

Abstract
Peroxisome proliferator-activated receptor alpha (PPARα) ligands are medications used to treat hyperlipidaemia and atherosclerosis. Increasing evidence suggests that these agents are immunosuppressive. In the following studies we demonstrate that WY14,643, a PPARα ligand, attenuates expression of anti-glomerular basement membrane disease (AGBMD). C57BL/6 mice were fed 0·05% WY14,643 or control food and immunized with the non-collagenous domain of the α3 chain of Type IV collagen [α3(IV) NC1] in complete Freund's adjuvant (CFA). WY14,643 reduced proteinuria and greatly improved glomerular and tubulo-interstitial lesions. However, the PPARα ligand did not alter the extent of IgG-binding to the GBM. Immunohistochemical studies revealed that the prominent tubulo-interstitial infiltrates in the control-fed mice consisted predominately of F4/80+ macrophages and WY14,643-feeding decreased significantly the number of renal macrophages. The synthetic PPARα ligand also reduced significantly expression of the chemokine, monocyte chemoattractant protein (MCP)-1/CCL2. Sera from mice immunized with AGBMD were also evaluated for antigen-specific IgGs. There was a significant increase in the IgG1 : IgG2c ratio and a decline in the intrarenal and splenocyte interferon (IFN)-γ mRNA expression in the WY14,643-fed mice, suggesting that the PPARα ligand could skew the immune response to a less inflammatory T helper 2-type of response. These studies suggest that PPARα ligands may be a novel treatment for inflammatory renal disease.
Keywords: anti-glomerular basement membrane disease, fibrates, glomerulonephritis, MCP-1, peroxisome proliferator-activated receptor
Introduction
Nuclear receptors, including the glucocorticoid, vitamin D and thyroid receptors regulate a wide variety of genes that control cellular differentiation, metabolism and inflammation. Peroxisome proliferator-activated receptors (PPARs), members of the nuclear receptor superfamily, are ligand-activated transcription factors that, in general, alter gene expression at the transcriptional level. The PPAR subfamily of receptors is encoded by three genes: α, δ (β and NUC1) and γ, each with distinct expression patterns and functions [1]. WY14,643, a fibric acid derivative, is a potent PPARα ligand. Other PPARα ligands, such as gemfibrozil and fenofibrate, are used clinically to treat hypertriglyceridaemia [2]. After PPARs are activated by their specific ligands, they bind to PPAR response elements in gene promoters and induce transcription. Alternatively, PPARs can modulate transcriptional events by binding and antagonizing other regulatory transcription factors or by sequestering key transcriptional co-activators or co-repressors [3].
PPARα ligands, such as gemfibrozil and fenofibrate, clearly reduce cardiovascular events in patients with atherosclerosis [4]. However, despite their proven efficacy in lowering triglycerides, there is accumulating evidence that fibrates are anti-inflammatory [5]. In our studies in mixed splenocyte cultures, fibrates potently increase interleukin (IL)-4, a key immunoregulatory cytokine [6]. In addition, in vivo WY14,643, a synthetic PPARα ligand, induces splenocyte depletion and alters production of antigen-specific immunoglobulins [7].
Anti-glomerular basement membrane disease (AGBMD) was considered originally the prototypical antibody-mediated autoimmune disease. However, studies suggest that cell-mediated mechanisms of immunity also cause renal injury [8–10]. As innate, adaptive, humoral and cell-mediated immune mechanisms are participatory in this disease, it provides an excellent model system to study anti-inflammatory agents and their mechanisms of action [11].
To investigate whether a PPARα ligand could abrogate expression of a renal inflammatory disease, mice were fed WY14,643 or control food and immunized to induce AGBMD. By multiple measures WY14,643 attenuated expression of AGBMD. WY14,643 reduced proteinuria and greatly improved glomerular and tubulo-interstitial lesions. However, the PPARα ligand did not alter the extent of IgG-binding to the GBM. Immunohistochemical studies revealed that the prominent tubulo-interstitial infiltrates in the control-fed mice consisted predominately of F4/80+ macrophages, and WY14,643-feeding decreased significantly the number of renal macrophages. Monocyte chemoattractant protein (MCP)-1/CCL2 is a major chemokine that directs the migration of macrophages and lymphocytes into the kidney. WY14,643 reduced significantly the expression of this chemokine. Sera from mice immunized with AGBMD were also evaluated for antigen-specific IgGs. There was a significant increase in the IgG1 : IgG2c ratio in the WY14,643-fed mice. WY14,643 treatment was also associated with lower intrarenal and splenocyte interferon (IFN)-γ mRNA expression, suggesting that the PPARα ligand could skew the immune response to a less inflammatory T helper 2 (Th2)-type of response. These studies support the concept that PPARα ligands are anti-inflammatory.
Materials and methods
Mice
C57BL/6 mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA). The mice were immunized at 6 weeks of age and followed for 6 months after immunization. Mice were housed and handled in accordance with Veterans Affairs (VA) and National Institute of Health (NIH) guidelines under Institutional Animal Care and Use Committee (IACUC) approved protocols.
Reagents
Incomplete Freund's adjuvant (IFA) and Mycobacterium tuberculosis (M.Tb) were obtained from Difco (Detroit, MI, USA). Complete Freund's adjuvant (CFA) was prepared as 4 mg/ml M.Tb and emulsifed 1 : 1 with IFA. WY14,643 was obtained from ChemSyn Laboratories (Lenexa, KS, USA).
Preparation of the human α3 NC1 domain of Type IV collagen [α3(IV) NC1]
Recombinant-α3(IV) NC1 was prepared as described previously [12]. 293EBNA cells were transfected with the expression plasmid pCEP-Pu containing a BM40 signal peptide, FLAG® tag and the human α3(IV) NC1 domain (including the last 10 amino acids of the collagenous domain) [13]. The transfected cells were grown in 10% fetal bovine serum (FBS)-Dulbecco's modified Eagle's medium (DMEM) containing 2 mM l-glutamine and 100 units/ml penicillin G and 100 μg/ml streptomycin. Cell lines expressing recombinant-α3(IV) NC1 were selected using 0·75 μg/ml puromycin. To isolate the α3(IV) NC1 protein, 293EBNA (α3(IV) NC1) cells were grown in DMEM (without FBS) for 48 h, the medium was collected and run over FLAG®-M2 agarose columns (Sigma-Aldrich, St. Louis, MO, USA). Protein was eluted with 0·1 M glycine, pH 3·5, concentrated with Amicon Microcon® Centrifugal Filter Devices (Millipore) and stored at −70°C.
Induction of AGBMD
To induce AGBMD, 32 male C57BL/6 mice were immunized with 20 μg of α3(IV) NC1 in CFA subcutaneously (s.c., base tail) and intradermally (i.d., footpad); each animal received 50% of antigen s.c. and 50% i.d. On day 30 mice were boosted with 20 μg α3(IV) NC1 in IFA. Mice were fed either control or standard rodent chow (7001) with 0·05% WY14,643 (Harlan-Teklad, Madison, WI, USA). Urinary albumins and creatinines were assessed weekly with the colorimetric albumin reagent bacille Calmette–Guérin (BCG) and creatinine assays (Sigma-Aldrich).
Assessment of α3(IV) NC1-specific immunoglobulins (Igs)
Serum was collected from mice by terminal cardiac puncture. Briefly, 96-well Maxisorp™ microtitre plates were coated with α3(IV) NC1 (1 μg/ml in 0·1 M carbonate buffer, pH 9·5) overnight at 4°C. Plates were blocked for 2 h with phosphate-buffered saline (PBS) containing 4% bovine serum albumin (BSA) (Sigma) and 0·05% Tween-20 (Sigma) and then incubated with serum samples for 2 h at room temperature (RT). Plates were developed with rabbit anti-mouse IgG (Calbiochem, San Diego, CA, USA), goat anti-mouse IgG1 (Caltag, Burlingame, CA, USA) or goat anti-mouse-IgG2c (Jackson ImmunoResearch, West Grove, PA, USA) alkaline phosphatase conjugates at RT for 2 h. Plates were incubated with p-nitrophenylphosphate disodium (Sigma, 1 mg/ml in 0·1 M carbonate buffer, pH 9·5) at RT and after 30 min incubation, optic densities at 405 nm were evaluated in a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Histology
After mice were euthanized, kidneys were removed and fixed in 10% buffered formalin. Alternatively, kidneys were frozen in Tissue-Tek OCT (optimal cutting temperature) compound (Sakura Finetek, Torrence, CA, USA) in liquid nitrogen. Light microscopic samples were embedded in paraffin, sectioned (4 μM) and stained with haematoxylin and eosin (H&E). Using the criteria described in Table 1, sections were graded by a blinded observer [14]. For immunofluorescence studies, 4 μM cryostat sections from OCT frozen samples were stained with fluorescein isothiocyanate (FITC)-goat-anti-mouse–IgG (1 : 1000; Caltag Laboratories). Fluorescence intensity was graded qualitatively by a blinded observer from 0 to 4 with 0·5 increments [8]. FITC–IgG-stained glomeruli were also graded in a quantitative manner. Images of at least five random glomeruli/mouse were captured at high power using a Jenalumar fluorescent microscope. Tagged image file format (TIFF) images were taken with a Nikon D50 digital camera with a set shutter speed (i.e. 2 s) and downloaded onto an Apple Macintosh Powerbook G4. Using Adobe Photoshop 7·0, images were selected and mean fluorescence intensity (MFI) and the number of pixels were evaluated in each glomerular tuft. Background values were subtracted and values were expressed as MFI (arbitrary units, AU) per glomerulus [15]. Glomerular size was also assessed (pixels) and MFI/pixels was evaluated.
Table 1.
Light microscopic grading criteria.*
| 0 | Normal |
| 1 | Minimal focal hypercellularity |
| 2 | Moderate hypercellularity, early tubular dilatation |
| 3 | Diffuse hypercellularity, tubular dilatation, early crescents and sclerosis |
| 4 | Maximum lesion, diffuse hypercellularity, marked tubular dilatation, crescents, sclerosis and glomerular obsolescence |
Based on grading system developed by Luo et al. Sections are graded in increments of 0·5.
Tubulo-interstitial and glomerular infiltrates were assessed by immunohistochemistry (IHC; University of California, San Diego Histology Core, CA, USA). Frozen sections (5 μM) were incubated with 0·03% H2O2, washed with PBS, incubated with 0·1% avidin, then 0·01% biotin (avidin–biotin blocking kit, Vector Laboratories, Burlingame, CA, USA) and blocked with 1% BSA/PBS. Slides were then stained with biotinylated anti-F4/80 (1 : 50; Caltag), biotinylated anti-CD3 (1:200; BD Pharmingen, San Diego, CA, USA) and anti-CD19 (1 : 100; BD Pharmingen) and then streptavidin horseradish peroxidase (HRP) (1 : 500; Jackson Immunoresearch, West Grove, PA, USA). Slides were washed and incubated with 3-amino-9-ethylcarbazole substrate (AEC; Vector Laboratories) and counterstained with Mayer's haematoxylin. For CD19 staining, slides were also treated with biotinylated anti-rat IgG (1 : 100). For F4/80, sections were incubated with streptavidin alkaline phosphatase (1 : 500, Jackson Immunoresearch) and Vector Blue (Vector Laboratories) and counterstained with nuclear Fast Red. Mononuclear cell infiltration was graded qualitatively based on previously published criteria [15] as follows: background staining: 0; lowest clearly positive staining: 1; mild staining: 2; moderate staining: 3; intense staining: 4. Also positively stained cells were counted in a minimum of 10 glomeruli per animal and results are expressed as cells/glomerular cross-section.
Real-time polymerase chain reaction (RT–PCR)
Whole kidney and spleen sections were stored in RNAlater™ (Qiagen, Valencia, CA, USA) as per the manufacturer's instructions. RNA was prepared with the RNeasy® Mini Kit (Qiagen) followed by a Turbo DNase™ (Ambion, Austin, TX, USA) treatment. cDNA was prepared with the Superscript II® First Strand Synthesis System (Invitrogen, Carlsbad, CA, USA). For quantification of IFN-γ, IL-4 and MCP-1, TaqMan® gene expression assays (IFN-γ: Mm00801778_m1 IL-4: Mm 00445259_m1 and MCP-1: Mm00441242_m1) were performed with TaqMan® Universal PCR Master Mix (10 min at 95°C with 50 cycles of 15 s at 95°C and 1 min at 60°C) in a Chromo4™ RT–PCR detector (MJ Research and Biorad, Hercules, CA, USA). Amplification efficiencies were normalized against RPL19 (forward: 5′-TGCTCAGGCTACAGAAGAGGCTTG-3′, reverse: 5′-GGAGTTGGCATTGGCGATTTC-3′) and relative fold increases were calculated using the Pfaffl technique of relative quantification, which accounts for real-time efficiencies. Each experiment was performed in triplicate and the data was log-transformed.
Statistics
Results were assessed by Student's t-tests or analyses of variance (anovas) with repeated measures with tests for linear and quadratic trends. Qualitative histological data were evaluated by the Mann–Whitney U-test and results were described with frequency distributions. Analysis was performed with spss version 11·0 (Chicago, IL, USA) with P < 0·05 used as the requirement for significance.
Results
Development of AGBMD
C57BL/6 mice were immunized on day 0 with α3(IV) NC1/CFA and boosted with α3(IV) NC1/IFA on day 30. Three days prior to immunization and throughout the study, mice were fed either control food or standard mouse chow with 0·05% WY14,643. Urinary albumins and creatinines were assessed weekly to follow the course of the disease. After 2·5 months control mice developed proteinuria. After this time-point and throughout the course of the disease the mean group urine albumin/creatinine ratio was always lower in the WY14,643-treated mice (Fig. 1a). In a similar manner, when mice developed urinary albumin/creatinine ratios of ≥ 3 they were considered to have high-grade proteinuria. Ninety days after immunization, the first control mice developed urinary albumin/creatinines of ≥ 3 and by day 189, 81% of control mice had high-grade proteinuria compared with 33% in the WY14,643-fed group. Therefore, proteinuria-free survival was significantly lower in the WY14,643-treated group of mice compared with the animals fed control food (Fig. 1b). Of note, mice immunized with α3(IV) NC1/CFA and fed WY14,643 do not gain weight over time when compared with the control-fed group (Fig. 2).
Fig. 1.
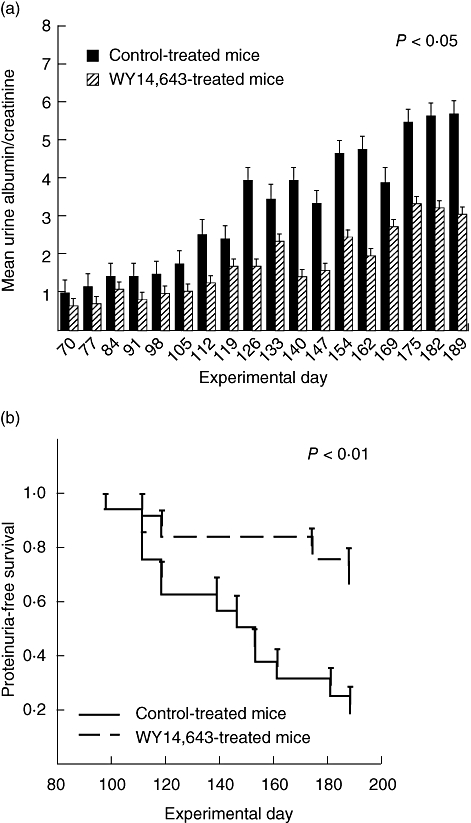
WY14,643 decreases proteinuria in anti-glomerular basement membrane disease (AGBMD). AGBMD was induced in C57BL/6 mice. Three days prior to immunization and throughout the time–course of the study mice were treated with control or WY14,643 (0·05% in the food). (a) Throughout the course of the disease mean group urine albumin/creatinine ratios were lower in the WY14,643-treated mice. (P < 0·05, 2 by 18 analysis of variance with repeated measures and tests for linear and quadratic trends). (b) Mice that developed albumin/creatinine ratios of ≥ 3 were considered to have high-grade proteinuria. Accordingly, proteinuria-free survival is significantly higher in the WY14,643-treated mice when compared with the control-fed mice. P < 0·01, Kaplan–Meier survival analysis.
Fig. 2.
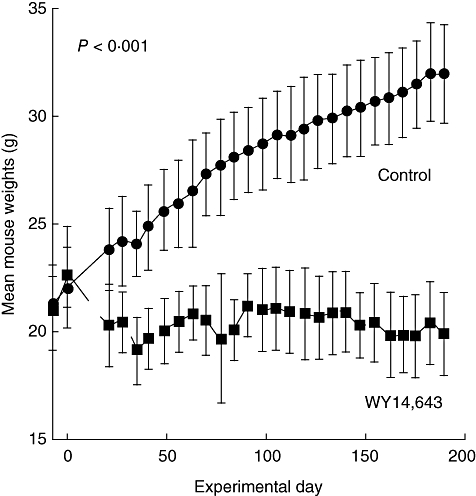
Mice fed WY14,643 do not gain weight over time. C57BL/6 mice were fed 0·05% WY14,643 or control food and immunized with α3(IV)NC1/CFA. Average weight in each group is represented with standard deviations. At all time-points except on the day of immunization, the mice weights differ in the control and WY14,643-fed groups.
Histology
Six months after induction of AGBMD, mice were euthanized and kidneys were prepared for histological evaluation. Representative H&E sections from the two groups are depicted in Fig. 3. H&E sections were graded qualitatively by a blinded observer using a using 0–4 criteria (with 0·5 increments) as described by Luo et al. (Table 1) [14]. These criteria are well suited to grade the histological manifestations in C57BL/6 mice, as the major findings associated with disease in this strain of mice include tubular dilatation, inflammatory tubulo-interstitial infiltrates and crescents [9]. WY14,643-treated animals had significantly less severe histological changes. The median score in the WY14,643-treated group was 1 and in the control-treated group was 3 (Fig. 3e).
Fig. 3.
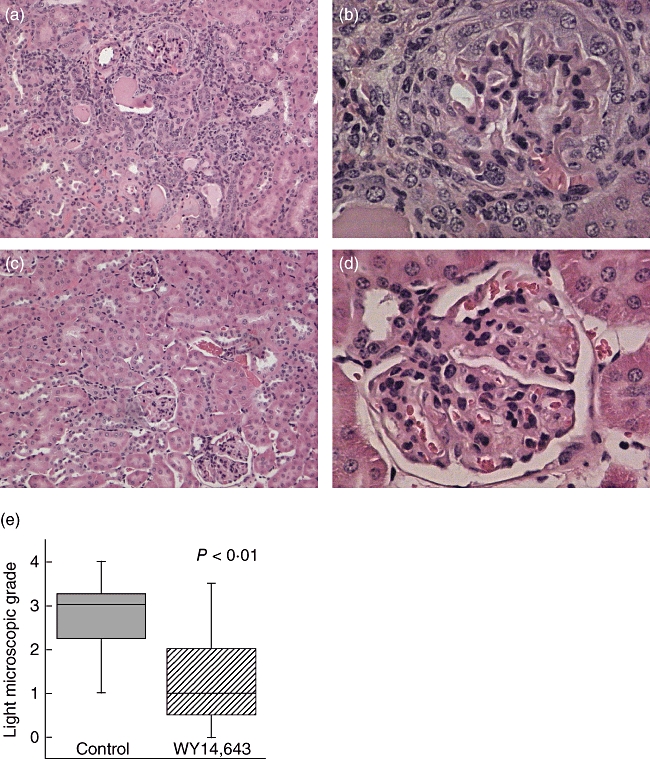
WY14,643 feeding improves renal histology in anti-glomerular basement membrane disease (AGBMD). C57BL/6 mice were fed 0·05% WY14,643 or control food and then immunized to induce AGBMD. Six months after induction of disease kidney sections were stained with haematoxylin and eosin. (a and b) Control-treated animals versus (c and d) WY14,643-treated animals (a, c, 100×; b, d, 400×). (e) Kidney sections were graded qualitatively by a blinded observer using 0–4 criteria (Table 1). There was a significant difference in control versus WY14,643-fed mice. P < 0·01 by the Mann–Whitney U-test, N = 26.
To determine whether WY14,643 interferes with induction of disease by altering binding of anti-glomerular basement membrane IgGs to the glomerular basement membrane, direct immunofluorescence of renal sections was performed. Mouse IgG was distributed in a linear manner along the glomerular basement membrane (Fig. 4a). Fluorescence intensity was graded qualitatively by a blinded observer from 0 to 4 in 0·5 increments [8]. There was no significant difference in the grade in the two groups (Fig. 4b). FITC–IgG-stained glomeruli were also graded in a quantitative manner as described above. MFI and number of pixels were evaluated in each glomerular tuft. Background values were subtracted and values were expressed as MFI (AU) per glomerulus [15,16]. Glomerular size was also assessed (pixels) and MFI/pixels was evaluated (unpublished observations). By both quantitative and qualitative measures WY14,643-feeding did not alter antibody binding to the GBM (Fig. 4b and c).
Fig. 4.
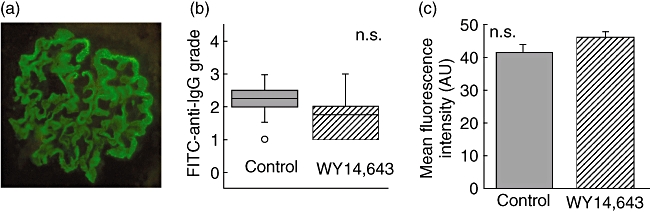
WY14,643 feeding does not alter IgG binding to the glomerular basement membrane (GBM). C57BL/6 mice were fed 0·05% WY14,643 or control food and then immunized to induce anti-glomerular basement membrane disease (AGBMD). Six months after induction of disease, kidneys were fixed and stained with fluorescein isothiocyanate-anti-mouse IgG. (a) Representative glomerulus. (b) Sections were graded qualitatively on a scale from 0 to 4 and there was no difference in the two treatment groups. (c) Fluorescence was measured quantitatively and there was no difference in IgG binding to the GBM. AU: arbitrary units; n.s.: not significant; N = 24.
Immunohistochemistry
H&E sections of the kidneys revealed significant tubulo-interstitial infiltration of cells in diseased kidneys; therefore, immunohistochemistry was performed to identify infiltrating cells. F4/80 is a membrane-bound glycoprotein that is expressed by a variety of macrophage subsets and a few dendritic cell subpopulations [17]. There were dramatic differences in cortical and medullary staining of F4/80 in the two treatment groups (Fig. 5). Quantitative and qualitative evaluations revealed significantly less F4/80+-stained cells in the PPARα ligand-treated animals (Fig. 5e and f). In a similar manner, staining was performed for T and B cells. Lymphocytes were distributed in glomeruli and perivascular locations; however, there were far fewer T and B cells than F4/80+-stained cells. Moreover, there was no significant difference in the number of glomerular or tubulo-interstitial B or T cells in the WY14,643-treated animals (unpublished observations).
Fig. 5.
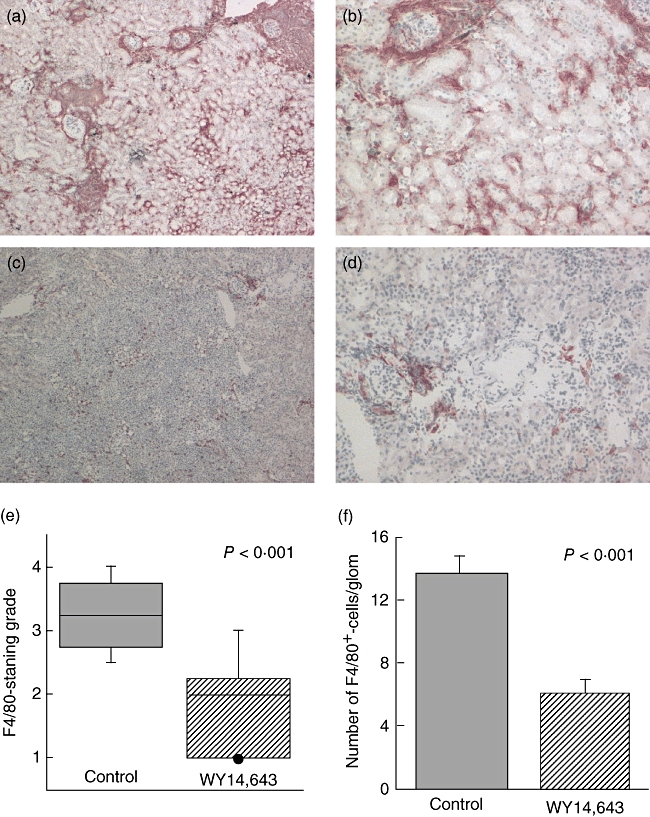
(a) A PPARα ligand decreases renal macrophage infiltration. Frozen sections were stained with the macrophage marker F4/80 and immunohistochemistry was performed. (a) and (b) Representative sections of F4/80-staining of control and (c) and (d) sections of WY14,643-treated mice (a and c, 40×; b and d, 100×). (e) The histological grade was significantly lower in the PPARα-ligand-treated animals (P < 0·001, Mann–Whitney U-test; N = 27). (f) The number of F4/80+-staining cells per glomerulus was also determined and there was a significant decline in the number of intraglomerular macrophages in the WY14,643-treated mice. P < 0·001, Student's t-test; N = 27.
α3(IV) NC1-specific IgGs
Six months after induction of AGBMD, antigen-specific IgGs [anti-α3(IV) NC1 IgGs] were assessed by enzyme-linked immunosorbent assay (ELISA). In the WY14,643-treated animals there were significant increases in antigen-specific IgG and IgG1 with complementary decrements in IgG2c (C57BL/6 mice express IgG2c in place of IgG2a [18]) when compared with the control-treated animals (Fig. 6). The increase in antigen-specific IgG1 expression over IgG2c suggests that PPARα ligands may induce a Th2-type of inflammatory response.
Fig. 6.
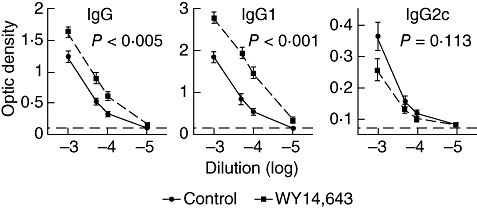
WY14,643 alters the IgG1 and IgG response to α3(IV) NC1. Six months after induction of anti-glomerular basement membrane disease (AGBMD) sera was isolated and tested for anti-α3(IV) NC1-specific antibodies. These antigen-specific IgG enzyme-linked immunosorbent assays (ELISAs) generate relative curves that are not quantitative. Serum from a naive, unimmunized C57BL/6 mouse was also evaluated by ELISA and the horizontal dashed-lines represent the background of this assay (optic density of naive mouse serum). There was a significant increase in titres of antigen-specific IgG1 and IgG with a trend for a decline in titres of IgG2c in the WY14,643-treated mice. Also the IgG1/IgG2c ratio was significantly higher in the PPARα ligand-treated animals suggesting that WY14,643 induces a Th2-type of immune response. Values were assessed by analysis of variance with repeated measures and bars represent standard error of the mean (N = 27).
Cytokines
RNA was prepared from the spleens of 16 random mice from the above study (eight controls and eight PPARα ligand-treated mice). There was a trend for higher expression of IFN-γ mRNA in the spleens from the control-treated mice when compared with the PPARα ligand-treated mice (Fig. 7b). We also evaluated splenic IL-4 mRNA expression in these mice. Figure 7c demonstrates that splenic IL-4 was not elevated in the WY14,643-treated mice. To evaluate cytokine expression at an earlier time-point in this study, mice were immunized with α3(IV) NC1/CFA and boosted 30 days later. Sixty days after the initial immunization, prior to appreciable renal injury, kidneys were removed and RNA was extracted. Intrarenal IFN-γ mRNA expression was higher in the kidneys of the control-treated mice compared with the WY14,643-treated mice (0·98 versus 0·6, P = 0·034, Fig. 7a). In these studies, IL-4 was not detectable in the kidneys. These data suggest that WY14,643 treatment is associated with lower IFN-γ expression.
Fig. 7.
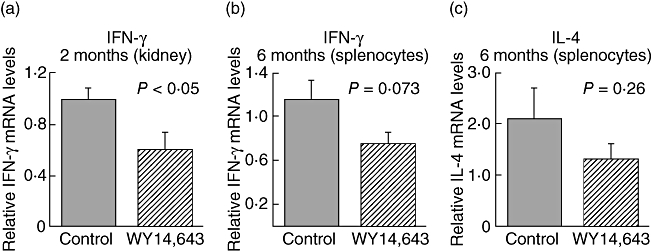
WY14,643 treatment is associated with lower splenocyte and intrarenal interferon (IFN)-γ expression. (a) C57BL/6 mice were fed WY14,643 or control food, immunized with α3(IV) NC1/complete Freund's adjuvant on day 0 and boosted 30 days later. On day 60 the mice were killed and kidneys were removed for assessment of intrarenal IFN-γ mRNA expression. Interleukin (IL)-4 expression was not detectable in the kidneys at this time-point (N = 8). (b) Mice were immunized to induce anti-glomerular basement membrane disease (AGBMD) and spleens were isolated 6 months after immunization. Real-time polymerase chain reaction was performed to detect splenocyte IFN-γ and IL-4 mRNA expression (N = 16).
Chemokines
MCP-1 is a major chemokine that directs the migration of macrophage/monocytes and T cells into the kidney. RNA was isolated from the kidneys described in the above studies and RT–PCR was performed to determine the relative intrarenal MCP-1 mRNA levels. There was a 10-fold reduction in MCP-1 mRNA expression in the WY14,643-treated animals. Proteinuria can induce production of MCP-1 directly [19], therefore MCP-1 mRNA expression was assessed in mice prior to the onset of proteinuria and overt renal histological changes. Mice were immunized with α3(IV) NC1/CFA and boosted 30 days later. Sixty days after the initial immunization and prior to the onset of proteinuria, macrophage influx or histopathological changes (unpublished observations), there was still a five- to sixfold decrease in renal MCP-1 mRNA expression in the PPARα ligand-treated animals. Five months after induction of AGBMD (experimental day 154), urine was collected and urinary MCP-1 was evaluated by ELISA (OptEIA™, BD Pharmingen). There was a significant decline in urine MCP-1 protein expression in the PPARα ligand-treated mice. It is of interest that in individual animals the urine MCP-1 protein levels correlated closely with the renal mRNA MCP-1 expression. Results were analysed with the Spearman's correlation coefficient (r = 0·7, P = 0·025). These studies suggest that PPARα ligands may directly decrease MCP-1 mRNA.
Discussion
In this study we demonstrate that WY14,643, a PPARα ligand, significantly inhibits proteinuria and expression of experimental autoimmune glomerulonephritis. This complements studies that support the efficacy of PPARα ligands in inflammatory diseases (reviewed in [5]) [20–22]. WY14,643 is not approved for clinical use; however, unlike many experimental anti-inflammatory agents, PPARα ligands such as gemfibrozil and fenofibrate are already used clinically to treat hyperlipidaemia and have clearly established safety profiles. Moreover, these agents have been used successfully in humans with rheumatoid arthritis, inflammatory liver disease and lipoprotein glomerulopathy.
The mechanism underlying the PPARα ligand-induced amelioration of AGBMD is probably multi-factorial. The cytokine milieu in which T helper (Th, CD4+) cells develop can influence their differentiation into distinct subtypes [23]. IFN-γ and IL-12 promote Th1 differentiation, which drives cell-mediated immunity. Conversely, IL-4 has the greatest influence in promoting Th2 differentiation. Unbalanced Th1 reactions (low IL-4 expression) may precipitate organ-specific autoimmunity, whereas enhanced Th2 responses may be therapeutically efficacious in inflammatory diseases including renal transplantation [24] and crescentic glomerulonephritis (GN) [25–28].
We and others have shown that PPARα ligands potently increase IL-4 [6,29] and it is possible that WY14,643-induces a Th2-type of immune response. The increased IgG1/IgG2c levels in PPARα ligand-treated mice supports this hypothesis. However, in the current studies at various time-points (10 days, 60 days and 189 days after immunization) there was no evidence of augmented IL-4 mRNA or protein levels. This could be related to the paracrine nature of cytokines, making it difficult to measure levels in vivo, and it is possible that we did not select the correct time-points in which to measure cytokine production. However, the PPARα ligand-treated mice had lower splenic and intrarenal IFN-γ mRNA expression compared with the control-fed mice. These findings support further our general hypothesis that PPARα ligands induce a Th2-type of immune response in a chronic inflammatory renal disease.
It is notable that in this study there was an increase in antigen-specific IgG and IgG1 in the WY14,643-treated mice. These findings were unexpected given our early studies in the experimental allergic encephalomyelitis (EAE) model, in which WY14,643 greatly diminished all subclasses of antigen-specific IgGs. In the EAE model we saw significant T and B cell depletion associated with WY14,643 treatment and hypothesized that B cell depletion could have contributed to the decline in antigen-specific IgGs. In the current studies, we also saw significant declines in T and B cell numbers (50% normal values) at 10 days and 2 months after immunization (data not shown). Despite this reduction in B cell numbers there was an increase in antigen-specific IgG and IgG1. Therefore it is unlikely that reduced B cell numbers altered immunoglobulin production. It is possible that the different antigen-specific IgG responses in WY14,643-fed mice was related to the immunized antigens. Notably, myelin oligodendrocyte glycoprotein (MOG) is highly immunogenic compared with the α3(IV) NCI peptide. In keeping with our current studies, Daynes and colleagues have shown that WY14,643 enhances antigen-specific IgG and IgA in aged animals, they postulate, by ameliorating dysregulated cytokine production [30]. Interestingly, their studies also showed that WY14,643 feeding was associated with reduced splenocyte IFN-γ production, which parallels our studies [31]. Therefore we postulate that the differences in IgG levels in the EAE model compared with the AGBMD model are related to the immunogenicity and composition of MOG compared with α3(IV) NC1.
The reduction in glomerular and tubulo-interstitial macrophage infiltration was the most dramatic histological finding in this study and probably contributes to the protective effect of WY14,643. Equally dramatic were the differences in MCP-1 expression in kidneys isolated from mice 2 months and 6 months after induction of AGBMD. In general, reduced MCP-1 levels attenuate expression of inflammatory renal diseases [32–34]. MCP-1 is produced by mesangial and tubular epithelial cells in response to immune complexes, proteinuria, hyperglycaemia, IL-1β, TNFα and IFN-γ (reviewed in [35]). Thus, it is possible that 6 months after induction of disease, WY14,643 reduced MCP-1 indirectly. Proteinuria in the control group could have induced production of intrarenal MCP-1, which in turn promoted monocyte/macrophage infiltration into the glomeruli and tubulo-interstitium. Because there was less proteinuria in the WY14,643-treated animals, this could have resulted in lower MCP-1 expression. However, 2 months after induction of AGBMD, prior to onset of proteinuria and renal histopathological changes, there was a significant reduction in MCP-1 mRNA expression in the PPARα ligand-treated animals (Fig. 8). This parallels findings in a model of lupus nephritis, where elevated chemokine expression preceded the onset of proteinuria and cytokine elaboration [36] (reviewed in [35]). This suggests that WY14,643 may have directly reduced MCP-1.
Fig. 8.
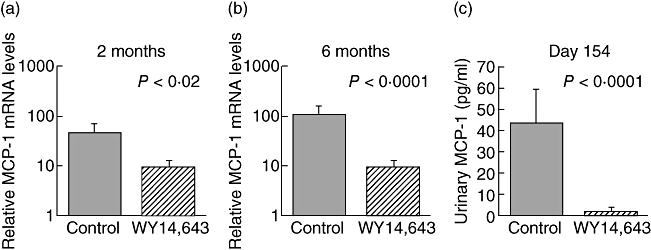
Mice treated with WY14,643 have lower renal expression of monocyte chemoattractant protein (MCP)-1. Mice were immunized to induce anti-glomerular basement membrane disease (AGBMD) and kidneys were harvested (a) 2 months (N = 8) and (b) 6 months after immunization (N = 20). RNA was prepared from kidneys and real-time polymerase chain reaction determined relative MCP-1/CCL2 mRNA expression. At both time-points there was a significant reduction in the expression of MCP-1 mRNA. (c) Five months after induction of AGBMD, urine was collected and MCP-1 protein levels were evaluated by enzyme-linked immunosorbent assay. There was a significant difference in MCP-1 urine values in control compared with WY14,643-treated mice (N = 29).
MCP-1 can direct migration of both T cells and macrophages into the kidney; however, in our study there was no difference in the number of T cells in the glomeruli and tubulo-interstitium. These findings are somewhat similar to those of Tesch et al. [33]. In their study MCP-1 knock-out mice with lupus had fewer T cells in the interstitium, but similar numbers of intraglomerular T cells compared with their wild-type controls [33]. Several studies in a variety of inflammatory diseases have shown that PPARα ligands decrease the infiltration of macrophages into inflammatory tissues [29,37–39]. The reduction in macrophage infiltration probably resulted in less intrarenal production of nitric oxide, tumour necrosis factor (TNF)-α and reactive oxygen species, thereby causing less renal injury and proteinuria.
There are conflicting data on whether PPARα ligands decrease MCP-1 mRNA directly. Some studies have shown reductions in MCP-1 associated with PPARα ligand treatment [37,38,40]. In contrast, in human airway smooth muscle cells 5 and 10 μM WY14,643 did not lower MCP-1 promoter activity [41]. Moreover, in human aortic endothelial cells, Lee et al. showed that WY14,643 increased MCP-1 [42]. Our preliminary studies demonstrate that WY14,643 reduces MCP-1 mRNA expression directly at the transcriptional level (unpublished observations), and this remains an active area of investigation. However, it is likely that the effect of PPARα ligands on MCP-1 production is both cell- and stimulus-specific [41].
In our studies mice immunized with α3(IV) NC1/CFA and fed WY14,643 do not gain weight over time when compared with the control-fed group (Fig. 2). In the United States there is an epidemic of obesity, and one would consider these findings desirable. However, it is possible that the weight loss could have confounded our results, as protein-calorie malnutrition may ameliorate inflammatory renal disease [43–46]. Interestingly, we have shown that PPARα ligands augment expression of TRB3 [47], a protein that alters phosphorylation of AKT/Protein Kinase B [48] and mediates ubiquitination of acetyl-CoA carboxylase, the rate-limiting enzyme of fatty acid synthesis [49]. Recent studies have shown that mice with enhanced TRB3 expression in the fat have lower body weights than their controls [49]. We propose that the weight loss in the WY14,643-treated mice may be related to enhanced TRB3 expression. Lower weights may also have been related to lower intakes in the PPARα ligand-fed mice [50] (and unpublished observations). Despite these observations, we believe it is important to determine whether the clinically used fibrates, such as gemfibrozil or fenofibrate, ameliorate expression of inflammatory kidney disease.
This study supports a growing body of literature supporting the hypothesis that PPARα ligands are anti-inflammatory in renal diseases. PPARα ligands ameliorate renal ischaemia/reperfusion-induced injury [20,51] and are protective in a mouse model of cisplatin-induced acute renal failure (ARF). Portilla et al. [21] have shown that in ARF, treatment of mice with PPARα ligands is associated with lower expression of a wide range of cytokines, chemokines and adhesion molecules including TNF-α, IL-6, IFN-γ, vascular cell adhesion molecule (VCAM)-1 and intercellular adhesion molecule (ICAM)-1. Moreover, bezafibrate, a dual-acting PPAR ligand, which activates both PPARδ and PPARα, attenuates serum nephrotoxic glomerulonephritis [22,39]. PPARα ligands also improve diabetic nephropathy in db/db mice and attenuate diabetic kidney disease in apolipoprotein E knock-out mice [50,52]. In the current study we have shown that a PPARα ligand attenuates the expression of AGBMD, probably by reducing IFN-γ expression and by reducing MCP-1 production. The current study supports the concept that PPARα ligands are anti-inflammatory and suggests that these agents may be therapeutically efficacious in inflammatory disease processes.
Acknowledgments
The authors thank Vickie Pei-Chun Cheng for her excellent technical support. We thank Dr Carolyn Kelly for her invaluable support on this project. We also would like to thank Dr Raghu Kalluri for providing us with the 293EBNA cells stably transfected with the human α3(IV) NC1 construct and Dr Nissi Varki and the UCSD Histology Core for their help with preparing and analysing the histological sections. The studies were performed with the support of the Department of Veterans Affairs Advanced Career Development grant for R. C. The National Kidney Foundation's Clinical Scientist Award (John Bower MD Foundation) and the American Society of Nephrology's Carl W. Gottschalk Research Scholar Award also provided generous support for Dr Cunard.
References
- 1.Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha-beta, and -gamma in the adult rat. Endocrinology. 1996;137:354–66. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 2.Issemann I, Prince RA, Tugwood JD, Green S. The peroxisome proliferator-activated receptor: retinoid X receptor heterodimer is activated by fatty acids and fibrate hypolipidaemic drugs. J Mol Endocrinol. 1993;11:37–47. doi: 10.1677/jme.0.0110037. [DOI] [PubMed] [Google Scholar]
- 3.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–41. [PubMed] [Google Scholar]
- 4.Rubins HB, Robins SJ, Collins D, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999;341:410–18. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- 5.Cunard R. The potential use of PPARalpha agonists as immunosuppressive agents. Curr Opin Invest Drugs. 2005;6:467–72. [PubMed] [Google Scholar]
- 6.Cunard R, Ricote M, DiCampli D, et al. Regulation of cytokine expression by ligands of peroxisome proliferator activated receptors. J Immunol. 2002;168:2795–802. doi: 10.4049/jimmunol.168.6.2795. [DOI] [PubMed] [Google Scholar]
- 7.Cunard R, DiCampli D, Archer DC, et al. WY14,643, a PPAR alpha ligand, has profound effects on immune responses in vivo. J Immunol. 2002;169:6806–12. doi: 10.4049/jimmunol.169.12.6806. [DOI] [PubMed] [Google Scholar]
- 8.Bolton WK, Tucker FL, Sturgill BC. New avian model of experimental glomerulonephritis consistent with mediation by cellular immunity. Nonhumorally mediated glomerulonephritis in chickens. J Clin Invest. 1984;73:1263–76. doi: 10.1172/JCI111328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalluri R, Danoff TM, Okada H, Neilson EG. Susceptibility to anti-glomerular basement membrane disease and Goodpasture syndrome is linked to MHC class II genes and the emergence of T cell-mediated immunity in mice. J Clin Invest. 1997;100:2263–75. doi: 10.1172/JCI119764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu J, Hicks J, Ou C, Singleton D, Borillo J, Lou YH. Glomerulonephritis induced by recombinant collagen IV alpha 3 chain noncollagen domain 1 is not associated with glomerular basement membrane antibody: a potential T cell-mediated mechanism. J Immunol. 2001;167:2388–95. doi: 10.4049/jimmunol.167.4.2388. [DOI] [PubMed] [Google Scholar]
- 11.Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med. 2003;348:2543–56. doi: 10.1056/NEJMra022296. [DOI] [PubMed] [Google Scholar]
- 12.Hopfer H, Maron R, Butzmann U, Helmchen U, Weiner HL, Kalluri R. The importance of cell-mediated immunity in the course and severity of autoimmune anti-glomerular basement membrane disease in mice. FASEB J. 2003;17:860–8. doi: 10.1096/fj.02-0746com. [DOI] [PubMed] [Google Scholar]
- 13.Neilson EG, Kalluri R, Sun MJ, et al. Specificity of Goodpasture autoantibodies for the recombinant noncollagenous domains of human type IV collagen. J Biol Chem. 1993;268:8402–5. [PubMed] [Google Scholar]
- 14.Luo AM, Fox JW, Chen L, Bolton WK. Synthetic peptides of Goodpasture's antigen in antiglomerular basement membrane nephritis in rats. J Lab Clin Med. 2002;139:303–10. doi: 10.1067/mlc.2002.123623. [DOI] [PubMed] [Google Scholar]
- 15.Dean EG, Wilson GR, Li M, et al. Experimental autoimmune Goodpasture's disease: a pathogenetic role for both effector cells and antibody in injury. Kidney Int. 2005;67:566–75. doi: 10.1111/j.1523-1755.2005.67113.x. [DOI] [PubMed] [Google Scholar]
- 16.Lehr HA, Mankoff DA, Corwin D, Santeusanio G, Gown AM. Application of photoshop-based image analysis to quantification of hormone receptor expression in breast cancer. J Histochem Cytochem. 1997;45:1559–65. doi: 10.1177/002215549704501112. [DOI] [PubMed] [Google Scholar]
- 17.Hume DA, Gordon S. Mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80. Identification of resident macrophages in renal medullary and cortical interstitium and the juxtaglomerular complex. J Exp Med. 1983;157:1704–9. doi: 10.1084/jem.157.5.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin RM, Lew AM. Is IgG2a a good Th1 marker in mice? Immunol Today. 1998;19:49. doi: 10.1016/s0167-5699(97)87499-3. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Chen J, Chen L, Tay YC, Rangan GK, Harris DC. Induction of monocyte chemoattractant protein-1 in proximal tubule cells by urinary protein. J Am Soc Nephrol. 1997;8:1537–45. doi: 10.1681/ASN.V8101537. [DOI] [PubMed] [Google Scholar]
- 20.Portilla D, Dai G, Peters JM, Gonzalez FJ, Crew MD, Proia AD. Etomoxir-induced PPARalpha-modulated enzymes protect during acute renal failure. Am J Physiol Renal Physiol. 2000;278:F667–75. doi: 10.1152/ajprenal.2000.278.4.F667. [DOI] [PubMed] [Google Scholar]
- 21.Li S, Gokden N, Okusa MD, Bhatt R, Portilla D. Anti-inflammatory effect of fibrate protects from cisplatin-induced ARF. Am J Physiol Renal Physiol. 2005;289:F469–80. doi: 10.1152/ajprenal.00038.2005. [DOI] [PubMed] [Google Scholar]
- 22.Saga D, Sakatsume M, Ogawa A, et al. Bezafibrate suppresses rat antiglomerular basement membrane crescentic glomerulonephritis. Kidney Int. 2005;67:1821–9. doi: 10.1111/j.1523-1755.2005.00280.x. [DOI] [PubMed] [Google Scholar]
- 23.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–57. [PubMed] [Google Scholar]
- 24.Kist-van Holthe JE, Gasser M, Womer K, et al. Regulatory functions of alloreactive Th2 clones in human renal transplant recipients. Kidney Int. 2002;62:627–31. doi: 10.1046/j.1523-1755.2002.00469.x. [DOI] [PubMed] [Google Scholar]
- 25.Tipping PG, Kitching AR, Huang XR, Mutch DA, Holdsworth SR. Immune modulation with interleukin-4 and interleukin-10 prevents crescent formation and glomerular injury in experimental glomerulonephritis. Eur J Immunol. 1997;27:530–7. doi: 10.1002/eji.1830270226. [DOI] [PubMed] [Google Scholar]
- 26.Tam FW, Smith J, Karkar AM, Pusey CD, Rees AJ. Interleukin-4 ameliorates experimental glomerulonephritis and up-regulates glomerular gene expression of IL-1 decoy receptor. Kidney Int. 1997;52:1224–31. doi: 10.1038/ki.1997.447. [DOI] [PubMed] [Google Scholar]
- 27.Kluth DC, Ainslie CV, Pearce WP, et al. Macrophages transfected with adenovirus to express IL-4 reduce inflammation in experimental glomerulonephritis. J Immunol. 2001;166:4728–36. doi: 10.4049/jimmunol.166.7.4728. [DOI] [PubMed] [Google Scholar]
- 28.Holdsworth SR, Kitching AR, Tipping PG. Th1 and Th2 T helper cell subsets affect patterns of injury and outcomes in glomerulonephritis. Kidney Int. 1999;55:1198–216. doi: 10.1046/j.1523-1755.1999.00369.x. [DOI] [PubMed] [Google Scholar]
- 29.Lovett-Racke AE, Hussain RZ, Northrop S, et al. Peroxisome proliferator-activated receptor alpha agonists as therapy for autoimmune disease. J Immunol. 2004;172:5790–8. doi: 10.4049/jimmunol.172.9.5790. [DOI] [PubMed] [Google Scholar]
- 30.Enioutina EY, Visic VD, Daynes RA. Enhancement of common mucosal immunity in aged mice following their supplementation with various antioxidants. Vaccine. 2000;18:2381–93. doi: 10.1016/s0264-410x(00)00008-6. [DOI] [PubMed] [Google Scholar]
- 31.Poynter ME, Daynes RA. Age-associated alterations in splenic iNOS regulation: influence of constitutively expressed IFN-gamma and correction following supplementation with PPARalpha activators or vitamin E. Cell Immunol. 1999;195:127–36. doi: 10.1006/cimm.1999.1525. [DOI] [PubMed] [Google Scholar]
- 32.Tesch GH, Schwarting A, Kinoshita K, Lan HY, Rollins BJ, Kelley VR. Monocyte chemoattractant protein-1 promotes macrophage-mediated tubular injury, but not glomerular injury, in nephrotoxic serum nephritis. J Clin Invest. 1999;103:73–80. doi: 10.1172/JCI4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR. Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Fas (lpr) mice. J Exp Med. 1999;190:1813–24. doi: 10.1084/jem.190.12.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okada H, Moriwaki K, Kalluri R, et al. Inhibition of monocyte chemoattractant protein-1 expression in tubular epithelium attenuates tubulointerstitial alteration in rat Goodpasture syndrome. Kidney Int. 2000;57:927–36. doi: 10.1046/j.1523-1755.2000.00909.x. [DOI] [PubMed] [Google Scholar]
- 35.Kelley VR, Rovin BH. Chemokines: therapeutic targets for autoimmune and inflammatory renal disease. Springer Semin Immunopathol. 2003;24:411–21. doi: 10.1007/s00281-003-0124-4. [DOI] [PubMed] [Google Scholar]
- 36.Perez de Lema G, Maier H, Nieto E, et al. Chemokine expression precedes inflammatory cell infiltration and chemokine receptor and cytokine expression during the initiation of murine lupus nephritis. J Am Soc Nephrol. 2001;12:1369–82. doi: 10.1681/ASN.V1271369. [DOI] [PubMed] [Google Scholar]
- 37.Ichihara S, Obata K, Yamada Y, et al. Attenuation of cardiac dysfunction by a PPAR-alpha agonist is associated with down-regulation of redox-regulated transcription factors. J Mol Cell Cardiol. 2006;41:318–29. doi: 10.1016/j.yjmcc.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 38.Delayre-Orthez C, Becker J, Guenon I, et al. PPARalpha downregulates airway inflammation induced by lipopolysaccharide in the mouse. Respir Res. 2005;6:91. doi: 10.1186/1465-9921-6-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saga D, Sakatsume M, Kaneko Y, et al. The effect of bezafibrate in crescentic glomerulonephritis. Nephrology (Carlton) 2005;10(Suppl. 6):A432. [Google Scholar]
- 40.Dragomir E, Tircol M, Manduteanu I, Voinea M, Simionescu M. Aspirin and PPAR-alpha activators inhibit monocyte chemoattractant protein-1 expression induced by high glucose concentration in human endothelial cells. Vascul Pharmacol. 2006;44:440–9. doi: 10.1016/j.vph.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 41.Nie M, Corbett L, Knox AJ, Pang L. Differential regulation of chemokine expression by peroxisome proliferator-activated receptor gamma agonists: interactions with glucocorticoids and beta2-agonists. J Biol Chem. 2005;280:2550–61. doi: 10.1074/jbc.M410616200. [DOI] [PubMed] [Google Scholar]
- 42.Lee H, Shi W, Tontonoz P, et al. Role for peroxisome proliferator-activated receptor alpha in oxidized phospholipid-induced synthesis of monocyte chemotactic protein-1 and interleukin-8 by endothelial cells. Circ Res. 2000;87:516–21. doi: 10.1161/01.res.87.6.516. [DOI] [PubMed] [Google Scholar]
- 43.Fernandes G, Friend P, Yunis EJ, Good RA. Influence of dietary restriction on immunologic function and renal disease in (NZB × NZW) F1 mice. Proc Natl Acad Sci USA. 1978;75:1500–4. doi: 10.1073/pnas.75.3.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friend PS, Fernandes G, Good RA, Michael AF, Yunis EJ. Dietary restrictions early and late: effects on the nephropathy of the NZB × NZW mouse. Lab Invest. 1978;38:629–32. [PubMed] [Google Scholar]
- 45.Fernandes G, Yunis EJ, Miranda M, Smith J, Good RA. Nutritional inhibition of genetically determined renal disease and autoimmunity with prolongation of life in kdkd mice. Proc Natl Acad Sci USA. 1978;75:2888–92. doi: 10.1073/pnas.75.6.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neugarten J, Feiner HD, Schacht RG, Baldwin DS. Amelioration of experimental glomerulonephritis by dietary protein restriction. Kidney Int. 1983;24:595–601. doi: 10.1038/ki.1983.199. [DOI] [PubMed] [Google Scholar]
- 47.Selim E, Frkanec JT, Cunard R. Fibrates upregulate TRB3 in lymphocytes independent of PPARalpha by augmenting CCAAT/enhancer-binding proteinbeta (C/EBPbeta) expression. Mol Immunol. 2007;44:1218–29. doi: 10.1016/j.molimm.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 48.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–7. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 49.Qi L, Heredia JE, Altarejos JY, et al. TRB3 links the E3 ubiquitin ligase COP1 to lipid metabolism. Science. 2006;312:1763–6. doi: 10.1126/science.1123374. [DOI] [PubMed] [Google Scholar]
- 50.Park CW, Zhang Y, Zhang X, et al. PPARalpha agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney Int. 2006;69:1511–17. doi: 10.1038/sj.ki.5000209. [DOI] [PubMed] [Google Scholar]
- 51.Sivarajah A, Chatterjee PK, Hattori Y, et al. Agonists of peroxisome-proliferator activated receptor-alpha (clofibrate and WY14643) reduce renal ischemia/reperfusion injury in the rat. Med Sci Monit. 2002;8:BR532–9. [PubMed] [Google Scholar]
- 52.Calkin AC, Giunti S, Jandeleit-Dahm KA, Allen TJ, Cooper ME, Thomas MC. PPAR-alpha and -gamma agonists attenuate diabetic kidney disease in the apolipoprotein E knockout mouse. Nephrol Dial Transplant. 2006;21:2399–405. doi: 10.1093/ndt/gfl212. [DOI] [PubMed] [Google Scholar]