfaces were exposed to 2 mM MgATP, ∼2 min after patch excision. But both WT and S768A channels were activated when first a low (55 nM), and then a high (550 nM), concentration of PKA catalytic subunit was added to the MgATP. The fractional current activated by the low [PKA], relative to that subsequently elicited by 550 nM PKA, was evidently larger for S768A (Fig. 5, B and C; 0.57 ± 0.06, n = 5) than for WT channels (Fig. 5, A and C; 0.35 ± 0.03, n = 7). For both WT and S768A channels, 550 nM PKA was almost a saturating concentration, since fractional activation was already high at 220 nM PKA (I220/I550 values were 0.80 ± 0.03, n = 3 for WT, 0.86 ± 0.09, n = 3 for S768A).
At low [PKA], S768A channels were also activated more rapidly than WT CFTR channels, and after a shorter delay (Fig. 5, A and B). To compare activation rates, single exponentials were fitted to the later phases of the more-or-less sigmoid time courses of macroscopic current increase following step applications of 55 nM (green fit lines, Fig. 5, A and B) or 550 nM PKA (blue fit lines). The resulting time constants for WT (gray bars) and S768A (black bars) are summarized in Fig. 5 D. On exposure to 55 nM PKA, the current increase was twice as fast, on average, for S768A (τrelax = 25 ± 5 s, n = 5) as for WT CFTR (τrelax = 56 ± 12 s, n = 5; Fig. 5 D, top). However, when [PKA] was then raised to 550 nM, the current relaxed with similar rates for both mutant (τrelax = 8 ± 2 s, n = 9) and WT CFTR (τrelax = 8 ± 2 s, n = 11; Fig. 5 D, bottom).
Longer Burst Durations Underlie Higher Open Probability of S768A Channels
Channel gating characteristics underlying the differences in macroscopic currents were investigated in excised patches with fewer channels; currents from patches containing four WT and five S768A channels are shown in Fig. 6, A and B, respectively. Single-channel gating transitions were observed in 2 mM MgATP with 55 nM, followed by 550 nM, PKA, and then active channels were counted by locking them in the open-burst state by exposure to 2 mM pyrophosphate with 0.1 mM MgATP. Kinetic analysis yielded the mean open probabilities (Po), and interburst (closed), and open-burst, durations plotted in Fig. 6, C–E. The fractional increase in Po on increasing [PKA] from 55 to 550 nM replicated the observed macroscopic current ratios (approximately threefold for WT, Fig. 6 C, gray bars, <2-fold for S768A, Fig. 6 C, black bars; c.f. Fig. 5 C). However, absolute Po of S768A channels was at least 50% higher than that of WT at 550 nM PKA (0.48 ± 0.05, n = 6 for S768A vs. 0.29 ± 0.02, n = 9 for WT) and approximately threefold higher at 55 nM PKA (0.25 ± 0.03, n = 4 for S768A vs. 0.07 ± 0.01, n = 4 for WT; Fig. 6 C). At 55 nM PKA, interburst durations were somewhat shorter for S768A (2681 ± 534 ms, n = 4; Fig. 6 D, black bar) than for WT (3805 ± 454 ms, n = 4; Fig. 6 D, gray bar), a difference not apparent at 550 nM PKA (824 ± 62 ms, n = 6 for S768A vs. 958 ± 71 ms, n = 9 for WT; Fig. 6 D). Open burst durations of S768A channels were ≥2-fold longer (Fig. 6 E, black bars) than those of WT channels (Fig. 6 E, gray bars) both at 55 nM (941 ± 282 ms, n = 4 vs. 335 ± 49 ms, n = 6) and at 550 nM PKA (1036 ± 376 ms, n = 6 vs. 435 ± 38 ms, n = 10).
Interestingly, the open burst duration of S768A channels was reduced at least threefold (to τb = 234 ± 28 ms, n = 5) when measured shortly after withdrawal of PKA (see Fig. S1, available at http://www.jgp.org/cgi/content/full/jgp200409076/DC1), just as we have previously reported for WT CFTR channels (Csanády et al., 2000; Vergani et al., 2003). Also as we have found for WT CFTR, macroscopic current in excised patches containing phosphorylated S768A channels (just after PKA removal) was half-maximally activated by roughly 50 μM MgATP (Michaelis fit yielded K0.5 = 40 ± 2 μM MgATP; see Fig. S2, available at http://www.jgp.org/cgi/content/full/jgp200409076/DC1).
The enhanced fractional activation at low [PKA] of S768A channels relative to WT CFTR channels, evident in comparisons of amplitudes of macroscopic current (Fig. 5 C) or of Po (Fig. 6 C), implies that the mutant channels display a higher apparent “affinity” for PKA (at least, as assayed by channel activity). The implication is borne out by the summary of Po values plotted against [PKA] for S768A and WT channels (Fig. 7). Half-maximal activation of Po requires ∼150 nM PKA for WT channels, but only ∼70 nM PKA for S768A channels. The Hill coefficient was 1.5 for WT and 1.8 for S768A, suggesting that more than one site on a CFTR channel must be phosphorylated before its Po becomes measurable; this is also consistent with the sigmoid time courses of current activation observed at low [PKA] (Fig. 5, A and B).
Comparable Phosphorylation Time Courses of Recombinant WT and S768A R-domain Peptides
Our mass spectrometric and functional analysis of WT CFTR expressed in oocytes (Figs. 3 and 4) revealed in vivo phosphorylation of serines involved in channel activation as well as of the inhibitory Ser 768. That demonstrable phosphorylation of other sites notwithstanding, a possible explanation for the observed inhibitory effect of phosphoserine 768 on the activity of WT CFTR channels at low [PKA] (Fig. 7) is that early phosphorylation of Ser 768 impairs subsequent phosphorylation of serines in stimulatory sites. We addressed this possibility by examining the phosphorylation kinetics of His-tagged WT and mutant S768A R-domain protein incubated with PKA and γ32P-MgATP to see whether differences were discernible. In addition, to verify the inferred link between the major mobility shift of the R domain and phosphorylation of Ser 737 (Fig. 2), the other candidate inhibitory serine (Wilkinson et al., 1997), we studied phosphorylation of His-tagged R-domain proteins containing the single mutation S737A, or the double mutation S737A-S768A. The incremental mobility shifts already seen in Figs. 1 and 2 as phosphorylation of the R domain progressed were recapitulated in the His-tagged WT peptide (Fig. 8 A), and were not substantially altered by the S768A mutation in either the WT or S737A background (Fig. 8, B and D vs. A and C). As anticipated, however, the S737A mutation abolished the large mobility shift of both WT and S768A R-domain peptides (Fig. 8, C and D vs. A and B).
Figure 8.
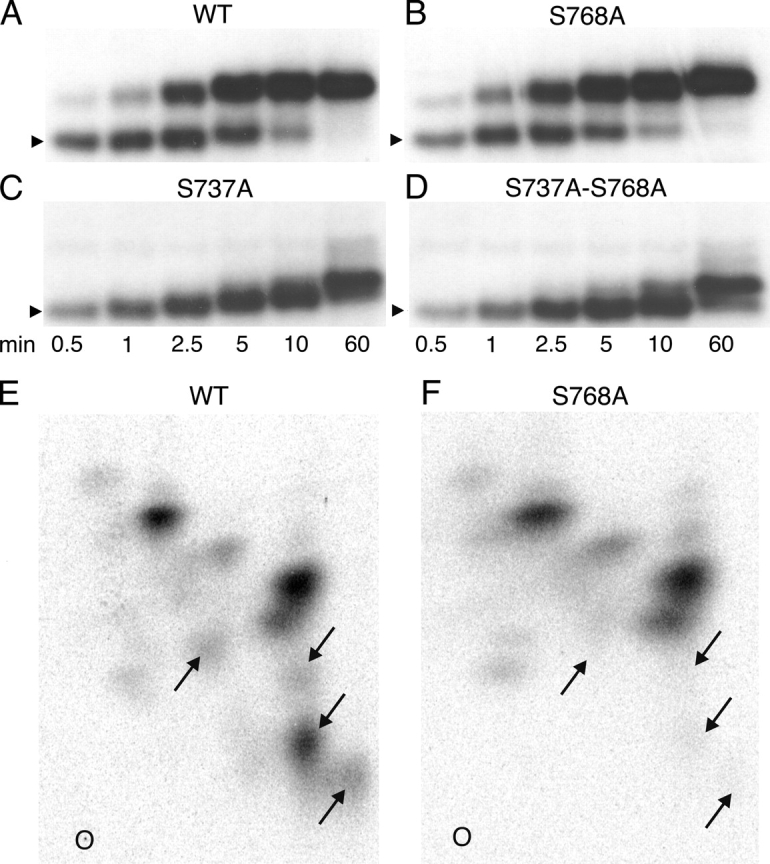
(A–D) Time courses of in vitro phosphorylation of R-domain peptides with COOH-terminal His tags. WT (A) and mutant R-domain peptides, with Ser768 replaced by alanine (S768A; B), Ser737 replaced by alanine (S737A; C), or Ser737 and Ser768 both replaced by alanine (S737A-S768A; D), were phosphorylated with 10 nM PKA and 5 μM γ32P-MgATP for 0.5–60 min as indicated. Samples were subjected to SDS-PAGE and analyzed by autoradiography; the arrowheads indicate relative molecular mass of 28 kD. The major mobility shift (to band 3; Figs. 1 and 2) was seen in the WT and S768A peptides, but not in the S737A or S737A-S768A peptides. The kinetics of R-domain phosphorylation, as demonstrated by the mobility shifts, was little altered in the two S768A mutants. (E and F) Two-dimensional tryptic phosphopeptide maps of His-tagged WT and S768A R-domain peptides phosphorylated in vitro as in A and B, but for 0.5 min with 10 nM PKA and 50 μM γ32P-MgATP, and then subjected to SDS-PAGE and analyzed by autoradiography. The lower radioactive bands were excised, digested overnight with 50 μg/ml TPCK-trypsin, and the digests separated on thin layer cellulose plates by electrophoresis at pH 3.5 in the first dimension and ascending chromatography in the second dimension. O, 0rigin; left, positive; right, negative. Four spots (arrows) in the WT R domain map are absent from the S768A map, but no similarly striking differences are seen in the pattern or intensity of other spots.
The separation of phosphoforms by SDS PAGE in Fig. 8, A–D, does not provide detailed information about phosphorylation levels of individual serines. To more closely investigate the possibility that phosphoserine 768 impairs phosphorylation of specific sites, 2-D phosphopeptide maps were prepared of WT and S768A R-domain peptides phosphorylated in vitro as in Fig. 8, A and B. If phosphoserine 768 inhibits or delays phosphorylation of other serines, then its absence from the mutant S768A R domain might result in new spots, or spots with higher intensity, in the S768A map. On the contrary, however, the principal difference between the two phosphopeptide maps of WT and S768A R-domain samples obtained from lower gel bands, after 30 s of phosphorylation in the presence of 50 μM MgATP, is the omission from the S768A map of four spots in the WT map (arrows, Fig. 8, E and F) that can therefore be presumed to contain phosphoserine 768. In particular, the strongest spots in the phosphopeptide map of phosphorylated S768A R domain are also seen in the WT map; the S768A map contains no obvious novel or intensified spots, and so provides no clear evidence for major enhancement of phosphorylation of other R-domain serines. Similar observations were made using phosphopeptide maps of either lower or upper bands from SDS-PAGE gels of R-domain samples obtained later during the course of phosphorylation (unpublished data).
DISCUSSION
By examining the kinetics of phosphorylation of purified R-domain peptide by PKA at low or high [MgATP], we could identify six distinct phosphoforms from their different mobilities in SDS-PAGE gels. Mass spectrometric analysis showed Ser 768 to be the most easily phosphorylated residue, and Ser 737 to be the site of the phosphorylation causing the major mobility shift. Mass spectrometry also revealed phosphorylation of Ser 768 in vivo in resting oocytes. We found that mutation of Ser 768 to alanine enhanced average CFTR current in excised patches at all levels of [PKA], but especially at low [PKA], confirming the inhibitory role of Ser 768 proposed earlier (Wilkinson et al., 1997). The principal mechanism of inhibition by phosphoserine 768 is a reduction of the open burst duration of CFTR channels, evident at both low and high [PKA]. The overall influence of phosphoserine 768 on WT CFTR channels is to shift their requirement for activation toward higher [PKA] levels, favoring their activation by only the strongest stimuli.
Ready Phosphorylation of Ser 768 Both In Vitro and In Vivo
Phosphorylation of Ser 768 in full-length CFTR treated with PKA in vitro has been shown by analysis of 2-D phosphopeptide maps (Cheng et al., 1991) or of mass spectra (Neville et al., 1997). Also, kinetic analysis of phosphorylation of several small synthetic peptides containing R-domain consensus sites revealed that the decapeptide containing Ser 768 was the preferred substrate for PKA (Picciotto et al., 1992). Possibly, the Ser-768 site (RRRQSV in human CFTR) is an excellent substrate because it is tribasic, allowing more extensive favorable interaction with the large negatively charged patch in the substrate-binding site on PKA catalytic subunit (Grant et al., 1996). Accordingly, our mass spectrometry of entire R domain (amino acids 645–835) revealed Ser 768 to be its most readily phosphorylated residue, as only phosphopeptides containing Ser 768 could be found in tryptic digests of the fastest-running band (band 1), the first to appear of the six phosphorylated bands eventually detectable in SDS-PAGE gels of phosphorylated R domain (Figs. 1 and 2). Because the mobility of band 1, though clearly phosphorylated, was indistinguishable from that of unphosphorylated R domain (band 0, Figs. 1 and 2), we may also conclude that any conformational change associated with phosphorylation of Ser 768 causes only a negligible alteration of the mobility of the R domain during SDS-PAGE (in contrast to our finding with Ser 737, see below).
In addition to Ser 768, we found (inferred for Ser 660) phosphoserines at positions 660, 700, 712, 737, and 795, by mass spectrometry of full-length CFTR isolated from resting, unstimulated oocytes (Fig. 4; summarized in Fig. 9). The finding corresponds well with the combined results from previous studies of in vivo phosphorylation of CFTR in transfected Cos cells (Cheng et al., 1991), or of native CFTR in T84 cells (Picciotto et al., 1992), in both cases after stimulation of PKA with forskolin, which showed that 2-D phosphopeptide maps contained phosphoserines 660, 700, 737, 795, 813, and perhaps 768.
Figure 9.

Summary of R-domain serines found to be phosphorylated by mass spectrometric analysis of trypic digests of bands (1, 2, 3, 4, 6 as indicated) from SDS-PAGE gels after in vitro phosphorylation (data from Fig. 2) or of full-length WT CFTR isolated from resting oocytes (in vivo; data from Fig. 4). Ser 768 is the first site phosphorylated, and is found phosphorylated in oocytes at rest; the major mobility shift to band 3 is associated with phosphorylation of Ser 737.
The stoichiometry of phosphorylation of the sites found phosphorylated in CFTR in resting oocytes could not be estimated from the ratios of signal intensities of tryptic peptides differing in mass by 80 D, because phosphorylation of the serines interferes, to varying degrees (Boyle et al., 1991; Neville et al., 1997), with trypsin's ability to cut next to the Arg and Lys residues common to PKA consensus sequences. Nevertheless, the sites phosphorylated by basal PKA activity in oocytes (serines 660, 700, 712, 737, 768, and 795) match fairly well the pattern observed in band 4 (serines 700, 712, 737, 768, and 795; Fig. 9), representing moderately phosphorylated R domain. This correspondence between phosphorylation patterns of the R domain in full-length CFTR in living cells and of an R-domain peptide purified after expression in bacteria, supports the assumption that the isolated R domain adopts a physiologically relevant conformation (Picciotto et al., 1992; Dulhanty and Riordan, 1994; Neville et al., 1997; Ostedgaard et al., 2000). Moreover, the similar pattern and kinetics of phosphorylation-induced mobility shifts observed in purified (Fig. 1 and Fig. 2 A), and in refolded (Fig. 8, A-D), isolated R-domain peptides, suggests that those two proteins interact with PKA in a similar manner and undergo the same conformational changes in response to phosphorylation, and, therefore, that they also share the same overall fold. This bolsters confidence in the R-domain peptide as a useful model for studies of regulatory phosphorylation of CFTR.
Inhibitory Influence of Phosphoserine 768 in WT CFTR
Mutational analyses of R-domain serines have generally assessed CFTR Cl− channel function after strong activation by PKA. Under those conditions, at high enough [MgATP], the Po of the mutants turned out to be little reduced (<2-fold) compared with that of WT CFTR channels, regardless of whether the mutations were single Ser-Ala substitutions at positions 660, 737, 795, or 813 (Winter and Welsh, 1997), or all four substitutions together (4SA; Winter and Welsh, 1997; Mathews et al., 1998), or even 10 simultaneous substitutions (10SA), including the same four sites plus serines at positions 422, 686, 700, 712, and 768, and Thr 788 (Mathews et al., 1998). In only one previous study was the influence of Ser-Ala mutations on the sensitivity of CFTR channels to activation by PKA examined (Wilkinson et al., 1997). These authors mutated (to alanine) 10 serines (at positions 641, 660, 670, 686, 700, 712, 737, 768, 795, and 813), individually and in selected combinations of two, three, or four, then expressed the mutant CFTR channels in Xenopus oocytes, and compared their sensitivity to activation by increasing [IBMX] in the presence of 10 μM forskolin with that of WT CFTR. Sensitivity was little altered by the mutations at 641, 686, or 712, but was diminished by mutation of serines 660, 670, 700, 795, or 813, implying that phosphorylation at the latter sites in WT CFTR enhances activation, at least at lower levels of phosphorylation. Importantly, a single Ser-Ala mutation at residue 737 or 768 increased sensitivity to activation by IBMX, suggesting that phosphoserines 737 and 768 play an inhibitory role in WT CFTR. The increase in sensitivity was largest (approximately fivefold) for S768A mutant CFTR (Wilkinson et al., 1997).
We confirm that inhibitory influence of phosphoserine 768 here by directly showing in excised patches that the sensitivity to activation by PKA catalytic subunit is shifted to lower [PKA] for S768A channels compared with WT CFTR channels (Figs. 5–7). Accordingly, we found that the basal level of [PKA] (expected to be low) in resting oocytes caused a greater activation of S768A channels than of WT channels (Fig. 3). Strictly, in the absence of structural information, we cannot rule out the possibility that the enhanced sensitivity of S768A channels results not from loss of an inhibitory PKA phosphorylation site but from a structural change caused by the Ser-Ala mutation itself. However, our evidence shows that Ser 768 is indeed phosphorylated, both in isolated R domain (Fig. 2) and in intact CFTR (Fig. 4), and unless phosphorylation of this conserved consensus serine is entirely without effect it must be either inhibitory or stimulatory. If the latter, any postulated stimulatory influence of the Ser-Ala mutation per se would have to be much larger than that of the stimulatory phosphorylation lost because of that mutation. It seems more reasonable to assume that, in the absence of phosphorylation, CFTR channel structure is not discernibly altered by this replacement of an exposed hydroxymethyl side chain with a methyl group, and hence that Ser 768 is an inhibitory phosphorylation site. If that assumption is correct, a corollary of our observation of a larger chloride conductance in unstimulated oocytes expressing S768A channels than in those expressing WT CFTR (Fig. 3) is that Ser 768 should be phosphorylated in WT channels in resting oocytes. This predicted phosphorylation of Ser 768 was verified by our mass spectrometric analysis of WT CFTR protein immunoprecipitated from membranes of resting oocytes (Fig. 4).
We can be sure that the large conductance observed for S768A mutant CFTR in resting oocytes (Fig. 3) was phosphorylation dependent, and did not reflect constitutive channel activity induced simply as a consequence of the Ser-Ala mutation itself, because the conductance was abolished by injection of the PKA inhibitor RpcAMPS. That high resting conductance of S768A channels thus depended on continuous phosphorylation of stimulatory sites by the relatively low PKA activity in the unstimulated oocytes, consistent with the enhanced sensitivity of S768A channels to PKA we found in excised patches (Fig. 7). The deactivation of the S768A channels upon kinase inhibition by RpcAMPS means that phosphatases must also have been continuously active in the resting oocytes. In support of this interpretation, 2 mM MgATP caused negligible opening of S768A channels in excised patches, ∼2 min after excision, before application of exogenous PKA, just as found for WT channels (Fig. 5, A and B), suggesting that, even in the excised patch, membrane-associated phosphatases dephosphorylate at least the stimulatory sites and so deactivate both WT and S768A CFTR channels (compare Chan et al., 2000; Csanády et al., 2000).
Mechanism of Inhibitory Influence of Phosphoserine 768 in CFTR Channels
Is phosphoserine 768 inhibitory because it impairs the phosphorylation of stimulatory sites, or does it dampen the stimulatory influence of those sites once they are phosphorylated, or does it more directly influence CFTR channel gating? Direct comparison of the gating kinetics of S768A and WT CFTR in excised patches showed that the Po of S768A channels was greater than that of WT channels at all [PKA] tested, and was at least 50% greater at saturating, 550 nM, PKA (Fig. 7), largely because the open burst duration of S768A channels was roughly double that of WT channels at 550 nM PKA. The burst durations of the two channel types were similarly different at 55 nM PKA, although Po was then much smaller (Fig. 6). This suggests that in WT channels Ser 768 is already phosphorylated at relatively low [PKA], and that phosphoserine 768 acts to somehow speed channel closure from open bursts. That influence is also evident at higher levels of phosphorylation, and hence of Po, and therefore likely reflects a direct effect on channel gating, rather than an indirect effect mediated via altered phosphorylation of other serine residues.
Such a direct effect of phosphoserine 768 to reduce burst duration would be expected, by itself, to result in a reduction in apparent affinity for activation of channel Po by PKA, as we observed (Fig. 7). Thus, assuming that the rate of channel closing from a burst, rOC, is independent of [PKA] (above a threshold level; consistent with Fig. 6), but that the rate of opening to a burst, rCO, increases with [PKA] along a hyperbolic curve characterized by Kr and rCO,max, then Po will also show a hyperbolic dependence on [PKA], with Po,max = rCO,max/(rCO,max + rOC) and KP = Kr(1 − Po,max) (compare Csanády et al., 2000). In that case, the observed ∼50% increase in Po,max of S768A channels (Fig. 7) alone would suffice to reduce the half-maximally activating [PKA], KP, by about one third compared with that for WT channels, without any influence of phosphoserine 768 on phosphorylation of other sites (and hence on Kr). However, the measured change in the apparent affinity for Po activation by PKA caused by the S768A mutation appeared larger than that, around twofold (Fig. 7), and so an additional indirect influence of this mutation on channel phosphorylation cannot be ruled out.
But we were unable to obtain clear evidence that phosphoserine 768 impaired phosphorylation of other WT R-domain serines. In our tests with isolated R-domain proteins, the time courses of mobility shifts that report progress of phosphorylation were not markedly different in the presence or absence of phosphoserine 768 (Fig. 8, A–D). Nor could we find in 2-D phosphopeptide maps any peptides phosphorylated to obviously higher stoichiometry in S768A than in WT R domain at early times during the phosphorylation time course (Fig. 8, E and F). Possible explanations for these failures to biochemically identify phosphorylation sites that might be influenced by phosphoserine 768 could be that those sites might not reside in the R domain, or that the influence might depend on the presence of another part of CFTR. With regard to the former possibility, serine 422 has been suggested to play a functional role in CFTR channel regulation (Chang et al., 1993) and can be phosphorylated in an NBD1-R peptide (Neville et al., 1997; Lewis et al., 2004), though it has never been shown to be phosphorylated in intact CFTR in vivo or in vitro. Also, the constitutive channel activity of split CFTR lacking the whole R domain (634–836) is further increased on exposure to PKA catalytic subunit (Csanády et al., 2000), implying a role for phosphorylation sites outside the R domain in controlling channel gating.
Our finding that, at low [PKA], the somewhat sigmoid macroscopic current activation was slower for WT than for S768A channels and occurred after a longer delay (Fig. 5) is consistent with an inhibitory influence of phosphoserine 768 on subsequent phosphorylation of other, activating, sites in WT CFTR. However, the left-shifted Po vs. [PKA] curve of S768A channels (Fig. 7) could by itself provide an explanation for their faster current activation, even if phosphorylation of all other serines were unaffected by the S768A mutation. In that case, for both WT and S768A channels, the [PKA] axis of the steady-state Po vs. [PKA] curves (Fig. 7) could be rescaled to read the same steady level of phosphorylation. Then, by hypothesis, on exposure to a given [PKA] the time course of phosphorylation would be the same for WT and S768A channels, equivalent to moving to the right at the same speed along the now rescaled abscissa of Fig. 7. This would result in a faster climb of the ordinate for S768A CFTR, and hence faster current rise, as observed (Fig. 5).
In summary, we have demonstrated a direct influence of phosphoserine 768 to speed closure from bursts in WT CFTR channels which, in principle, offers at least a qualitative explanation for every other effect we observe, including their reduced sensitivity to activation by PKA and slower activation time course, relative to S768A CFTR. We cannot rule out that phosphoserine 768 also plays an additional inhibitory role, by impairing phosphorylation of stimulatory sites, but we cannot argue convincingly that our observations demand any additional influence, nor could we obtain evidence for it in biochemical tests with isolated R domain.
Phosphorylation of Ser 737 Causes Principal Slowing of Mobility of R-domain Protein
Phosphorylation by PKA of R-domain (Picciotto et al., 1992; Borchardt et al., 1996; Kole et al., 1998) or NBD1-R-domain (Neville et al., 1998) proteins has previously been noted to cause conformational changes that slow their mobility in SDS-PAGE gels. We demonstrate here that PKA-mediated phosphorylation of an increasing number of sites in purified R-domain protein (Fig. 2 B) is accompanied by incremental reductions in mobility during SDS-PAGE that allow several phosphorylated bands to be discerned (Fig. 1 and Fig. 2 A). As addition of negative charge per se would tend to shift bands in the opposite direction, we can conclude that the bands represent distinct conformations of the R domain, each phosphorylated to a different degree (Fig. 2 B). Mass spectrometry and site-directed mutagenesis revealed that the major mobility shift was linked to phosphorylation of Ser 737 both at low and high [MgATP] (Fig. 2), and this requirement was confirmed by the absence of the large mobility shift after mutation of Ser 737 to Ala in either WT or S768A R-domain peptide (Fig. 8, A–D; see also Borchardt et al., 1996; Kole et al., 1998). The large mobility shift upon phosphorylation is also seen in full-length native CFTR in epithelial cells (Seibert et al., 1999), and in CFTR purified from insect cells (Kole et al., 1998). In contrast, phosphorylation of Ser 768 was accompanied by no discernible shift in R-domain mobility (Fig. 2), and mutating Ser 768 to Ala did not obviously alter the pattern of mobility shifts upon phosphorylation of the R domain (Fig. 8, A–D). Though we did not examine its consequences for channel gating, the S737A mutation has been reported to leave burst duration unchanged from that of WT CFTR (Winter and Welsh, 1997), whereas we found the S768A mutation to roughly double burst duration (Fig. 6).
We conclude that phosphoserines 737 and 768 each have qualitatively distinct, specific effects on the structure and function of CFTR channels, despite the fact that both might be inhibitory (Wilkinson et al., 1997). Such specific actions refute earlier conclusions that regulation of CFTR channels by PKA reflects relatively nonspecific accumulation of negative charge (Rich et al., 1993; Seibert et al., 1999) on an unstructured R domain (Ostedgaard et al., 2000), as does the finding that CFTR channels bearing combinations of mutations at inferred inhibitory (serines 737 or 768) and stimulatory (serines 660, 795, or 813) sites displayed sensitivities to activation by IBMX intermediate between those of the individual mutations (Wilkinson et al., 1997). Indeed, substantial structural differences have been demonstrated between nonphosphorylated and phosphorylated R-domain proteins (residues 595–831; Dulhanty and Riordan, 1994). Most likely, when PKA phosphorylates CFTR channels, there is an accumulation of both small and large structural changes that may differentially alter the manner in which nucleotide-dependent events at the NBDs are linked to the disposition of the gates that control Cl− ion flow through the transmembrane pore.
Control of Open Burst Duration by PKA
We have previously described PKA-induced changes in the open burst duration of native CFTR channels in patches excised from cardiac myocytes (Hwang et al., 1994), or of CFTR channels in patches from oocytes expressing human epithelial CFTR (Csanády et al., 2000; Vergani et al., 2003). Upon withdrawal of PKA, mean burst duration (which is independent of [MgATP]) quickly declines at least twofold (Csanády et al., 2000; Vergani et al., 2003), an effect we have attributed to rapid partial dephosphorylation, by membrane-bound phosphatases, of a certain site (or sites) that, when phosphorylated, stabilizes the open burst state (e.g., Gadsby and Nairn, 1999). Which are those sites? We show here that, although the Po of WT CFTR channels differed markedly during steady-state phosphorylation by low (55 nM) or high (550 nM) [PKA] (Fig. 6 C), confirming that CFTR's overall phosphorylation status was also different, the open burst durations were similar (Fig. 6 E), implying that the hypothesized burst-stabilizing sites were already phosphorylated at low [PKA].
Ser 768 cannot be the responsible site because the burst duration of S768A CFTR channels, like that of WT, was reduced at least twofold following withdrawal of PKA (Fig. S1). Our mass spectrometric analyses of isolated R domain (Fig. 2), and of CFTR in unstimulated oocytes (Fig. 4), suggested that other readily phosphorylated serines include those at positions 660, 700, 712, 737, and 795 (Fig. 9). However, individual mutation of serines 660, 737, 795, or 813 was reported to not change the burst duration of CFTR channels exposed to PKA (Winter and Welsh, 1997) and, in the presence of PKA, even the burst duration of 4SA or 10SA CFTR mutants was found to be like that of WT (Mathews et al., 1998). These findings would appear to narrow candidates for burst duration control to serines 670 and/or 753 in monobasic consensus sites.
An alternative possibility is that, by binding to its substrate sites in CFTR, PKA catalytic subunit itself stabilizes the channel open state, and that the stabilizing effect is lost upon PKA dissociation. A similar suggestion was offered to explain PKA activation of 10SA CFTR mutants (Chang et al., 1993). Interestingly, burst duration was short, and little affected by PKA withdrawal, in split CFTR channels, either simply severed after the R domain (between residues 835 and 837), or also severed at residue 633 and hence lacking the entire R domain (Csanády et al., 2000). So, whether PKA stabilizes burst duration by phosphorylation or binding, covalent attachment of the R domain to the COOH-terminal transmembrane domain seems to be required.
Why Inhibitory Phosphorylation Sites?
Because the Po of phosphorylated CFTR channels in excised patches is half maximal at <100 μM MgATP (e.g., Csanády et al., 2000; Vergani et al., 2003), the millimolar [MgATP] levels in living cells ought to maximally activate them, and so phosphorylation likely represents the major step controlling chloride conductance. What, then, is the consequence of inhibitory phosphorylation sites? We did not examine the suggested inhibitory effect of Ser 737 (Wilkinson et al., 1997), but the influence of phosphoserine 768 is evident in the steady-state Po vs. [PKA] plots of Fig. 7. The ready phosphorylation of Ser 768 in WT CFTR channels results in a somewhat greater reduction of Po at low than at high [PKA], so shifting the dose–response curve for WT to the right relative to that for S768A CFTR channels. Although it remains unclear how the reduced current at high [PKA] attributable to phosphoserine 768 could be beneficial, a possible advantage of the shifted activation curve is that responses to strong stimuli would be preserved whereas those to weak stimuli would be disfavored. The result would be an improved signal-to-noise ratio in the stimulation pathway.
Supplemental Material
Acknowledgments
We thank Atsuko Horiuchi and Peter Hoff for excellent technical assistance.
This work was supported by National Institutes of Health (NIH) DK51767 and TW05761 (to D.C. Gadsby) and by NIH Research Resource Grant RR00862 (to B.T. Chait). D.T. McLachlin was supported by a postdoctoral fellowship from the Canadian Institutes of Health Research.
Luis Reuss served as guest editor.
D. Seto-Young's present address is Division of Endocrinology, Beth Israel Medical Center, New York, NY 10003.
K.W. Chan's present address is Department of Physiology and Biophysics, Case Western Reserve University, Cleveland, OH 44106.
J. Qin's present address is Department of Biochemistry and Molecular Biology, Baylor College of Medicine, Houston, TX 77030.
D.T. McLachlin's present address is BioMedCom Consultants Inc., Montreal, Canada.
Abbreviations used in this paper: ABC, ATP-binding casette; CFTR, cystic fibrosis transmembrane conductance regulator; NBD, nucleotide binding domain; R, regulatory; WT, wild type.
References
- Aleksandrov, A.A., X.-B. Chang, L. Aleksandrov, and J.R. Riordan. 2000. The non-hydrolytic pathway of cystic fibrosis transmembrane conductance regulator ion channel gating. J. Physiol. 528:259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrov, L., A. Mengos, X. Chang, A. Aleksandrov, and J.R. Riordan. 2001. Differential interactions of nucleotides at the two nucleotide binding domains of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 276:12918–12923. [DOI] [PubMed] [Google Scholar]
- Aleksandrov, L., A.A. Aleksandrov, X.B. Chang, and J.R. Riordan. 2002. The first nucleotide binding domain of cystic fibrosis transmembrane conductance regulator is a site of stable nucleotide interaction, whereas the second is a site of rapid turnover. J. Biol. Chem. 277:15419–15425. [DOI] [PubMed] [Google Scholar]
- Basso, C., P. Vergani, A.C. Nairn, and D.C. Gadsby. 2003. Prolonged nonhydrolytic interaction of nucleotide with CFTR's NH2-terminal nucleotide binding domain and its role in channel gating. J. Gen. Physiol. 122:333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchardt, R., J. Kole, and J.A. Cohn. 1996. Phosphorylation of CFTR Ser-737 by protein kinase A. Pediatr. Pulmonol. Suppl. 13:212. [Google Scholar]
- Boyle, W.J., P. van der Geer, and T. Hunter. 1991. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 201:110–149. [DOI] [PubMed] [Google Scholar]
- Chan, K.W., L. Csanády, D. Seto-Young, A.C. Nairn, and D.C. Gadsby. 2000. Severed molecules functionally define the boundaries of the cystic fibrosis transmembrane conductance regulator's NH2-terminal nucleotide binding domain. J. Gen. Physiol. 116:163–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, X.-B., J.A. Tabcharani, Y.-X. Hou, T.J. Jensen, N. Kartner, N. Alon, J.W. Hanrahan, and J.R. Riordan. 1993. Protein kinase A (PKA) still activates CFTR chloride channel after mutagenesis of all 10 PKA consensus phosphorylation sites. J. Biol. Chem. 268:11304–11311. [PubMed] [Google Scholar]
- Chappe, V., D.A. Hinkson, L.D. Howell, A. Evagelidis, J. Liao, X.B. Chang, J.R. Riordan, and J.W. Hanrahan. 2004. Stimulatory and inhibitory protein kinase C consensus sequences regulate the cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA. 101:390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J., G. Lu, J. Lin, A.L. Davidson, and F.A. Quiocho. 2003. A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol. Cell. 12:651–661. [DOI] [PubMed] [Google Scholar]
- Cheng, S.H., D.P. Rich, J. Marshall, R.J. Gregory, M.J. Welsh, and A.E. Smith. 1991. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 66:1027–1036. [DOI] [PubMed] [Google Scholar]
- Csanády, L. 2000. Rapid kinetic analysis of multichannel records by a simultaneous fit to all dwell-time histograms. Biophys. J. 78:785–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády, L., K.W. Chan, D. Seto-Young, D.C. Kopsco, A.C. Nairn, and D.C. Gadsby. 2000. Severed channels probe regulation of gating of cystic fibrosis transmembrane conductance regulator by its cytoplasmic domains. J. Gen. Physiol. 116:477–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulhanty, A.M., and J.R. Riordan. 1994. Phosphorylation by cAMP-dependent protein kinase causes a conformational change in the R domain of the cystic fibrosis transmembrane conductance regulator. Biochemistry. 33:4072–4079. [DOI] [PubMed] [Google Scholar]
- Dulhanty, A.M., X.B. Chang, and J.R. Riordan. 1995. Mutation of potential phosphorylation sites in the recombinant R domain of the cystic fibrosis transmembrane conductance regulator has significant effects on domain conformation. Biochem. Biophys. Res. Commun. 206:207–214. [DOI] [PubMed] [Google Scholar]
- Gadsby, D.C., and A.C. Nairn. 1999. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol. Rev. 79:S77–S107. [DOI] [PubMed] [Google Scholar]
- Grant, B.D., I. Tsigelny, J.A. Adams, and S.S. Taylor. 1996. Examination of an active-site electrostatic node in the cAMP-dependent protein kinase catalytic subunit. Protein Sci. 5:1316–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner, K.P., A. Karcher, D.S. Shin, L. Craig, L.M. Arthur, J.P. Carney, and J.A. Tainer. 2000. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 101:789–800. [DOI] [PubMed] [Google Scholar]
- Hwang, T.C., G. Nagel, A.C. Nairn, and D.C. Gadsby. 1994. Regulation of the gating of cystic fibrosis transmembrane conductance regulator Cl channels by phosphorylation and ATP hydrolysis. Proc. Natl. Acad. Sci. USA. 91:4698–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara, H., and M.J. Welsh. 1997. Block by MOPS reveals a conformational change in the CFTR pore produced by ATP hydrolysis. Am. J. Physiol. 273:C1278–C1289. [DOI] [PubMed] [Google Scholar]
- Kaczmarek, L.K., K.R. Jennings, F. Strumwasser, A.C. Nairn, U. Walter, F.D. Wilson, and P. Greengard. 1980. Microinjection of catalytic subunit of cyclic AMP-dependent protein kinase enhances calcium action potentials of bag cell neurons in cell culture. Proc. Natl. Acad. Sci. USA. 77:7487–7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole, J., R. Borchardt, L.D. Howell, L. Chien, and J.A. Cohn. 1998. Regulation of the CFTR R-domain by phosphorylation of serine-737. Pediatr. Pulmonol. Suppl. 17:200. [Google Scholar]
- Krutchinsky, A.N., W. Zhang, and B.T. Chait. 2000. Rapidly switchable matrix-assisted laser desorption/ionization and electrospray quadrupole-time-of-flight mass spectrometry for protein identification. J. Am. Soc. Mass Spectrom. 11:493–504. [DOI] [PubMed] [Google Scholar]
- Lewis, H.A., S.G. Buchanan, S.K. Burley, K. Conners, M. Dickey, M. Dorwart, R. Fowler, X. Gao, W.B. Guggino, W.A. Hendrickson, et al. 2004. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 23:282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher, K.P., A.T. Lee, and D.C. Rees. 2002. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 296:1091–1098. [DOI] [PubMed] [Google Scholar]
- Mathews, C.J., J.A. Tabcharani, X.-B. Chang, T.J. Jensen, J.R. Riordan, and J.W. Hanrahan. 1998. Dibasic protein kinase A sites regulate bursting rate and nucleotide sensitivity of the cystic fibrosis transmembrane conductance regulator chloride channel. J. Physiol. 508:365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville, D.C., C.R. Rozanas, E.M. Price, D.B. Gruis, A.S. Verkman, and R.R. Townsend. 1997. Evidence for phosphorylation of serine 753 in CFTR using a novel metal-ion affinity resin and matrix-assisted laser desorption mass spectrometry. Protein Sci. 6:2436–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville, D.C., C.R. Rozanas, B.M. Tulk, R.R. Townsend, and A.S. Verkman. 1998. Expression and characterization of the NBD1-R domain region of CFTR: evidence for subunit-subunit interactions. Biochemistry. 37:2401–2409. [DOI] [PubMed] [Google Scholar]
- Ostedgaard, L.S., O. Baldursson, D.W. Vermeer, M.J. Welsh, and A.D. Robertson. 2000. A functional R domain from cystic fibrosis transmembrane conductance regulator is predominantly unstructured in solution. Proc. Natl. Acad. Sci. USA. 97:5657–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostedgaard, L.S., O. Baldursson, and M.J. Welsh. 2001. Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by its R domain. J. Biol. Chem. 276:7689–7692. [DOI] [PubMed] [Google Scholar]
- Picciotto, M.R., J.A. Cohn, G. Bertuzzi, P. Greengard, and A.C. Nairn. 1992. Phosphorylation of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 267:12742–12752. [PubMed] [Google Scholar]
- Qin, J., and B.T. Chait. 1997. Abstract identification and characterization of posttranslational modifications of proteins by MALDI ion trap mass spectrometry. Anal. Chem. 69:4002–4009. [DOI] [PubMed] [Google Scholar]
- Rich, D.P., R.J. Gregory, M.P. Anderson, P. Manavalan, A.E. Smith, and M.J. Welsh. 1991. Effect of deleting the R domain on CFTR-generated chloride channels. Science. 253:205–207. [DOI] [PubMed] [Google Scholar]
- Rich, D.P., H.A. Berger, S.H. Cheng, S.M. Travis, M. Saxena, A.E. Smith, and M.J. Welsh. 1993. Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by negative charge in the R domain. J. Biol. Chem. 268:20259–20267. [PubMed] [Google Scholar]
- Riordan, J.R., J.M. Rommens, B.S. Kerem, N. Alon, R. Rozmahel, Z. Grzelczak, J. Zielinski, S. Lok, N. Plavsic, J.L. Chou, et al. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 245:1066–1073. [DOI] [PubMed] [Google Scholar]
- Seibert, F.S., X.B. Chang, A.A. Aleksandrov, D.M. Clarke, J.W. Hanrahan, and J.R. Riordan. 1999. Influence of phosphorylation by protein kinase A on CFTR at the cell surface and endoplasmic reticulum. Biochim. Biophys. Acta. 1461:275–283 (Review). [DOI] [PubMed] [Google Scholar]
- Sheppard, D.N., and M.J. Welsh. 1999. Structure and function of the CFTR chloride channel. Physiol. Rev. 79:S23–S45. [DOI] [PubMed] [Google Scholar]
- Smith, P.C., N. Karpowich, L. Millen, J.E. Moody, J. Rosen, P.J. Thomas, and J.F. Hunt. 2002. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell. 10:139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani, P., A.C. Nairn, and D.C. Gadsby. 2003. On the mechanism of MgATP-dependent gating of CFTR Cl− channels. J. Gen. Physiol. 121:17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson, D.J., T.V. Strong, M.K. Mansoura, D.L. Wood, S.S. Smith, F.S. Collins, and D.C. Dawson. 1997. CFTR activation: additive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am. J. Physiol. 273:L127–L133. [DOI] [PubMed] [Google Scholar]
- Winter, M.C., and M.J. Welsh. 1997. Stimulation of CFTR activity by its phosphorylated R domain. Nature. 389:294–296. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
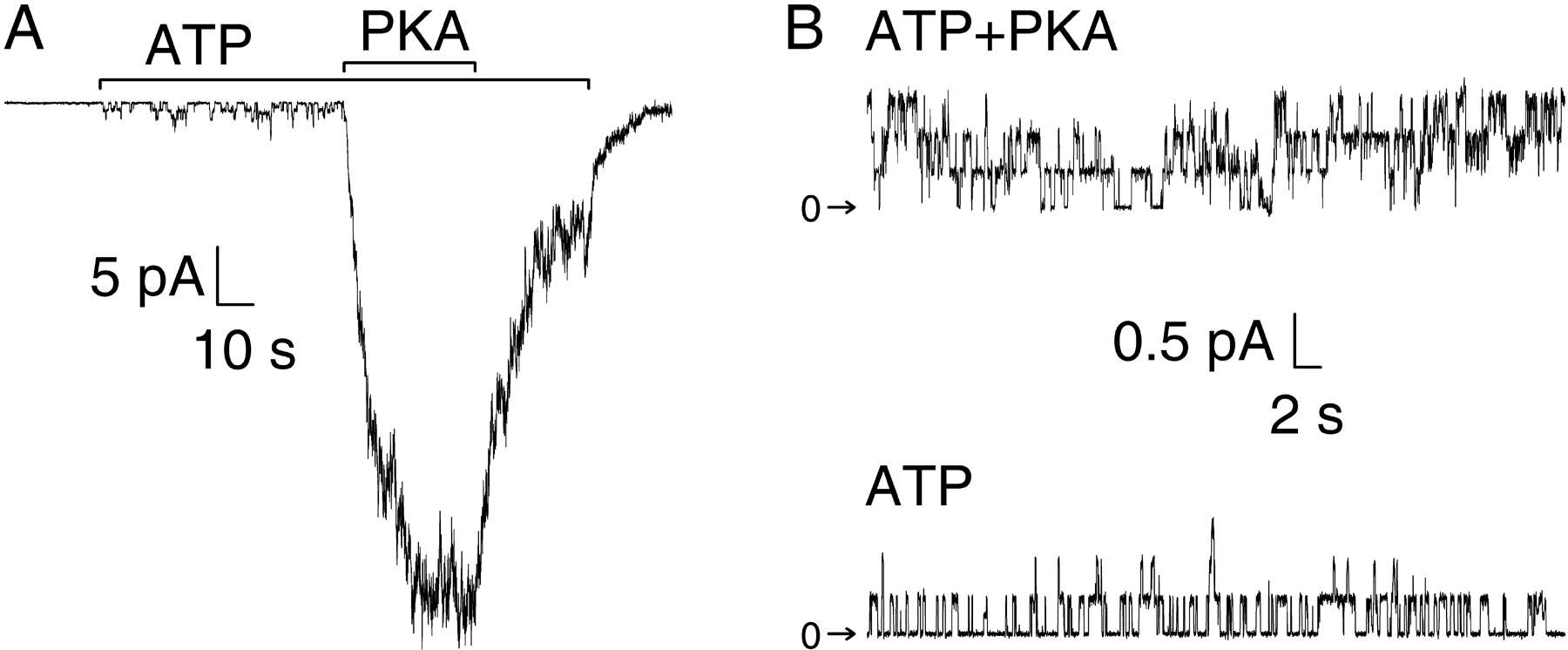{kind=link}
{kind=link}
{kind=link}
{kind=link}