

SUMMARY
Phosphorylation of endogenous inhibitor proteins specific for type-1 Ser/Thr phosphatase (PP1) provides a mechanism for reciprocal coordination of kinase and phosphatase activities. Phosphorylation of Thr38 in the inhibitor protein CPI-17 transduces G-protein-mediated signaling into a > 1000-fold increase of inhibitory potency toward myosin phosphatase. We show here the solution NMR structure of phospho-T38-CPI-17 with r. m. s. d. of 0.36 ± 0.06 Å for the backbone secondary structure, which reveals how phosphorylation triggers a conformational change and exposes the PP1 inhibitory surface. This active conformation is stabilized by the formation of a hydrophobic core of intercalated side-chains, which is not formed in a phospho-mimetic D38 mutant form of CPI-17. Thus, the profound increase in potency of CPI-17 arises from phosphorylation, conformational change and hydrophobic stabilization of a rigid structure that poses the phosphorylated residue on the protein surface and restricts its hydrolysis by myosin phosphatase. Our results provide structural insights into transduction of kinase signals by PP1 inhibitor proteins.
INTRODUCTION
Type-1 Ser/Thr phosphatase (PP1) is conserved among all eukaryotes and is responsible for regulation of a plethora of cellular functions,. A specific multisubunit form of PP1, called myosin phosphatase, is a ubiquitous enzyme that functions in various signaling circuits (Hartshorne, et al, 2004). Myosin phosphatase is a trimeric holoenzyme consisting of a catalytic subunit of PP1 and a myosin targeting MYPT1 subunit with an accessory M21 subunit (Hartshorne, et al, 2004). The N-terminal ankyrin-repeat domain of MYPT1, including a PP1 binding site, functions as an allosteric regulator of the catalytic subunit. On the other hand, C-terminal domain is phosphorylated by multiple kinases, ROCK, ZIPK and ILK, that is involved in the inhibition of myosin phosphatase in response to G-protein activation (Trinkle-Mulcahy, et al, 1995). In smooth muscle, agonist-induced activation of G-protein enhances the activity of Ca2+/calmodulin-dependent myosin light-chain kinase, and coincidently suppresses the activity of myosin phosphatase via the activation of kinases ROCK and PKC (Kitazawa, et al, 1991; Somlyo, et al, 2003). The inhibition of myosin phosphatase is required for robust and sustained myosin phosphorylation in response to agonist stimuli (Dimopoulos, et al, 2007). The inhibition of myosin phosphatase is also involved in regulation of cytoskeletal reorganization during cell migration (Kawano, et al, 1999) and cytokinesis (Matsumura, 2005).
In addition to direct phosphorylation of the MYPT1 subunit myosin phosphatase is regulated by a specific inhibitor protein, CPI-17, that is predominantly expressed in smooth muscles and neurons (Eto, et al, 1997; Eto, et al, 2002; Woodsome, et al, 2001). Phosphorylation of CPI-17 at Thr38 is necessary and sufficient to convert the protein into a potent inhibitor of myosin phosphatase (Eto, et al, 1995; Kitazawa, et al, 2000; Koyama, et al, 2000; Pang, et al, 2005). Other PP1 holoenzymes in cells are insensitive to phospho-CPI-17 (P-CPI-17) (Senba, et al, 1999). One example is a glycogen-bound form of PP1 holoenzyme, which binds P-T38-CPI-17 but dephosphorylates it. In contrast, with myosin phosphatase there is binding of P-T38-CPI-17, but the hydrolysis step is arrested, resulting in formation of inactive complex. Thus, P-T38-CPI-17 can be a substrate or an inhibitor of different forms of PP1, depending on regulatory subunits (Eto, et al, 2004). In smooth muscle cells, rapid phosphorylation of CPI-17 at Thr38 occurs in parallel to phosphorylation of myosin via activation of Ca2+-dependent PKC, followed by prolonged phosphorylation, maintained via RhoA/ROCK signal (Dimopoulos, et al, 2007). Phosphorylation of CPI-17 is reduced through a cGMP-dependent pathway in response to nitric oxide release, in parallel to muscle relaxation (Bonnevier, et al, 2004; Etter, et al, 2001). Thus, in smooth muscles phosphorylation of CPI-17 functions to inhibit myosin phosphatase and thereby promote myosin phosphorylation and contraction.
Fluctuation in the expression and the phosphorylation levels of CPI-17 is associated with smooth muscle-related diseases, such as intestinal bowel disease (Ohama, et al, 2003), asthma (Sakai, et al, 2005), pulmonary hypertension (Dakshinamurti, et al, 2005), and diabetic dysfunction of smooth muscle (Chang, et al, 2006; Xie, et al, 2006). In Purkinje neurons, CPI-17 is essential to maintain G-protein-mediated AMPA receptor internalization and generate long-term synaptic depression in response to stimulation(Eto, et al, 2002). In addition, the upregulation of CPI-17 was found in tumor cells, which causes hyperphosphorylation of tumor suppressor merlin (NF2) and transformation of cells (Jin, et al, 2006). Therefore, CPI-17 poses a potential target of pharmaceutical approaches for these diseases.
Determination of the three-dimensional (3D) structure of unphospho-CPI-17 by NMR revealed a 2×2 pairing of a four-helix bundle, forming a unique V-shape structure (Ohki, et al, 2001). The phosphorylation site, Thr38, is located in a loop structure (P-loop) at the N-terminus, sitting in a cavity between helices (Ohki, et al, 2001). When Thr38 is substituted for Asp to mimic phosphorylation, the P-loop becomes exposed to solvent, suggesting a conformational change of the P-loop upon activation of CPI-17 (Ohki, et al, 2003). However, structural analysis of the basis for enhanced inhibition of myosin phosphatase by CPI-17 was limited because the D38-form of CPI-17 is significantly less potent, compared with phospho (P)-T38-CPI-17 (Ohki, et al, 2003). We concluded that despite a conformational change in the D38 protein, this structure was not the same as that of the highly potent and fully activated P-T38-CPI-17. Therefore we prepared CPI-17 protein fully phosphorylated with PKC and determined the 3D structure using multidimensional NMR techniques. The 3D structure indicates a global conformational switch upon phosphorylation of CPI-17 at Thr38, shows differences relative to the phospho-mimetic D38 structure and exposes novel interactions between P-CPI-17 and myosin phosphatase.
RESULTS
Phosphorylation of CPI-17(22–120)
The inhibitory domain of CPI-17 comprised of residues 22–120 is conserved among the CPI-17 family of PP1 inhibitor proteins, such as PHI-1, KEPI and GBPI (Figure 1A). The recombinant 13C/15N-labeled 22–120 protein was phosphorylated to completion by extended incubation with purified PKC (Supplementary Figure S1), and subjected to a series of multidimensional NMR experiments (Supplementary Figure S2, and S3). Figure 1B shows part of the 1H-15N HSQC spectrum of 13C/15N-labeled P-CPI-17 (red) superimposed on the previous spectra of unphosphorylated (gray) and Asp-substituted D38-CPI-17 (blue) (22–120) proteins (Ohki, et al, 2003). The backbone amide resonance of Thr38 is shifted to downfield upon phosphorylation (Figure 1B). This phosphorylation-dependent downfield shift is consistent with a previous report on phosphorylation of the tau protein (Landrieu, et al, 2006). In addition, the phosphorylation at Thr38 induces a relatively large chemical shift change of amide resonances from the neighbor residues in the phosphorylation loop (P-loop), such as V39 and Y41. The chemical shifts of these residues in P-T38-CPI-17 did not match those in the D38-form of CPI-17 (blue). This suggests that the conformation of the phosphorylated protein differs from the conformation of the protein with a “phosphomimetic” mutation to introduce an Asp at the same residue. Overall the profiles of HSQC spectra in the full-scale chart (Supplementary Figure S2) are similar in all three forms of CPI-17 examined by NMR. Furthermore, there was no evidence of multiple conformation states in the spectrum of P-T38-CPI-17.
Figure 1. Inhibitory domain of CPI-17 family.
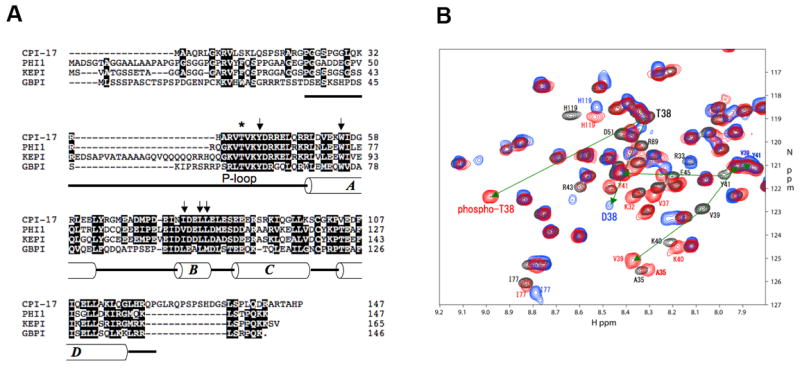
(A) Amino acid sequence of CPI-17 family of PP1 inhibitor proteins. Asterisk indicates Thr38, and arrows indicate Trp55, Ile77, Leu80, and Leu81. Boxes show regions in helices A to D. (B) 1H-15N HSQC spectrum of 13C/15N-labeled P-CPI-17 (1 mM). Red, gray and blue indicate amide resonances of P-, U-, and D38-CPI-17(22–120), respectively. Resonance peaks of T38, V39 and V41 were connected by arrows, from U-form to P- or D38-form.
Structure of phospho-T38-CPI-17
The 3D structure of P- T38-CPI-17 was calculated using 1,518 structural restraints obtained from the NMR data (Supplementary Figure S3), summarized in Supplementary Table S1. Overall the topology of P-T38-CPI-17 is composed of a long loop with the phosphorylation site, Thr38, followed by a left-handed 4-helix bundle (termed A to D from the N-terminus) (Figure 2). The four helices are arranged in an anti-parallel orientation. The structure of residues 22 to 31 at the N terminus of the truncated protein was in a flexible conformation, seen by lack of long-range NOE. This conclusion is supported by very small heteronuclear 15N-{1H} NOE values (Supplementary Figure S4) and the fast H-D exchange rates (data not shown) of these10 residues. In contrast, the conserved region of residues 32 to 40 with phospho-Thr38 (P-Thr38), termed the P-loop, converged into a single conformation with a backbone r.m.s.d. of 1.18 ± 0.27 Å, with relatively higher heteronuclear 15N-{1H} NOE values (Supplementary Figure S4, red). The P-Thr38 side chain is exposed to solvent, and the P-loop lays on the surface of the four-helix bundle. The side chains of Val37 and Val39 face into the protein, making contacts with Ile56 and Tyr41/Val52/Ile77, respectively. These two Val residues function to anchor the P-loop to the four-helix bundle by hydrophobic interactions of the aliphatic side chains.
Figure 2. Solution NMR structure of phospho-CPI-17(22–120).
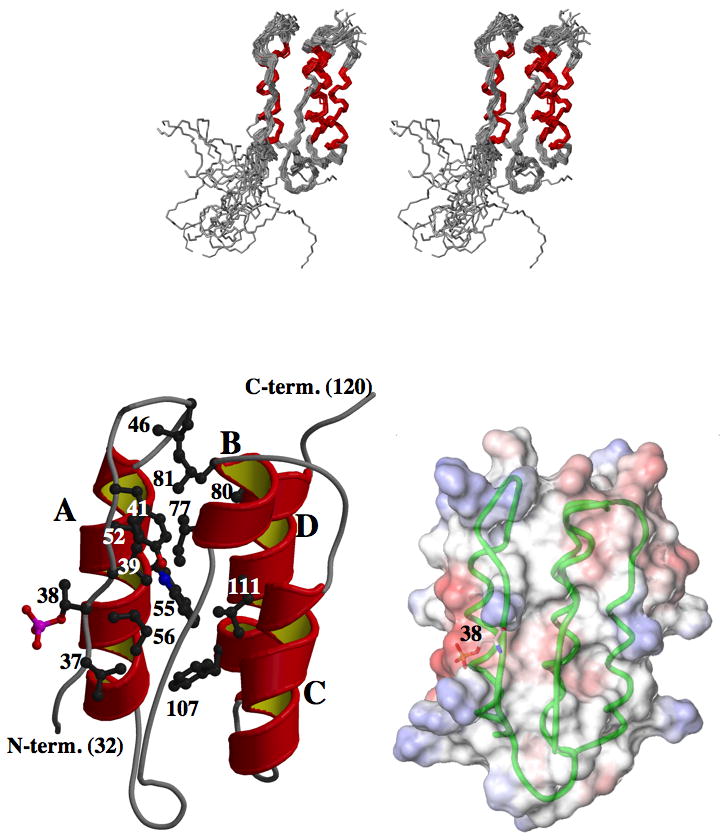
(A) Stereo view of the backbone of 20 superimposed structures obtained by NMR analysis. Red indicates the region of α-helix. (B) Ribbon representation of an energy-minimized average structure. In the ribbon model, the flexible N-terminal 31 residues are not depicted. T38 and residues involved in hydrophobic clustering are drawn with ball-and-stick side chains labeled with residue numbers to facilitate identification, and the helices are labeled with upper case letters. (C) Space-filling representation with molecular surface potential, superimposed on backbone structure (green). Red and blue on surface indicate regions of negative and positive charges, respectively. A side chain of P-T38 is drawn as ball-and-stick.
Comparison between unphospho-, phospho-, and Asp-substituted CPI-17 structures
Comparison of P-T38-CPI-17 structure with that in the unphospho-form (U-CPI-17) and D38-form (Ohki, et al, 2003) reveals global conformational change in response to phosphorylation (Figure 3) (Table S2). The most remarkable difference is the position of the key residue, Thr38, whose side chain comes out of a cavity between A and B helices upon phosphorylation. When structures are superimposed using the A/D helix pair for alignment (Figure 3, bottom), the phosphorylation at Thr38 is seen to trigger a swinging motion of the P-loop around the A-helix, resulting in 8.1 Å movement of residue 38. The swing of the P-loop is coupled with a right-handed rotation of A-helix by 29 degrees, along with a complementary rotation of the D-helix. These rotations expose new surfaces of both helices that become available for binding to myosin phosphatase. Similar to U-CPI-17, the A/D helix-pair in P-T38-CPI-17 is stabilized by hydrophobic residues as shown in Figure 3. In concert with motion of the A-helix, the B/C helix-pair becomes close to the A/D helix-pair. This aligns the four helices into an anti-parallel bundle (Table S3). The average distance between the four helices becomes 15 % shorter (Table S3), and the compaction of the structure causes the overall surface area of the protein to be reduced from 7,221 to 6,374 Å2. Substitution of T38 to Asp results in the P-loop becoming exposed to solvent, consistent with changes in P-form. However, the re-alignment of four helices is not evident in the D38 protein relative to the U-CPI-17, and the overall structure remains V-shape (Figure 3, center).
Figure 3. Conformational transition of CPI-17 upon phosphorylation.
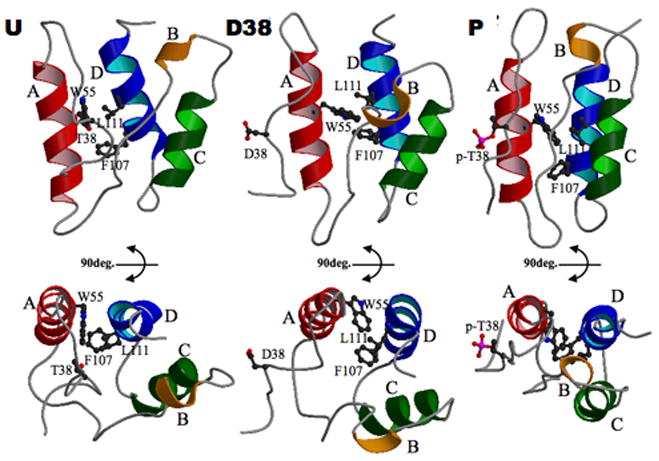
U-CPI-17 (1J2M) (left) D38-CPI-17 (1J2N) (middle) and P-T38-CPI-17 (right) are displayed as ribbon structures. Side chains of hydrophobic residues that tether Helix A to D are shown as ball-and-stick for emphasis.
Formation of hydrophobic core
The phosphorylation-induced compression of the four helices gathers Tyr41 in the P-loop, Leu46/Val52 in A-helix, and Ile77/Leu80/Leu81 in B-helix into a stable hydrophobic core (Figure 4A). For example, the distance between Val 52 and Ile77 Cα atoms shortens from 19.0 to 6.4 Å. Such hydrophobic clustering likely functions to stabilize the anti-parallel-aligned four-helix bundle of P-CPI-17. This stabilization reflects in higher heteronuclear NOE value of P-T38-CPI-17, compared with U- and D38-CPI-17 (Supplementary Figure S3). We assayed thermal stabilities of U-and P-T38-CPI-17, and indeed, phosphorylation increased the mean melting temperature Tm from 59 °C (U-CPI-17) to 64 °C (P-T38-CPI-17), showing an effect on the thermodynamic stability of the protein. We attribute this change to the hydrophobic core in P-T38-CPI-17 (Figure 4B). The hydrophobic core is not well-defined in the D38-CPI-17 structure (Figure 4A, middle). Consistent with this observation, we found the thermal stability of D38-CPI-17 was lower (Tm = 54 °C), compared to P-T38-CPI-17 (Figure 4B). Overall, the structure and thermal stability are consistent with the idea that phosphorylation but not mutation to an acidic residue triggers condensation of the protein into a rigid structure, with a surface topography distinctly different from the unphosphorylated protein.
Figure 4. Formation of the hydrophobic core in P-CPI-17.
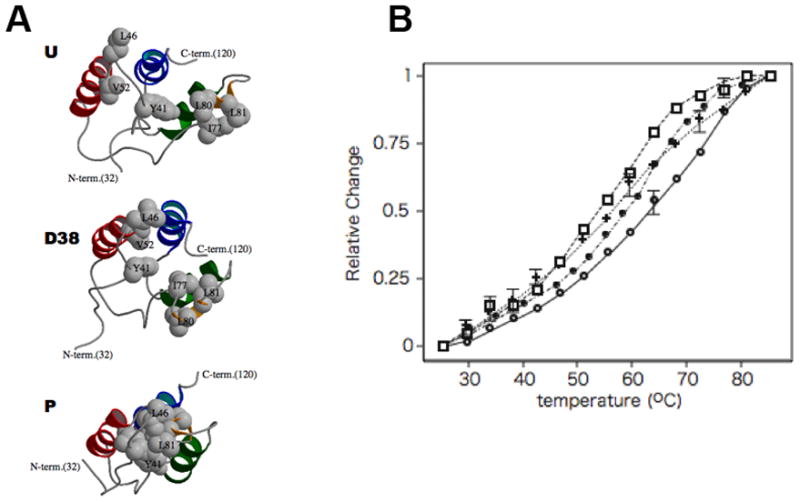
(A) Top, middle, and bottom panels represent the structures of U-, D38-, and P-T38-CPI-17, respectively. Hydrophobic side chains responsible for the condensed conformation are displayed as Corey, Pauling and Koltun colored model. (B) Thermal stability of CPI-17 proteins monitored by circular dichroism (CD). Vertical axis shows relative change of the molar ellipticity at 222 nm at indicated temperature. CD spectrum was measured with 20 μM protein; P-T38-CPI-17 (open circle), U-CPI-17 (closed circle), D38-CPI-17 (Rectangle), and P-T38-CPI-17 (Y41A) (cross), in 50 mM potassium phosphate buffer, pH 7.0. Mean values from triplicate assays are shown.
The hydrophobic core of P-T38-CPI-17
We examined effects of mutation of residues in the hydrophobic core on the inhibition of purified myosin phosphatase, using wild type CPI-17 and Y41A, W55A, D73A/E74A, and I77A/L80A/L81A mutants. All these recombinant CPI-17, were equally phosphorylated by purified PKC, suggesting that the mutations do not impair reaction with Thr38 (Table S4). However, the phosphorylated mutant CPI-17 showed different inhibitory potency (Figure 5A). Mutation at either Trp55 or Asp73/Glu74 caused moderate reduction in inhibitory potency, and the triple mutation of Ile77/Leu80/Leu81 showed greatly reduced potency, to an IC50 of around 100 nM. Our previous data showed that an Ala-mutation at Tyr41 (Y41A) eliminated the inhibitory activity of CPI-17 (Hayashi, et al, 2001). Thermal stability of the Y41A protein (Tm = 56 °C) was lower than that of wild type (Figure 4B, cross). We concluded that the hydrophobic clustering of Y41 and Ile77/Leu80/Leu81 was required for CPI-17 to act as an inhibitor for myosin phosphatase. The function of CPI-17 mutated in the hydrophobic core was examined using beta-escin-permeabilized vas deferens smooth muscle tissues (Figure 5B). Because CPI-17 is present at only negligible levels in vas deferens smooth muscle, phorbol ester (PDBu, a PKC activator) stimulation does not induce force production at limiting Ca2+ concentrations (Figure 5B, arrowhead) (Woodsome, et al, 2001). Doping of recombinant U-CPI-17 into a permeabilized vas deferens smooth muscle strip restored PDBu-induced force production at clamped Ca2+ concentration, suggesting that PKC induced the phosphorylation of CPI-17 causing inhibition of the endogenous myosin phosphatase in the strip (Figure 5B, double arrowhead). The triple-mutant CPI-17, I77A/L80A/L81A or Y41A (Figure 5B), did not induce PKC-mediated smooth muscle contraction, suggesting that the formation of hydrophobic core is necessary for CPI-17 to inhibit myosin phosphatase in tissues. On the other hand, CPI-17 mutants D73A/E74A and W55A supported PDBu-induced force production. The maximum extent and the rate of force development were slightly lower with these mutant proteins, compared with wild type -17. We note that D73, E74 and W55 are conserved among CPI-17 from various species, so changes in these residues have not survived probably because of the changes in properties. .
Figure 5. Effects of the mutation in the hydrophobic core on the inhibitory potency of CPI-17.
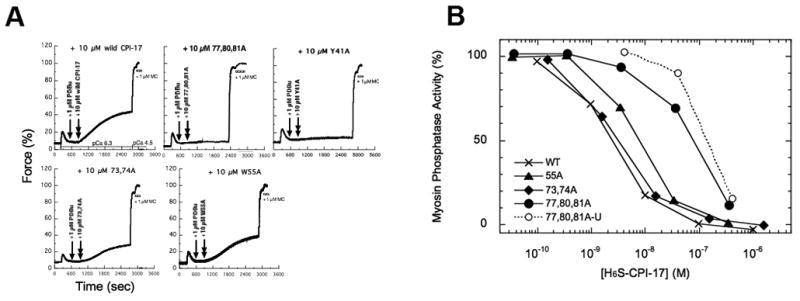
(A) Inhibition of myosin phosphatase purified from pig aorta. The myosin phosphatase activity without inhibitor proteins was set as 100 %. Data are averaged from two independent assays done in duplicate. (B) Force measurement of vas deferens smooth muscle strip. Panels represent the time-dependent force trace with U-CPI-17: wild type, D73A/E74A, I77A/L80A/L81A and W55A. The rabbit vas deferens smooth muscle strips were permeabilized with beta-escin. The CPI-17 proteins were doped into the tissue after the addition of PDBu. The extent of contraction in the presence of 1 μM Ca2+ is set as 100 % value.
Modeling of the interaction between P-T38-CPI-17 and myosin phosphatase
The phospho-pivot computer modeling method was used to examine hypothetical docking of P-T38-CPI-17 with myosin phosphatase (Eto, et al, 2004; Matsuzawa, et al, 2005a; Matsuzawa, et al, 2005b). The phosphate group of P-T38-CPI-17 was set into the active site of PP1δ•MYPT1(1–299) complex (Terrak, et al, 2004), and 186,624 in-silico complexes were generated by rotating P-T38-CPI-17 around the phosphorus atom as a pivot. Each model complex was scored based on only atomic distances between two proteins. The modeling yields one converged structure of P-CPI-17 on myosin phosphatase (Figure 6A). In this complex, the P-loop fits in the active site groove of PP1, from the cavity of ankyrin-repeat domain toward the N-terminal alpha-helical segment of MYPT1 (Terrak, et al, 2004). This model suggests that four Arg side chains in P-loop, Arg 33, 36, 43, and 44, are involved in the interaction with myosin phosphatase, in addition to phospho-Thr38. This is consistent to our previous results that an Ala mutation of CPI-17 at Arg 43 and Arg 44 impairs the inhibitory potency over 40 fold (Hayashi, et al, 2001). Interestingly, this model positions Asp5 on N-terminal segment of MYPT1 in close proximity to Arg44 of P-T38-CPI-17 (Figure 6A). Experimental testing of this model using immobilized peptides corresponding to MYPT1 segments (1–19) and (24–41) showed weak binding of P-T38-CPI-17 to MYPT1(1–19), but not to MYPT1(24–41) (Figure 6B). Thus, the N-terminal segment of MYPT1 (1–19) is possibly involved in the interaction with P-CPI-17.
Figure 6. Interaction of P-CPI-17 with PP1•MYPT1 complex.
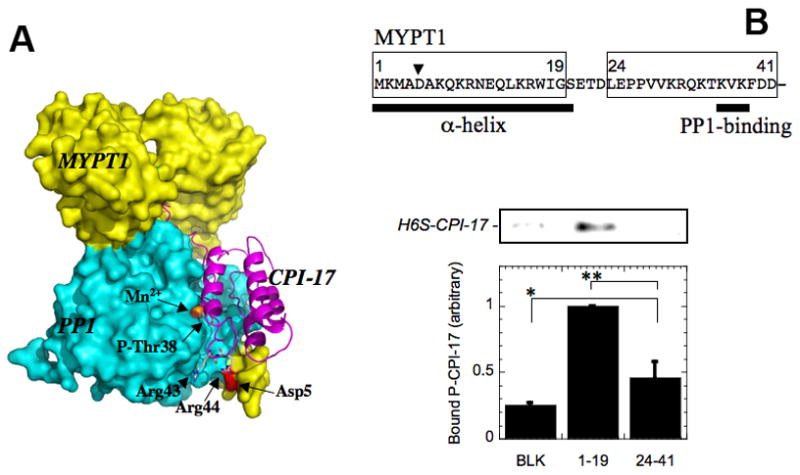
(A) The coordinates of MYPT1(1–299)•PP1 complex (1S70) in Protein Data Bank were used for computer modeling of the complex. MYPT1 (yellow) and PP1 (cyan) are depicted in a surface model, and P-T38-CPI-17 is drawn as a ribbon structure. The positive charge of MYPT1 Asp5 is shown as red, and the side chains of phospho-Thr38 and Arg43/44 in CPI-17 are shown as sticks. (B) Binding assay of P-T38-CPI-17 was performed with synthetic peptides corresponding to MYPT1 segments (1–19) and (24–41), immobilized on agarose beads. Asp5 is indicated by arrowhead in the sequence. Bound P-T38-CPI-17 was quantified by immunoblotting with anti-CPI-17 antibody (top). Mean values from 3 independent experiments are represented as a bar graph. Student’s t test was used to assess significance and * and ** indicate p > 0.15 and P < 0.03 respectively.
DISCUSSION
From comparison of the structures of the inactive U-CPI-17 and the active P-T38-CPI-17 we identify a conformational transition upon phosphorylation of CPI-17 at Thr38. The phosphorylation induces a swing of the P-loop and a formation of the hydrophobic core to lock and stabilize a condensed active conformation of P-T38-CPI-17. We conclude that this swing-and-lock mechanism of CPI-17 is responsible for potent and specific inhibition of myosin phosphatase to switch phosphorylation levels of myosin and merlin in response to G-protein activation. All PP1 specific inhibitor proteins are regulated by phosphorylation, and five of them are in the CPI-17 family, based on the similarity of the sequences including residues in the hydrophobic core identified here by NMR, so these results should apply to other CPI-17 family members. On the other hand, phosphorylation of another class of PP1 inhibitors, inhibitor-1 and DARPP-32, apparently only induces a minor conformational change in the molecule, despite over 1000-fold increase in the inhibitory potency (Endo, et al, 1996; Neyroz, et al, 1993). The phosphorylation-induced conformational switch seems to be a unique feature in the CPI-17 family of proteins.
Asp- or Glu-substitution of Ser or Thr is often used for mimicking phosphorylation of proteins. One example is the activation loop of protein kinases. Phosphorylation of protein kinases in the activation loop is required for full activity, and it can be mimicked by substitution with Asp or Glu (Zhang, et al, 1995). The phosphate group in the activation loop of kinases directly forms salt bridges with Arg residues in N-terminal domain (Canagarajah, et al, 1997; Johnson, et al, 1996) and the effectiveness of Glu or Asp suggests these residues can form salt bridges with the same Arg. In contrast the phosphate of P-T38-CPI-17 is exposed to solvent and interacts instead with the active site of PP1 in myosin phosphatase.. Asp-substitution at Thr38 induces a swing of the P-loop, but not the same re-alignment of helices or formation of the hydrophobic core in the protein as is formed when T38 is phosphorylated (Ohki, et al, 2003). The failure of hydrophobic clustering in D38-CPI-17 is evidenced by lower thermal stability. Without the hydrophobic core there is a 100-fold lower inhibitory potency of D38-CPI-17 compared to P-T38-CPI-17. Thus, the negative charge of a carboxyl group is insufficient for completion of the conformational switching in CPI-17. This gives an example of where an acidic side chain is not phosphomimetic, offering a note of caution to the common practice of using properties of mutated proteins to make conclusions about effects of phosphorylation.
Currently, about 150 protein structures with phospho-Thr are listed in PDB database. In most cases, the phosphate group is directly tethered to other residues in the same protein via salt bridges and/or hydrogen bonds. In this way the phosphate drives formation of a new protein conformation that is stabilized by these intramolecular interactions, as seen in glycogen phosphorylase-a and protein kinases (Johnson, et al, 1996; Sprang, et al, 1988). Another example is the NMR structure of a 44-residue P-Thr peptide mimicking the forkhead-associated (FHA) domain of human Ki67 protein in the complex with its receptor, the human nucleolar protein hNIHK, where the phosphate functions as a ligand (Byeon, et al, 2005). Our studies reveal a different mechanism of phosphorylation-induced conformational change. The phospho conformation of the CPI-17 P-loop is stabilized without a hydrogen bond or salt bridge to the phosphate group from other residues in the protein. Based on heteronuclear NOE data, the P-loop of unphosphorylated CPI-17 is highly flexible and this presumably exposes Thr38 to kinases, such as PKC and ROCK. Upon the phosphorylation of T38 the side chains of neighboring V37, V39 and Y41 anchor the P-loop to the hydrophobic core formed between the helices that restrains the overall solution structure. The rigid structure of phosphorylated P-loop is likely critical for the potent inhibition of the phosphatase by positioning the phosphate relative to other residues in CPI-17 that form contacts with MYPT1 and PP1. When the hydrophobic core of P-T38-CPI-17 is perturbed by Ala-substitution of Tyr41, the P-Thr38 residue is readily dephosphorylated by myosin phosphatase (Hayashi, et al, 2001). We propose that the rigidity of the structure due to the hydrophobic core is key to preventing hydrolysis of the P-T38, making P-T38-CPI-17 an inhibitor instead of a substrate. Thus, the swing-and-lock mechanism of CPI-17 activation restricts flexibility of the P-loop and protects P-Thr38 from being hydrolyzed at the active site of myosin phosphatase. Interestingly, P-T38 is hydrolyzed by other PP1 holoenzymes, such as glycogen phosphatase (a heterodimer of PP1 plus GM subunit), so phospho-CPI-17 is both a substrate and an inhibitor, depending on the form of PP1 it encounters.
Previously, phospho-pivot modeling with D38-CPI-17 (PDB: 1J2N) and monomeric PP1 alpha isoform catalytic subunit (1IT6) predicted critical roles for residues at the active site of PP1δ, namely D137, D193, R220, Y271, and E274 Indeed, these residues are necessary for the binding of PP1δ with P-T38-CPI-17 in an in vitro assay (Matsuzawa, et al, 2005a). These same residues at the PP1 active site seem to be involved in the model of the P-T38-CPI-17•PP1δ•MYPT1(1–299) complex. However, the best fitting model was obtained when the pivot phosphorus atom was set at a position 0.5 Å away from the active site. Thus, the phosphate group is not in an optimum position for hydrolysis in this model. We speculate that weak interactions of the MYPT1 subunit with P-T38-CPI-17, perhaps involving the MYPT1 N-terminal (1–19) segment, displaces phospho-Thr38 from being in an optimum position at the PP1 active site for dephosphorylation. Some involvement of MYPT1 seems to be needed to account for the inhibitor vs. substrate reaction of P-T38-CPI-17 with different PP1 holoenzymes.
It is noteworthy that the phospho-pivot model positions the unstructured N-terminal tail of CPI-17 near the MYPT1 ankyrn repeat domain. Other CPI-17 family members, PHI-1 and KEPI, also inhibit myosin phosphatase, but with different potencies. The IC50 for PHI-1 (50 nM) is 50-fold higher than that for CPI-17, whereas KEPI inhibits myosin phosphatase with IC50 of 0.1 nM (Erdodi, et al, 2003; Eto, et al, 1999). It is possible that the N-terminal tail of the different CPI-17 family members functions as a sensor for different regulatory subunits of PP1, This gives us a future direction for studying functional diversity in the CPI-17 family. Despite a focus on the MYPT1 N terminal domain we cannot completely rule out involvement of the MYPT1 C-terminal domain and/or the M21 subunit in specific recognition of P-CPI-17. Other PP1 inhibitor proteins are known to contact both PP1 catalytic and regulatory subunits to form trimeric complexes that account for their selective inhibition of PP1 in different contexts. One example is inhibitor-1 binding to GADD-34 that is bound to PP1, and other examples are inhibitor-2 binding to either neurabin or KPI-2 that both bind PP1 at independent sites. (Connor, et al, 2001; Terry-Lorenzo, et al, 2002; Wang, et al, 2002). Inhibitor-2 also binds to PP1 that is engaged with an VxF motif in Nek2A, forming a heterotrimer with PP1 as the bridging protein (Eto, 2002 and Li 2007) Direct contact between regulatory subunits and PP1 inhibitor proteins is an attractive idea to provide signaling specificity for controlling individual PP1 complexes. The structure of an activated PP1 inhibitor gives general insight into phosphorylation-dependent conformational changes and regulation of cellular PP1, and opens a door to understanding mechanisms of signaling crosstalk between kinases and phosphatases.
METHODS
Proteins
The cDNA of CPI-17 was cloned from pig aorta smooth muscle library. Recombinant CPI-17 (22–120) for NMR was prepared using pET30 bacterial expression vector with the minimal medium containing 15N-ammnonium chloride and/or 13C-glucose as the sole source of nitrogen and/or carbon. This method yields the uniformly stable-isotope labeled (> 95 %) sample for NMR spectroscopy (Ohki, et al, 2001; Ohki, et al, 2003). CPI-17 proteins for other assays are expressed as full-length versions with N-terminal His6-and S-tag™ sequences (Eto, et al, 2003). Phosphorylation of the protein was performed with an active fragment of PKC. The active fragment of PKC was extracted from human red cells with hypotonic buffer and purified by ammonium sulfate precipitation, and sequencial column chromatographies using DEAE-Sepharose, Phenyl-Sepharose, Q-Sepharose and Protamine-agarose. Stoichiometric phosphorylation of CPI-17 was verified by urea-PAGE analysis (Figure S1), as described previously (Eto, et al, 2003). Anti-P-T38-CPI-17 IgY was prepared using synthetic phospho-peptide as an antigen and purified by affinity chromatography, as described previously (Kitazawa, et al. 2000) (Aves Lab, Tigard OR).
NMR structure determination
All NMR data for ~1 mM P-T38-CPI-17 (in 50 mM phosphate buffer pH 6.8, 100 mM KCl, 1 mM DTT, and 0.02 % NaN3) were recorded on a Varian INOVA750 spectrometer. The sample temperature was kept at 25.0 °C. A set of two-and three-dimensional NMR data was obtained for resonance assignments (Bax, et al, 1992). Homonuclear 2D-NOESY, 15N-edited NOESY, and 13C-edited NOESY with mixing time of 100 msec were recorded to collect distance information. To obtain the information about hydrogen bonding, amide proton H-D exchange was monitored by 1H-15N HSQC. 2D-HMQC-J spectra were also recorded to obtain dihedral angle constraints. All NMR data were processed using nmrPipe/nmrDraw (Delaglio, et al, 1995) and were analyzed with PIPP (Garrett, 1991). Structure calculation was carried out using X-PLOR version 3.851 (Brunger, et al, 1998; Kleywegt, et al, 1998). Quality of each coordinate was examined by using AQUA/procheck-NMR (Laskowski, et al, 1996). The Ramachandran plot analysis of the final structures showed that 1.7 ± 1.1% of non-glycine and non-proline residues were in the disallowed regions.
Structural analysis
All structural images were drawn with MOLMOL (Koradi, et al, 1996), Molscript (Kraulis, 1991), and Raster3D (Merrit, et al, 1997). Vector geometry mapping and calculations of surface potential and area were done using an algorithm of Yap, et al. (Yap, et al, 2002) and PyMol Molecular Graphic System (http://www.pymol.org), respectively. The rotation angle of A-helix was estimated from mean value of differences on Calpha-Cbeta angle of each residue.
Phospho-pivot modeling
An in silico model of the ternary complex, P-T38-CPI-17/PP1 delta/MYPT1(1–299) was obtained using the phospho-pivot modeling method, as described previously (Matsuzawa, et al, 2005a; Matsuzawa, et al, 2005b). Structural data of P-T38-CPI-17 (present study) and the myosin phosphatase complex of MYPT1(1–299) and PP1 delta (Terrak, et al, 2004) were used for the modeling. The best result was obtained when the phosphate group of P-CPI-17 was placed at a point 0.5 Å-away from the putative phosphorus position. The atomic distance violation was set with the threshold of 3.5 Å (Ca-Ca), or 2.0 Å (N-O, N-N, O-O). Models without distance violation between main chain atoms, identified as 68 out of 186,624 models, were subjected to an energy minimization process with permitted of structural adjustments on both molecules, using AMBER force field on SYBYL/BIOPOLYMER (Tripos Inc.). In this modeling atoms within 8 Å of CPI-17 were used for the energy calculation. The results of energy minimization were shown in Table S5. The orientation of P-CPI-17 against MYPT1•PP1 complex is the same in 4 of the best 5 models. Both electrostatic and hydrogen-bonding energy values dominantly contribute to the total binding force. The best model (shown in Figure 6A) includes 15 atomic violations.
Assays
CD spectrum measurements were performed on a JASCO 720 spectropolarimeter at various temperatures (20–95 °C) with 20 μM U- and H6S-P-T38-CPI-17 full-length proteins in phosphate-buffered saline. Thermal stability of CPI-17 was assessed via the melting temperature (Tm) from the plot of molar ellipticity at 222 nm against the temperature. Other assays were carried out at 20 °C. The H6S-tag does not affect the CD spectrum (data not shown). The inhibitory potency of CPI-17 proteins was measured using myosin phosphatase purified from pig aorta smooth muscle with 0.5 μM 32P-labeled phospho-myosin light chain as a substrate, at 20 °C (Eto, et al, 2003). The Ca2+ sensitizing effect of CPI-17 protein was examined at 20 °C in vas deferens smooth muscle strips permeabilized with beta-escin (Masuo, et al, 1994). This tissue preparation s depleted to a minimum amount of endogenous CPI-17, but retains other regulatory proteins, such as myosin phosphatase, PKC and ROCK (Woodsome, et al, 2001). Binding assay of P-CPI-17 was performed with synthetic segments of MYPT1 (1–19) and (24–41) (GenScript, Piscataway, NJ). MYPT1(1–19) and (24–41) peptides were immobilized onto Sulfo-link beads (Pierce) via a C-terminal Cys residue added for cross-linking, and then excess reactive groups on the beads were quenched with 50 mM L-Cys. Blank beads for controls were treated with 50 mM L-Cys without peptide conjugation. H6S-P-T38-CPI-17 (0.1 μM) was mixed for 30 min at 23 °C, with 10 μL of slurry in 150 μL of 50 mM MOPS-NaOH, pH 7.0 including 0.1 M NaCl, 1 mM EGTA, 0.1 % Tween 20, 5 % glycerol, 0.4 mM Pefabloc™, and 0.5 mM TCEP. The beads were washed with 100 μL of the binding buffer 3 times, and bound CPI-17 was detected by immunobloting with anti-CPI-17 using FluoroChemSP CCD imaging system (Alpha-Innotech). Relative amount of bound CPI-17 was obtained from three independent experiments by quantifying the band intensity using AlphaEace FC imaging software.
Acknowledgments
This work was supported in part by a grant from CNMT, JAIST (to S.O.), NHLBI HL070881 (to T.K.), NIGMS GM56362 (to D.L.B.), and AHA Scientist Development Grant, PA Department of Health C.U.R.E. grant, and NHLBI HL083261 (to M.E.).
Footnotes
Data deposition: The Chemical shift table and atomic coordinates are deposited in the BioMagResoBank and Protein Data Bank.
Accession codes in Protein Data Bank and BioMagResBank
The atomic coordinates and the chemical shift table of P-T38-CPI-17(22–120) protein are deposited in the Protein Data Bank (2RLT) and BioMagResoBank (15428), respectively.
References
- Bax A, Pochapsky SS. Optimized recording of heteronuclear multidimensional NMR spectra using pulsed field gradients. J Magn Reson. 1992;99:638–643. [Google Scholar]
- Bonnevier J, Arner A. Actions downstream of cyclic GMP/protein kinase G can reverse protein kinase C-mediated phosphorylation of CPI-17 and Ca(2+) sensitization in smooth muscle. J Biol Chem. 2004;279:28998–29003. doi: 10.1074/jbc.M404259200. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Byeon IJ, Li H, Song H, Gronenborn AM, Tsai MD. Sequential phosphorylation and multisite interactions characterize specific target recognition by the FHA domain of Ki67. Nat Struct Mol Biol. 2005;12:987–993. doi: 10.1038/nsmb1008. [DOI] [PubMed] [Google Scholar]
- Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- Chang S, Hypolite JA, DiSanto ME, Changolkar A, Wein AJ, Chacko S. Increased basal phosphorylation of detrusor smooth muscle myosin in alloxan-induced diabetic rabbit is mediated by upregulation of Rho-kinase beta and CPI-17. Am J Physiol Renal Physiol. 2006;290:F650–6. doi: 10.1152/ajprenal.00235.2005. [DOI] [PubMed] [Google Scholar]
- Connor JH, Weiser DC, Li S, Hallenbeck JM, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol Cell Biol. 2001;21:6841–650. doi: 10.1128/MCB.21.20.6841-6850.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakshinamurti S, Mellow L, Stephens NL. Regulation of pulmonary arterial myosin phosphatase activity in neonatal circulatory transition and in hypoxic pulmonary hypertension: a role for CPI-17. Pediatr Pulmonol. 2005;40:398–407. doi: 10.1002/ppul.20290. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Dimopoulos A, Semba S, Kitazawa K, Eto M, Kitazawa T. Ca2+-dependent rapid Ca2+ sensitization of contraction in arterial smooth muscle. Circ Res. 2007;100:121–129. doi: 10.1161/01.RES.0000253902.90489.df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo S, Zhou X, Connor J, Wang B, Shenolikar S. Multiple structural elements define the specificity of recombinant human inhibitor-1 as a protein phosphatase-1 inhibitor. Biochemistry (N Y) 1996;35:5220–528. doi: 10.1021/bi952940f. [DOI] [PubMed] [Google Scholar]
- Erdodi F, Kiss E, Walsh MP, Stefansson B, Deng JT, Eto M, Brautigan DL, Hartshorne DJ. Phosphorylation of protein phosphatase type-1 inhibitory proteins by integrin-linked kinase and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Comm. 2003;306:382–387. doi: 10.1016/s0006-291x(03)00976-8. [DOI] [PubMed] [Google Scholar]
- Eto M, Bock R, Brautigan DL, Linden DJ. Cerebellar long-term synaptic depression requires PKC-mediated activation of CPI-17, a myosin/moesin phosphatase inhibitor. Neuron. 2002;36:1145–1158. doi: 10.1016/s0896-6273(02)01107-8. [DOI] [PubMed] [Google Scholar]
- Eto M, Karginov A, Brautigan DL. A novel phosphoprotein inhibitor of protein type-1 phosphatase holoenzymes. Biochemistry. 1999;38:16952–16957. doi: 10.1021/bi992030o. [DOI] [PubMed] [Google Scholar]
- Eto M, Kitazawa T, Brautigan DL. Phosphoprotein inhibitor CPI-17 specificity depends on allosteric regulation of protein phosphathase-1 by regulatory subunits. Proc Natl Acad Sci USA. 2004;101:8888–8893. doi: 10.1073/pnas.0307812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M, Leach C, Tountas NA, Brautigan DL. [19] Phosphoprotein Inhibitors of Protein Phosphatase-1. Meth Enzymol. 2003;366:243–260. doi: 10.1016/s0076-6879(03)66019-2. [DOI] [PubMed] [Google Scholar]
- Eto M, Ohmori T, Suzuki M, Furuya K, Morita F. A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. Isolation from porcine aorta media and characterization. J Biochem. 1995;118:1104–1107. doi: 10.1093/oxfordjournals.jbchem.a124993. [DOI] [PubMed] [Google Scholar]
- Eto M, Senba S, Morita F, Yazawa M. Molecular cloning of a novel phosphorylation-dependent inhibitory protein of protein phosphatase-1 (CPI17) in smooth muscle: Its specific localization in smooth muscle. FEBS Lett. 1997;410:356–360. doi: 10.1016/s0014-5793(97)00657-1. [DOI] [PubMed] [Google Scholar]
- Etter EF, Eto M, Wardle RL, Brautigan DL, Murphy RA. Activation of Myosin Light Chain Phosphatase in Intact Arterial Smooth Muscle during Nitric Oxide-induced Relaxation. J Biol Chem. 2001;276:34681–34685. doi: 10.1074/jbc.M104737200. [DOI] [PubMed] [Google Scholar]
- Garrett DSea. A commonsense approach to peak picking in two-, three-, and four-dimensional spectra using automatic computer analysis of contour diagrams. J Magn Reson. 1991;95:214–220. doi: 10.1016/j.jmr.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Hartshorne DJ, Ito M, Erdodi F. Role of protein phosphatase type 1 in contractile functions: myosin phosphatase. J Biol Chem. 2004;279:37211–37214. doi: 10.1074/jbc.R400018200. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Senba S, Yazawa M, Brautigan DL, Eto M. Defining the Structural Determinants and a Potential Mechanism for Inhibition of Myosin Phosphatase by the Protein Kinase C-potentiated Inhibitor Protein of 17 kDa. J Biol Chem. 2001;276:39858–39863. doi: 10.1074/jbc.M107302200. [DOI] [PubMed] [Google Scholar]
- Jin H, Sperka T, Herrlich P, Morrison H. Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature. 2006;442:576–579. doi: 10.1038/nature04856. [DOI] [PubMed] [Google Scholar]
- Johnson LN, Noble MEM, Owen DJ. Active and inactive protein kinases: Structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/s0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, Matsumura F, Inagaki M, Kaibuchi K. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. J Cell Biol. 1999;147:1023–138. doi: 10.1083/jcb.147.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa T, Eto M, Woodsome TP, Brautigan DL. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J Biol Chem. 2000;275:9897–9900. doi: 10.1074/jbc.275.14.9897. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Masuo M, Somlyo AP. G protein-mediated inhibition of myosin light-chain phosphatase in vascular smooth muscle. Proc Natl Acad Sci U S A. 1991;88:9307–910. doi: 10.1073/pnas.88.20.9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA. Databases in protein crystallography. Acta Crystallogr D Biol Crystallogr. 1998;54:1119–1131. doi: 10.1107/s0907444998007100. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–5. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Koyama M, Ito M, Feng J, Seko T, Shiraki K, Takase K, Hartshorne DJ, Nakano T. Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Lett. 2000;475:197–200. doi: 10.1016/s0014-5793(00)01654-9. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots pf protein structures. J Appl Crystallog. 1991;24:946–950. [Google Scholar]
- Landrieu I, Lacosse L, Leroy A, Wieruszeski J, Trivelli X, Sillen A, Sibille N, Schwalbe H, Saxena K, Langer T, Lippens G. NMR analysis of a Tau phosphorylation pattern. Journal of the American Chemical Society. 2006;128:3575–3583. doi: 10.1021/ja054656+. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–86. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Masuo M, Readon S, Ikebe M, Kitazawa T. A novel mechanism for the Ca2+-sensitizating effect of protein kinase C on vascular smooth muscle: Inhibition of myosin light chain phosphatase. J Gen Physiol. 1994;104:265–286. doi: 10.1085/jgp.104.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura F. Regulation of myosin II during cytokinesis in higher eukaryotes. Trends in Cell Biology. 2005;15:371–377. doi: 10.1016/j.tcb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Matsuzawa F, Aikawa S, Ohki S, Eto M. Phospho-pivot modeling predicts specific interactions of protein phosphatase-1 with a phospho-inhibitor protein CPI-17. J Biochem. 2005a;137:633–641. doi: 10.1093/jb/mvi077. [DOI] [PubMed] [Google Scholar]
- Matsuzawa F, Ohki S, Aikawa S, Eto M. Computational simulation for interactions of nano-molecules: The phospho-pivot modeling algorithm for prediction of interactions between a phospho-protein and its receptor. Science and Technology of Advanced Materials. 2005b;6:463–467. [Google Scholar]
- Merrit EA, Bacon DJ. Raster 3D: photorealistics molecular graphics. In Methods Enzymol. 1997:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- Neyroz P, Desdouits F, Benfenati F, Knutson JR, Greengard P, Girault JA. Study of the conformation of DARPP-32, a dopamine- and cAMP-regulated phosphoprotein, by fluorescence spectroscopy. J Biol Chem. 1993;268:24022–24031. [PubMed] [Google Scholar]
- Ohama T, Hori M, Sato K, Ozaki H, Karaki H. Chronic treatment with interleukin-1beta attenuates contractions by decreasing the activities of CPI-17 and MYPT-1 in intestinal smooth muscle. J Biol Chem. 2003;278:48794–48804. doi: 10.1074/jbc.M310166200. [DOI] [PubMed] [Google Scholar]
- Ohki S, Eto M, Kariya E, Hayano T, Hayashi Y, Yazawa M, Brautigan D, Kainosho M. Solution NMR structure of the myosin phosphatase inhibitor protein CPI-17 shows phosphorylation-induced conformational changes responsible for activation. J Mol Biol. 2001;314:839–849. doi: 10.1006/jmbi.2001.5200. [DOI] [PubMed] [Google Scholar]
- Ohki S, Eto M, Shimizu M, Takada R, Brautigan DL, Kainosho M. Distinctive solution conformation of phosphatase inhibitor CPI-17 substituted with aspartate at the phosphorylation-site threonine residue. J Mol Biol. 2003;326:1539–1547. doi: 10.1016/s0022-2836(03)00048-2. [DOI] [PubMed] [Google Scholar]
- Pang H, Guo Z, Su W, Xie Z, Eto M, Gong MC. RhoA-Rho kinase pathway mediates thrombin- and U-46619-induced phosphorylation of a myosin phosphatase inhibitor, CPI-17, in vascular smooth muscle cells. Am J Physiol- Cell Physiology. 2005;289:C352–C360. doi: 10.1152/ajpcell.00111.2005. [DOI] [PubMed] [Google Scholar]
- Sakai H, Chiba Y, Hirano T, Misawa M. Possible involvement of CPI-17 in augmented bronchial smooth muscle contraction in antigen-induced airway hyper-responsive rats. Mol Pharmacol. 2005;68:145–151. doi: 10.1124/mol.104.004325. [DOI] [PubMed] [Google Scholar]
- Senba S, Eto M, Yazawa M. Identification of trimeric myosin phosphatase (PP1M) as a target for a novel PKC-potentiated protein phosphatase-1 inhibitory protein (CPI17) in porcine aorta smooth muscle. J Biochem. 1999;125:354–362. doi: 10.1093/oxfordjournals.jbchem.a022294. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Sprang SR, Acharya KR, Goldsmith EJ, Stuart DI, Varvill K, Fletterick RJ, Madsen NB, Johnson LN. Structural changes in glycogen phosphorylase induced by phosphorylation. Nature. 1988;336:215–221. doi: 10.1038/336215a0. [DOI] [PubMed] [Google Scholar]
- Terrak M, Kerff F, Langsetmo K, Tao T, Dominguez R. Structural basis of protein phosphatase 1 regulation. Nature. 2004;429:780–784. doi: 10.1038/nature02582. [DOI] [PubMed] [Google Scholar]
- Terry-Lorenzo RT, Elliot E, Weiser DC, Prickett TD, Brautigan DL, Shenolikar S. Neurabins Recruit Protein Phosphatase-1 and Inhibitor-2 to the Actin Cytoskeleton. J Biol Chem. 2002;277:46535–46543. doi: 10.1074/jbc.M206960200. [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L, Ichikawa K, Hartshorne DJ, Siegman MJ, Butler T. Thiophosphorylation of the 130-kDa subunit is associated with a decreased activity of myosin light chain phosphoatase in a-toxin-permeabilized smooth muscle. J Biol Chem. 1995;270:18191–18194. doi: 10.1074/jbc.270.31.18191. [DOI] [PubMed] [Google Scholar]
- Wang H, Brautigan DL. A novel transmembrane Ser/Thr kinase complexes with protein phosphatase-1 and inhibitor-2. J Biol Chem. 2002;277:49605–49612. doi: 10.1074/jbc.M209335200. [DOI] [PubMed] [Google Scholar]
- Woodsome TP, Eto M, Everett A, Brautigan DL, Kitazawa T. Expression of CPI-17 and myosin phosphatase correlates with Ca2+ sensitivity of protein kinase C-induced contraction in rabbit smooth muscle. J Physiol. 2001;535:553–564. doi: 10.1111/j.1469-7793.2001.t01-1-00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Su W, Guo Z, Pang H, Post SR, Gong MC. Up-regulation of CPI-17 phosphorylation in diabetic vasculature and high glucose cultured vascular smooth muscle cells. Cardiovasc Res. 2006;69:491–501. doi: 10.1016/j.cardiores.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Yap KL, Ames JB, Swindells MB, Ikura M. Vector geometry mapping. A method to characterize the conformation of helix-loop-helix calcium-binding proteins. Methods Mol Biol. 2002;173:317–324. doi: 10.1385/1-59259-184-1:317. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang F, Ebert D, Cobb MH, Goldsmith EJ. Activity of the MAP kinase ERK2 is controlled by a flexible surface loop. Structure. 1995;3:299–307. doi: 10.1016/s0969-2126(01)00160-5. [DOI] [PubMed] [Google Scholar]