

Abstract
Overexpression of one or more membrane-bound complement regulatory proteins (mCRPs) protects tumour cells against complement-mediated clearance by the autologous humoral immune response and is also considered as a barrier for successful immunotherapy with monoclonal anti-tumour antibodies. Neutralization of mCRPs by blocking antibodies, enzymatic removal or cytokine-mediated down-regulation has been shown to sensitize tumour cells to complement attack. In our study we applied, for the first time, anti-sense phosphorothioate oligonucleotides (S-ODNs) to knock down the expression of the mCRPs CD55 and CD46 with the aim of exploiting complement more effectively for tumour cell damage. Potent anti-sense oligonucleotides against CD55 and CD46 were identified by screening various target sequences (n = 10) for each regulator. S-ODN anti-CD55(687) reduced CD55 protein expression up to 84% and CD46 protein expression was inhibited up to 76% by S-ODN anti-CD46(85). Reverse transcription–polymerase chain reaction (RT–PCR) analysis revealed a similar reduction of the CD55 and CD46 mRNA levels, which argues for an RNAse H-dependent anti-sense mechanism. T47D, A549 and PC3 cells, representing breast, lung and prostate carcinoma, were used for functional studies. Dependent on the particular cell line, anti-sense-based inhibition of mCRP expression enhanced complement-dependent cytolysis (CDC) up to 42% for CD55 and up to 40% for CD46, and the combined inhibition of both regulators yielded further additive effects in T47D cells. C3 opsonization of CD55/CD46-deficient tumour cells was also clearly enhanced upon mCRP suppression. Due to the clinical applicability of S-ODNs, the anti-sense approach described in this study may offer an additional alternative to improve the efficacy of antibody- and complement-based cancer immunotherapy.
Keywords: cancer, complement, therapy/immunotherapy
Introduction
Clinical and experimental data support a potential role of complement in the control of neoplastic cells. In cancer patients, endogenous complement activation has been demonstrated with subsequent deposition of complement components on tumour tissue [1, 2]. Certain monoclonal antibodies (mAbs), which are applied in cancer immunotherapy, were also shown to deposit complement on malignant cells in vitro and in vivo[3–5]. However, complement resistance of tumour cells is a main hindrance for the efficiency of complement-dependent tumouridical effects.
Normal cells employ several protective strategies to control an inappropriate complement attack and these mechanisms also confer complement resistance on neoplastic cells [6, 7]. Most attention was focused upon the expression of the membrane-bound complement regulatory proteins (mCRPs) CD55, CD46 and CD59, which are often detected at elevated levels on tumour cells [7, 8]. Increased mCRP expression on cancer cells compared to the corresponding normal tissue may be the consequence of a selective force, which is caused by multiple events of complement attack during neoplastic transformation [7].
The mCRPs CD55 (DAF) and CD46 (MCP) are regulators of the early complement pathway, controlling complement activation at the level of C3. CD46 has co-factor activity for factor I-mediated cleavage of C3b and C4b [9], whereas CD55 induces the decay of C3/C5 convertases [10]. CD55 and CD46 not only prevent complement-dependent cytolysis (CDC), but also limit both the amount of C3 opsonins deposited on the cell surface and generation of the soluble anaphylatoxins C3a and C5a. Opsonization of the target cell and inflammation induced by C3a/C5a support the recruitment of cell-mediated immune defence mechanisms.
CD59 (protectin) interferes with the terminal pathway of complement activation. Via binding to C9 it prevents the assembly of the membrane attack complex (MAC) and therefore specifically inhibits CDC [11].
Neutralization of mCRPs should sensitize tumour cells significantly to complement attack. In the past this has been demonstrated by multiple approaches: (i) use of blocking antibodies against CD55, CD46 and CD59 [3, 12, 13]; (ii) removal of CD55 and CD59 by phosphatidylinositol–phospholipase C (PI-PLC) [14]; and (iii) down-regulation of mCRP expression by applying cytokines [15].
So far, the efficacy of anti-sense oligonucleotides (ODN) to specifically knock down the expression of selected complement regulatory proteins on tumours has not been demonstrated. Anti-sense technology is well established and fairly widespread [16–18]. Complementary binding of short synthetic oligonucleotides to the mRNA of the selected target protein suppresses its translation either by steric hindrance of the ribosome movement or by activation of RNAse H, which degrades RNA/DNA hybrids. Phosphorothioate oligonucleotides (S-ODNs), which carry sulphur instead of oxygen in their phosphodiester backbone, are best studied and characterized by high nuclease resistance [16, 18].
To exploit more effectively the biological potency of complement activation in the eradication of tumour cells, this study aimed at reducing complement resistance of neoplastic cells by the use of anti-sense technology. The mCRPs CD55 and CD46 were selected as targets for the anti-sense approach to improve the efficiency of CDC as well as to support cell-mediated anti-tumour responses.
Materials and methods
Cell culture
Tumour cell lines of different origin were selected for the experiments: T47D, breast carcinoma cells, which were maintained in Dulbecco's modified Eagle's medium (DMEM)/Hams F12 (Biochrom AG, Berlin, Germany), A549, lung carcinoma cells, which were cultured in DMEM (Cambrex, Verviers, Belgium) and prostate carcinoma cells PC3, which were grown in RPMI-1640 (Cambrex). All media were supplemented with 10% heat-inactivated fetal calf serum (FCS; Invitrogen, Karlsruhe, Germany).
Antibodies, buffers and other reagents
Rabbits were used to prepare polyclonal anti-tumour antibodies specific for T47D and PC3 cells. The animals were immunized by three intravenous injections of 1 × 106 intact cells. Rabbit serum was ready for use after heat inactivation (56°C, 30 min) [3, 13].
The following antibodies were purchased: mouse anti-CD55, IgG1, clone Bric 110 (International Blood Group Reference Laboratory, IBGRL, Birmingham, UK); mouse anti-CD46, IgG1, clone J4-48 (Dianova, Hamburg, Germany); mouse anti-C3d, IgG1, clone A207 (Quidel, San Diego, CA, USA); rabbit anti-actin (Sigma, St Louis, MO, USA); mouse IgG1 isotype control, 15H6 (Southern Biotech, Birmingham, AL, USA); fluorescein isothiocyanate (FITC)-conjugated F(ab′)2 goat anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and F(ab′)2 goat anti-rabbit IgG (H+L) PO (Dianova).
Fluorescence-activated cell sorter (FACS) buffer [1% bovine serum albumin (BSA), 0·1% NaN3 in phosphate-buffered saline (PBS; PAA Laboratories, Cölbe, Germany)] was applied as indicated. Complement activation was induced in veronal buffered sodium (VBS++) containing 5 mM sodium barbital (pH 7·4), 0·15 mM CaCl2, 1 mM MgCl2, 150 mM NaCl and 0·1% BSA.
Normal human serum (NHS), freshly prepared from healthy blood donors and subsequently stored at −70°C, was used as a source of complement.
S-ODN sequences
CD55- and CD46-specific oligonucleotide sequences were selected using GenBank Accession number M31516 for CD55 and X59410 for CD46. All S-ODNs were specified by the number according to the position of the first nucleotide of the respective target sequence. A blast (Basic Local Alignment Search Tool) search was performed for each selected S-ODN sequence to avoid significant complementary binding to mRNAs of additional genes and therefore to minimize ‘off-target’ effects. The sequence of S-ODN anti-CD55(687), which was identified to be most effective in reduction of CD55 expression, was 5′-CTCCACTGGACAGAGCTGCC-3′. CD46 expression was most efficiently reduced by S-ODN anti-CD46(85) (5′-GCGCGGCGCGGAAGACGCTG-3′) (see Results). A random oligonucleotide was used as S-ODN control (5′-CGACAGGTCTGGAGTCATC-3′). Uptake studies were performed with a 5′ fluorescein-labelled control S-ODN (5′-GTTCATGATCCTGACTGAC-3′).
All oligomers were purchased from Hermann GbR (Freiburg, Germany) and high performance liquid chromatography (HPLC) was used to purify S-ODNs.
S-ODN transfection
Lipofectin- and oligofectamine-transfection of S-ODNs was performed according to the manufacturer's recommendations, with minor modifications.
Briefly, to transfect tumour cells of one well of a six-well plate, each 500 pmol S-ODNs and 10 μl lipofectin (1 mg/ml) (Invitrogen, Karlsruhe, Germany) were diluted in 750 μl OptiMEM (Invitrogen). After preincubation of the lipofectin solution for 45 min at 37°C, both solutions were mixed and incubated for additional 15 min at room temperature. The lipofectin/S-ODN mixture was subsequently overlaid onto the cells and incubated for 2 h. Finally, 1 ml growth medium (20% FCS) per well was added for further cultivation of the tumour cells.
Oligofectamine (Invitrogen) transfection was performed with 350 pmol S-ODNs and 5 μl oligofectamine (4 h).
For the combined transfection of two different oligonucleotides, each S-ODN was applied using the same amount as for the single transfection.
In general, cells were seeded 24 h before transfection and were grown to 40–60% confluency by the time of transfection. Tumour cells were transfected on three following days and functional assays were performed 72 h after the first transfection.
Flow cytometry
Tumour cells were removed from culture plates by treatment with trypsin/ethylenediamine tetraacetic acid (EDTA) solution (PAA Laboratories, Cölbe, Germany), washed and resuspended in FACS buffer; 105 cells/100 μl buffer were incubated with 1 μg of the first antibody for 30 min at 4°C. After washing the cells three times with FACS buffer, the appropriate second FITC-conjugated antibody (1 μg) was applied for 30 min at 4°C. Finally, cells were washed and fixed in 1% paraformaldehyde/PBS before cytofluorometric analysis (FACScalibur, Becton Dickinson, Heidelberg, Germany).
QIFIKIT (Dako, Glostrup, Denmark) was used to quantify the molecules analysed by FACS staining (CD55, CD46 and C3d). Beads coated with different amounts of mouse monoclonal IgG molecules allow the construction of a calibration curve.
Real-time reverse transcription–polymerase chain reaction (RT–PCR)
A total of 0·5 × 106 tumour cells were collected in 300 μl lysis buffer from the MagnaPure mRNA Isolation Kit I (RAS, Mannheim, Germany) and mRNA was isolated subsequently with the MagnaPure-LC device according to the mRNA-I standard protocol. cDNA was synthesized with avian myeloblastosis virus (AMV)–RT and oligo-dT as primer (First Strand cDNA synthesis kit; RAS).
Target sequences were amplified by PCR with the LightCycler FastStart DNA Sybr GreenI kit (RAS). CD55- and CD46-specific primer sets were developed and provided by SEARCH-LC GmbH, Heidelberg. Specificity of the amplification products was verified by melting curve analysis.
RNA input was normalized by average expression of the two housekeeping genes β-actin and cyclophilin B. Values were thus given as input-adjusted copy numbers per μl cDNA.
Complement-mediated cytotoxicity
A non-radioactive cytotoxicity assay (Europium assay) [19], which is based on time-resolved fluorometry, was used to measure CDC. As described previously [13], 106 tumour cells/ml culture medium were incubated for 20 min at 37°C with 10–20 μM of the fluorescence-enhancing ligand BATDA [bis(acetoxymethyl)2,2′:6′,2′-terpyridine-6,6′-dicarboxylic acid; Wallac, Turku, Finland]. Intracellular esterases generate the membrane-impermeable TDA from BATDA. Labelled tumour cells were washed and adjusted subsequently to 105 cells/ml VBS buffer. Fifty μl of these BATDA-labelled tumour cells were transferred to a round-bottomed 96-well plate (Greiner, Frickenhausen, Germany) and mixed subsequently with 50 μl of the respective complement-activating anti-tumour antibody (30 min, 37°C); 100 μl/well of 20% NHS/VBS buffer was added as a source of complement and incubated for an additional 60 min at 37°C. To determine spontaneous release NHS was replaced by buffer, and for evaluation of maximal release complete cell lysis was induced by digitonin (20 μg/ml final concentration).
Following incubation, plates were centrifuged (5 min, 500 g) and 20 μl supernatant from each well were transferred to a flat-bottomed 96-well plate (Greiner). Finally, 200 μl Europium solution (Wallac) was added to each well and fluorescence of the EuTDA chelates was measured in a time-resolved fluorometer (Victor, Wallac). The percentage of specific release was calculated as 100 × (experimental release − spontaneous release/maximal release − spontaneous release). All tests were performed in triplicate.
Alternatively, propidium iodide staining (PrI) (Sigma, St Louis, MO, USA) was applied to determine CDC [20]. Complement was activated on tumour cells, as described for C3d binding studies. Cells were stained with 5 μg PrI/ml FACS buffer (15 min, room temperature) and uptake of the dye was finally analysed by flow cytometry.
C3 binding studies
To initiate complement activation, cancer cells were first incubated for 30 min at 4°C with different concentrations of polyclonal rabbit anti-tumour antibodies (105 cells/100 μl VBS buffer); 50 μl NHS per well were added as a source of complement (30 min at 37°C, 5% CO2). Complement activation was terminated by washing cells once with ice-cold EDTA solution (20 mM EDTA/FACS buffer) and two additional times with FACS buffer. Flow cytofluorometrical analysis, as described above, was used to quantify C3d binding, chosen as surrogate marker for opsonizing C3b and iC3b molecules (1.ab: mouse anti-C3d; 2.ab: FITC goat anti-mouse).
Statistical analysis
Most results are presented as means ± standard deviation (s.d.). Differences between various data sets were tested for significance using Student's t-test and P-values of less than 0·05 were considered significant (*P < 0·05; **P < 0·01; ***P < 0·001).
Results
Design of different CD55- and CD46-specific anti-sense oligonucleotides
To identify potent CD55- and CD46-specific S-ODNs, we designed 10 different oligonucleotides for each regulator (Fig. 1).
Fig. 1.
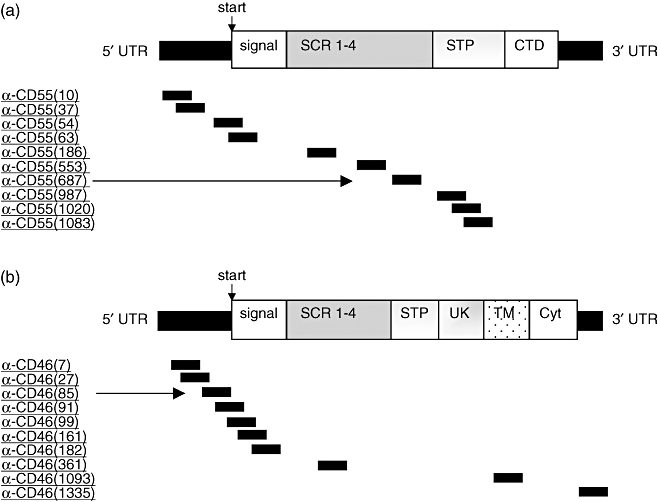
A schematic diagram of (a) CD55 and (b) CD46 mRNA with selected phosphorothioate oligonucleotides (S-ODNs) hybridizing to different target regions. Numbers of S-ODNs in parentheses correspond to the position of the first nucleotide of the respective target sequence. S-ODNs indicated by arrows reveal most potent anti-sense activity (see Fig. 1). 5′-UTR, 5′-untranslated region; signal, signal peptide; SCR, short consensus repeat; STP, serine–threonine–proline-rich region; CTD, C-terminal domain; UK, area of unknown significance; TM, transmembrane region; Cyt, cytoplasmic domain; 3′-UTR, 3′-untranslated region.
Several S-ODN target sequences were chosen around the start codon, because this area is known for its strong ‘anti-sense-activity’[21]. The presence of ‘activity-enhancing sequence motifs’[22] and predicted stabilities of anti-sense-oligonucleotide/target mRNA duplexes [23] were also considered for S-ODN design.
Target sequences were not selected from mRNA segments, which are known to differ between existing isoforms of CD55 and CD46. The variable part of CD55 is the C-terminal domain (CTD), whereas CD46 isoforms vary mainly in the serine–threonine–proline-rich region (STP) as well as in the cytoplasmic domain (Cyt) [24–26].
S-ODN uptake studies
FITC-labelled oligonucleotides were used to optimize transfection conditions for the selected tumour cell lines in order to minimize toxicity of the transfection procedure and to achieve high penetration of oligonucleotides into the tumour cells.
Cytotoxic effects were determined by trypan blue staining and intracellular uptake, and localization of FITC-labelled oligonucleotides was evaluated by confocal laser microscopy (data not shown).
Using lipofectin for T47D and PC3 cells and oligofectamine for A549 cells yielded high transfection efficiency at low toxicity of the transfection procedure. Microscopic analysis further revealed localization of FITC-labelled oligonucleotides predominantly in the nucleus, which is considered the major location of anti-sense action [27, 28].
S-ODN mediated inhibition of CD55 and CD46 expression
To evaluate the selected CD55- and CD46-specific S-ODNs for their functional efficiency, each oligonucleotide was transfected in T47D cells and 72 h later the level of the respective target protein was analysed by flow cytometry.
S-ODN anti-CD55(687), hybridizing to the short consensus repeats (SCR) of the CD55 mRNA, was identified as the most potent inhibitor of CD55 synthesis (−84%) (Figs 1a and 2a). Surprisingly, six of 10 selected CD46-specific S-ODNs revealed anti-sense activity. The S-ODN anti-CD46(85), complementary to the 5′ non-coding region of CD46 mRNA, suppressed CD46 expression most effectively (−76%) (Figs 1b and 2b).
Fig. 2.
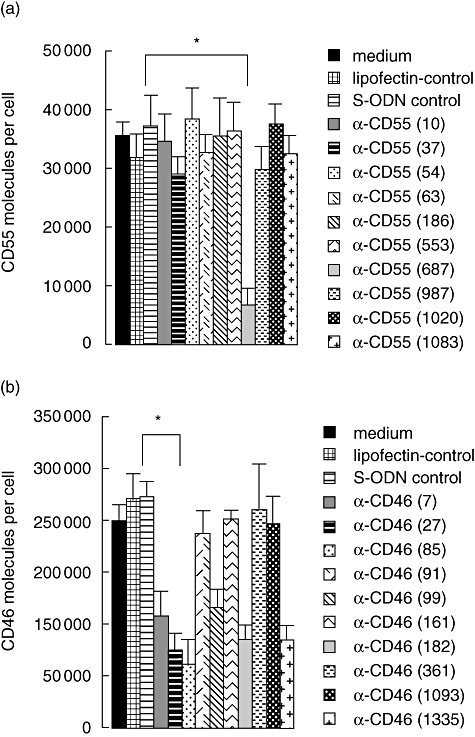
Screening for anti-sense activity. Selected oligonucleotides (see Fig. 1) were transfected in T47D cells on three subsequent days. Numbers of (a) CD55 and (b) CD46 molecules per cell were determined 72 h after the first oligonucleotide transfection by fluorescence activated cell sorter analysis and subsequent calculation based on QIFIKIT. Phosphorothioate oligonucleotide (S-ODN) anti-CD55(687) and S-ODN anti-CD46(85) suppress CD55/CD46 expression most effectively. Data are presented as means ± standard deviation of two independent experiments. Student's t-test: specific S-ODNs versus S-ODN control, respectively. *P < 0·05
As revealed by Western blot analysis, S-ODN transfection did not alter the expression of the housekeeping gene actin. This confirms that neither CD55 nor CD46 expression was reduced by general inhibition of protein synthesis (data not shown).
To demonstrate common applicability of the anti-sense-mediated knock-down of CD55 and CD46 expression, S-ODN anti-CD55(687) and S-ODN anti-CD46(85) were also transfected in A549 and PC3 cells. Approximately 60% inhibition of CD55 synthesis was achieved in both cell lines and CD46 expression could be reduced by approximately 70% (Fig. 3). We also analysed whether S-ODN anti-CD55(687) and S-ODN anti-CD46(85) inhibit target protein expression via reduction of the respective mRNA. CD55 and CD46 mRNA transcript numbers of T47D cells were quantified by real-time RT–PCR 72 h after the first S-ODN transfection. mRNA expression was reduced by 77% for CD55 and by 67% for CD46, which corresponds with our findings on protein level (Fig. 4).
Fig. 3.
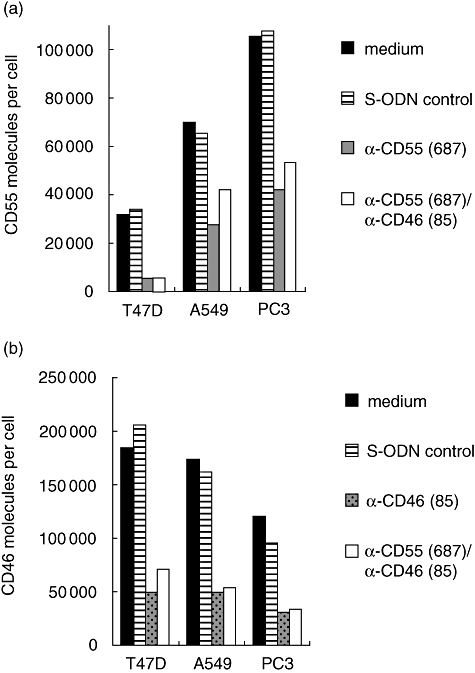
Phosphorothioate oligonucleotide (S-ODN) anti-CD55(687) and S-ODN anti-CD46(85) exert anti-sense activity in different tumour cell lines. Both oligonucleotides were transfected either individually or combined in T47D, A549 and PC3 cells. Cell lines were analysed for (a) CD55 and (b) CD46 expression by flow cytometry. Data are shown from one representative experiment (of three).
Fig. 4.
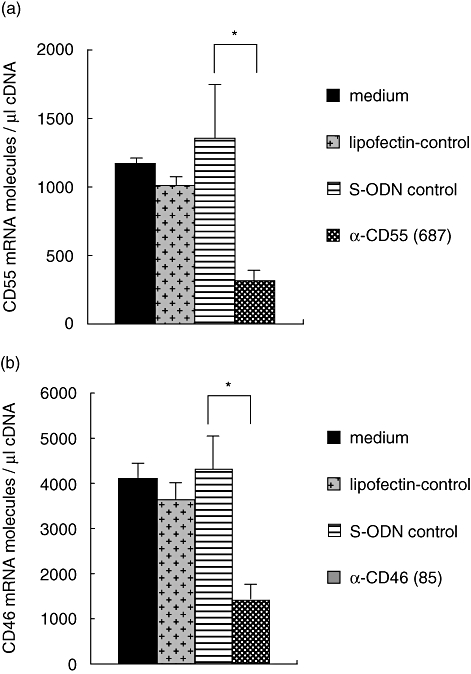
Phosphorothioate oligonucleotide (S-ODN) anti-CD55(687) and S-ODN anti-CD46(85) induce degradation of their corresponding mRNA. (a) CD55 and (b) CD46 mRNA expression of T47D cells was analysed by real-time reverse transcription–polymerase chain reaction 72 h after the first transfection of S-ODNs. Data represent the number of CD55/CD46 transcripts per μl of input cDNA, normalized to the housekeeping genes β-actin and cyclophilin B. Data are presented as means ± standard deviation of three independent experiments. Student's t-test: specific S-ODNs versus S-ODN control, respectively. *P < 0·05.
Dose–response evaluation of S-ODN anti-CD55(687) and S-ODN anti-CD46(85)
Specific anti-sense activity of S-ODN anti-CD55(687) and S-ODN anti-CD46(85) was dose-dependent in T47D cells (data not shown). Increasing quantities of S-ODNs applied during transfection correlated with the percentage of CD55/CD46 knock-down. The mean inhibitory concentration (IC50) for S-ODN anti-CD55(687) was approximately 80 nM and approximately 40 nM for S-ODN anti-CD46(85). For both oligonucleotides, concentrations higher than 200 nM did not increase further their inhibitory potential.
Functional analysis of S-ODN-mediated inhibition of CD55 and/or CD46 expression
CDC induced by tumour-directed antibodies was measured after pretreatment of tumour cells with S-ODN anti-CD55(687) and/or S-ODN anti-CD46(85).
Reduction of CD55 expression was found to sensitize significantly T47D and PC3 cells to complement attack. CDC of CD55-deficient T47D cells was augmented by 42% and CDC of CD55-deficient PC3 cells increased by 17%. Inhibition of CD46 synthesis was almost ineffective in A549 and PC3 cells, whereas CDC of T47D cells was enhanced by about 40% following knock-down of CD46 (Fig. 5).
Fig. 5.
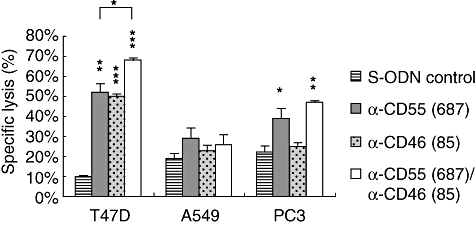
Complement-dependent cytolysis of CD55- and CD46-deficient T47D, A549 and PC3 cells. Tumour cells were transfected with phosphorothioate oligonucleotides (S-ODNs), as indicated in the diagram; 72 h later, complement-dependent cytolysis (CDC) of T47D, A549 and PC3 cells was measured. Data are presented as means ± standard deviation of triplicates of one representative experiment (of three). Student's t-test: specific S-ODNs versus S-ODN control, respectively. *P < 0·05; **P < 0·01; ***P < 0·001; T47D: S-ODN anti-CD55(687) versus S-ODN anti-CD55(687)/S-ODN anti-CD46(85) *P < 0·05.
A further significant augmentation of CDC could be demonstrated in T47D cells upon simultaneous transfection of S-ODN anti-CD55(687) and S-ODN anti-CD46(85). Compared to CDC following knock-down of CD55 (most potent single effect), a combined inhibition of CD55 and CD46 expression further enhanced significantly CDC of T47D cells by 16% (Fig. 5).
Because CD55 and CD46 are regulators of the early complement pathway, their knock-down was also expected to improve C3 opsonization of tumour cells. Therefore, C3 split product deposition (measured as C3d) was analysed on CD55- and/or CD46-deficient tumour cells following complement activation. All cell lines showed improved C3 opsonization upon mCRP suppression, even A549 cells, where down-regulation of CD55 and CD46 did not enhance CDC significantly (Fig. 6).
Fig. 6.
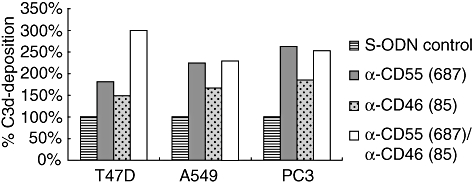
Deposition of opsonizing C3 split products on CD55- and/or CD46-deficient tumour cells (measured as common C3d moiety). Tumour cells were transfected with phosphorothioate oligonucleotides (S-ODNs), as indicated in the diagram. Following complement activation by polyclonal tumour-specific antibodies, deposited C3d molecules were quantified by flow cytometry. Data are shown from one representative experiment (of three).
Discussion
Complement activation is targeted to cancer tissue by either endogenous anti-tumour antibodies [29] or by immunotherapeutically applied mAbs directed against a tumour-specific antigen [30]. However, due to the expression of membrane-bound complement regulatory proteins (CD55, CD46 and CD59), complement deposition on neoplastic cells is limited and therefore not sufficient to induce potent tumour cell killing [3, 31]. Neutralization of mCRPs has been shown to sensitize tumour cells to complement attack [7]. Therefore, we applied an anti-sense strategy to inhibit specifically the expression of CD55 and CD46 aiming at better employment of complement for tumour cell destruction.
Although mCRP-blocking experiments in vitro suggest a prominent role of CD59 in the protection of tumour cells against CDC [6, 32], there are in vivo data which emphasize the role of early complement pathway regulation by CD55 and CD46. Caragine et al. [33] underline the importance of recruiting cell-mediated effector mechanisms for the eradication of tumour cells in vivo by generation of C3 opsonins and by the release of inflammatory activation fragments (C3a, C5a).
Approximately one in 10 tested anti-sense oligonucleotides is thought to down-regulate specifically target protein expression [34]. In good agreement with this prediction, one in 10 selected CD55-specific oligonucleotides was found to be active. The complement-regulatory protein CD46 was much more accessible for S-ODN-mediated knock-down. Six of 10 CD46-specific anti-sense oligonucleotides suppressed translation of their target protein at least by 40%.
S-ODN anti-CD55(687) and S-ODN anti-CD46(85), both identified as the most efficient, inhibit CD55 expression up to 84% and CD46 expression up to 76%, dependent upon the cell line.
Co-transfection of the various tumour cell lines with S-ODN anti-CD55(687) and S-ODN anti-CD46(85) often appeared to reduce the suppressive effect of the individual S-ODN (Fig. 3). For this reason different transfection conditions were tested, but results of single S-ODN transfection were continuously superior to combined use (data not shown). It could be speculated that there is a limited availability of cellular components, which are implicated in the anti-sense-induced target mRNA degradation.
RT–PCR analysis revealed a reduction of CD55 and CD46 mRNA levels following S-ODN transfection (comparable to the protein level). This indicates that both oligonucleotides decrease the expression of their target protein via activation of the nuclear enzyme RNAse H, which binds to RNA/DNA hybrids and induces degradation of the mRNA. Alternatively, anti-sense oligonucleotides, which do not change the mRNA level, prevent target protein synthesis by steric hindrance of the ribosome movement [18].
Functional studies were performed to evaluate complement resistance of tumour cells after anti-sense-mediated knock-down of CD55 and/or CD46 expression. The most striking effects were found for T47D cells, where CDC was increased about 40% after inhibition of either CD55 or CD46; 17% increased CDC was apparent in CD55-deficient PC3 cells. In former studies, blocking antibodies were used to neutralize mCRPs on these cell lines [3, 12]. In contrast to the anti-sense approach, blocking CD55 by antibodies had only a weak effect on CDC of T47D and PC3 cells and CD46-specific antibodies had no effect at all. It therefore appears that, in certain tumours, anti-sense-mediated neutralization of CD55 and CD46 may be more effective.
It could be assumed that during antibody blocking intracellular regulators may surface and support resistance. On the other hand, as discussed by Donin et al. [12], inefficiency of mCRP-blocking antibodies may be due simply to a low inhibitory capacity.
According to former experiments with blocking antibodies, the role of CD46 in particular may have been underestimated. Blok et al. [2] found that the expression of CD46 correlated with tumour stage and therefore they proposed a decisive role of CD46 for the protection of renal tumour cells against CDC in vivo. This was in contrast to preceding in vitro experiments, where C3d deposition on renal tumour cell lines could not be enhanced by the use of blocking antibodies against CD46 [35].
The significance of CD46 in the protection of tumour cells against CDC is also supported by ongoing experiments in our laboratory using a CD46-specific siRNA (manuscript in preparation).
To our surprise, the lung carcinoma cell line A549 was not sensitized significantly to CDC following knock-down of CD55 and CD46 by S-ODNs. Similar results were reported by Varsano et al. [36]. They analysed two lung carcinoma cell lines for mCRP expression and their resistance to CDC. In comparison with normal epithelial cells of the respiratory tract, lung carcinoma cells were extremely resistant to CDC and blocking antibodies against CD55 and CD46 were entirely ineffective. Existence of additional complement resistance mechanisms, such as the secretion of soluble complement inhibitors or soluble forms of mCRPs, respectively, into the microenvironment [7], expression of sialic acid [8] or complement cleaving proteases [7] may account for the finding.
The concomitant blocking of CD55 and CD46 expression further reduced significantly complement resistance of the breast carcinoma cell line T47D. Using an artificial GPI-anchored form of CD46 and naturally glycosyl phosphatidy-inositol (GPI)-anchored CD55 on rabbit erythrocytes, Brodbeck et al. [37] also demonstrated that both regulators act in a co-operative fashion on the cell surface to prevent deposition of activated complement proteins. Therefore, synergism of CD55 and CD46 should be considered as an additional aspect for evasion of tumour cells from complement attack. Reduced expression of CD55 and/or CD46 after S-ODN treatment clearly enhanced the binding of opsonizing C3 split products on the surface of all three tested tumour cell lines following antibody-induced complement activation. This was apparent even for the lung carcinoma cells A549, where the effect on CDC was marginal. The obvious discrepancy between C3 opsonization and CDC might be due to the special character of the lung carcinoma cells (see above). Additional complement regulatory mechanisms may interfere with the late complement lytic pathway.
C3 fragments bound to the cell surface are known to increase the efficiency of antibody-dependent cellular cytotoxicity (ADCC) by employment of the CR3 receptor on macrophages and polymorphonuclear leucocytes [31].
Because S-ODNs are qualified for in vivo use [16], the described anti-sense approach may be applied in the clinic as an adjuvant to support the efficiency of tumour-directed immunotherapies. In the past, similar approaches to use anti-sense drugs for the modulation of tumour-specific immune reactions have been reported. For example, anti-sense-based suppression of tumour-derived transforming growth factor (TGF)-β prevented local immunosuppression induced by this cytokine [38, 39] and down-regulation of the Ii protein was shown to enhance the immunogenicity of tumour cell major histocompatibility complex (MHC) class II presented antigens [40, 41].
Lack of cell-specific delivery methods for oligonucleotides is still a key obstacle to their clinical application. However, promising strategies are being developed currently to overcome the targeting problem. Mier et al. [42] prepared oligonucleotide/somatostatin conjugates to enhance the uptake of the anti-sense molecule via the somatostatin receptor, which is overexpressed on many tumour cells. A very popular method for oligonucleotide-targeting is the use of immunoliposomes, but recently encouraging data have also come from experiments with a fusion protein, composed of a tumour-specific antibody and the naturally occurring DNA-binding protein protamine [43, 44].
In summary, we were able to identify potent anti-sense oligonucleotides for efficient down-regulation of CD55 and CD46 expression on tumour cells. Knock-down of both surface-regulators clearly sensitized tumour cells to complement attack. This has been proved by analysis of CDC and also by investigation of C3d-deposition.
Acknowledgments
This study was supported by the DKFZ (Germany)-MOST (Israel) cooperation programme in Cancer Research.
References
- 1.Lucas SD, Karlsson-Parra A, Nilsson B, et al. Tumor-specific deposition of immunoglobulin G and complement in papillary thyroid carcinoma. Hum Pathol. 1996;27:1329–35. doi: 10.1016/s0046-8177(96)90346-9. [DOI] [PubMed] [Google Scholar]
- 2.Blok VT, Daha MR, Tijsma OM, et al. A possible role of CD46 for the protection in vivo of human renal tumor cells from complement-mediated damage. Lab Invest. 2000;80:335–44. doi: 10.1038/labinvest.3780038. [DOI] [PubMed] [Google Scholar]
- 3.Jurianz K, Maslak S, Garcia-Schuler H, et al. Neutralization of complement regulatory proteins augments lysis of breast carcinoma cells targeted with rhumAb anti-HER2. Immunopharmacology. 1999;42:209–18. doi: 10.1016/s0162-3109(99)00006-5. [DOI] [PubMed] [Google Scholar]
- 4.Idusogie EE, Wong PY, Presta LG, et al. Engineered antibodies with increased activity to recruit complement. J Immunol. 2001;166:2571–5. doi: 10.4049/jimmunol.166.4.2571. [DOI] [PubMed] [Google Scholar]
- 5.Di Gaetano N, Cittera E, Nota R, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171:1581–7. doi: 10.4049/jimmunol.171.3.1581. [DOI] [PubMed] [Google Scholar]
- 6.Jurianz K, Ziegler S, Garcia-Schuler H, et al. Complement resistance of tumor cells: basal and induced mechanisms. Mol Immunol. 1999;36:929–39. doi: 10.1016/s0161-5890(99)00115-7. [DOI] [PubMed] [Google Scholar]
- 7.Fishelson Z, Donin N, Zell S, et al. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol Immunol. 2003;40:109–23. doi: 10.1016/s0161-5890(03)00112-3. [DOI] [PubMed] [Google Scholar]
- 8.Gorter A, Meri S. Immune evasion of tumor cells using membrane-bound complement regulatory proteins. Immunol Today. 1999;20:576–82. doi: 10.1016/s0167-5699(99)01537-6. [DOI] [PubMed] [Google Scholar]
- 9.Kojima A, Iwata K, Seya T, et al. Membrane cofactor protein (CD46) protects cells predominantly from alternative complement pathway-mediated C3-fragment deposition and cytolysis. J Immunol. 1993;151:1519–27. [PubMed] [Google Scholar]
- 10.Nicholson-Weller A, Wang CE. Structure and function of decay accelerating factor CD55. J Lab Clin Med. 1994;123:485–91. [PubMed] [Google Scholar]
- 11.Meri S, Morgan BP, Davies A, et al. Human protectin (CD59), an 18 000–20 000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology. 1990;71:1–9. [PMC free article] [PubMed] [Google Scholar]
- 12.Donin N, Jurianz K, Ziporen L, et al. Complement resistance of human carcinoma cells depends on membrane regulatory proteins, protein kinases and sialic acid. Clin Exp Immunol. 2003;131:254–63. doi: 10.1046/j.1365-2249.2003.02066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jurianz K, Ziegler S, Donin N, et al. K562 erythroleukemic cells are equipped with multiple mechanisms of resistance to lysis by complement. Int J Cancer. 2001;93:848–54. doi: 10.1002/ijc.1406. [DOI] [PubMed] [Google Scholar]
- 14.Brasoveanu LI, Altomonte M, Fonsatti E, et al. Levels of cell membrane CD59 regulate the extent of complement-mediated lysis of human melanoma cells. Lab Invest. 1996;74:33–42. [PubMed] [Google Scholar]
- 15.Blok VT, Gelderman KA, Tijsma OH, et al. Cytokines affect resistance of human renal tumour cells to complement-mediated injury. Scand J Immunol. 2003;57:591–9. doi: 10.1046/j.1365-3083.2003.01265.x. [DOI] [PubMed] [Google Scholar]
- 16.Agrawal S. Antisense oligonucleotides: towards clinical trials. Trends Biotechnol. 1996;14:376–87. doi: 10.1016/0167-7799(96)10053-6. [DOI] [PubMed] [Google Scholar]
- 17.Chan JH, Lim S, Wong WS. Antisense oligonucleotides: from design to therapeutic application. Clin Exp Pharmacol Physiol. 2006;33:533–40. doi: 10.1111/j.1440-1681.2006.04403.x. [DOI] [PubMed] [Google Scholar]
- 18.Askari FK, McDonnell WM. Antisense-oligonucleotide therapy. N Engl J Med. 1996;334:316–18. doi: 10.1056/NEJM199602013340508. [DOI] [PubMed] [Google Scholar]
- 19.Blomberg K, Hautala R, Lovgren J, et al. Time-resolved fluorometric assay for natural killer activity using target cells labelled with a fluorescence enhancing ligand. J Immunol Methods. 1996;193:199–206. doi: 10.1016/0022-1759(96)00063-4. [DOI] [PubMed] [Google Scholar]
- 20.Marchbank KJ, van den Berg CW, Morgan BP. Mechanisms of complement resistance induced by non-lethal complement attack and by growth arrest. Immunology. 1997;90:647–53. doi: 10.1046/j.1365-2567.1997.00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaroszewski JW, Syi JL, Ghosh M, et al. Targeting of antisense DNA: comparison of activity of anti-rabbit beta-globin oligodeoxyribonucleoside phosphorothioates with computer predictions of mRNA folding. Antisense Res Dev. 1993;3:339–48. doi: 10.1089/ard.1993.3.339. [DOI] [PubMed] [Google Scholar]
- 22.Matveeva OV, Tsodikov AD, Giddings M, et al. Identification of sequence motifs in oligonucleotides whose presence is correlated with antisense activity. Nucleic Acids Res. 2000;28:2862–5. doi: 10.1093/nar/28.15.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stull RA, Taylor LA, Szoka FC., Jr Predicting antisense oligonucleotide inhibitory efficacy: a computational approach using histograms and thermodynamic indices. Nucleic Acids Res. 1992;20:3501–8. doi: 10.1093/nar/20.13.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Post TW, Liszewski MK, Adams EM, et al. Membrane cofactor protein of the complement system: alternative splicing of serine/threonine/proline-rich exons and cytoplasmic tails produces multiple isoforms that correlate with protein phenotype. J Exp Med. 1991;174:93–102. doi: 10.1084/jem.174.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caras IW, Davitz MA, Rhee L, et al. Cloning of decay-accelerating factor suggests novel use of splicing to generate two proteins. Nature. 1987;325:545–9. doi: 10.1038/325545a0. [DOI] [PubMed] [Google Scholar]
- 26.Purcell DF, Russell SM, Deacon NJ, et al. Alternatively spliced RNAs encode several isoforms of CD46 (MCP), a regulator of complement activation. Immunogenetics. 1991;33:335–44. doi: 10.1007/BF00216692. [DOI] [PubMed] [Google Scholar]
- 27.Crooke ST, Bennett CF. Progress in antisense oligonucleotide therapeutics. Annu Rev Pharmacol Toxicol. 1996;36:107–29. doi: 10.1146/annurev.pa.36.040196.000543. [DOI] [PubMed] [Google Scholar]
- 28.Wagner RW. Gene inhibition using antisense oligodeoxynucleotides. Nature. 1994;372:333–5. doi: 10.1038/372333a0. [DOI] [PubMed] [Google Scholar]
- 29.Niculescu F, Rus HG, Retegan M, et al. Persistent complement activation on tumor cells in breast cancer. Am J Pathol. 1992;140:1039–43. [PMC free article] [PubMed] [Google Scholar]
- 30.Houghton AN, Mintzer D, Cordon-Cardo C, et al. Mouse monoclonal IgG3 antibody detecting GD3 ganglioside: a phase I trial in patients with malignant melanoma. Proc Natl Acad Sci USA. 1985;82:1242–6. doi: 10.1073/pnas.82.4.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gelderman KA, Tomlinson S, Ross GD, et al. Complement function in mAb-mediated cancer immunotherapy. Trends Immunol. 2004;25:158–64. doi: 10.1016/j.it.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 32.Coral S, Fonsatti E, Sigalotti L, et al. Overexpression of protectin (CD59) down-modulates the susceptibility of human melanoma cells to homologous complement. J Cell Physiol. 2000;185:317–23. doi: 10.1002/1097-4652(200012)185:3<317::AID-JCP1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 33.Caragine TA, Okada N, Frey AB, et al. A tumor-expressed inhibitor of the early but not late complement lytic pathway enhances tumor growth in a rat model of human breast cancer. Cancer Res. 2002;62:1110–15. [PubMed] [Google Scholar]
- 34.Bennett CF, Cowsert LM. Antisense oligonucleotides as a tool for gene functionalization and target validation. Biochim Biophys Acta. 1999;1489:19–30. doi: 10.1016/s0167-4781(99)00144-x. [DOI] [PubMed] [Google Scholar]
- 35.Blok VT, Daha MR, Tijsma O, et al. A bispecific monoclonal antibody directed against both the membrane-bound complement regulator CD55 and the renal tumor-associated antigen G250 enhances C3 deposition and tumor cell lysis by complement. J Immunol. 1998;160:3437–43. [PubMed] [Google Scholar]
- 36.Varsano S, Rashkovsky L, Shapiro H, et al. Human lung cancer cell lines express cell membrane complement inhibitory proteins and are extremely resistant to complement-mediated lysis; a comparison with normal human respiratory epithelium in vitro, and an insight into mechanism(s) of resistance. Clin Exp Immunol. 1998;113:173–82. doi: 10.1046/j.1365-2249.1998.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brodbeck WG, Mold C, Atkinson JP, et al. Cooperation between decay-accelerating factor and membrane cofactor protein in protecting cells from autologous complement attack. J Immunol. 2000;165:3999–4006. doi: 10.4049/jimmunol.165.7.3999. [DOI] [PubMed] [Google Scholar]
- 38.Park JA, Wang E, Kurt RA, et al. Expression of an antisense transforming growth factor-beta1 transgene reduces tumorigenicity of EMT6 mammary tumor cells. Cancer Gene Ther. 1997;4:42–50. [PubMed] [Google Scholar]
- 39.Wu RS, Kobie JJ, Besselsen DG, et al. Comparative analysis of IFN-gamma B7.1 and antisense TGF-beta gene transfer on the tumorigenicity of a poorly immunogenic metastatic mammary carcinoma. Cancer Immunol Immunother. 2001;50:229–40. doi: 10.1007/s002620100197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu M, Lu X, Kallinteris NL, et al. Immunotherapy of cancer by antisense inhibition of Ii protein, an immunoregulator of antigen selection by MHC class II molecules. Curr Opin Mol Ther. 2004;6:160–5. [PubMed] [Google Scholar]
- 41.Lu X, Kallinteris NL, Li J, et al. Tumor immunotherapy by converting tumor cells to MHC class II-positive, Ii protein-negative phenotype. Cancer Immunol Immunother. 2003;52:592–8. doi: 10.1007/s00262-003-0404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mier W, Eritja R, Mohammed A, et al. Preparation and evaluation of tumor-targeting peptide-oligonucleotide conjugates. Bioconjug Chem. 2000;11:855–60. doi: 10.1021/bc000041k. [DOI] [PubMed] [Google Scholar]
- 43.Rodriguez M, Coma S, Noe V, et al. Development and effects of immunoliposomes carrying an antisense oligonucleotide against DHFR RNA and directed toward human breast cancer cells overexpressing HER2. Antisense Nucleic Acid Drug Dev. 2002;12:311–25. doi: 10.1089/108729002761381294. [DOI] [PubMed] [Google Scholar]
- 44.Song E, Zhu P, Lee SK, et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23:709–17. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]