

Abstract
Recently we reported that monocyte phagocytosis and chemotaxis are impaired in X-linked agammaglobulinaemia (XLA) and common variable immunodeficiency (CVI) patients. Few data exist on the in vivo expression of receptors for the constant region of immunoglobulin (IgG) (FcγR) and complement receptors (CR) in these patients. The objective of this study was to investigate the expression of FcγR and CR on monocytes from XLA and CVI patients and compare it to that of healthy controls. Whole blood samples were obtained from 10 patients with XLA, 12 with CVI and 18 healthy controls. Monocyte phenotype was determined by flow cytometry with gating on CD14+ cells. Surface expression of FcγRI (CD64), FcγRII (CD32) and FcγRIII (CD16), CR1 (CD35) and CR3 (CD11b and CD18) was measured by determination of the proportion of CD14+ cells positive for each receptor and by receptor density. Compared to controls, a significantly higher percentage of CD16 and CD35+ monocytes from XLA (P = 0·002 and P = 0·007, respectively) were observed. The relative fluorescence intensity (RFI) expression of FcyRII (CD32) and FcyRIII (CD16) were significantly lower on CVI monocytes compared to controls (P = 0·001 and P = 0·035, respectively). XLA patients, who have a reduction of Bruton's tyrosine kinase (Btk), showed normal or increased percentages of monocytes expressing Fcy and complement receptors. CVI patients, who have normal expression of Btk, showed reduced expression of CD16 and CD32 on monocytes. Inefficient chemotaxis and phagocytosis, reported previously in XLA patients, could be due to defects of cytoplasmatic transduction mechanisms.
Keywords: CR1 (CD35); CR3 (CD11b, CD18) receptors; monocytes; Fcγ; flow cytometry; common variable immunodeficiency; X-linked agammaglobulinaemia
Introduction
X-linked agammaglobulinaemia, or Bruton's disease, is caused by mutations in cytoplasmic Bruton's tyrosine kinase (Btk), which are associated with a reduction in Btk mRNA, Btk protein and kinase activity [1–3]. This defect results in a blockage of the expansion of pre-B cells into later B cell stages or incomplete differentiation of B cell precursors to pre-B cells [3]. XLA is characterized by recurrent bacterial and enteroviral infections with a paucity of circulating B cells (≤ 1%) and a marked reduction in serum levels of all immunoglobulin isotypes.
Of note is that in normal individuals Btk is expressed in most haematopoietic lineages, except for T cells and plasma cells [4–9]. Despite its crucial role in B cell receptor (BCR)-dependent calcium mobilization, Btk functions in all cells, except B cells, remain defined incompletely [10]. Btk is likely to play analogous functions during the development and functioning of the myeloid lineage, affecting the outcome of many infectious as well as non-infectious inflammatory events [11].
Common variable immunodeficiency (CVI) is due probably to the block in B cell differentiation, which leads to insufficient serum IgG and IgA levels. In half the patients IgM levels are also reduced [12, 13]. CVI is characterized by recurrent infections and normal numbers of B cells in peripheral blood, but the basic immunological and molecular defects of the majority of patients remain unknown. The genetic predisposition and multiple defects in both innate and adaptive compartments of the immune system might thus account cumulatively for the heterogeneous symptoms displayed in CVI.
Monocytes from CVI patients can exhibit chronic hyperactivity and enhanced oxidative stress due to abnormalities in the tumour necrosis factor (TNF)–interleukin (IL)-12–interferon (IFN)-γ circuit, which might contribute to autoimmune disorders, granuloma formation, impaired T cell proliferation and natural killer (NK) cell-mediated cytotoxicity [14].
Previously, we demonstrated that monocytes obtained from XLA and CVI patients displayed a decreased chemotaxis and defective Fcγ, complement receptor (CR) CR-1 and CR3-mediated phagocytosis compared to monocytes obtained from healthy donors [15]. All these receptors are very effective in binding, internalizing and destroying opsonized particles [16]. They differ in their structure, binding affinity for IgG, cell distribution and downstream signalling [17]. Btk function in phagocytosis signalling pathways is not yet clarified [18]. Moreover, monocytes from CVI patients exhibit normal concentration of Btk, suggesting that other signalling pathways may have a role in these phagocytosis and chemotaxis deficiencies.
Few or no data exist on the expression of specific Fcγ and complement receptors in phagocytes of XLA and CVI patients. In order to investigate whether the observed alteration in phagocytosis was attributable to differences in the number of these receptors, we investigated their expression on monocytes from XLA and CVI patients compared to healthy controls.
Materials and methods
Patients and controls
Ten patients with XLA and 12 with CVI, followed at the Pediatric Immunology Division at the State University of Campinas Medical School Hospital, and 18 healthy volunteers participated in the study.
The diagnosis of CVI was carried out according to the criteria of the World Health Organization expert group for primary immunodeficiency diseases [19]. Diagnosis of XLA was based on the decreased expression of Btk proteins in monocytes, markedly reduced levels of all major classes of immunoglobulins and circulating B cell numbers (< 1%). At the time of the study, all patients were receiving replacement therapy with intravenous immunoglobulin (IVIg). The blood samples were collected before the administration of IVIg. There was no evidence of autoimmune disorders, granulomatous diseases or neoplasias in any patient. At the time of the investigation, none of them had acute infection.
Cell isolation and flow cytometry analysis of Btk expression in monocytes
Btk expression was evaluated to confirm the diagnosis of XLA. Peripheral blood mononuclear cells (PBMC) were separated from heparinized venous blood by a Ficoll-Hypaque gradient. PBMC were washed three times in phosphate-buffered saline (PBS) and stained with phycoerythrin (PE)-labelled anti-CD14 monoclonal antibody (mAb) DakoCytomation, Glostrup, Denmark for 20 min on ice to discriminate monocytes from other cells. The cells were fixed (4% paraformaldehyde in PBS for 15 min at room temperature) and permeabilized [0·1% Triton X-100 in Tris-buffered saline pH 7·4, with 1 mg/ml bovine serum albumin (BSA) for 5 min]. These cells were reacted with 2 μg/ml of anti-Btk (48–2H) or control IgG1 (Dako) mAb for 20 min on ice, washed and incubated further with a 1 : 2000 dilution of fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG1 antibody (Southern Biotechnology Associates, Inc., Birmingham, AL, USA) for 20 min on ice. Stained cells were analysed by two-colour flow cytometry (Epics XL-MCL®; Coulter Corp., Hialeah, FL, USA) [3].
Flow cytometry analysis of Fcγ and complement receptors (CR) expression in monocytes
Venous blood was collected into ethylenediamine tetraacetic acid (EDTA) tubes for detection of surface antigens. To analyse the CRs whole blood (WB) was incubated first with 20 μl of human IgG for 5 min and then 100 μl of WB per tube were labelled with the following fluorochrome-conjugated mAbs: anti-CD14 PeCy5, anti-CD64 PE, anti-CD32 FITC, anti-CD16 FITC, anti-CD 18 PE, anti-CD35 FITC and anti-CD11b FITC. Appropriate isotype controls were used to define positive and negative cell populations. After incubation for 20 min on ice red blood cells were lysed (Optlyse®; Coulter) and washed twice in PBS with 1% fetal calf serum (FCS) and 0·1% sodium azide. All fluorochrome-conjugated mAbs and isotype controls were obtained from Immunotech (Marseille, France).
Monocyte phenotype was determined by flow cytometric analysis of whole blood samples, with gating on CD14-positive cells. Surface expression of FcγRI (CD64), RII (CD32) and RIII (CD16), complement receptors CR1 (CD35) and CR3 (Cd11b/CD18) was determined. During flow cytometric analysis, monocyte autofluorescence was detected on the FL1 channel, without differences between XLA, CVI or control samples. Cytometer settings were adjusted with the aim of keeping unstained or isotype control-stained monocytes (from a monocyte gate set by forward and lateral light scatter) in the first decade of the logarithmic scale for each fluorescence channel. As the settings were established, they were kept the same for all study samples (patients and controls). Receptor expression was measured by determination of the proportion of CD14+ monocyte positive for each of the receptors above. Receptor density was determined by calculation of relative fluorescence intensity (RFI), the division of the median fluorescence intensity (MFI) of each positive cell population by the MFI of the negative control sample. Data were acquired on a Coulter Epics XL® flow cytometer. At least 5 × 103 of CD14+ events were acquired for each sample.
Statistical analysis
Statistical analysis was performed using spss software version 7.5.1 (SPSS Inc., Chicago, IL, USA). Data were expressed as medians and extremes. Statistical estimation of the difference between disease groups and normal controls was performed by Mann–Whitney test (significance level, P < 0·05).
The study was approved by the Committee for Ethics in Research of the State University of Campinas Medical School. Informed consent was obtained from the patients and healthy controls.
Results
Clinical and laboratory features of patients
All 10 patients with XLA were male, with ages varying from 2·6 to 28·3 years (median, 19·6). The CVI group had eight female and four male patients, with ages varying from 10·6 to 57·9 years (median, 38·3). The ages of the 18 control subjects varied from 22 to 58 years (median, 29), and 11 were male. The main clinical and immunological characteristics of the patients are reported in Tables 1 and 2.
Table 1.
Clinical and laboratory characteristics of X-linked agammaglobulinaemia patients.
| Patient | Age in years at diagnosis | Serum IgM mg/dl* | Serum IgG mg/dl* | Serum IgA mg/dl* | B cells CD19 % | Patient Btk expression monocytes% | Mother Btk expression monocytes % |
|---|---|---|---|---|---|---|---|
| EOB | 6 | 15 | 109 | 7 | 0·0 | 25·6 | 51·1 |
| AHO | 14 | 15 | 203 | 53·9 | 0·04 | 70·2 | 58·5 |
| AAAJ | 7 | 10 | 240 | 0·6 | 0·0 | 26·3 | 49 |
| RCJ | 8 | 3·4 | 54 | 0·0 | 0·19 | 13·3 | 39·8 |
| RJF | 2 | 2 | 22 | 2 | 0·0 | 97 | 97 |
| AJAV | 10 | 0·0 | 150 | 20 | 0·0 | 9·0 | 22·4 |
| FFL | 6 | 0·0 | 0·0 | 3·0 | 0·04 | 9·0 | 53·8 |
| MHPF | 1 | 18 | 198 | 0·0 | 0·09 | 39 | 96 |
| MD | 4 | 26·8 | 86·2 | < 23·4 | 0·0 | 4·5 | 29·2 |
| DSM | 4 | 182 | < 30 | 0·0 | 1·0 | 86·4 | 97·9 |
Serum immunoglobulin levels at the age of diagnosis. Btk: Bruton's tyrosine kinase.
Table 2.
Clinical and laboratory characteristics of common variable immunodeficiency patients.
| Patient | Sex | Age in years at diagnosis | Serum IgM mg/dl* | Serum IgG mg/dl* | Serum IgA mg/dl* | B cells CD19% |
|---|---|---|---|---|---|---|
| VAM | F | 17 | 8·3 | 256 | 8·2 | 10·45 |
| FJO | M | 13 | 1·8 | 100 | 0·0 | 11·93 |
| JMC | M | 27 | 2·0 | 85 | 0·0 | 18·0 |
| AFB | F | 22 | 10 | 566 | < 6·0 | 16·7 |
| MRC | F | 16 | 14 | 797 | < 6·0 | 3·6 |
| RSB | F | 19 | 10 | 400 | 0·0 | 10·1 |
| LMD | F | 26 | 11·6 | 477 | < 6·6 | 20 |
| VDC | F | 32 | 24 | 284 | < 22 | 10·8 |
| JDC | M | 23 | 14·4 | 817 | < 6·6 | 22·9 |
| APM | F | 14 | 16 | 722 | < 25 | 7·74 |
| CDFM | M | 12 | 9·2 | 536 | < 6·0 | 9·3 |
| RLGF | F | 5 | 13 | 428 | < 6·0 | 13 |
Serum immunoglobulin levels at the age of diagnosis. F, female, M, male.
Surface expression of Fcγ and CR on monocytes from XLA and CVI patients
Comparing XLA with normal controls, there was a higher percentage of CD16+ (median, 11·9% versus 6%, P = 0·002), CD35+ (median, 95·7% versus 84·2%, P = 0·007) and CD11b+ (median, 100% versus 99·8%, P = 0·001) positive monocytes in XLA. However, there were no significant differences between XLA and controls or XLA and CVI regarding receptor density as measured by RFI. Patients with XLA showed a higher percentage of CD11b+ (median, 100% versus 99·8%, P = 0·008) in relation to CVI. Patients with CVI, compared to normal controls, showed a lower receptor density for CD16+ (median RFI 9 versus 14·42, P = 0·03) and CD32+ (median, RFI 38·5 versus 67·8, P = 0·001) positive monocytes. There were no significant differences between CVI and controls regarding the percentages of monocytes expressing FcγR and CR. The results above are summarized on Figs 1 and 2.
Fig. 1.
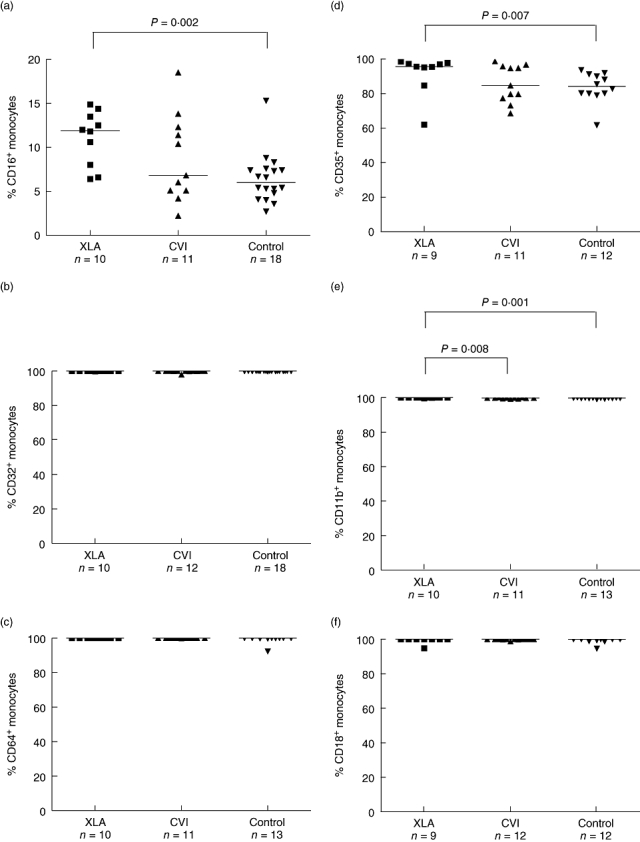
Comparison of Fcγ (a–c) and complement (d–f) receptor expression, measured by percentages of positive monocytes, among patients with X-linked agammaglobulinaemia, common variable immunodeficiency and normal controls.
Fig. 2.
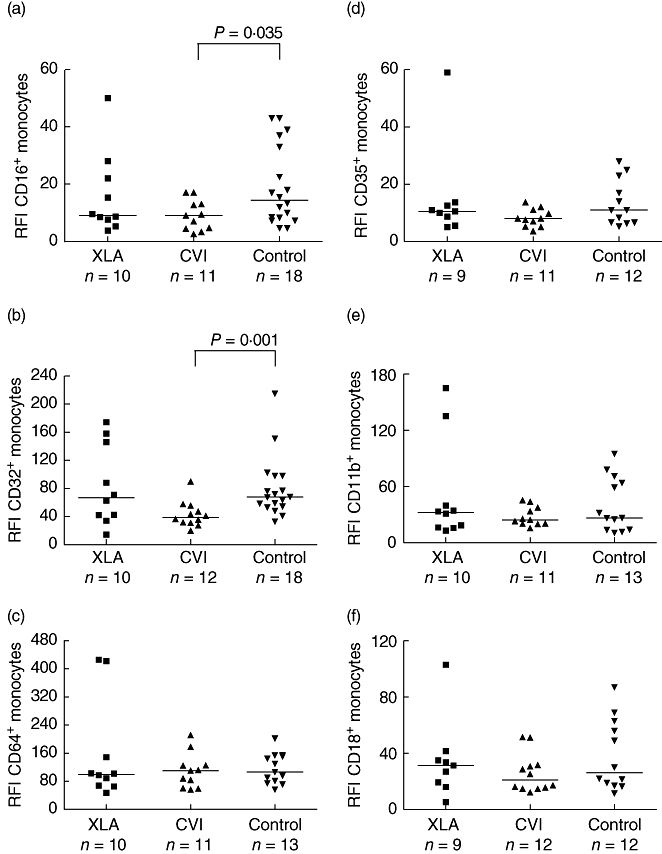
Comparison of Fcγ (a–c) and complement (d–f) receptor expression, measured by relative fluorescence intensity, among patients with X-linked agammaglobulinaemia, common variable immunodeficiency and normal controls.
Discussion
In this study, we carried out a phenotypic analysis of monocytes using flow cytometry, comparing cells from XLA and CVI patients to those of healthy controls. The XLA patients showed a significantly higher percentage of CD14CD16+ and CD14CD35+ monocytes in relation to the controls (Fig. 1). However, the receptor density was similar among the groups (Fig. 2).
Peripheral blood monocytes are a heterogeneous population of circulating precursors for tissue macrophages and dendritic cells (DCs). In humans, classical CD14+CD16− monocytes express CCR2, CD64 (FcγRI) and CD62L (l-selectin), whereas ‘non-classical’ CD14low CD16+ monocytes lack CCR2 and have higher levels of MHC-II and FcγRII (CD32) [20].
However, a third subset has been described, which is characterized as being CD14+CD64+CD16+. Although we did not perform simultaneous multicolour staining, as virtually 100% of CD14+ monocytes are also CD64+, it is reasonable to assume that CD16+ monocytes in our patient samples were also CD14+CD64+. This subset of monocytes has been described as combining features of typical dendritic cells [high IL-12 production and expression of human leucocyte antigen D-related (HLA-DR)] and monocytes (monocytic morphology, high phagocytic activity) [21, 22].
The presence of this population in XLA, with normal or increased numbers, reinforces previous findings that the Btk defect is not involved in DC differentiation and maturation [23]. On the other hand, we observed that chemotaxis and phagocytosis of monocytes appeared reduced in XLA patients [15], suggesting that these findings could be due to defects in Btk-mediated cytoplasmatic transduction mechanisms. The activation of tyrosine kinases has been recognized as an early step [24], although the role of individual kinases in phagocytosis signalling is still to be determined [25]. Btk can induce cytoskeletal remodeling in stimulated cells, further suggesting that cytoskeletal regulation mediated by Btk may be of physiological relevance for phagocytosis and chemotaxis [26, 27]. All FcγR and CR induce rearrangements in the actin cytoskeleton that lead to the internalization of the particle. Accordingly, the cells that are impaired or deficient in Btk activation have been shown to lack BCR-dependent intracellular calcium mobilization almost totally [28].
Here we demonstrated that the percentages of monocytes expressing CD35 were also normal or increased in XLA patients. This finding has not been reported previously. The integrity of CD35 expression may be important in avoiding the incidence of immune-complex diseases in XLA patients, as the transcription of CD35 has been reported to be related inversely to circulating immune complexes in patients with systemic lupus erythematosus [29]. In contrast to XLA, the CVI patients showed a lower receptor density for CD16+ and CD32+ monocytes in relation to controls (Fig. 2). However, an increase in the expression of CD14+CD16+ monocytes from CVI patients was reported, a phenomenon that could be related to immunoglobulin therapy in these individuals. The expression of CD64, CD32 and CD16 is linked to their state of activation [17]. Nevertheless, CD16 cell surface expression levels diminish after stimulation because of receptor internalization and shedding of the soluble receptor from the cell surface [17]. The patients in our study were under monthly immunoglobulin therapy but the blood samples were collected immediately before the intravenous immunoglobulin infusion. Although we did not measure IgG levels, we can suppose that they must have been very low, potentially interfering with monocyte activation.
The antigen-presenting function of DCs has been reported to be normal in CVI [30, 31], although a deep alteration in the distribution of myeloid DC subsets in the peripheral blood of CVI patients was found [32]. Myeloid dendritic cells from these patients displayed severely perturbed differentiation and maturation with decreased expression of the co-stimulatory molecules CD80, CD86 and HLA-DR. Impaired IL-12 production with T helper 1 (Th1)-biased cytokine environment, unfavourable to antibody production, was also noticed [14, 33, 34]. The processes are complex with positive and negative feedback between cells, including the regulation of surface-signalling molecules [35] and the production of cytokines. Evidence of dysregulation in the monocyte/T cell interactions involving IL-12 and IFN-γ in CVI has been described [33].
Although the percentages of CD11b+ monocytes (CR3) were quite different between XLA, CVI and the control group, we considered this to have no physiological significance because the values were very similar, all close to 100%.
The results reported above, showing a general pattern of normality of Fc and CRs, agree with the clinical observation that XLA and CVI patients have good outcomes under intravenous immunoglobulin (IVIg) replacement. The reported actions of IVIg mediated by the variable regions F(ab′)2, actions of Fc on a range of Fc receptors (FcR) and actions mediated by complement binding within the Fc fragment are possible because the patients are expressing FcR and CR.
In conclusion, the current work offers an important insight into monocyte subsets by providing evidence that XLA patients, who have Btk reduction, show normal or increased percentages of monocytes expressing Fcy and complement receptors. These findings provide support for the inference that inefficient chemotaxis and phagocytosis, reported previously in XLA patients, could be due to defects in cytoplasmatic transduction mechanisms. CVI patients, who have a normal Btk expression, showed reduced CD16 and CD32 membrane expression, although the percentage of positive monocytes for these receptors was not reduced.
Acknowledgments
We are grateful to all participating patients and also to Mrs Noemia Orii and Simone Corte for their technical assistance. This work was supported by ‘Fundação de Amparo à Pesquisa do Estado de São Paulo’ (FAPESP) and ‘Coordenação de Aperfeiçoamento de Pessoal de Nível Superior’ (CAPES), Brazil.
References
- 1.Tsukada S, Saffran DC, Rawlings DJ, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72:279–90. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 2.Vetrie D, Vorechovsky I, Sideras P, et al. The gene involved in X-linked agammaglobulinemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361:226–33. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 3.Futatani T, Miyawaki T, Tsukada S, et al. Deficient expression of Bruton's tyrosine kinase in monocytes from X-linked agammaglobulinemia as evaluated by a flow cytometric analysis and its clinical application to carrier detection. Blood. 1998;91:595–602. [PubMed] [Google Scholar]
- 4.Smith CI, Baskin B, Humire-Greiff P, et al. Expression of Bruton's agammaglobulinemia tyrosine kinase gene, Btk, is selectively down-regulated in T lymphocytes and plasma cells. J Immunol. 1994;152:557–65. [PubMed] [Google Scholar]
- 5.Genevier HC, Hinshelwood S, Gaspar HB, et al. Expression of Bruton's tyrosine kinase protein within the B cell lineage. Eur J Immunol. 1994;24:3100–5. doi: 10.1002/eji.1830241228. [DOI] [PubMed] [Google Scholar]
- 6.Quek LS, Bolen J, Watson SP. A role for Bruton's tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8:1137–40. doi: 10.1016/s0960-9822(98)70471-3. [DOI] [PubMed] [Google Scholar]
- 7.Futatani T, Watanabe C, Baba Y, Tsukada S, Ochs HD. Bruton's tyrosine kinase is present in normal platelets and its absence identifies patients with X-linked agammaglobulinaemia and carrier females. Br J Haematol. 2001;114:141–9. doi: 10.1046/j.1365-2141.2001.02905.x. [DOI] [PubMed] [Google Scholar]
- 8.Cunningham-Rundles C. Hematologic complications of primary immune deficiencies. Blood Rev. 2002;16:61–4. doi: 10.1054/blre.2001.0185. [DOI] [PubMed] [Google Scholar]
- 9.Kanegane H, Taneichi H, Nomura K, Futatani T, Miyawaki T. Severe neutropenia in Japanese patients with X-linked agammaglobulinemia. J Clin Immunol. 2005;25:490–5. doi: 10.1007/s10875-005-5370-x. [DOI] [PubMed] [Google Scholar]
- 10.Brunner C, Müller B, Wirth T. Bruton's tyrosine kinase is involved in innate and adaptive immunity. Histol Histopathol. 2005;20:945–55. doi: 10.14670/HH-20.945. [DOI] [PubMed] [Google Scholar]
- 11.Mangla A, Khare A, Vineeth V, et al. Pleiotropic consequences of Bruton tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood. 2004;104:1191–7. doi: 10.1182/blood-2004-01-0207. [DOI] [PubMed] [Google Scholar]
- 12.Hammarström L, Vorechovsky I, Webster D. Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID) Clin Exp Immunol. 2000;120:225–31. doi: 10.1046/j.1365-2249.2000.01131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapel H, Geba R, Rosen F IUIS PID (Primary Immunodeficiency) Classification Committee. Primary immunodeficiency diseases: an update. Clin Exp Immunol. 2003;132:9–15. doi: 10.1046/j.1365-2249.2003.02110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayry J, Hermine O, Webster DA, Levy Y, Kaveri SV. Common variable immunodeficiency: the immune system in chaos. Trends Mol Med. 2005;11:370–6. doi: 10.1016/j.molmed.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Amoras AL, Kanegane H, Miyawaki T, Vilela MM. Defective Fc-, CR1- and CR3-mediated monocyte phagocytosis and chemotaxis in common variable immunodeficiency and X-linked agammaglobulinemia patients. J Invest Allergol Clin Immunol. 2003;13:181–8. [PubMed] [Google Scholar]
- 16.Aderen A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 17.Heijnen IAFM, van de Winkel JGJ. Human IgG Fc receptors. Int Rev Immunol. 1997;16:29–55. doi: 10.3109/08830189709045702. [DOI] [PubMed] [Google Scholar]
- 18.Lindvall JM, Blomberg KE, Valiaho J, et al. Bruton's tyrosine kinase: cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling. Immunol Rev. 2005;203:200–15. doi: 10.1111/j.0105-2896.2005.00225.x. [DOI] [PubMed] [Google Scholar]
- 19.WHO Scientific Group. Primary immunodeficiency diseases. Clin Exp Immunol. 1999;118(Suppl. 1):1–28. doi: 10.1046/j.1365-2249.1999.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tacke F, Randolph GJ. Migratory fate and differentiation of blood monocyte subsets. Immunobiology. 2006;211:609–18. doi: 10.1016/j.imbio.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 21.Grage-Griebenow E, Flad HD, Ernst M. Heterogeneity of human peripheral blood monocyte subsets. J Leukoc Biol. 2001;69:11–20. [PubMed] [Google Scholar]
- 22.Gordon S, Taylor P. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 23.Gagliardi MC, Finocchi A, Orlandi P, et al. Bruton's tyrosine kinase defect in dendritic cells from X-linked agammaglobulinaemia patients does not influence their differentiation, maturation and antigen-presenting cell function. Clin Exp Immunol. 2003;133:115–22. doi: 10.1046/j.1365-2249.2003.t01-1-02178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hackam DJ, Rotstein OD, Schreiber A, Zhang WJ, Grinstein S. Rho is required for the initiation of calcium signaling and phagocytosis by Fc gamma receptors in macrophages. J Exp Med. 1997;186:955–66. doi: 10.1084/jem.186.6.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki T, Kono H, Hirose N, Okada M, Yamamoto K, Honda Z. Differential involvement of Src family kinase in Fc gamma receptor-mediated phagocytosis. J Immunol. 2000;165:473–82. doi: 10.4049/jimmunol.165.1.473. [DOI] [PubMed] [Google Scholar]
- 26.Nore BF, Vargas L, Mohamed AJ, et al. Redistribution of Bruton's tyrosine kinase by activation of phosphatidylinositol 3-kinase and Rho-family GTPases. Eur J Immunol. 2000;30:145–54. doi: 10.1002/1521-4141(200001)30:1<145::AID-IMMU145>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 27.Inabe K, Ishiai M, Scharenberg AM, Freshney N, Downward J, Kurosaki T. Vav3 modulates B cell receptor responses by regulating phosphoinositide 3-kinase activation. J Exp Med. 2002;195:189–200. doi: 10.1084/jem.20011571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fluckiger AC, Li ZM, Kato RM, et al. Btk/Tec kinases regulate sustained increases in intracellular Ca2+ following B-cell receptor activation. EMBO J. 1998;17:1973–85. doi: 10.1093/emboj/17.7.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verma J, Arora V, Marwaha V, Kumar A, Das N. Association of leukocyte CR1 gene transcription with the disease severity and renal involvement in systemic lupus erythematosus. Lupus. 2005;14:273–9. doi: 10.1191/0961203305lu2074oa. [DOI] [PubMed] [Google Scholar]
- 30.Thon V, Eggenbauer H, Wolf HM, et al. Antigen presentation by common variable immunodeficiency (CVID) B cells and monocytes is unimpaired. Clin Exp Immunol. 1997;108:1–8. doi: 10.1046/j.1365-2249.1997.d01-989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fischer MB, Hauber I, Wolf HM, Vogel E, Mannhalter JW, Eibl MM. Impaired TCR signal transduction, but normal antigen presentation, in a patient with common variable immunodeficiency. Br J Haematol. 1994;88:520–6. doi: 10.1111/j.1365-2141.1994.tb05068.x. [DOI] [PubMed] [Google Scholar]
- 32.Viallard JF, Camou F, Andre M, et al. Altered dendritic cell distribution in patients with common variable immunodeficiency. Arthritis Res Ther. 2005;7:1052–5. doi: 10.1186/ar1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cambronero R, Sewell WA, North ME, Webster AD, Farrant J. Up-regulation of IL-12 in monocytes: a fundamental defect in common variable immunodeficiency. J Immunol. 2000;164:488–94. doi: 10.4049/jimmunol.164.1.488. [DOI] [PubMed] [Google Scholar]
- 34.Braig DU, Schäffer AA, Glocker E, et al. Linkage of autosomal dominant common variable immunodeficiency to chromosome 5p and evidence for locus heterogeneity. Hum Genet. 2003;112:369–78. doi: 10.1007/s00439-002-0890-4. [DOI] [PubMed] [Google Scholar]
- 35.Gollob JA, Li J, Kawasaki H, et al. Molecular interaction between CD58 and CD2 counter-receptors mediates the ability of monocytes to augment T cell activation by IL-12. J Immunol. 1996;157:1886–93. [PubMed] [Google Scholar]