

Abstract
Cyclooxygenase-2 (COX-2) gene expression in the lung is induced in pathological conditions such as asthma and pneumonia; however, the exact impact of COX-2 gene expression in the airway in regulating inflammatory and immunological response in the lung is not understood. To define a physiological role of inducible COX-2 in airway epithelial cells, we developed a novel line of transgenic mice, referred to as CycloOxygenase-2 TransActivated (COTA) mice, that overexpress a COX-2 transgene in the distribution of the CC-10 promoter in response to doxycycline. In response to doxycycline treatment, COX-2 expression was increased in airway epithelium of COTA mice and whole lung tissue contained a three- to sevenfold increase in prostaglandin E2 (PGE2), prostaglandin D2 (PGD2) thromboxane B2 (TXB2) and 6-Keto prostaglandin F2α (PGF2α) compared to wild-type and untreated COTA mice. Interestingly, primary mouse tracheal epithelial cells from COTA mice produced only PGE2 by doxycycline-induced COX-2 activation, providing an indication of cellular specificity in terms of mediator production. In the ovalbumin model, in which doxycycline was given at the sensitization stage, there was an increase in interleukin (IL)-4 level in lung tissue from COTA mice compared to untreated COTA and wild-type mice. In addition, COTA mice that were treated with doxycycline had impaired clearance of Pseudomonas aeruginosa pneumonia compared to wild-type mice. COX-2 gene expression in airway epithelial cells has an important role in determining immunological response to infectious and allergic agents.
Keywords: asthma, bronchial epithelial cells, cyclooxygenase-2, PGE2, Pseudomonas aeruginosa
Introduction
There is increasing evidence indicating that airway epithelial cells act not only as a physical barrier but also play an active role in regulating inflammatory and immunological responses to inhaled environmental stimuli. Airway epithelial cells possess pathogen recognition receptors (e.g. Toll-like receptors) and are one of the first cells that contact inhaled antigen, prior to the professional immune cells [1,2]. Airway epithelial cells express low basal levels of cyclooxygenase-2 (COX-2) that liberate active prostanoid mediators into the airway and alveolar space [3]. COX-2 is induced in airway epithelial cells in response to inhaled allergen challenge and airborne infection with bacteria [4].
Prostanoids are enzymatic products of COX that are potent regulatory lipid mediators involved in numerous physiological and pathological processes. Prostanoids act as autocrine and paracrine lipid mediators in the vicinity of their production site to maintain local homeostasis. COX is the rate-limiting enzyme in the metabolism of arachidonic acid which is then acted upon further by specific isomerases and oxidoreductases to catalyse the production of various bioactive prostaglandin isomers. Differential cellular gene expression of the enzymes that are involved in prostanoid biosynthesis, as well as differences in the distribution of specific prostanoid synthases within cells, determine the profile of prostanoid production and thereby their effects on neighbouring cells. Thus, there is a spectrum of eicosanoid production that differs from one cell type to another.
The pattern of eicosanoid production in bronchoalveolar lavage (BAL) fluid is altered in many pulmonary disease states [5–7]. Because inflammatory cells in the airway also release prostanoids and contribute to the prostanoid balance, the specific contribution of airway epithelial cells in producing COX-2 that results in cell-to-cell interaction is unknown [8]. We hypothesize that COX-2 gene expression and prostanoid production by airway epithelial cells are critical determinants of immune response in the lung. To investigate this hypothesis, we developed novel conditional inducible transgenics that express COX-2 specifically in the airway epithelium. We examined the response of these mice with and without induction of COX-2 gene expression in the tracheal–bronchial epithelium in both an allergic and a pneumonia disease model. The ovalbumin-sensitized airway inflammation model represents a T helper 2 (Th2)-type immune reaction and the Pseudomonas aeruginosa pneumonia model examines the effect of airway prostanoid production in regulating host defence.
Materials and methods
Genotyping the transgenic mice
All transgenic mice were generated on the FVB strain background under specific pathogen-free condition at Vanderbilt University, Nashville, TN. The presence or absence of the transgene was evaluated initially using Southern blot analysis and polymerase chain reaction (PCR). DNA prepared from tail biopsies was used for genotyping. PCR for the COX-2 transgene was performed using primers which were designed not to detect endogenous COX-2 gene: 5′-CAGCAAATCCTTGCTGTTCC-3′, 5′-TTCCAAGGGCATCGGTAAACATCTG-3′. Mice transgenic for CC10-tetracycline-controlled transcriptional silencer (tTS)/(tet-O)7-COX2 were mated with CC10-rtTA homozygous mice to obtain transgenic mice carrying all three transgenes, designated CycloOxygenase-2 TransActivated (COTA) mice (Fig. 1a).
Fig. 1.
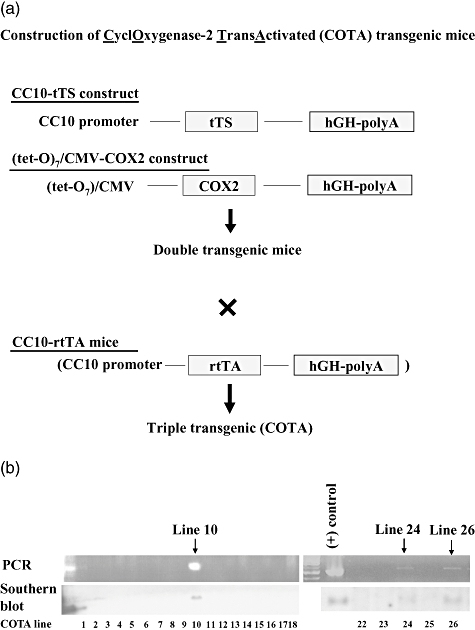
(a) Schematic for construction of CycloOxygenase-2 TransActivated (COTA) transgenic mice. (b) Genotyping to detect cyclooxygenase-2 (COX-2) transgene in DNA from tail biopsies. Transgenic founders were identified by Southern blot and confirmed by polymerase chain reaction (PCR) analysis using COX-2 primers. We identified total three double-positive lines by PCR and Southern blot and used both lines 10 and 26 mice in the following experiments.
COX-2 expression in lung tissue
Lung tissue homogenates were prepared as described previously [4]. Immunostains for COX-2 were performed with lung homogenates. The anti-mouse COX-2 antibody (Cayman Chemical, MI, USA) was applied at a 1/100 dilution. For COX-2 immunohistochemistry in lung tissue, mice were perfused with saline. The lungs were inflated with 1 ml of 10% neutral-buffered formalin. The Vectastain ABC Elite (Vector Laboratories, Burlingame, CA, USA) system was used to produce localized, visible staining of COX-2 protein.
Assessment of prostaglandins production in lung
Mouse lung was harvested after flushing with saline and stored at −80°C until measured. The total lung homogenate and culture supernatant was used for measurement. Prostaglandins were determined by gas chromatography in conjunction with mass spectrometry (GC/MS), as described previously [4].
Mouse tracheal epithelial cell (MTEC) isolation
For isolating primary MTEC, we followed a previously published protocol with minimal modifications [9]. Briefly, the tracheas were opened longitudinally, incubated in 1·5 mg/ml pronase (Roche Molecular Biochemicals, Indianapolis, IN, USA) for 18 h at 4°C. Epithelial cells were dislodged from tracheas, collected by centrifugation and resuspended in DNase solution to avoid clumping together. These cells were centrifuged and resuspended in MTEC basic media [Dulbecco's modified Eagle's medium (DMEM)-Ham's F-12 (1 : 1 v/v), 15 mM HEPES, 3·6 mM sodium bicarbonate, 4 mM l-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin and 0·25 µg/ml fungizone]. After incubating in tissue culture plates (Primera; Becton-Dickinson Labware, Franklin Lakes, NJ, USA) for 3–4 h at 37°C, floating epithelial cells were collected by centrifugation. MTEC cells were seeded on a 12-mm diameter, 0·4 µm pore size polycarbonate semipermeable membrane (Transwell, Corning Costar, Cambridge, MA, USA). The cells were cultured for 5 days with the culture medium MTEC plus, which is MTEC basic media supplemented with 10 µg/ml insulin, 5 µg/ml transferrin, 0·1 µg/ml cholera toxin, 25 ng/ml epidermal growth factor (Becton-Dickinson, Bedford, MA, USA), 30 µg/ml bovine pituitary extract, 5% fetal bovine serum (FBS) and 0·01 µM retinoic acid. Media was changed every 2 days until the transmembrane resistance (Rt) is > 1000 Ω cm2, as measured by an epithelial Ohm-voltmeter (World Precision Instruments, Sarasota, FL, USA). Media was removed from the upper chamber to establish an air–liquid interface (ALI), and the lower chambers only were provided with fresh MTEC/NS media, which is MTEC basic media supplemented with 2% Nu Serum (Becton-Dickinson) and 0·01 µm retinoic acid every 2 days.
COX-2 expression in primary MTEC
The transwell membrane of MTEC was fixed and permeabilized. After incubating with primary anti-mouse COX-2 (1 : 100 dilutions) and secondary antibody, the membrane was mounted on slides in mounting media with diamidino phenyl indole (DAPI) (Vector Laboratories). COX-2-immunostained MTEC was imaged using the LSM 510 laser scanning confocal microscope (Carl Zeiss, Jena, Germany).
Scanning electron microscopy
The transwell membrane of MTEC was fixed with 2·5% glutaraldehyde and processed and visualized on a Hitachi S-3000 N microscope (Tokyo, Japan).
Allergen sensitization protocol
Mice were injected intraperitoneally with 0·1 ml (10 µg) of ovalbumin (Sigma, St Louis, MO, USA) as shown in a previous report [4]. On days 14–15, the mice were exposed to aerosols of 1% ovalbumin diluted in sterile phosphate-buffered saline, using an ultrasonic nebulizer (Ultraneb 99; DeVilbiss, Somerset, PA, USA). We performed the ovalbumin aerosol challenge only twice, on days 14 and 15, in order to minimize ovalbumin-induced COX-2 induction in epithelial cells [4]. On day 16, the mice were harvested and analysed.
Quantification of interleukin (IL)-4, IL-5 and IL-13 in lung tissues
Levels of cytokines in lung tissues of mice were measured with the Bio-plex mouse cytokine kit (Bio-Rad, Hercules, CA, USA), following the manufacturer's instructions.
P. aeruginosa pneumonia mouse model
COTA mice weighing 20–30 g were used for this experiment. After sedation with ketamine/xylazine, mice were treated with intratracheal (IT) administration of P. aeruginosa (strain PA103), as reported by our group [10]. After the indicated times, lungs were harvested and serial dilutions of lung homogenates were made, and 10 µl of each dilution were plated in soy base blood agar plates (Difco, Detroit, MI, USA). The plates were incubated at 37°C and the number of colonies was counted.
Statistical analysis
Our statistical analyses were performed with GraphPad InStat (GraphPad Software, San Diego, CA, USA), using an unpaired t-test and analysis of variance (anova).
Results
Generation of transgenic mice with inducible activation of COX-2 in airway epithelium
To generate inducible COX-2 expression using the tet-on system, we placed murine COX-2 under the control of the (tet-O)7-CMV promoter. To prevent basal leakiness of transgene expression, a construct expressing a tTS under the control of the Clara cell-specific CC10 promoter (obtained from Dr Jack Elias, Yale University, with the permission of Andrew Farmer, BD Clontech, Palo Alto, CA, USA) was co-injected with (tet-O)7-COX-2 to generate double transgenic mice [11]. Unbound tTS interacts with tet-O sites and functions as a transcriptional repressor; however, binding of doxycline to tTS results in dissociation from DNA, allowing rtTA binding and promoter activation [12,13]. The tTS construct was co-injected with (tet-O)7-COX-2 to generate double transgenic mice (Fig. 1a). Founder mice were identified by Southern analysis of tail DNA and confirmed by PCR (Fig. 1b). Double transgenic mice were bred with transgenic mice expressing rtTA under the control of the rat CC10 promoter (obtained from Dr Jeffrey Whitsett, University of Cincinnati) to generate triple transgenic mice, designated COTA (Fig. 1a). COTA mice from two separate founder lines (designated 10 and 26) were developed and despite differences in the integrated copy numbers of transgenes induced equivalent levels of COX-2 response to doxycycline treatment (Fig. 2b) Both lines were used for these studies.
Fig. 2.
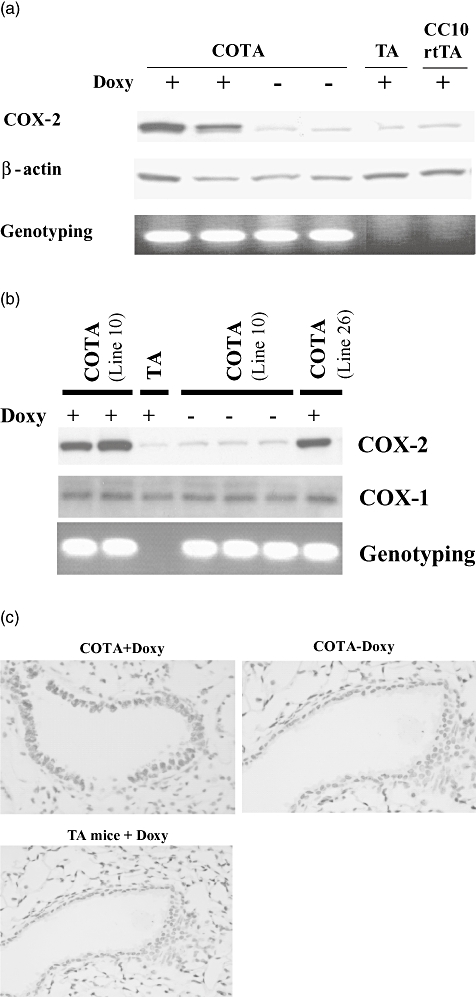
(a) Doxycycline induced the expression of cyclooxygenase-2 (COX-2) in the lungs of CycloOxygenase-2 TransActivated (COTA) transgenics. COTA mice (line 26) and transgene-negative littermate (TA mice) were treated with doxy for 7 days. Control mice were untreated. Western blot analysis for COX-2 expression demonstrated that inducible COX-2 is detected only in the lung of COTA mice following doxy treatment. β-actin was used as a protein control. Mice were genotyped by polymerase chain reaction using the primers for COX-2 transgene. (b) COX-1 expression in the lung was analysed from COTA mice and control littermates. Unlike COX-2 expression, COX-1 was not induced by doxy treatment. We obtained equivalent results from both COTA lines. (c) Immunohistochemistry for COX-2 in lung tissue from an untreated COTA mouse or a COTA mouse treated with doxy for 7 days. COX-2 staining (brown) is localized in airway epithelial cells in doxy-treated COTA mice.
Inducible expression of transgene COX-2 in airway epithelial cells
COTA mice were treated with doxycycline in drinking water (2 g/l) for 7 days. Immunoreactive COX-2 protein was increased in whole lung tissue of COTA mice in response to doxycycline treatment relative to COX-2 transgene-negative littermates (TA mice). In the absence of doxycycline, the background expression of COX-2 was comparable to those in transgene-negative littermates (Fig. 2a,b). However, COX-1, a protein that is expressed constitutively, was not changed by doxycycline treatment (Fig. 2b). To assess the cellular localization of doxycycline-induced COX-2 protein, we performed immunohistochemistry for COX-2 on lung sections from doxycycline-untreated and -treated COTA mice and control littermates (TA mice). After 7 days of doxycycline treatment, COX-2 expression was clearly visible in airway epithelium in the proximal and distal airways in COTA mice, but not in lung tissue from the two control groups (Fig. 2c).
Overproduction of prostaglandins in lung by elevation of COX-2 levels in airway epithelial cells
Next, we measured the enzymatic products of COX-2 in lung homogenate. Figure 3 demonstrates increased production of prostaglandins in the lung following doxycycline treatment. There was no statistical difference between wild-type mice treated with doxy and COTA mice without doxycycline in the level of prostaglandins in lung tissue. However, the lung homogenate from doxycycline-treated COTA mice contained a three- to sevenfold increase in prostaglandin E2 (PGE2) (121 ± 19 versus 36 ± 27 ng/g), prostaglandin D2 (PGD2) (74 ± 13 versus 11 ± 7 ng/g), thromboxane B2 (TXB2) (34 ± 5 versus 10 ± 7 ng/g) and 6-Keto prostaglandin F2α (PGF2α) (219 ± 24 versus 46 ± 25 ng/g) compared to untreated COTA mice (Fig. 3). The prostaglandin profile of BAL fluid was similar to that of the lung tissue. Specifically, PGE2 (1·02 ± 0·49 versus 0·073 ± 0·047) and PGD2 (0·91 ± 0·47 versus 0·064 ± 0·044 ng/g) were elevatedsignificantly in COTA mice treated with doxycycline compared to COTA without doxy. However, TXB2 was also increased in BAL fluid, even though it was elevated insignificantly in lung tissue. In contrast, although increased in lung tissue, 6-keto-PGF1α and PGF2α were not increased in the BAL fluid of COTA mice that were treated with doxycycline.
Fig. 3.
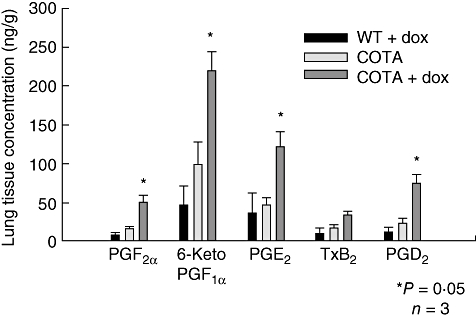
Profile of prostaglandin production in lung homogenates of CycloOxygenase-2 TransActivated (COTA) mice and controls. Mice were treated with doxycycline for 7 days. Measurements were performed by gas chromatography in conjunction with mass spectrometry (GC/MS). *P < 0·05 compared with doxy-untreated group.
PGE2 is a major product of COX-2 induction in primary bronchial epithelial cells
The different profiles of prostaglandin products determined in lung homogenate and BAL fluid prompted us which prostanoid products of COX-2 were produced specifically by airway epithelial cells. We cultured primary MTEC in transwell double chambers. To differentiate them into highly differentiated airway epithelium we treated the cells with retinoic acid, a differentiating agent, in air–liquid interface conditions. These cultured MTEC cells were then treated with or without doxycycline (0·5 µg/ml) in culture media. The scanning electron microscopy images showed a well-differentiated ciliated and microciliated epithelial monolayer (Fig. 4a). Aliquots of the culture supernatant were taken sequentially from day 1 to day 3 and analysed for PGE2 and PGD2 by liquid chromatography and mass spectrometry.
Fig. 4.
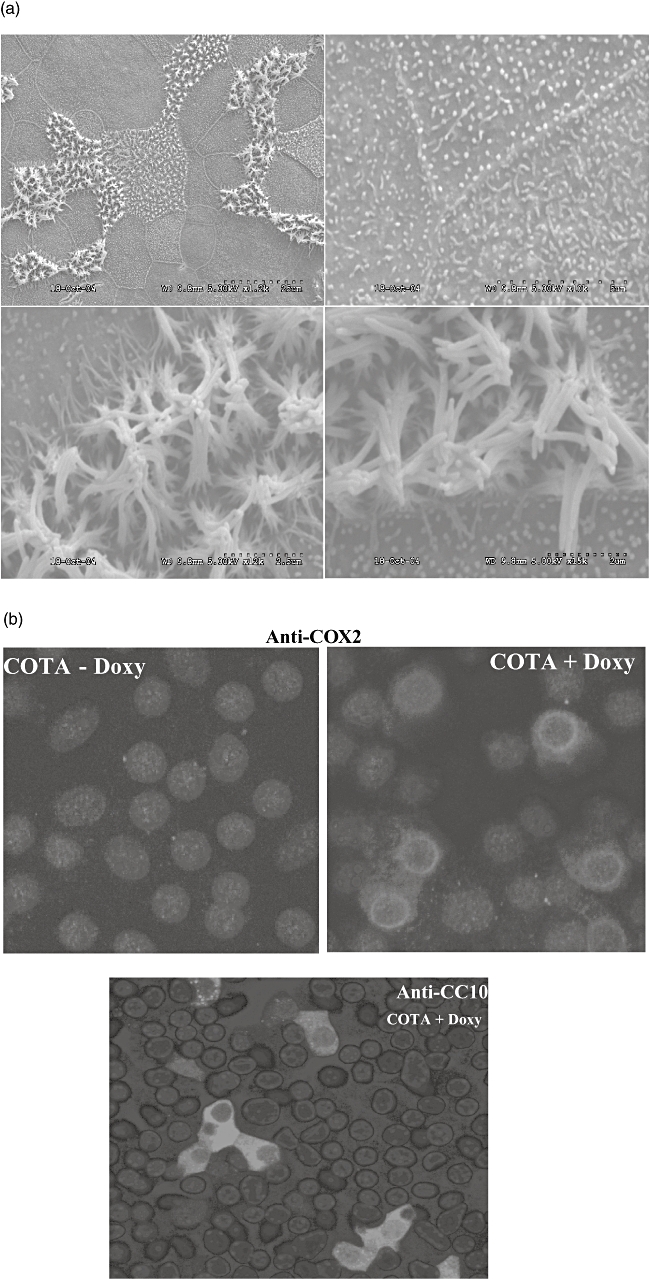
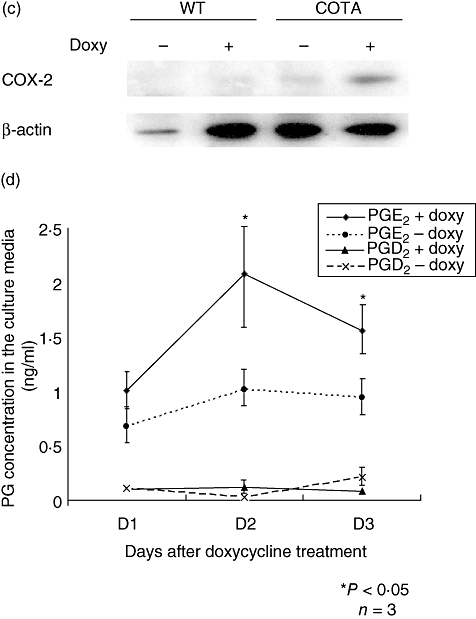
Primary culture of airway epithelial cells and measurement of inducible cyclooxygenase-2 (COX-2) and prostaglandin production. Primary airway epithelial cells from wild-type and CycloOxygenase-2 TransActivated (COTA) mice were cultured as described in Methods. (a) The cell morphology was analysed by scanning electron microscopy (SEM) and the intracellular location of COX-2 and CC10 expression was examined by immunostaining with confocal microscopy (b). Although we were unable to co-localize CC10 protein and COX-2 because of technical issues (both antibodies were made in the same species), a similar percentage of positive cells was detected. Western blot analysis for COX-2 showed doxycycline-induced COX-2 expression in COTA cells (c). The production of prostaglandin D2 (PGD2) and PGE2 was measured from culture supernatant after doxycycline treatment by liquid chromatography in conjunction with mass spectrometry. In this and other experiments, PGE2 was abdundant whereas levels of PGD2 were at the lower limits of detection (d).
Immunoreactive COX-2 protein was induced by doxycyline treatment in MTEC cells from COTA mice but not in wild-type controls or without doxycyline treatment (Fig. 4c). Because PGE2 and PGD2 were the major prostaglandins detected in BAL fluid, we measured them in culture supernatant from MTEC cells. From day 1 after doxycycline, the PGE2 levels began to increase (1·01 versus 0·68 ng/ml) and peaked at day 2 (2·08 versus 1·02 ng/ml). However, PGD2 levels were consistently very low-level at the beginning, providing no evidence of induction (Fig. 4c). The transwell membrane was cut and used for immunofluorescent staining to determine the distribution and intracellular location of COX-2. As the CC10 promoter was used to drive expression of COX-2 we also determined the distribution of CC10 protein staining. As expected, immunofluorescent staining suggested that the pattern of CC10 positive cells was patchy and that the protein was mostly cytoplasmic (Fig. 4b). The distribution of the COX-2 transgene, in terms of the cell population expressing detectable protein, was similar to that of CC10 protein. However, in contrast to CC10 protein that is expressed homogeneously in cytoplasm, the COX-2 protein is localized to the nuclear membrane and perinuclear region, as has been described by others for endogenous COX-2 protein [14].
Elevated production of prostaglandins in the airway epithelium during a sensitization period increases IL-4 production
Next, we examined the phenotype of COTA mice in an ovalbumin-sensitized allergic airway disease model. Doxycycline was administrated in the drinking water 7 days prior to an intraperitoneal injection of ovalbumin. Doxycycline treatment was then maintained throughout the experimental protocol. Mice were challenged with ovalbumin on two separate days (days 14 and 15) because this is prior to the time when endogenous epithelial COX-2 gene expression occurs in response to an aerosolized ovalbumin challenge (Fig. 5a). PGE2 level was measured in BAL fluid on day 16. PGE2 was elevated in COTA plus doxy group relative to controls (0·247 ± 0·206 and 0·032 ± 0·030 ng/ml) (Fig. 5b), although levels were much less than after 7 days of doxycycline treatment. As shown in Fig. 5c, there was no significant difference in total serum IgE levels between the groups. Cytokine concentrations were measured in whole lung homogenates on day 16. There was an increase in IL-4 level in doxy-treated COTA, compared to untreated COTA mice and doxy-treated TA (77·26 ± 7·79 versus 58·06 ± 7·96 and 52·30 ± 14·39, P < 0·05). However, there was no difference in IL-5 and IL-13 levels in both groups (Fig. 5d). We did not detect differences in IL-4, IL-5 or IL-13 concentrations in lung when treatment with doxycycline occurred exclusively during the challenging stage (from day 9 to day 16) (data not shown).
Fig. 5.
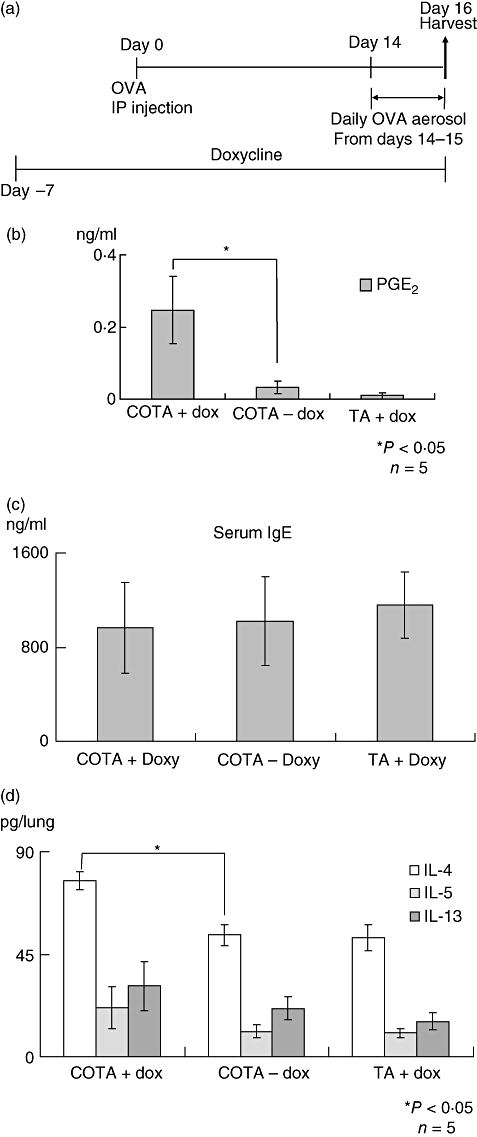
Allergen sensitization model. (a) The mice were sensitized and challenged with ovalbumin as described in the protocol. There were three experimental groups, CycloOxygenase-2 TransActivated (COTA) with doxy, COTA without doxy and TA with doxy. (b) Prostaglandin E2 (PGE2) in bronchoalveolar lavage (BAL) fluid was also measured on day 16 from the mice of each group. (c) Total IgE level was measured in serum and (d) the cytokines was measured from lung homogenate on day 16 using a Bio-Plex system (Bio-Rad).
Overproduction of airway prostaglandins impairs bacterial clearance from the lung
Others have shown that prostanoids can modulate immune and inflammatory responses [15,16]. PGE2, in particular, has an immunosuppressive effect on the phagocytic capacity of alveolar macrophages [17]. We tested the phenotype of COTA mice in a P. aeruginosa-induced pneumonia model. To determine whether overexpression of COX-2 in airway epithelial cells alters bacterial clearance after P. aeruginosa infection, both wild-type and COTA mice were treated with doxycycline in the drinking water (2 g/l) for 7 days, a concentration that is not expected to be bactericidal for P. aeruginosa [18]. On day 7 the mice were given an intratracheal bacterial inoculation. Twenty-hour h after the inoculation, we measured bacterial colony counts from the lung. There was significant impairment of bacterial clearance in COTA mice (Fig. 6b) compared to wild-type mice [75·8 ± 11·5 versus 10·5 ± 3·0 × 103 colony-forming units (CFU)/lung]. No difference in the total cell and differential count of BAL fluid was detected (data not shown).
Fig. 6.
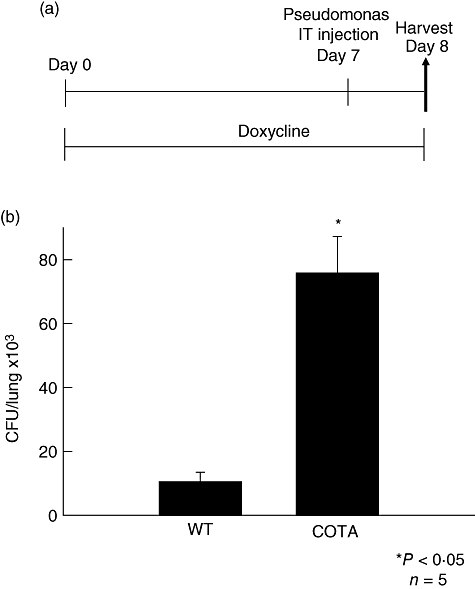
Impaired bacterial clearance in CycloOxygenase-2 TransActivated (COTA) mice. (a) Wild-type and COTA mice were challenged with intratracheal P. aeruginosa (105 CFU) at 7 days after doxy treatment. Mice were harvested at 24 h after intratracheal instillation. (b) Bacterial colony counts in lung tissue were measured as described in Methods. *P < 0·05 compared to wild-type mice.
Discussion
COX-2 gene expression is increased in the airway epithelium in both asthma and pneumonia models of mice [4,19,20]. Although some studies suggest that epithelial cell expression of COX-2 and production of eicosanoids contributes to the regulation of immune-inflammatory effector responses using in vitro cell line models [20–22], the role of COX-2 production by intact airway epithelium has not been defined in biologically relevant animal models. In spite of the limitation of the artificial overexpression system, which might differ from physiological activation, to our knowledge this is the first report of cell-specific overexpression of COX-2 enzyme using an inducible COX-2 transgenic animal model. Previous studies have examined COX-2 inhibition in animal models using selective or non-selective COX inhibitors or COX-2 gene knock-out mice. While these models have provided interesting data, the modulation of COX-2 expression is not targeted specifically to the airway epithelial cells and, thus, they have not defined the role of COX-2 in this specific cell type in lung disease. We have developed transgenic mice that express COX-2 under the regulation of the epithelial cell-specific CC10 promoter in response to treatment with doxycycline. This model overcomes many of the inherent limitations in constitutive overexpression or knock-out models such as activation of compensatory mechanisms, developmental alteration in fetal life and substrate depletion [11].
In addition to COX expression, the availability of arachidonic acid is a determining factor for prostaglandin production [23]. We found that there was significant decreased prostaglandin production when we overexpressed COX-2 for 4 weeks, compared to 1-week doxycycline treatment (data not shown).
In this study, we found that primary MTEC that overexpress COX-2 in response to treatment with doxycyline produce PGE2 almost exclusively. This is consistent with other reports which find that COX-2 and microsomal PGE synthase are induced simultaneously in certain inflammatory conditions [24]. Our studies in the COTA mice, however, show that both PGE2 and PGD2 are major components in BAL fluid. This suggests that PGD2 is produced by other cell types in the lungs [25]. One possible source for PGD2 are macrophages which can produce PGD2 through induction of haematogenous or lipocalin PGD synthase (H- or L-PGDS). PGE2 has also been shown to induce COX-2 expression in other cell types by directly inducing COX-2 gene expression or modulating mRNA stability [25–27]. PGE2 binds its EP receptors and increases intracellular c-AMP level, which results in increased COX-2 expression that is mediated through a functionally active cAMP response element (CRE) binding site in the COX-2 promoter. Therefore, one mechanism by which the elevated production of PGD2 in the BAL fluid of induced COTA mice occurs may be that the airway epithelial cells produce PGE2 that binds to receptors on macrophages and stimulates them to produce PGD2.
Airway epithelium has an important role in homeostasis by regulating the composition of the airway surface liquid and the airway response to injury [28]. Prostanoids are secreted locally, where they communicate with other cell types in the microenvironment of the airway [8,26]. By recognition of airborne allergen and pathogens, epithelial cells are capable of COX-2 gene expression and production of biologically active prostanoids into the airspace. In this study, we show that airway epithelial cells produce PGE2 by induction of COX-2, which results subsequently in alteration of the allergic and innate immune phenotype. By interaction with other immune cells through prostanoid production, airway epithelial COX-2 polarizes the immune response toward a Th2-type phenotype, suggesting that epithelial COX-2 plays an important role in determining the direction of the immune response in the airspace milieu. In previous studies, inhibition of COX-2 with indomethacin during the allergen sensitization stage resulted in an increase in IL-4 protein levels in lung [4]. We used the opposite approach, the overexpression of COX-2 in the airway, and discovered paradoxically that IL-4 levels were also increased. One possible explanation for this difference is that indomethacin is a potent ligand for the DP-2 (also called CRTH2) receptor that mediates the effects of PGD2 [29]. It is possible that indomethacin activates the DP-2 receptor and generates IL-4 in a COX-2-independent manner. We suggest that PGE2 production by airway epithelial cells stimulated production of PGD2 by macrophages and this, in turn, mediates the production of IL-4 and the subsequent TH2-type allergic inflammation.
There is dispute regarding the role of PGE2 in inflammation because of discrepancies between in vitro and in vivo studies [30]. In vitro studies show that PGE2 promotes antigen stimulated mast cell degranulation and inhibits IL-12 production by macrophages [31,32], which leads to Th2 types of immune response. However, in vivo data suggest the opposite direction of immune response. PGE2 inhibits ovalbumin-induced airway inflammation and suppresses T cell proliferation, which is an anti-asthmatic property [33]. The complexity of the role of PGE2 is due probably to its multiple epithelial receptors and multiple cells types that are involved. Most of the major cells that are involved in inflammation, including T and B lymphocytes and dendritic cells, have four epithelial receptors. In addition, structural cells may also be involved in regulation of immune and inflammatory process. However, our study shows that epithelial COX-2 plays an important role in determining the immunological response to both allergic and pathogenic challenge of the airspace.
Prostanoids have emerged as potent modulators of innate immunity [34]. It has been shown that PGE2 inhibits the production of cytokines such as tumour necrosis factor (TNF)-α in macrophages through the EP4 receptor [35]. PGE2 also suppresses anti-microbial activity of alveolar macrophages via the EP2 receptor [17]. Our data suggest that epithelial cells are the source of endogenous PGE2 and that COX-2 activation in epithelial cells suppresses the bacterial clearance of P. aeruginosa. This finding supports the emerging hypothesis that the function of alveolar macrophages is regulated negatively by PGE2 in the airspace [36,37].
Further studies are required to clarify the role of non-epithelial cell-derived prostanoids in the pathophysiology of airway diseases, but our data indicate that airway epithelial cells produce PGE2 exclusively, which mediates a shift of the immunological balance in favour of a Th2 response that counterbalances the inhibition of bactericidal activity.
Acknowledgments
This work was supported by the Department of Veterans Affairs and National Institutes of Health grants HL 075557 and HL 66196. The authors thank Jason Morrow (Vanderbilt) and Hongmei Cao (University of Illinois) for expert technical support with gas/liquid chromatography/mass spectrometry.
References
- 1.Severgnini M, Takahashi S, Rozo LM, et al. Activation of the STAT pathway in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1282–92. doi: 10.1152/ajplung.00349.2003. [DOI] [PubMed] [Google Scholar]
- 2.Sadikot RT, Zeng H, Joo M, et al. Targeted immunomodulation of the NF-kappaB pathway in airway epithelium impacts host defense against Pseudomonas aeruginosa. J Immunol. 2006;176:4923–30. doi: 10.4049/jimmunol.176.8.4923. [DOI] [PubMed] [Google Scholar]
- 3.Watkins DN, Peroni DJ, Lenzo JC, Knight DA, Garlepp MJ, Thompson PJ. Expression and localization of COX-2 in human airways and cultured airway epithelial cells. Eur Respir J. 1999;13:999–1007. doi: 10.1034/j.1399-3003.1999.13e12.x. [DOI] [PubMed] [Google Scholar]
- 4.Stokes Peebles R, Jr, Hashimoto K, Morrow JD, et al. Selective cyclooxygenase-1 and -2 inhibitors each increase allergic inflammation and airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 2002;165:1154–60. doi: 10.1164/ajrccm.165.8.2106025. [DOI] [PubMed] [Google Scholar]
- 5.Zehr BB, Casale TB, Wood D, Floerchinger C, Richerson HB, Hunninghake GW. Use of segmental airway lavage to obtain relevant mediators from the lungs of asthmatic and control subjects. Chest. 1989;95:1059–63. doi: 10.1378/chest.95.5.1059. [DOI] [PubMed] [Google Scholar]
- 6.Baughman RP, Gallon LS, Barcelli U. Prostaglandins and thromboxanes in the bronchoalveolar lavage fluid: possible immunoregulation in sarcoidosis. Am Rev Respir Dis. 1984;130:933–6. doi: 10.1164/arrd.1984.130.5.933. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz DA, Galvin JR, Frees KL, et al. Clinical relevance of cellular mediators of inflammation in workers exposed to asbestos. Am Rev Respir Dis. 1993;148:68–74. doi: 10.1164/ajrccm/148.1.68. [DOI] [PubMed] [Google Scholar]
- 8.Ko SC, Chapple KS, Hawcroft G, Coletta PL, Markham AF, Hull MA. Paracrine cyclooxygenase-2-mediated signalling by macrophages promotes tumorigenic progression of intestinal epithelial cells. Oncogene. 2002;21:7175–86. doi: 10.1038/sj.onc.1205869. [DOI] [PubMed] [Google Scholar]
- 9.You Y, Richer EJ, Huang T, Brody SL. Growth and differentiation of mouse tracheal epithelial cells: selection of a proliferative population. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1315–21. doi: 10.1152/ajplung.00169.2002. [DOI] [PubMed] [Google Scholar]
- 10.Sadikot RT, Zeng H, Yull FE, et al. p47phox deficiency impairs NF-{kappa}B activation and host defense in Pseudomonas pneumonia. J Immunol. 2004;172:1801–8. doi: 10.4049/jimmunol.172.3.1801. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Z, Ma B, Homer RJ, Zheng T, Elias JA. Use of the Tetracycline-controlled transcriptional silencer (tTS) to eliminate transgene leak in inducible overexpression transgenic mice. J Biol Chem. 2001;276:25222–9. doi: 10.1074/jbc.M101512200. [DOI] [PubMed] [Google Scholar]
- 12.Deuschle U, Meyer W, Thiesen H. Tetracycline-reversible silencing of eukaryotic promoters. Mol Cell Biol. 1995;15:1907–14. doi: 10.1128/mcb.15.4.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forster K, Helbl V, Lederer T, Urlinger S, Wittenburg N, Hillen W. Tetracycline-inducible expression systems with reduced basal activity in mammalian cells. Nucl Acids Res. 1999;27:708–10. doi: 10.1093/nar/27.2.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morita I, Schindler M, Regier MK, et al. Different intracellular locations for prostaglandin endoperoxide H synthase-1 and -2. J Biol Chem. 1995;270:10902–8. doi: 10.1074/jbc.270.18.10902. [DOI] [PubMed] [Google Scholar]
- 15.Canning BJ, Hmieleski RR, Spannhake EW, Jakab GJ. Ozone reduces murine alveolar and peritoneal macrophage phagocytosis. the role of prostanoids. Am J Physiol. 1991;261:L277–82. doi: 10.1152/ajplung.1991.261.4.L277. [DOI] [PubMed] [Google Scholar]
- 16.Laegreid WW, Liggitt HD, Silflow RM, Evermann JR, Taylor SM, Leid RW. Reversal of virus-induced alveolar macrophage bactericidal dysfunction by cyclooxygenase inhibition in vitro. J Leukoc Biol. 1989;45:293–300. doi: 10.1002/jlb.45.4.293. [DOI] [PubMed] [Google Scholar]
- 17.Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J Immunol. 2004;173:559–65. doi: 10.4049/jimmunol.173.1.559. [DOI] [PubMed] [Google Scholar]
- 18.Timurkaynak F, Can F, Azap OK, Demirbilek M, Arslan H, Karaman SO. In vitro activities of non-traditional antimicrobials alone or in combination against multidrug-resistant strains of Pseudomonas aeruginosa and Acinetobacter baumannii isolated from intensive care units. Int J Antimicrob Agents. 2006;27:224–8. doi: 10.1016/j.ijantimicag.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Taha R, Olivenstein R, Utsumi T, et al. Prostaglandin H synthase 2 expression in airway cells from patients with asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161:636–40. doi: 10.1164/ajrccm.161.2.9811063. [DOI] [PubMed] [Google Scholar]
- 20.N'Guessan PD, Hippenstiel S, Etouem MO, et al. Streptococcus pneumoniae induced p38 MAPK- and NF-kappaB-dependent COX-2 expression in human lung epithelium. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1131–8. doi: 10.1152/ajplung.00383.2005. [DOI] [PubMed] [Google Scholar]
- 21.Mizumura K, Hashimoto S, Maruoka S, et al. Role of mitogen-activated protein kinases in influenza virus induction of prostaglandin E2 from arachidonic acid in bronchial epithelial cells. Clin Exp Allergy. 2003;33:1244–51. doi: 10.1046/j.1365-2222.2003.01750.x. [DOI] [PubMed] [Google Scholar]
- 22.Jahn HU, Krull M, Wuppermann FN, et al. Infection and activation of airway epithelial cells by Chlamydia pneumoniae. J Infect Dis. 2000;182:1678–87. doi: 10.1086/317608. [DOI] [PubMed] [Google Scholar]
- 23.Scott KF, Bryant KJ, Bidgood MJ. Functional coupling and differential regulation of the phospholipase A2-cyclooxygenase pathways in inflammation. J Leukoc Biol. 1999;66:535–41. doi: 10.1002/jlb.66.4.535. [DOI] [PubMed] [Google Scholar]
- 24.Mancini JA, Blood K, Guay J, et al. Cloning, expression, and up-regulation of inducible rat prostaglandin E synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J Biol Chem. 2001;276:4469–75. doi: 10.1074/jbc.M006865200. [DOI] [PubMed] [Google Scholar]
- 25.Bonazzi A, Bolla M, Buccellati C, et al. Effect of endogenous and exogenous prostaglandin E2 on interleukin-1{beta}-induced cyclooxygenase-2 expression in human airway smooth-muscle cells. Am J Respir Crit Care Med. 2000;162:2272–7. doi: 10.1164/ajrccm.162.6.2003127. [DOI] [PubMed] [Google Scholar]
- 26.Caughey GE, Cleland LG, Gamble JR, James MJ. Up-regulation of endothelial cyclooxygenase-2 and prostanoid synthesis by platelets. Role of thromboxane A2. J Biol Chem. 2001;276:37839–45. doi: 10.1074/jbc.M010606200. [DOI] [PubMed] [Google Scholar]
- 27.Minghetti L, Polazzi E, Nicolini A, Creminon C, Levi G. Up-regulation of cyclooxygenase-2 expression in cultured microglia by prostaglandin E2, cyclic AMP and non-steroidal anti-inflammatory drugs. Eur J Neurosci. 1997;9:934–40. doi: 10.1111/j.1460-9568.1997.tb01444.x. [DOI] [PubMed] [Google Scholar]
- 28.Busse W, Elias J, Sheppard D, Banks-Schlegel S. Airway remodeling and repair. Am J Respir Crit Care Med. 1999;160:1035–42. doi: 10.1164/ajrccm.160.3.9902064. [DOI] [PubMed] [Google Scholar]
- 29.Hata AN, Lybrand TP, Marnett LJ, Breyer RM. Structural determinants of arylacetic acid nonsteroidal anti-inflammatory drugs necessary for binding and activation of the prostaglandin D2 receptor CRTH2. Mol Pharmacol. 2005;67:640–7. doi: 10.1124/mol.104.007971. [DOI] [PubMed] [Google Scholar]
- 30.Park GY, Christman JW. Involvement of cyclooxygenase-2 and prostaglandins in the molecular pathogenesis of inflammatory lung diseases. Am J Physiol Lung Cell Mol Physiol. 2006;290:L797–805. doi: 10.1152/ajplung.00513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fedyk ER, Phipps RP. Prostaglandin E2 receptors of the EP2 and EP4 subtypes regulate activation and differentiation of mouse B lymphocytes to IgE-secreting cells. Proc Natl Acad Sci USA. 1996;93:10978–83. doi: 10.1073/pnas.93.20.10978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Pouw Kraan TC, Boeije LC, Smeenk RJ, Wijdenes J, Aarden LA. Prostaglandin-E2 is a potent inhibitor of human interleukin 12 production. J Exp Med. 1995;181:775–9. doi: 10.1084/jem.181.2.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pavord ID, Wong CS, Williams J, Tattersfield AE. Effect of inhaled prostaglandin E2 on allergen-induced asthma. Am Rev Respir Dis. 1993;148:87–90. doi: 10.1164/ajrccm/148.1.87. [DOI] [PubMed] [Google Scholar]
- 34.Tilley SL, Coffman TM, Koller BH. Mixed messages. modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108:15–23. doi: 10.1172/JCI13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nataraj C, Thomas DW, Tilley SL, et al. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J Clin Invest. 2001;108:1229–35. doi: 10.1172/JCI13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. 2002;23:144–50. doi: 10.1016/s1471-4906(01)02154-8. [DOI] [PubMed] [Google Scholar]
- 37.Goodwin JS, Webb DR. Regulation of the immune response by prostaglandins. Clin Immunol Immunopathol. 1980;15:106–22. doi: 10.1016/0090-1229(80)90024-0. [DOI] [PubMed] [Google Scholar]