

Abstract
The Na+ channel is the primary target of anticonvulsants carbamazepine, phenytoin, and lamotrigine. These drugs modify Na+ channel gating as they have much higher binding affinity to the inactivated state than to the resting state of the channel. It has been proposed that these drugs bind to the Na+ channel pore with a common diphenyl structural motif. Diclofenac is a widely prescribed anti-inflammatory agent that has a similar diphenyl motif in its structure. In this study, we found that diclofenac modifies Na+ channel gating in a way similar to the foregoing anticonvulsants. The dissociation constants of diclofenac binding to the resting, activated, and inactivated Na+ channels are ∼880 μM, ∼88 μM, and ∼7 μM, respectively. The changing affinity well depicts the gradual shaping of a use-dependent receptor along the gating process. Most interestingly, diclofenac does not show the pore-blocking effect of carbamazepine on the Na+ channel when the external solution contains 150 mM Na+, but is turned into an effective Na+ channel pore blocker if the extracellular solution contains no Na+. In contrast, internal Na+ has only negligible effect on the functional consequences of diclofenac binding. Diclofenac thus acts as an “opportunistic” pore blocker modulated by external but not internal Na+, indicating that the diclofenac binding site is located at the junction of a widened part and an acutely narrowed part of the ion conduction pathway, and faces the extracellular rather than the intracellular solution. The diclofenac binding site thus is most likely located at the external pore mouth, and undergoes delicate conformational changes modulated by external Na+ along the gating process of the Na+ channel.
Keywords: diclofenac, inactivation, permeation, Na+, use-dependent inhibitor
INTRODUCTION
Voltage-gated Na+ channels are rapidly activated and then inactivated upon membrane depolarization, producing transient inward Na+ currents that initiate action potentials in most physiological conditions. Na+ channels constitute the primary target of many local anesthetic and anticonvulsant drugs, which typically have much higher binding affinity to the inactivated channel than to the resting channel and therefore show a use (or holding voltage)-dependent inhibitory effect on the Na+ currents (Bean et al., 1983; Matsuki et al., 1984; Butterworth and Strichartz, 1990; Kuo and Bean, 1994; Xie et al., 1995; Kuo and Lu, 1997; Kuo et al., 1997). Recently we showed that the binding rates of these use-dependent inhibitors to the open Na+ channel are even faster than those to the inactivated channel, and thus the favorable drug binding conformation of the receptor is probably made during channel activation (Yang and Kuo, 2002). However, a complete quantitative characterization of the changing affinity toward different gating states (at least the resting, activated, and inactivated states) of the Na+ channel has never been reported for any of the use-dependent inhibitors of this kind. A picturesque biophysical demonstration of the shaping of a use-dependent receptor (from an unfavorable into a favorable drug binding site) along the basic gating process of the Na+ channel thus is still lacking.
We have proposed that anticonvulsants phenytoin, carbamazepine, and lamotrigine probably bind to the same receptor area located in the ion conduction pathway of Na+ channels with a common diphenyl structural motif (Kuo et al., 2000; Yang and Kuo, 2002). The exact location of the binding site(s) for the use-dependent inhibitors is of great pharmacological and physiological interests, because it may shed light on the molecular mechanism underlying not only the actions of many clinically important drugs but also some essential gating conformational changes of the Na+ channel. However, there have been different ideas on whether the use-dependent drug binding site(s) face the extracellular or intracellular side in the pore. It was proposed that the local anesthetics and anticonvulsants bind to the cytoplasmic side of the Na+ channel, chiefly based on two lines of experimental findings. First, point mutation of a few presumably intracellular residues in the sixth transmembrane segment of domain IV (IVS6) reduced the affinity of local anesthetic and anticonvulsant drugs to the inactivated Na+ channels (Ragsdale et al., 1994, 1996). Second, the permanently charged quaternary analogs of local anesthetic drugs could block neuronal Na+ channels only when applied to the intracellular side but not to the extracellular side (Frazier et al., 1970; Strichartz, 1973; Hille, 1977). However, because IVS6 is an important gating component involved in both Na+ channel activation and inactivation (McPhee et al., 1995; Doyle et al., 1998; Jiang et al., 2002), mutations in IVS6 might well alter the activated and inactivated Na+ channel conformations and thus allosterically reduce drug affinity. This could also be one of the reasons why a point mutation in IVS6 (F1762A) was shown to affect the binding of both internal and external quaternary ammoniums (Qu et al., 1995). It might also be noted that the blocking effect of internal and external quaternary ammoniums was Na+ channel isoform specific. In contrast to the foregoing findings in neuronal Na+ channels, membrane-impermeant quaternary ammoniums significantly blocked the native and cloned cardiac Na+ channels when applied to either side of the cell membrane, and the effect of external quaternary ammoniums was significantly altered by the other extracellular pore blockers such as tetrodotoxin or mutations of some isoform-specific residues located on the external side of the channel (Alpert et al., 1989; Qu et al., 1995; Sunami et al., 2000; Lee et al., 2001). Moreover, it has been shown that anticonvulsants phenytoin, carbamazepine, and lamotrigine are effective neuronal Na+ channel inhibitors when applied externally but not applied internally (with continuous perfusion of the external solution in an internally dialyzed cell; Kuo, 1998). The exact location of the binding site(s) for the use-dependent blocking agents thus remains unsettled and needs to be characterized in more detail.
Diclofenac is a widely prescribed anti-inflammatory cyclooxygenase inhibitor rather than an anticonvulsant, but it also contains a diphenyl structural motif similar to that in carbamazepine, phenytoin, and lamotrigine. Detailed analysis of the three–dimensional structure of these drugs with molecular modeling further indicates that the spatial orientation of the diphenyl motif in carbamazepine is even more similar to that in diclofenac than to that in phenytoin or lamotrigine (Fig. 1 A; Table I; Kuo et al., 2000). In this study, we show that diclofenac also inhibits Na+ currents in a holding potential–dependent (“use-dependent”) fashion like that of carbamazepine. We characterize the affinity between diclofenac and the Na+ channel not only for the resting and inactivated states but also for the activated (open) state of the channel, demonstrating the gradual shaping of a use-dependent receptor along the gating process. Most interestingly, diclofenac cannot mimic the pore-blocking action of carbamazepine in the presence of 150 mM external Na+, but is dramatically turned into an effective pore blocker of the Na+ channel when there is no external Na+. These findings suggest that diclofenac binds to a receptor whose conformation is finely tuned not only by channel gating but also by external Na+, and whose location is most likely at the junction of the widened external vestibule and the narrow part of the ion conduction pathway.
Figure 1.

Chemical formulae of the drugs and inhibition of neuronal Na+ currents by diclofenac. (A) Chemical formulae of carbamazepine and diclofenac. Both drugs contain the diphenyl structural motif (see Table I for more detailed comparison of the structural features). (B) Na+ currents in control, 10 μM, or 30 μM diclofenac were recorded in a neuron. The cell was held at −120 mV and then stepped to 0 mV to elicit Na+ currents. 10–30 μM diclofenac produces only equivocal inhibition of the Na+ current. The dotted line indicates the zero current level. (C) The same experiment was repeated in the same cell as in B except that the holding potential was changed to −70 mV. The control current is much smaller than that in B but is scaled to the same size for a better comparison (note the difference in the vertical scale bars). In contrast to the findings in part B, 10–30 μM diclofenac has a pronounced and dose-dependent inhibitory effect on the Na+ current. The dotted line indicates the zero current level.
TABLE I.
Configuration of the Two Benzene Rings in Diclofenac and Three Anticonvulsant Drugs a
| Stem bond angleb | Distancec | Distance | Distance | Torsion angled | |
|---|---|---|---|---|---|
| Degree | Å, center–center | Å, C1–C1 | Å, C4–C4 | Degree | |
| Diclofenac | 123.3 | 5.00 | 2.54 | 7.46 | −55.5, −29.2e |
| Carbamazepine | 118.5 | 4.93 | 2.35 | 7.47 | −63.1, 62.4 |
| Phenytoin | 111.0 | 4.84 | 2.52 | 7.12 | −57.4, −56.5 |
| Lamotrigine | NA | 4.38 | 1.50 | 7.10 | −59.9, 122.1 |
Models of the tertiary drug structure were built by the Macromodel version 4.5 program (Department of Chemistry, Columbia University, 1994). More than 200 conformations were picked, and energy minimization using the Monte Carlo method and MM2 force-field parameters was then exercised. Most of the data have been published before in a more comprehensive chart (Kuo et al., 2000).
In each drug molecule, the carbon or nitrogen atom that the two benzene rings directly connect to is designated as the pivotal atom. The C–C or N–C bond connecting the pivotal atom and the benzene ring is designated as the stem bond. The angle made by the two stem bonds at the pivotal atom is the stem bond angle. In lamotrigine, the two benzene rings are connected to each other directly, and thus no stem bond or stem bond angle could be defined.
The distance between the two benzene rings are measured between the centers (center–center), the two C1 atoms (C1–C1), or the two C4 atoms (C4–C4) of the rings. C1 is the carbon atom forming the stem bond with the pivotal atom, or in lamotrigine, it is the atom directly connected to the other benzene ring. C4 is the atom farthest to C1 in the ring.
The torsion angle defines rotation of the benzene ring. The stem bond is the rotating axis, and the plane formed by the two stem bonds is the reference plane. For lamotrigine, there is a hypothetical reference plane that contains both rings by neglecting ring rotation first. Each ring then rotates with the bond connecting the two rings being the rotating axis. Positive angle means clockwise rotation if one views the ring from the pivotal atom.
The benzene rings in diclofenac have more flexible torsion angles than the other drugs. There are more than five similarly low-energy conformers with torsion angles distributed between −55.5, −29.2 and 43.2, 53.2 degrees.
MATERIALS AND METHODS
Cell Preparation
CA1 region was dissected from coronal slices of the whole brain of 7- to 14-d-old Long-Evans rats and cut into small chunks. Tissue chunks were treated with 0.5 mg/ml type XI trypsin (Sigma-Aldrich) at 37°C for 5–10 min in dissociation medium (in mM, 82 Na2SO4, 30 K2SO4, 3 MgCl2, 5 HEPES, 0.001% phenol red indicator, and trypsin, pH 7.4,). Each time when cells were needed, two to three chunks were picked and triturated to release single neurons.
Whole-cell Recording
The dissociated neurons were put in a recording chamber containing Tyrode's solution (in mM, 150 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, and 10 HEPES, pH 7.4). Whole-cell voltage clamp recordings were obtained using fire-polished pipettes pulled from borosilicate micropipettes (OD 1.55–1.60 mm; Hilgenberg Inc.) and fire polished. The input resistance was 1–2 MΩ when filled with the standard “150 mM Cs+” (or “0 mM Na+”) internal solution containing (in mM) 75 CsCl, 75 CsF, 2.5 MgCl2, 5 HEPES, 2.5 EGTA, pH adjusted to 7.4. Some of the experiments were performed with the 150 mM Na+ (or “0 mM Cs+”) internal solutions containing the same component as the 150 mM Cs+ internal solution except that the 75 mM CsCl and 75 mM CsF were replaced by 75 mM NaCl and 75 mM NaF. Seal was formed and the whole-cell configuration obtained in Tyrode's solution. The cell was then lifted from the bottom of the chamber and moved in front of an array of flow pipes (Microcapillary from Hilgenberg Inc.; content 1 μl, length 64 mm) emitting either control or drug-containing external recording solutions. The standard external solution was Tyrode's solution, but in some experiments 150 mM Cs+ (or 0 mM Na+) external solution containing the same constituents of Tyrode's solution except that 150 mM NaCl was replaced by 150 mM CsCl was used. Carbamazepine and diclofenac (Sigma-Aldrich) were dissolved in dimethylsulfoxide to make 100 mM stock solutions, which were then diluted into Tyrode's solution to attain the final concentrations desired. Currents were recorded at room temperature (∼25°C) with an Axoclamp 200A amplifier, filtered at 5 kHz with four-pole Bessel filter, digitized at 50–200 μs intervals, and stored using a Digidata-1200 analogue/digital interface as well as the pCLAMP software (Axon Instruments).
Molecular Biology
The plasmid pNa200 containing the rat brain type IIA (rBIIA) Na+ channel α-subunit cDNA is a gift from A.L. Goldin (University of California, Irvine, CA). F1489Q point mutation (West et al., 1992) was done with pNa200 template and PCR-based method (QuikChange Mutagenesis kit; Stratagene). F1489Q/F1651A double mutation was made by the same method with the constructed F1489Q mutation-containing pNa200 as the template. DNA sequencing was performed, and two independent clones were tested to exclude effects of inadvertent mutations. The full-length cRNA transcript was synthesized from the pNa200 containing F1489Q or F1489Q/F1651A mutations using the T7 mMESSAGE mMACHINE transcription kit (Ambion). Defolliculated Xenopus oocytes (stage V–VI) were then injected with the cRNA transcript and maintained at 18°C for 1–7 d before electrophysiological studies.
Intracellular Recording
Macroscopic Na+ currents were examined by two-microelectrode voltage-clamp recordings in oocytes. During recording, the oocyte was continuously perfused with ND-96 solution (in mM, 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, pH 7.6) or ND-22 solution (in mM, 22 NaCl, 74 CsCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, pH 7.6) that did or did not contain the drugs. Both voltage-sensing and current-passing electrodes were filled with 3 M KCl and had a resistance of 0.1–0.8 MΩ. Membrane potential was controlled by a two-electrode voltage-clamp amplifier with a virtual ground circuit (model OC-725C; Warner Instrument). Currents were recorded at room temperature (∼25°C), filtered at 5 kHz, digitized at 20–100 μs interval, and stored using a Digidata-1200 analogue/digital interface as well as the pCLAMP software (Axon Instruments). All statistics in this study are given as mean ± SEM.
RESULTS
Different Inhibitory Effect of Diclofenac on Neuronal Na+ Currents Elicited from Different Holding Potentials
Fig. 1 B shows that 10–30 μM diclofenac produces negligible inhibition of the macroscopic neuronal Na+ currents elicited from a holding potential of −120 mV. However, the same concentrations of diclofenac significantly inhibits the currents elicited from a more depolarized holding potential of −70 mV, demonstrating a voltage (holding potential)-dependent inhibitory effect of diclofenac on Na+ channels.
Measurement of the Binding Affinity of Diclofenac to the Inactivated Neuronal Na+ Channel
We characterize the voltage-dependent effect of diclofenac in more detail by examination of the inactivation curve, which describes the voltage-dependent steady-state distribution of the Na+ channel between the resting (R) and the inactivated (I) states (Scheme 1;Fig. 2).
(SCHEME 1).

Figure 2.
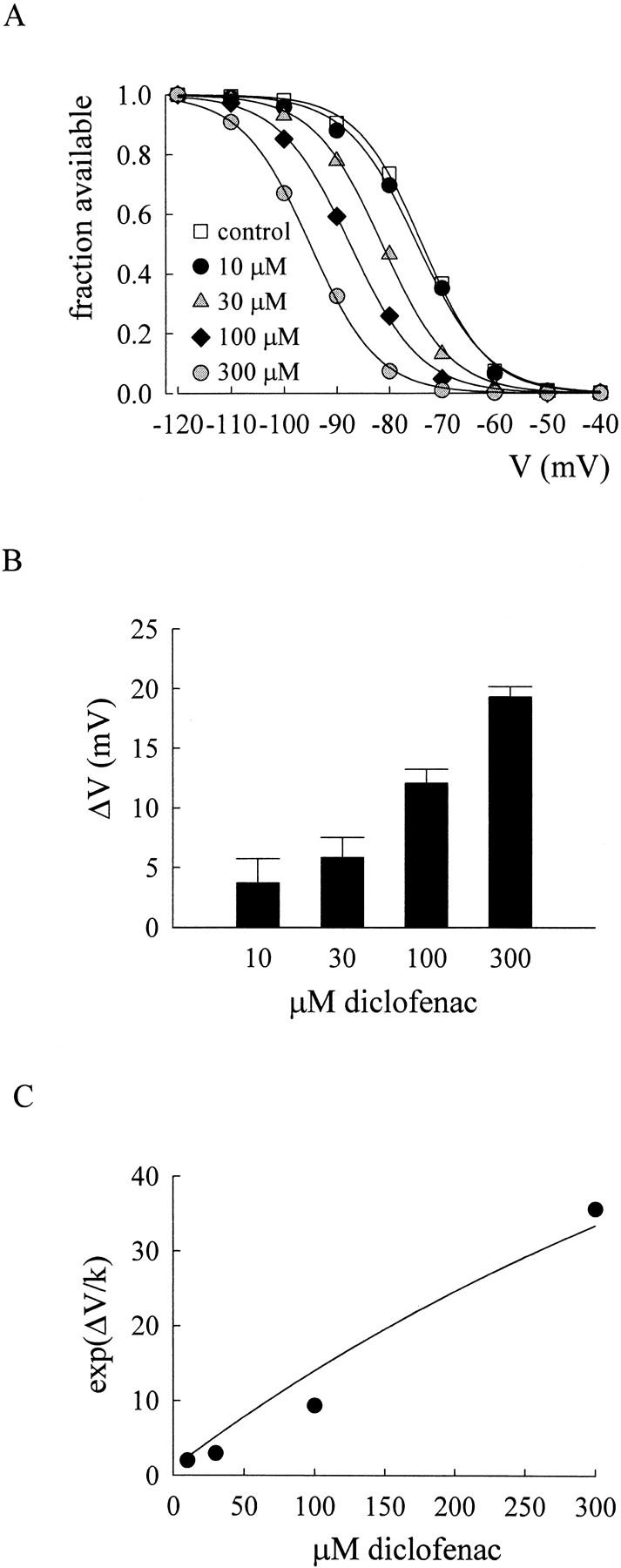
Shift of the inactivation curve of neuronal Na+ channels by diclofenac. (A) The inactivation curves are documented in the absence or presence of different concentrations of diclofenac. The neuron was held at −120 mV and stepped to the indicated inactivating prepulses for 9 s. The neuronal Na+ currents were then measured at 0 mV right after the 9-s prepulses. The available fraction is defined as the normalized current relative to the current elicited after a prepulse of −120 mV. The solid lines are the fits to each set of data with a Boltzmann function 1/(1 + exp((V − Vh)/k)), where the Vh values (in mV) are −73.7, −74.6, −81.4, −87.5, and −95.2 in 0 (control), 10, 30, 100, and 300 μM diclofenac, respectively. Because the slope of the fitting curves in different diclofenac concentrations always stay close to the slope in control, the above Vh values are obtained from the fits with a fixed k value of 5.4 (the mean of the k values in control). (B) The mean value of the shift of the inactivation curve by different concentrations of diclofenac. The shift (ΔV) is determined by the difference between the Vh values in control and in different concentrations of diclofenac. The mean ΔV values (in mV) are 3.7 ± 2.1 (n = 3), 5.8 ± 1.7 (n = 5), 12.1 ± 1.2 (n = 6), and 19.3 ± 0.9 (n = 12) for 10, 30, 100, and 300 μM diclofenac, respectively. (C) The mean ΔV values in B and a k value of 5.4 are used to calculate exp(ΔV/k), which is plotted against diclofenac concentrations to determine the dissociation constant of diclofenac binding to the inactivated channel. The solid line is the fit of the form (see Eq. 1 in the text): exp(ΔV/k) = [1 + (D/6.8)]/[1 + (D/880)], where a KR value of 880 μM is taken from Fig. 7 D, and D is the diclofenac concentration in μM.
We have seen in Fig. 1 B that diclofenac evidently inhibits Na+ currents elicited from a holding potential of −70 mV, where many channels would occupy the inactivated state, but not the currents elicited from −120 mV, where most channels shall stay in the resting state. Diclofenac thus probably binds much more tightly to the inactivated channel than to the resting channel. In other words, binding of diclofenac would favor redistribution of the channel to the inactivated state and consequently decrease the Na+ currents. Let K I and K R be the dissociation constants of diclofenac binding to the inactivated and resting channels, respectively, and D be the concentration of diclofenac. Based on the simplified gating Scheme 1, diclofenac should keep the shape (the slope factor k) of the inactivation curve unchanged but shift the inactivation curve leftwardly on the voltage axis, with the extent of the shift (ΔV) determined by K I, K R, and D according to the following equation (Bean et al., 1983; Bean, 1984):
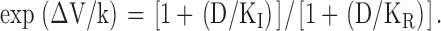 |
(1) |
Fig. 2 (A and B) shows that diclofenac indeed dose dependently shifts the inactivation curve without significant alteration of the shape of the curve. The exp(ΔV/k) values in different concentrations of diclofenac are fitted with Eq. 1 and a K R value of 880 μM (see Fig. 7 D). The dissociation constant between diclofenac and the inactivated Na+ channel (K I) obtained from the fit is ∼7 μM (Fig. 2 C), a value smaller than the K I of carbamazepine in the same preparation (i.e., ∼25 μM; Kuo et al., 1997). Because the K R value is probably >880 μM (see Fig. 7 D), diclofenac binding to the inactivated Na+ channel should be at least 100 times more tightly than to the resting Na+ channel.
Figure 7.
Diclofenac enhancement and carbamazepine inhibition of the F1489Q/F1651A double mutant current. The experimental protocol is the same as that in Fig. 5. (A) The inactivation phase of the macroscopic current in the double mutant is more completely abolished (i.e., the current decays even slower in the double mutant) than in the F1489Q mutant. Also, diclofenac shows more enhancement of the peak current evoked by a pulse at 0 mV in the double mutant than in the F1489Q mutant (Fig. 5 A). In sharp contrast, carbamazepine evidently reduces the double mutant current. The dotted line indicates the zero current level. (B) Diclofenac has an even stronger enhancement effect on the double mutant current evoked by a pulse at −20 mV in the same oocyte as that in A. (C) Another oocyte expressing the F1489Q/F1651A double mutant channel was exposed to repeated pulses to −20 mV for 100 ms to elicit a series of mutant Na+ currents (sweeps) with a 4-s interpulse interval at −120 mV. The black horizontal bars indicate the external solutions (ND-96 only or ND-96 with different concentrations of diclofenac) in which the oocyte stayed. The double mutant current peak is greatly enhanced by diclofenac in a concentration-dependent fashion. The diclofenac effect can be readily “washed out” when the external solution is switched back to the control ND-96 solution. (D) Estimation of the affinity of diclofenac binding to the resting Na+ channel by analysis of the increase of the F1489Q/F1651A double mutant current. The F1489Q/F1651A double mutant peak current (the maximal inward current) elicited by a −20 mV pulse in different concentrations of diclofenac is normalized to that in the control (no diclofenac) condition to obtain the relative peak current. The enhancement effect on the currents is diclofenac concentration dependent and can be well described by a one-to-one binding curve of the form: relative peak current = (1 + 2.7*D/880)/(1 + D/880), where D is the concentration of diclofenac in μM (see text for more details).
Mutual Exclusion of Diclofenac and Carbamazepine Binding to the Inactivated Na+ Channel
Diclofenac and carbamazepine share a diphenyl structural motif and both stabilize Na+ channel inactivation with much higher binding affinity to the inactivated channel than to the resting channel (Figs. 1 and 2). We therefore investigate the interaction between diclofenac and carbamazepine binding to the inactivated Na+ channel by examination of the shift of the inactivation curve in the simultaneous presence of both drugs (Fig. 3, see also Kuo, 1998). We have described that exp(ΔV/k) is equal to [1+(D/K I)]/[1+(D/K R)] (Eq. 1) in the presence of one drug. Because of the relatively large value of K R for either diclofenac or carbamazepine, D/K R could be neglected in equation 1 for simplicity. The equation then becomes
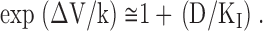 |
(2) |
Figure 3.

Shift of the inactivation curve of neuronal Na+ channels by carbamazepine and diclofenac. (A) Simplified diagrams describing the possible action of the drugs on different Na+ channel gating states. R and I denote the resting and the inactivated states of the steady-state channel, respectively. D1 and D2 denote two different drugs. V denotes that the distribution between R and I states is voltage dependent. The one-site model is illustrated in the upper panel, where D1 and D2 share the same receptor site and thus the inactivated channel can only be bound with one drug molecule at a time. For the sake of simplicity, we did not show a scheme illustrating that the two different drugs bind to two different inactivated conformations allosterically incompatible with each other. However, the basic quantitative analysis (e.g., Eq. 3 in the text) would be the same whether binding of one drug precludes binding of the other directly or allosterically. The lower panel represents the gating scheme of the two-site model, where the two drugs have separate and unrelated binding sites and thus could doubly occupy the inactivated channel. (B) The experimental protocol and the fitting procedures (with a fixed k value of 5.4) are similar to the case in Fig. 2 A. The Vh values of the inactivation curves are −48.5, −64.7, −69.2, and −67.3 mV for control, 300 μM carbamazepine, 300 μM diclofenac, and 150 μM carbamazepine plus 150 μM diclofenac, respectively. (C) Shift of the inactivation curve in the presence of diclofenac and/or carbamazepine. The shift (ΔV) is calculated by the difference between the Vh values in control and in the presence of different drugs. The mean values of ΔV (in mV) are 19.3 ± 0.9 (n = 12), 13.9 ± 1.4 (n = 4), and 15.9 ± 1.5 (n = 9) for 300 μM diclofenac, 300 μM carbamazepine, and 150 μM carbamazepine plus 150 μM diclofenac, respectively. The data of 300 μM diclofenac are from the same cells measured in Fig. 2 B. The predicted shift in the presence of both drugs by the one-site model (the striped bar) is much closer to the experimental data (the black bar) than the prediction by the two-site model (the white bar) (see text for more details).
In the presence of two different drugs, the channel may or may not be doubly occupied by the two different drug molecules. If both drugs bind to the same receptor in the inactivated channel, or if binding of one drug allosterically antagonizes binding of the other, then the channel cannot be doubly occupied (see the one-site model in Fig. 3 A; Kuo, 1998). Based on the one-site model and Eq. 2, the shift (ΔV) of the inactivation curve in the mixture of two drugs would be given by
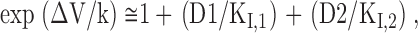 |
(3) |
where D1 and D2 are the concentrations of the two drugs, and K I,1 and K I,2 are the K I values of the two drugs, respectively. On the other hand, if the two drugs have separate and unrelated binding sites, then they may doubly occupy the channel (see the two-site model in Fig. 3 A; Kuo, 1998). In this case, there would be an extra state (i.e., ID1D2) signaling simultaneously double occupancy of the channel by the two drugs. The shift (ΔV) in the mixture of two drugs then would be given by
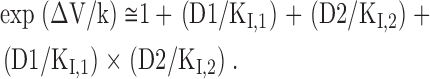 |
(4) |
It is evident that the one-site model and the two-site model predict very different shifts of the inactivation curve in a mixture of two drugs, especially in high drug concentrations. With a K I value of ∼7 μM for diclofenac (Fig. 2), a K I value of ∼25 μM for carbamazepine (Kuo et al., 1997), and a slope factor k of 5.4, the predicted shift (ΔV) by Eq. 2 in the presence of a single drug would be 20.4 mV and 13.9 mV for 300 μM diclofenac and 300 μM carbamazepine, respectively. Both values are very close to the experimental data (19.3 and 13.9 mV for diclofenac and carbamazepine, respectively; Fig. 3, B and C). On the other hand, in the presence of a mixture of 150 μM diclofenac and 150 μM carbamazepine, the predicted shift (ΔV) of the inactivation curve would be highly model dependent (18.1 mV by the one-site model and 27.3 mV by the two-site model). The experimental data (∼16 mV; Fig. 3 C) are evidently very much consistent with the prediction by the one-site model rather than the two-site model. This finding strongly argues against simultaneous (double) occupancy of the inactivated Na+ channel by diclofenac and carbamazepine. Diclofenac and carbamazepine thus cannot have completely unrelated binding sites in the inactivated Na+ channel. The two drugs must either bind to the same receptor site or bind to two separate sites whose conformational changes are allosterically and strictly interconnected with each other.
Lack of Pore-blocking Effect of Diclofenac on the Open Na+ Channel in 150 mM External Na+
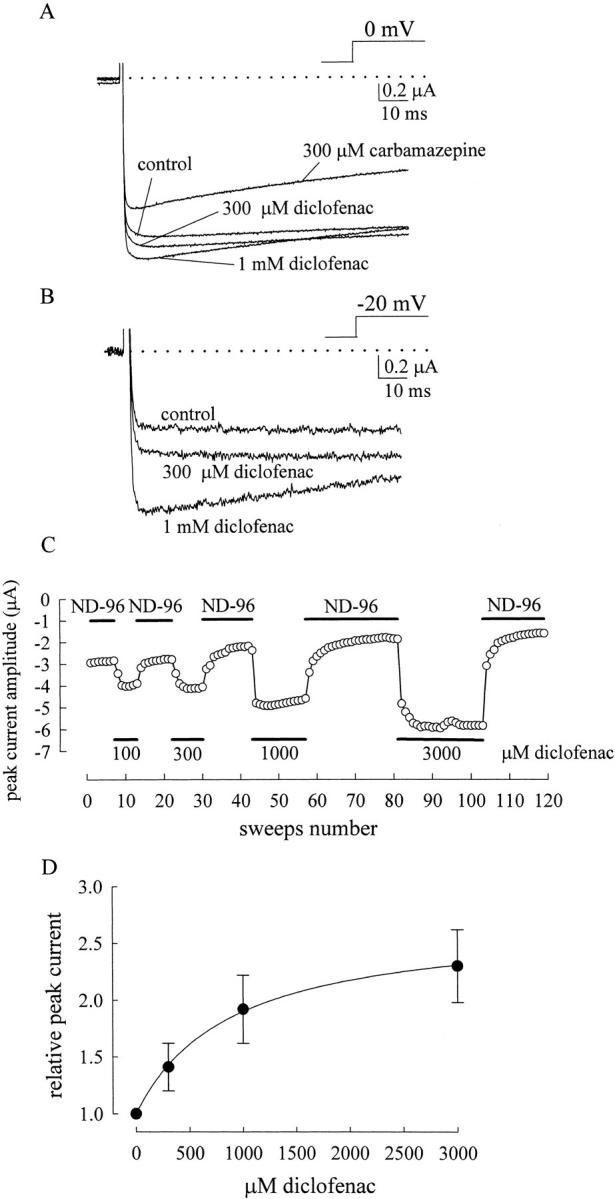
We have previously demonstrated that carbamazepine and imipramine accelerate macroscopic Na+ current decay in a linear dose-dependent fashion with macroscopic binding rate constants of ∼1.1 × 106 and ∼1.1 × 107 M−1s−1, respectively (Yang and Kuo, 2002). This finding strengthens the view that these use-dependent inhibitors bind to the Na+ channel with a one-to-one binding stoichiometry, and further suggests that carbamazepine and imipramine block the open Na+ channel pore in addition to stabilization of a nonconducting (or inactivated) state connected to the open state. This is because if only the latter case were true, then the forward rate (rate of genesis of this nonconducting state) must be much faster than 330 to 3300 s−1 to sustain the time constant of relaxation observed in the highest concentration (∼300 μM) of carbamazepine and imipramine (Yang and Kuo, 2002). Also, the corresponding backward rate must be quite sizable as compared with the forward rate to sustain the dose-dependent current decrease with time constants of relaxation linearly related to drug concentrations. It is thus hard to conceive such a nonconducting state, because it can be neither the fast inactivated state, which has a very slow backward rate (i.e., very high binding affinity of the inactivation lid to the open channel), nor the slow inactivated state, which usually takes a few hundred milliseconds or seconds to develop. If diclofenac and carbamazepine share the same binding site in Na+ channels, it is intuitive that diclofenac should also block the pore. Surprisingly, we found that diclofenac cannot block the open neuronal Na+ channel pore like what carbamazepine and imipramine do. With its even higher binding affinity to the inactivated channel than carbamazepine, diclofenac in concentrations of 300 μM to 1 mM (43- to 143-fold of K I) does not show any acceleration of the macroscopic current decay as that 300 μM (12-fold of K I) carbamazepine does (Fig. 4 A; Yang and Kuo, 2002). Actually, the current decay is slightly retarded rather than accelerated by diclofenac (Fig. 4, B–D). The significant and concentration-dependent retardation of current decay by diclofenac argues against the possibility that the lack of blocking effect is due to insignificant binding of diclofenac onto the open channel (e.g., the binding rate of diclofenac is too slow or the binding affinity is too low to the open channel). Instead, it seems that diclofenac significantly binds to the open channel and slightly retards channel inactivation without simultaneously blocking the pore. One may also note that the current peak is slightly shifted rightwardly (i.e., the time to current peak is slightly lengthened) and is reduced rather than enhanced despite of possible stabilization of the open channel by high concentrations of diclofenac (Fig. 4, A and B). The slightly rightwardly shifted and reduced peak implies that diclofenac, in such high concentrations, may bind to the resting Na+ channel and consequently slows the activation process upon depolarization, and/or that the diclofenac-bound open channel could still conduct Na+ currents but with a slightly decreased conductance. In any case, there must be significant diclofenac binding to the Na+ channel without blocking the channel pore, so that the gating alterations could be manifest.
Figure 4.
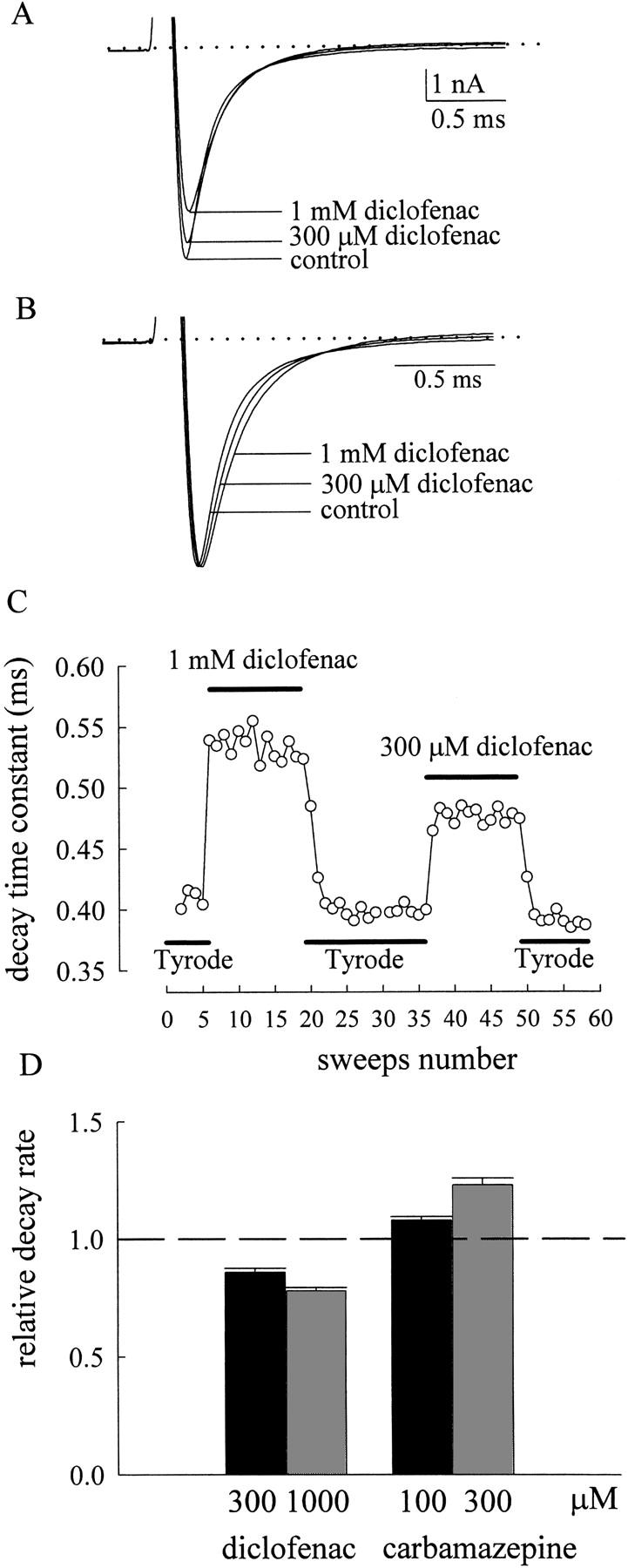
The effect of diclofenac on macroscopic neuronal Na+ currents. (A) The neuron was held at −120 mV and stepped to 0 mV to elicit Na+ currents. The external solution contains 150 mM NaCl, and the internal solution contains 150 mM CsCl (for details of the composition of the solutions, see materials and methods). 300 μM to 1 mM diclofenac mildly and dose dependently reduces the current peak as well as increases the time to peak without apparent acceleration of current decay. The dotted line indicates the zero current level. (B) The currents in A are superimposed and scaled to the same current size, showing that diclofenac shifts current peak rightwardly as well as slows current decay in a dose-dependent fashion. (C) The neuron was exposed to repeated pulses, which are stepped from −120 mV to 0 mV for 12 ms with an interpulse interval of 3 s to elicit a series of Na+ currents. The black horizontal bars indicate the external solutions (Tyrode's solution or Tyrode's solution with different concentrations of diclofenac) in which the neuron stayed. The time constant from a monoexponential fit to the macroscopic Na+ current decay is lengthened by diclofenac in a dose-dependent fashion. The diclofenac effect can be readily “washed out” when the external solution is switched back to the control Tyrode's solution. (D) Opposite effects of diclofenac and carbamazepine on the current decay rate. The decay rate is determined by the inverse of the time constant obtained from a monoexponential fit to the decay phase of the current. The relative decay rate is defined by the ratio between the decay rates in the presence of different drugs and in control, and are 0.86 ± 0.02 (n = 9, drug/control), 0.78 ± 0.01 (n = 11), 1.08 ± 0.02 (n = 6), 1.23 ± 0.03 (n = 9) for 300 μM diclofenac, 1 mM diclofenac, 100 μM carbamazepine, and 300 μM carbamazepine, respectively. The dashed line indicates the relative decay rate of 1.
Voltage-dependent Enhancement of the Inactivation-deficient F1489Q Mutant Na+ Currents by the Stabilization Effect of Diclofenac on the Activated Channel
To verify the effect of diclofenac on resting and open Na+ channels, we examined diclofenac action on the inactivation-deficient mutants (i.e., F1489Q mutant or F1489Q/F1651A double mutant RIIA Na+ channels expressed in oocytes). The F1489Q mutation is located at the intracellular loop connecting domains III and IV and presumably weakens binding of the inactivation “lid” to the open channel (West et al., 1992). Fig. 5 A shows that 300 μM and 1 mM diclofenac only mildly increase the peak of the inactivation-deficient F1489Q current elicited by a step depolarization to 0 mV from a holding potential of −120 mV. However, if the current is elicited by a less depolarized potential such as −20 mV, the peak current enhancement effect of diclofenac becomes much more evident (Fig. 5 B), making the currents as large as ∼1.5-fold of the original ones (Fig. 5 C). The following simplified Scheme 2 illustrates gating of the inactivation-deficient mutant channel. The inactivated state is disregarded in this scheme, where C, O, CD, and OD refer to the closed, open, drug-bound closed, and drug-bound open states, respectively, and V denotes the voltage-dependent gating processes.
Figure 5.
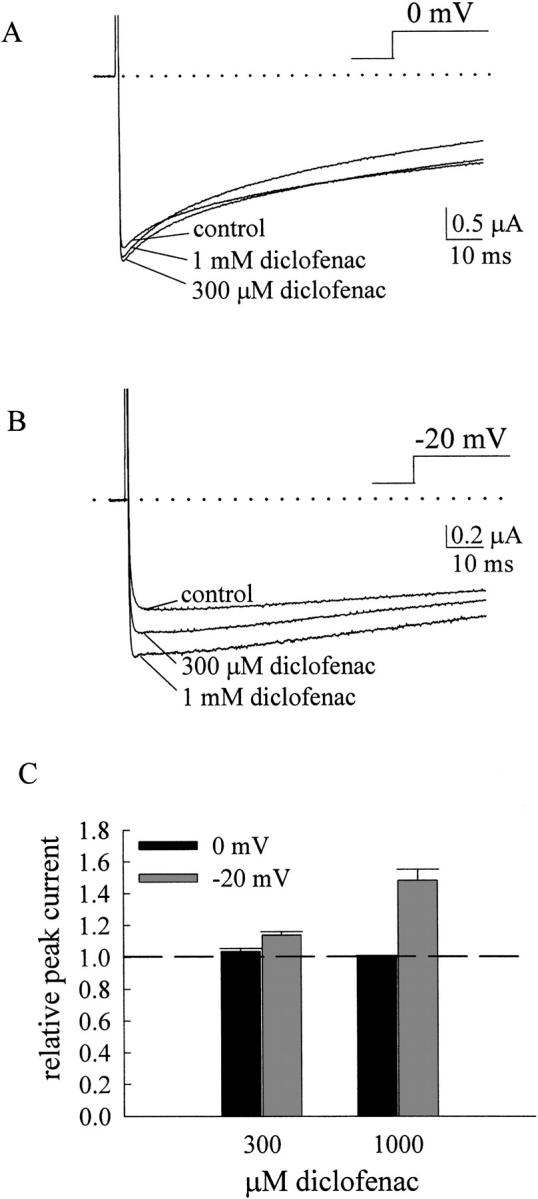
Diclofenac enhancement of the inactivation-deficient F1489Q mutant currents. (A) The oocyte expressing the F1489Q mutant channel was held at −120 mV and stepped to 0 mV to elicit currents. The control Na+ current recorded in ND-96 solution shows biphasic current decay (an initial fast phase followed by a slow phase of decay). 300 μM to 1 mM diclofenac only minimally or equivocally increases the peak current. The dotted line indicates the zero current level. (B) The same oocyte as that in A was stepped to −20 mV instead of 0 mV to elicit Na+ currents. The activation kinetics are consequently slower, but the enhancement effects of 300 μM and 1 mM diclofenac are stronger than that in A. (C) The F1489Q peak currents elicited by 0 (black bars) or −20 mV (gray bars) pulses in different concentrations of diclofenac are normalized to that in the control (no diclofenac) condition to obtain the relative peak current. The dashed line indicates the relative peak current of 1. The enhancement effect of diclofenac on peak currents is dependent on both the test pulse voltage and the diclofenac concentration.
(SCHEME 2).

Based on Scheme 2, the test-pulse voltage dependence of the enhancement effect of diclofenac in Fig. 5 would suggest that diclofenac enhances the currents by increasing the relative occupancy of the open state. In other words, the enhancement of the mutant currents can be explained by the ideas that both O and OD in Scheme 2 can conduct Na+ ions, and that diclofenac actually makes more distribution of channels to the open state (i.e., there is a higher affinity of diclofenac binding to the open than to the resting states). In that case, diclofenac binding should cause a leftward shift (in the voltage axis) of the “activation curve” describing distribution between the closed and the open states of the channel. We therefore examine the enhancement effect of diclofenac on peak F1489Q mutant currents over a wide range of test-pulse potentials (Fig. 6). Diclofenac causes a concentration-dependent leftward shift in the activation curve (the normalized conductance–voltage curve) of the F1489Q mutant channel (Fig. 6 A). Based on similar rationales to the shift in the inactivation curve (Eq. 1), the shift of the activation curve by a fixed concentration of diclofenac would be predicted by the following equation, where KO and KR denote the dissociation constants of diclofenac binding to the open and resting channels, respectively.
Figure 6.
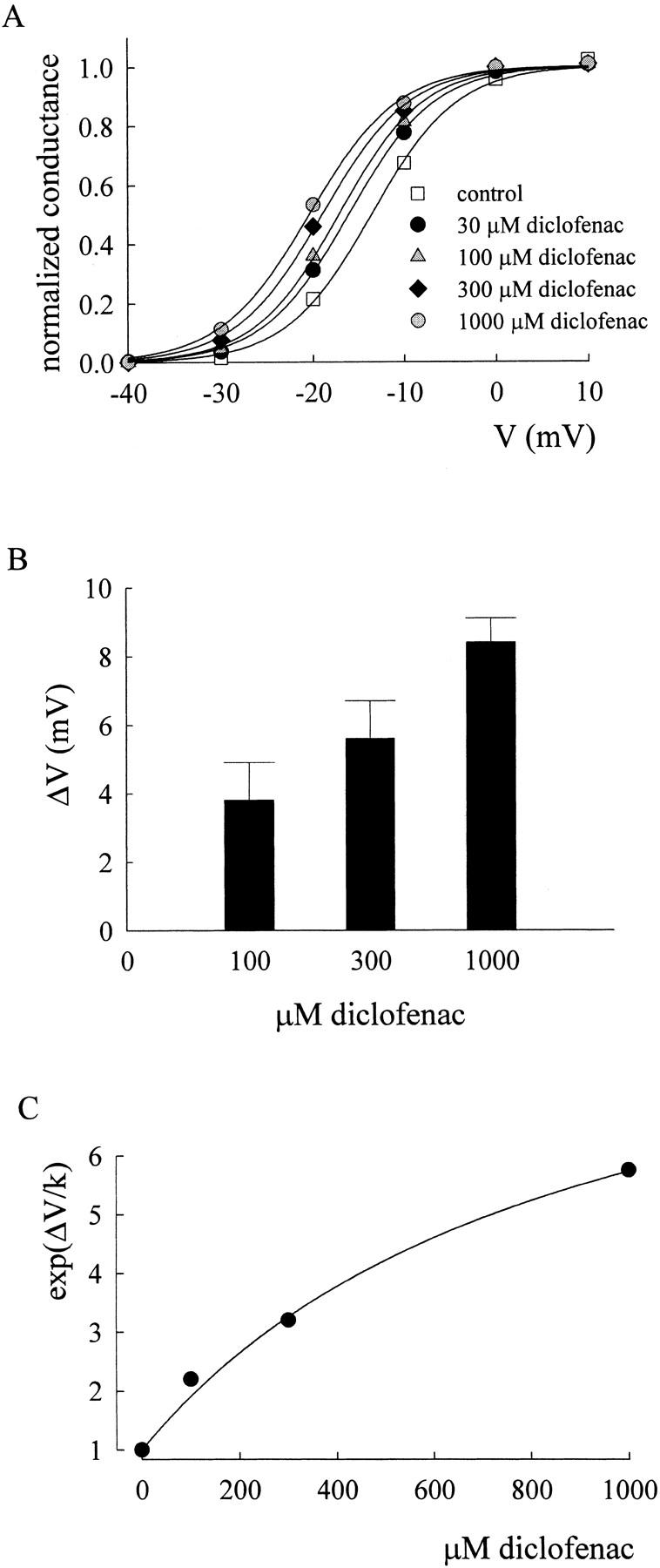
Shift of the normalized conductance–voltage curve of the F1489Q mutant channel by diclofenac. (A) The normalized conductance–voltage curve of the F1489Q mutant channel in different concentrations of diclofenac. The oocyte expressing F1489Q mutant Na+ channels was held at −120 mV and stepped to different test voltages to elicit Na+ currents. The maximal conductance (Gmax) is given by the slope of the linear regression fit to the data points between +10 and +40 mV in the peak current–voltage plot, and the reversal potential (Vrev, in mV) is estimated by extrapolating the foregoing linear regression to the zero current level. The normalized conductance is then determined by Ipeak/((V − Vrev)*Gmax), where Ipeak and V are the peak current amplitude and the test voltage (in mV), respectively. The normalized conductance is plotted against the test voltage, and then fitted with a Boltzmann function of the form: 1/(1 + exp ((V − Vh)/4.8)), where the Vh values (in mV) are −13.7, −16.9, −17.8, and −19 in 0 (control), 100, 300, and 1000 μM diclofenac, respectively. Because the slope of the fitting curves in diclofenac always stays close to the slope in control, the Vh values are obtained from the fits with a fixed k value of 4.8 (the mean of the k values in control) to facilitate the following quantitative analysis. (B) Mean shift of the normalized conductance–voltage curve by different concentrations of diclofenac. The shift (ΔV) is determined by the difference between the Vh values in control and in different concentrations of diclofenac. The mean ΔV values (in mV) are 3.8 ± 1.1 (n = 3), 5.6 ± 1.1 (n = 3), and 8.4 ± 0.7 (n = 4) for 100, 300, and 1000 μM diclofenac, respectively. (C) The mean ΔV values in B and a k value of 4.8 are used to calculate exp(ΔV/k), which is plotted against diclofenac concentrations to determine the dissociation constant between diclofenac and the activated channel. The solid line is the fit of the form (see Eq. 5 in the text) exp(ΔV/k) = [1 + (D/88)]/[1 + (D/880)], where D is the diclofenac concentration in μM, and a KR value of 880 μM is taken from Fig. 7 D.
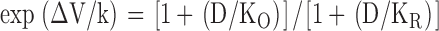 |
(5) |
We therefore have a KO of ∼88 μM with a KR value of 880 μM (Fig. 6, B and C, the KR value is taken from Fig. 7 D). These findings are well consistent with the idea that submillimolar diclofenac can effectively bind to the open Na+ channel without significantly blocking its pore (i.e., OD in Scheme 2 is still a conducting state).
Even Larger Enhancement of the Inactivation-deficient F1489Q/F1651A Double Mutant Na+ Currents by Diclofenac in High Concentrations of External Na+
The biphasic macroscopic current decay implies that there is still residual fast inactivation followed by a slow inactivation process in the F1489Q mutant channel (Lawrence et al., 1996; Nuss et al., 1996). To ensure more complete removal of fast inactivation, we make another mutant containing not only F1489Q but also F1651A mutations. The F1651A mutation is located at the S4–S5 linker in domain IV, presumably disrupting the “receptor” for the inactivation lid (Depp and Goldin, 1996; Lerche et al., 1997; McPhee et al., 1998). The F1489Q/F1651A double mutation almost completely abolishes fast inactivation in Na+ channels. The current-enhancing effect of diclofenac indeed is more evident in the double mutant channel (Fig. 7, A and B), presumably due to less “contamination” by residual fast inactivation. 300 μM to 1 mM diclofenac unequivocally increases the peak of the double mutant current elicited at 0 mV and dramatically enhances the current elicited at −20 mV. The current peak is augmented in a diclofenac concentration-dependent fashion (Fig. 7 C). The test-pulse voltage dependence of the enhancement effect of diclofenac in Fig. 7 again strongly supports the view that diclofenac increases the relative occupancy of the open state in Scheme 2, where the OD state remains unblocked and can readily conduct Na+. The occupancy of the open state is more significantly increased at weaker (e.g., −20 mV) than at stronger depolarization (e.g., 0 mV), probably because this occupancy is already saturating at 0 mV in the drug-free condition. On the other hand, carbamazepine is obviously an open pore blocker of the Na+ channel because it always accelerates the decay and reduces the peak of both native and inactivation-deficient mutant Na+ currents in the same experimental conditions (e.g., Fig. 4 D and Fig. 7 A).
Measurement of the Binding Affinity of Diclofenac to the Resting Na+ Channel
We have seen that diclofenac selectively binds to the activated and inactivated Na+ channel (e.g., Figs. 2 and 6). It would be desirable to estimate the binding affinity of diclofenac to the resting channel to have a more solid idea of the binding affinity changes along the whole gating process. However, an accurate estimate of diclofenac binding affinity to the resting Na+ channel may not be easy because the affinity presumably is very low and the resting channel itself is electrophysiologically silent (i.e., no observable ionic conductance through the resting channel). Diclofenac is not a pore blocker in this experimental configuration. However, binding of diclofenac evidently alters the C–O transition to augment Na+ currents in the inactivation-deficient mutants. We therefore try to have an estimate of the binding affinity of diclofenac to the resting Na+ channel based on Scheme 2 where a step depolarization will drive channels in states C (diclofenac-free resting state) and CD (diclofenac-bound resting state) to states O and OD to elicit Na+ currents. As a first approximation, we assume that the binding rate of diclofenac onto the channel is much slower than channel gating itself. The resting Na+ channel in states C and CD would then approximately be “separately” activated via the C–O route and the CD–OD route, respectively. The increase of the mutant current peak by diclofenac thus may serve as a rough estimate of the relative occupancy between states C and CD. The dissociation constant (KR) between diclofenac and the resting Na+ channel could therefore be measured. Fig. 7 D plots the increase of the double mutant current peak against the diclofenac concentration. The experimental data can be reasonably fitted with a one-to-one binding curve, giving a KR value of ∼880 μM. It should be noted that a KR value of ∼880 μM is very likely a slight underestimate because mild “contamination” by additional binding of diclofenac to the (partially and fully) activated channels may happen during the rising phase of the current.
Diclofenac as a Pore Blocker of the Na+ Channel in the Absence of External Na+
It is intriguing that diclofenac shares very similar features of Na+ channel gating modification to carbamazepine (Figs. 2 and 3; Kuo et al., 1997), but the effect on ion permeation is fundamentally different between these two drugs (Figs. 4 and 7; Yang and Kuo, 2002). In view of the possibility that the pore-blocking effect may be modulated by permeating ions, we examine the effect of Na+ on the action of carbamazepine and diclofenac. Surprisingly, we found that diclofenac is turned into an effective pore blocker and evidently accelerates the macroscopic neuronal Na+ current decay when the Na+ ions in the external solution are replaced by Cs+ (0 mM external and 150 mM internal Na+; Fig. 8, A and B). The onset of the effect is very rapid (Fig. 8 C), exactly analogous to the effect of carbamazepine in 150 mM external Na+ (Yang and Kuo, 2002). The linear correlation between the acceleration of current decay and the diclofenac concentration indicates a simple bimolecular reaction and gives a diclofenac binding rate constant of 4.1 × 105 M−1s−1 onto the open neuronal Na+ channel. This value is close to the previously reported binding rate constant of carbamazepine to the open channel (1.1 × 106 M−1s−1; Yang and Kuo, 2002). Actually the binding rate constants to the inactivated Na+ channel are also similar between these two drugs, and are ∼15–30 times slower than binding to the open channel in both cases (unpublished data; Kuo et al., 1997). In any case, it is clear that carbamazepine is always an open channel pore blocker and accelerates the macroscopic current decay irrespective of the external Na+ concentration, but diclofenac is a more versatile or an “opportunistic” pore blocker modulated by the Na+ ion in the external solution (Fig. 8 D). At first glance, the fundamentally different effects of diclofenac and carbamazepine on ion permeation in 150 mM external Na+ may seem to suggest two different sites for these two drugs. The mutual exclusion between binding of the two drugs then would be more likely ascribable to an allosteric rather than a direct interaction. However, the finding that diclofenac is turned into a pore blocker and shows an effect exactly analogous to carbamazepine in the absence of external Na+ would favor the possibility of a common binding site and a direct interaction between these two drugs again. In any case, the intriguing opportunistic pore-blocking effect strongly suggests that there is a binding site for the use-dependent Na+ channel inhibitor at the junction of the widened external vestibule and the narrowed part of the pore (see discussion and Fig. 11).
Figure 8.
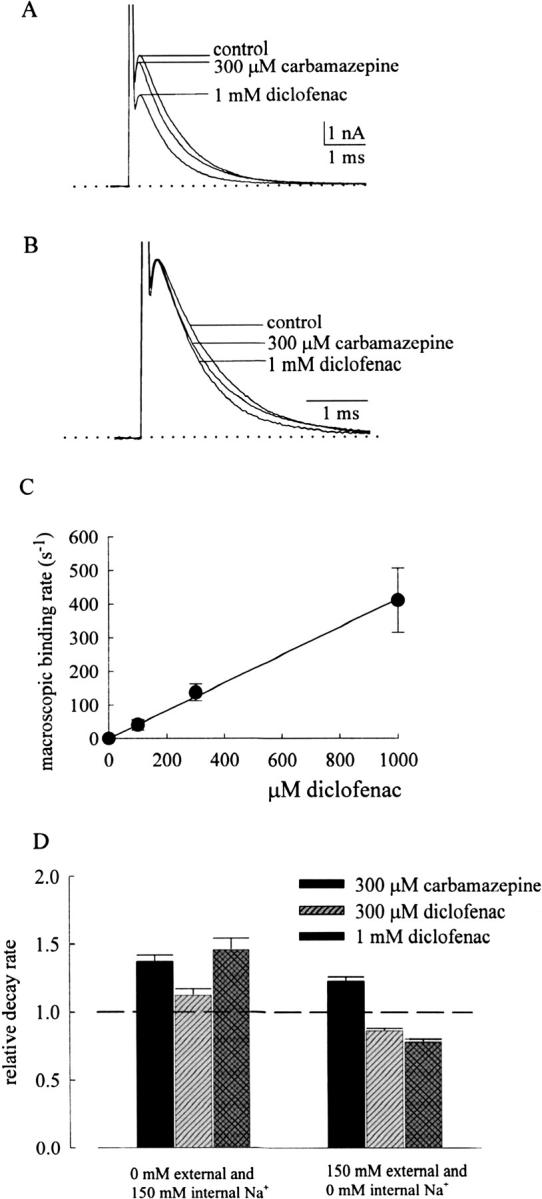
Diclofenac inhibition of outward neuronal Na+ currents in 0 mM external and 150 mM internal Na+. (A) The cell was held at −120 mV and stepped to 0 mV to elicit Na+ currents. The external solution contains no Na+ ions but 150 mM Cs+, and the internal solution contains 150 mM Na+ (for details of the composition of the solutions, see materials and methods). 1 mM diclofenac has a stronger inhibitory effect on the current than 300 μM carbamazepine, and both drugs accelerate current decay (see B). The dotted line indicates the zero current level. (B) The currents in A are superimposed and scaled to the same current size. It is evident that 300 μM carbamazepine accelerates current decay, and 1 mM diclofenac is even stronger in doing so. (C) In 0–1000 μM diclofenac, the decay phase of the macroscopic outward Na+ currents in the absence of external Na+ is fitted with a monoexponential function. The inverse of the time constant in control is subtracted from the inverse of the time constant in diclofenac to give the macroscopic drug binding rate to the open channel. The line is a linear regression fit to the macroscopic binding rates in different concentrations of diclofenac with a slope of 4.1 × 105 M−1s−1. (D) Comparison of the effects of diclofenac and carbamazepine on the macroscopic current decay rate in different ionic conditions. The neurons were held at −120 mV and stepped to 0 mV to elicit Na+ currents. The relative current decay rate is determined in the same way as that described in Fig. 4 D. The dashed line indicates the relative decay rate of 1. Diclofenac retards current decay in the presence of 150 mM external and 0 mM internal Na+ solutions but accelerates current decay in the presence of 0 mM external and 150 mM internal Na+ solutions, both in a diclofenac concentration–dependent fashion. On the other hand, carbamazepine always accelerates current decay irrespective of the Na+ concentration in the solutions.
Figure 11.

A schematic model describing one of the possible reasons why carbamazepine and diclofenac may bind to the same receptor area in the external pore mouth of the Na+ channel but produce different pore-blocking effects in the presence or absence of 150 mM external Na+. The diphenyl group in each drug is shown by two black squares, which in general have similar spatial orientation in these two drugs. However, the two phenyl groups are rigidly tied in carbamazepine but are more flexible (e.g., with more versatile torsion angles) in diclofenac (Table I). The shaded areas in the channel protein mark the location of the binding ligands for the diphenyl group. In the absence of external Na+, the two drugs both bind to the gray shaded areas in the external pore mouth to compromise the narrow part of the ion conduction pathway and block ion flow (left, 0 external Na+). When Na+ occupies the modulatory Na+ binding site in the external vestibule in 150 mM external Na+, local conformational changes happen with movement or reorientation of some potential binding ligands for the diphenyl group (right). A wider shaded area suitable for binding of the diphenyl group is thus formed in the external vestibule slightly farther away from the narrow part of the pore. With its more flexible diphenyl group, diclofenac can readily bind to this wider shaded area. The narrow part of the pore in the diclofenac-bound channel thus remains “patent” and permeable to Na+. In contrast, carbamazepine cannot effectively bind to the newly formed alternative receptive area because of its rigidly tied diphenyl group in the dibenzazepine tricyclic structure. Carbamazepine would therefore bind to the same area as that in the absence of Na+ and still blocks the pore. The modulatory Na+ binding site is probably located in the widened part of the external vestibule or even the external surface of the channel, so that its occupancy is correlated with only external but not internal Na+. On the other hand, the diclofenac binding site should be located at the junction of the narrow and widened parts of the external pore mouth (presumably the junction of the external vestibule and the single-file region of the pore), so that diclofenac could behave as an opportunistic pore blocker modulated by external Na+.
Less Enhancement Effect of Diclofenac on Inactivation-deficient Mutant Na+ Currents in Lower Concentration of External Na+
We also examined the effect of different concentrations of external Na+ on the action of diclofenac in the inactivation-deficient mutant channels. We have seen that 1 mM diclofenac markedly enhances the F1489Q mutant current elicited by a −20-mV pulse in ND-96 external solution (containing 96 mM Na+; Fig. 5 B). However, the same concentration of diclofenac can only barely enhance the F1489Q mutant current when the external Na+ concentration is decreased to 22 mM (in ND-22 solution; Fig. 9). It should be noted that the only difference in the experimental conditions here is the external Na+ ion concentration, as the internal Na+ concentration is unchanged and the macroscopic Na+ currents are always inward (e.g., the currents in Fig. 4 A and Fig. 8 A have opposite directions of current flow). The findings in Fig. 9 thus are not only consistent with Fig. 8, but further suggest that the major determinant of the blocking effect of diclofenac on the open Na+ channel pore is external Na+ rather than the direction of current flow or the internal ionic conditions (also see below).
Figure 9.

Comparison of diclofenac enhancement of the F1489Q mutant current in external solutions containing high and low concentrations of Na+. (A) The oocyte expressing the F1489Q mutant channel was held at −120 mV in the ND-96 solution (containing 96 mM Na+) or in the ND-22 solution (containing only 22 mM Na+, see materials and methods) and stepped to −20 mV for 100 ms to elicit the Na+ currents. 1 mM diclofenac significantly enhances the F1489Q mutant current measured in the ND-96 solution, but has only a negligible effect on the current measured in the ND-22 solution. The dotted line indicates the zero current level. (B) The relative peak current in the ND-96 and in the ND-22 solutions is determined by the same way as that in Fig. 5 C. The dashed line indicates the relative peak current of 1. Diclofenac enhancement of the F1489Q mutant current is evidently weaker with the lower concentration of external Na+.
Diclofenac as an “Opportunistic” Na+ Channel Pore Blocker Modulated by the External rather than the Internal Na+
Fig. 10 A further examines the effect of diclofenac on neuronal Na+ currents in symmetrical 150 mM Na+ solutions (both internal and external solutions contain 150 mM Na+). 1 mM diclofenac slightly reduces and rightwardly shifts the macroscopic current peak but does not accelerate the current decay in symmetrical Na+ solutions. This is similar to the case in 150 mM external and 0 mM internal Na+ (Fig. 4, A and B) but distinct from the case in 0 mM external and 150 mM internal Na+, where the current decay is accelerated and the current peak is reduced but not shifted by diclofenac (Fig. 8, A and B). This finding again supports the idea that the major factor modulating the diclofenac effect on Na+ currents is the external rather than the internal Na+ ions. Fig. 10 B compares the effect of diclofenac on the time to current peak in different internal and external solutions. No matter the internal solution contains 0 or 150 mM Na+, the time to peak current is always lengthened by diclofenac if the external solution contains 150 mM Na+. On the other hand, when the external solution contains no Na+, the current peak is never discernibly shifted by 10–3000 μM of diclofenac in any cell we examined (e.g., see the current traces in Fig. 8 B). It seems that high concentrations of diclofenac can significantly bind to the resting Na+ channel and modify subsequent gating (activation and inactivation) process of the channel, reminiscent of significant binding of high concentrations of diclofenac onto the resting inactivation-deficient channel expressed in oocytes (with a KR value of ∼880 μM; Fig. 7). This gating effect of diclofenac is manifest in the presence of 150 mM external Na+ where diclofenac does not block the channel pore. On the other hand, if binding of diclofenac to the resting channel blocks the channel pore (such as in the case of 0 mM external Na+), the diclofenac-bound resting channel could not conduct Na+ ion even when it is subsequently activated by a depolarization pulse, and thus no apparent gating effect on the macroscopic activation phase would be observed. These findings reinforce the argument that only external Na+, but not internal Na+, effectively alters the blocking action of diclofenac. In this case, the reduction of peak current in the absence of external Na+ would basically reflect the binding of diclofenac to the resting channel and serve as an estimate of the binding affinity between diclofenac and the resting channel in this experimental condition. Fig. 10 C plots the reduction of current peak against different concentrations of diclofenac in the “0 mM Na+” external solution. It is clear that the effect is far from saturation even with 1 mM of diclofenac, and a rough estimate of the dissociation constant of diclofenac binding to the resting channel (KR) in the absence of external Na+ is ∼1500 μM. The KR of diclofenac thus is probably not very different in 96 mM external Na+ (∼880 μM; Fig. 7 D) or in the absence of external Na+ (∼1500 μM; Fig. 10 C), and is approximately in the low millimolar range in both cases. External Na+ thus may only slightly alter the binding geometry and binding affinity of diclofenac onto the Na+ channel, but the slight alteration could already result in very different functional consequences in ion conduction through the pore.
Figure 10.

Lack of significant effect of internal Na+ on diclofenac action and an estimate of the binding affinity of diclofenac to the resting neuronal Na+ channel. (A) Diclofenac action on macroscopic neuronal Na+ currents in the presence of symmetrical 150 mM Na+ (150 mM Na+ internal solution and Tyrode's solution containing 150 mM Na+ extracellularly). The neuron was held at −120 mV and stepped to −10 mV to elicit inward Na+ currents. The dotted line indicates the zero current level. The currents in the top panel are rescaled to the same peak level in the bottom panel. 1 mM diclofenac slightly but significantly reduces current peak and increases the time to current peak. However, it does not accelerate the macroscopic current decay. (B) Effect of diclofenac on the time to current peak in different solutions. The current is elicited by a test pulse of 0 or −10 mV from a holding potential of −120 mV in 150 mM external and 0 or 150 mM internal Na+ (Ex150/In0 and Ex150/In150, respectively), or in 0 mM external and 150 mM internal Na+ (Ex0/In150). The test pulse is given beside each bar. In Ex150/In150 solution, the current cannot be measured at 0 mV and thus only that measured at −10 mV is shown. The time to peak in the presence of 1 mM diclofenac is normalized to the time to peak in control to give the relative time to peak. The dashed line indicates the relative time to peak of 1. When the external solution contains 150 mM external Na+, 1 mM diclofenac always increases the time to peak whether the internal solution contains 0 or 150 mM Na+. In contrast, the time to peak is always unchanged by 1 mM diclofenac if the external solution contains no Na+ (0 mM external Na+). (C) Estimation of the binding affinity of diclofenac to the resting Na+ channel in the absence of external Na+. The neuronal Na+ current is evoked by a pulse to 0 mV from a holding potential of −120 mV in 0 mM external and 150 mM internal Na+. The peak current in the presence of diclofenac is subtracted from the peak current in control and then normalized to the control current to give the relative reduction of peak current, which is plotted against the diclofenac concentration. The line is a fit to the data of the form: relative reduction of peak current = (D/1483)/[1 + (D/1483)], where D denotes the concentration of diclofenac in μM.
DISCUSSION
The Location of the Diclofenac Binding Site in the Na+ Channel
We have demonstrated that diclofenac selectively binds to the activated and inactivated rather than the resting Na+ channel (Figs. 2 and 6), very much analogous to the action of anticonvulsants carbamazepine, phenytoin, and lamotrigine (Kuo and Bean, 1994; Kuo and Lu, 1997; Kuo et al., 1997). Diclofenac thus is also a use-dependent inhibitor with one-to-one binding stoichiometry (Fig. 8 C) to the Na+ channel. However, diclofenac does not completely mimic the action of carbamazepine to block the open Na+ channel pore (Yang and Kuo, 2002). Diclofenac cannot block the pore in the presence of 150 mM external Na+ (Figs. 4–7), but is turned into an effective pore blocker with decreased external Na+ concentration (Figs. 8 and 10). We further show that the “opportunistic” pore-blocking effect of diclofenac is chiefly modulated by external Na+, but has little to do with internal Na+ or the direction of current flow (Figs. 8–10). Given its capability of blocking the pore at least in some experimental conditions, diclofenac should have its binding site in the ion conduction pathway of the Na+ channel. The findings that only external but not internal Na+ has an modulatory effect on the binding conformation of diclofenac would further limit the diclofenac binding site to a pore region that is effectively occupied by only external but not internal Na+ (see below). The diclofenac binding site thus must be located very “superficially” in the pore (i.e., at the external pore mouth). In the meanwhile, the opportunistic blocking effect would require diclofenac to bind to a site at the junction of a widened and a narrow part of the ion conduction pathway, so that its pore-blocking effect can be readily and drastically altered by some, presumably mild or modest, local conformational changes induced by Na+ binding (Fig. 11). Altogether these findings strongly indicate that diclofenac, a use-dependent inhibitor of the Na+ channel, binds to a site at the junction of the widened external vestibule and the narrow (possibly the selectivity filter, see below) part of the pore.
A Low Affinity Na+ Binding Site in the External Vestibule of the Na+ Channel Pore
The finding that diclofenac is an opportunistic pore blocker modulated by external but not internal Na+ is reminiscent of, but far more characteristic than, the observations that the kinetics and efficacy of many Na+ channel blockers might be influenced by external Na+ (Shapiro, 1977; Cahalan and Almers, 1979; O'Leary and Chahine, 2002). In Shaker K+ channels, the conformational change of the external vestibule near the selectivity filter during C-type inactivation is also found to be K+ dependent (Ikeda and Korn, 1995; Baukrowitz and Yellen, 1996; Immke et al., 1999). It is thus plausible that the permeating ions could modulate local conformation at the external pore mouth of an ion channel. There is a multi-ion single-file region at the external pore mouth of the Na+ channel, and this “narrow” single-file pore region presumably is connected to the external bulk solution by a wide vestibule (Ravindran et al., 1991; Heinemann et al., 1992; Teresa Perez-Garcia et al., 1997; Kuo et al., 2002). Moreover, external La3+ or other multivalent blocking ions binding to this single-file pore region may profoundly retard Na+ channel inactivation, indicating imperative gating conformational changes in this external pore mouth area (Kuo et al., 2004). Consistent with these views, our findings indicate that the gating-modifying (and gating-modified) diclofenac receptor is located at the junction of the narrow part of the ion conduction pathway and the widened vestibule in the external pore mouth. The experimental data further suggest that there is a Na+ site in the external vestibule of the pore, and Na+ binding to this site would effectively modulate the binding conformation of diclofenac (Fig. 11). Although this Na+-binding site is not too far away from the single-file pore region, it is already in the widened vestibule and thus can be effectively occupied by only external Na+ rather than internal Na+ (which readily dissipates into the external milieu when permeating to this point). The affinity of Na+ to this ion binding site probably is not high, considering that the effect of 22 mM external Na+ is still evidently different from 150 mM external Na+ (Fig. 9). However, it remains an interesting possibility that Na+ binding to this site may play a role in the molecular operation of the Na+ channel in physiological conditions, which usually dictate ∼150 mM Na+ in the extracellular fluid in mammals.
Continual Conformational Changes in the External Pore Mouth during Na+ Channel Activation and Inactivation
Diclofenac binds to the inactivated Na+ channel with a much higher affinity (KI of ∼7 μM; Fig. 2) than to the resting Na+ channel (KR of ∼880 and ∼1500 μM in the presence and absence of external Na+, respectively; Fig. 7 D and Fig. 10 C). With the “patent” ion conduction pathway in the diclofenac-bound Na+ channel in 150 mM external Na+, we were able to show that diclofenac binds to and stabilizes the open Na+ channel with an intermediate affinity (KO of ∼88 μM; Fig. 6). The effect of diclofenac on Na+ channels itself probably has no clinical significance, because the therapeutic concentration of diclofenac is too low (Willis et al., 1979) to make significant binding to the Na+ channel. However, quantitative characterization of the binding affinity of a use-dependent inhibitor to all three major gating states of the Na+ channel is unprecedented, and provides novel insight into the drug-channel interaction. These experimental data indicate that the binding affinity of diclofenac to the activated channel is ∼10-fold (∼880 μM vs. ∼88 μM) higher than its binding to the resting channel, and is ∼13-fold (∼88 μM vs. ∼7 μM) lower than its binding to the inactivated channel. In other words, there are probably conformational changes in the drug receptor during activation and subsequent inactivation gating process, causing roughly ∼2.3RT (∼1.3 kcal/mol) and ∼2.6RT (∼1.5 kcal/mol) changes in the total binding energy of diclofenac, respectively. The continually altered binding affinity of diclofenac to the Na+ channel during activation and inactivation implies continual conformational changes in the diclofenac receptor, which may become “better and better” for the binding of diclofenac along the gating process triggered by membrane depolarization. Because diclofenac has only two benzene rings but not a tertiary amine side chain (like that in local anesthetics) in structure, the findings in this study are also consistent with the view that the binding counterpart of the diphenyl motif in imipramine must undergo significant conformational changes in the gating process of Na+ channels (Yang and Kuo, 2002). In this regard, it is worthy to note that the fourth transmembrane segment (S4), presumably the major voltage sensor of the voltage-gated Na+, K+, and Ca2+ channels, is moved in close apposition to the extracellular end of the pore upon membrane depolarization (Elinder et al., 2001; Broomand et al., 2003; Gandhi et al., 2003). Moreover, the extent and kinetics of Na+ channel inactivation are tightly and delicately controlled by the movement of S4 in the fourth domain of the channel protein (Yang and Kuo, 2003). It seems plausible that the conformational changes in the diclofenac binding site at the external pore mouth are actually coupled to, or even caused by, the movement of S4, and thus the use-dependent inhibitor of the Na+ channel may have its binding affinity and binding kinetics well correlated with the gating status of the channel.
Mutual Exclusion of Diclofenac and Carbamazepine Binding to the Na+ Channel
There are two possible mechanisms underlying the strict mutual exclusion of diclofenac and carbamazepine binding to the Na+ channel (Fig. 3). The two drugs may share the same binding site, or they may have different binding sites but the conformations favorable for drug binding in these two sites are allosterically and strictly incompatible with each other. The different effects of diclofenac and carbamazepine on ion permeation (in 150 mM external Na+) seem to suggest different drug binding sites. However, this is not necessarily the case if one considers the more rigid torsion angles of the diphenyl motif in carbamazepine and consequently a more versatile binding conformation of diclofenac than carbamazepine (Fig. 11). On the other hand, the two drugs have similar structure and similar gating modification as well as pore-blocking effects (in 0 mM external Na+) on the Na+ channel. They also show similar changes in binding kinetics upon the activation–inactivation transition of the channel. These findings can be straightforward envisioned with a common binding site for diclofenac and carbamazepine. Although the possibility of two different sites cannot be completely excluded with these findings, the two sites must be both in the pore, and both undergo very similar conformational changes along channel activation and inactivation. Also, the conformations favorable for drug binding in each site must be allosterically and strictly incompatible with each other. If future data do support two separate sites for carbamazepine and diclofenac, then one should be aware of the existence of multiple mutually incompatible binding sites for the use-dependent inhibitors of the Na+ channel. On the other hand, if the two drugs indeed bind to the same site, then we may locate the common anticonvulsant receptor at the junction of the widened external vestibule and an acutely narrowed part of the Na+ channel pore, given the characteristic external Na+-modulated “opportunistic” pore-blocking effect of diclofenac.
Acknowledgments
This work is supported by grant NHRI-EX93-9105NN from the National Institute of Health, Taiwan, and grant NSC92-2320-B002-064 from National Science Council, Taiwan.
Lawrence G. Palmer served as editor.
References
- Alpert, L.A., H.A. Fozzard, D.A. Hanck, and J.C. Makielski. 1989. Is there a second external lidocaine binding site on mammalian cardiac cells? Am. J. Physiol. 257:H79–H84. [DOI] [PubMed] [Google Scholar]
- Baukrowitz, T., and G. Yellen. 1996. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+ channel. Science. 271:653–656. [DOI] [PubMed] [Google Scholar]
- Bean, B.P. 1984. Nitrendipine block of cardiac calcium channels: high-affinity binding to the inactivated state. Proc. Natl. Acad. Sci. USA. 81:6388–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean, B.P., C.J. Cohen, and R.W. Tsien. 1983. Lidocaine block of cardiac sodium channels. J. Gen. Physiol. 81:613–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broomand, A., R. Mannikko, H.P. Larsson, and F. Elinder. 2003. Molecular movement of the voltage sensor in a K+ channel. J. Gen. Physiol. 122:741–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth, J.F., and G.R. Strichartz. 1990. Molecular mechanisms of local anesthesia: a review. Anesthesiology. 72:711–734. [DOI] [PubMed] [Google Scholar]
- Cahalan, M.D., and W. Almers. 1979. Interaction between quaternary lidocaine, the sodium channel gates, and tetradotoxin. Biophys. J. 27:39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depp, M.R., and A.L. Goldin. 1996. Probing S4-S5 regions of the rat brain sodium channel for the fast inactivation particle receptor site. Biophys. J. 70:A317. [Google Scholar]
- Doyle, D.A., J. Morais-Cabral, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conductance and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Elinder, F., R. Mannikko, and H.P. Larsson. 2001. S4 charges move close to residues in the pore domain during activation in a K+ channel. J. Gen. Physiol. 118:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier, D.T., T. Narahashi, and M. Yamada. 1970. The site of action and active form of local anesthetics. II. Experiments with quaternary compounds. J. Pharmacol. Exp. Ther. 171:45–51. [PubMed] [Google Scholar]
- Gandhi, C.S., E. Clark, E. Loots, A. Pralle, and E.Y. Isacoff. 2003. The orientation and molecular movement of a K+ channel voltage-sensing domain. Neuron. 40:515–525. [DOI] [PubMed] [Google Scholar]
- Heinemann, S.H., H. Terlau, W. Stuhmer, K. Imoto, and S. Numa. 1992. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature. 356:441–443. [DOI] [PubMed] [Google Scholar]
- Hille, B. 1977. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69:497–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, S.R., and S.J. Korn. 1995. Influence of permeating ions on potassium channel block by external tetraethylammonium. J. Physiol. 486:267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immke, D., M. Wood, L. Kiss, and S.J. Korn. 1999. Potassium-dependent changes in the conformation of the Kv2.1 potassium channel pore. J. Gen. Physiol. 113:819–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Kuo, C.-C., and B.P. Bean. 1994. Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol. Pharmacol. 46:716–725. [PubMed] [Google Scholar]
- Kuo, C.-C., and L. Lu. 1997. Characterization of lamotrigine inhibition of Na+ channels in rat hippocampal neurons. Br. J. Pharmacol. 121:1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, C.-C., R.-S. Chen, L. Lu, and R.C. Chen. 1997. Carbamazepine inhibition of neuronal Na+ channels: quantitative distinction from phenytoin and possible therapeautic implications. Mol. Pharmacol. 51:1077–1083. [DOI] [PubMed] [Google Scholar]
- Kuo, C.-C. 1998. A common anticonvulsant binding site for phenytoin, carbamazepine, and lamotrigine in neuronal Na+ channels. Mol. Pharmacol. 54:712–721. [PubMed] [Google Scholar]
- Kuo, C.C., R.C. Huang, and B.S. Lou. 2000. Inhibition of Na+ current by diphenhydramine and other diphenyl compounds: molecular determinants of selective binding to the inactivated channels. Mol. Pharmacol. 57:135–143. [PubMed] [Google Scholar]
- Kuo, C.-C., T.-J. Lin, and C.-P. Hsieh. 2002. Effect of Na+ flow on Cd2+ block of tetradotoxin-resistant Na+ channels. J. Gen. Physiol. 120:159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, C.-C., W.-Y. Chen, and Y.-C. Yang. 2004. Block of tetrodotoxin-resistant Na+ channel pore by multivalent cations: gating modification and Na+ flow dependence. J. Gen. Physiol. 124:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, J.H., D.W. Orias, J.R. Balser, H.B. Nuss, G.F. Tomaselli, B. O'Rourke, and E. Marban. 1996. Single-channel analysis of inactivation-defective rat skeletal muscle sodium channel containing the F1304Q mutation. Biophys. J. 71:1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, P.J., A. Sunami, and H.A. Fozzard. 2001. Cardiac-specific external paths for lidocaine, defined by isoform-specific residues, accelerate recovery form use-dependent block. Circ. Res. 89:1014–1021. [DOI] [PubMed] [Google Scholar]
- Lerche, H., W. Peter, R. Fleischhauer, U. Pika-Hartlaub, T. Malina, N. Mitrovic, and F. Lehmann-Horn. 1997. Role in fats inactivation of the IV/S4-S5 loop of the human muscle Na+ channel probed by cysteine mutagenesis. J. Physiol. 505:345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki, N., F.N. Quandt, R.E. Ten Eick, and J.Z. Yeh. 1984. Characterization of the block of sodium channels by phenytoin in mouse neuroblastoma cells. J. Pharmacol. Exp. Ther. 228:523–530. [PubMed] [Google Scholar]
- McPhee, J.C., D.S. Ragsdale, T. Scheuer, and W.A. Catterall. 1995. A critical role for transmembrane segment IVS6 of the sodium channel α-subunit in fast inactivation. J. Biol. Chem. 270:12025–12034. [DOI] [PubMed] [Google Scholar]
- McPhee, J.C., D.S. Ragsdale, T. Scheuer, and W.A. Catterall. 1998. A critical role for the S4-S5 intracellular loop in domain IV of the sodium channel α-subunit in fast inactivation. J. Biol. Chem. 273:1121–1129. [DOI] [PubMed] [Google Scholar]
- Nuss, H.B., J.R. Balser, D.W. Orias, J.H. Lawrence, G.F. Tomaselli, and E. Marban. 1996. Coupling between fast and slow inactivation revealed by analysis of a point mutation (F1304Q) in μ1 rat skeletal muscle sodium channels. J. Physiol. 494:411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary, M.E., and M. Chahine. 2002. Cocaine binds to a common site on open and inactivated human heart (Nav 1.5) sodium channels. J. Physiol. 541:701–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu, Y., J. Rogers, T. Tanada, T. Scheuer, and W.A. Catterall. 1995. Molecular determinants of drug access to the receptor site for antiarrythmic drugs in the cardiac Na+ channel. Proc. Natl. Acad. Sci. USA. 92:11839–11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale, D.S., J.C. Mcphee, T. Scheuer, and W.A. Catterall. 1994. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 265:1724–1728. [DOI] [PubMed] [Google Scholar]
- Ragsdale, D.S., J.C. Mcphee, T. Scheuer, and W.A. Catterall. 1996. Common molecular determinants of local anesthetic, antiarrythmic, and anticonvulsant block of voltage gated Na+ channels. Proc. Natl. Acad. Sci. USA. 93:9270–9275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindran, A., L. Schild, and E. Moczydlowski. 1991. Divalent cation selectivity for external block of voltage-dependent Na+ channels prolonged by Batrachotoxin: Zn2+ induces discrete substates in cardiac Na channels. J. Gen. Physiol. 97:89–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, B.I. 1977. Effects of strychnine on the sodium conductance of the frog node of Ranvier. J. Gen. Physiol. 69:915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strichartz, G.R. 1973. The inhibition of sodium currents in myelinated nerve by quaternary derivatives of lidocaine. J. Gen. Physiol. 62:37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunami, A., I.W. Glasser, and H.A. Fozzard. 2000. A critical residue for isoform difference in tetradotoxin affinity in a molecular determinant of the external access path for local anesthetics in the cardiac sodium channel. Proc. Natl. Acad. Sci. USA. 97:2326–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teresa Perez-Garcia, M., N. Chiamvimonvat, R. Ranjan, J.R. Balser, D.F. Tomaselli, and E. Marban. 1997. Mechanisms of sodium/calcium selectivity in sodium channels probed by cysteine mutagenesis and sulfhydryl modification. Biophys. J. 72:989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, J.W., D.E. Patton, T. Scheuer, Y. Wang, A.L. Goldin, and W.A. Catteral. 1992. A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proc. Natl. Acad. Sci. USA. 89:10910–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis, J.V., M.J. Kendall, R.M. Flinn, D.P. Thornhill, and P.G. Welling. 1979. The pharmacokinetics of diclofenac sodium following intravenous and oral administration. Eur. J. Clin. Pharmacol. 16:405–410. [DOI] [PubMed] [Google Scholar]
- Xie, X., B. Lancaster, T. Peakman, and J. Garthwaite. 1995. Interaction of the antiepileptic drug lamotrigine with recombinant rat brain type IIA Na+ channels and with native Na+ channels in rat hippocampal neurons. Pflugers Arch. 430:437–446. [DOI] [PubMed] [Google Scholar]
- Yang, Y.C., and C.C. Kuo. 2002. Inhibition of Na+ current by imipramine and related compounds: different binding kinetics as an inactivation stabilizer and as an open channel blocker. Mol. Pharmacol. 62:1228–1237. [DOI] [PubMed] [Google Scholar]
- Yang, Y.C., and C.C. Kuo. 2003. The position of the fourth segment of domain 4 determines status of the inactivation gate in Na+ channels. J. Neurosci. 23:4922–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]