

Abstract
Cytotoxic T lymphocyte antigen 4 (CTLA4) is a potent inhibitory co-stimulatory molecule believed to be involved in type 1 diabetes and other autoimmune diseases. An association has been reported of both mRNA expression and serum levels of the soluble splice variant of CTLA4 (sCTLA4) with type 1 diabetes. Furthermore, recombinant fusion proteins CTLA4Ig and LEA29Y have been proposed as therapies for type 1 diabetes. We studied the role of (s)CTLA4 in islet autoimmunity. Binding capacity of the proteins to antigen-presenting cells was determined by flow cytometry in competition and binding assays. Functionality of sCTLA4 as well as the therapeutic inhibitory fusion proteins CTLA4Ig and LEA29Y was measured in a dose–response lymphocyte stimulation test, using a panel of diabetes-associated T cell clones reactive to islet autoantigens. As controls, mixed lymphocyte reactions (MLR) were performed to assess functionality of these proteins in a primary alloreactive setting. All three CTLA4 molecules were able to bind to antigen-presenting cells and inhibit the expression of CD80/CD86. sCTLA4 was able to suppress proliferation of different committed autoreactive T cell clones in a dose-dependent manner, whereas CTLA4Ig and LEA29Y were not. Conversely, CTLA4Ig and LEA29Y, rather than sCTLA4, were able to suppress naive alloreactive proliferation in a MLR. Our results indicate a differential role for sCTLA4, CTLA4Ig and LEA29Y proteins in memory versus primary immune responses with implications for efficacy in intervention therapy.
Keywords: alloimmunity, autoimmunity, CTLA4Ig, soluble CTLA4, type 1 diabetes
Introduction
The cytotoxic T lymphocyte antigen 4 molecule (CTLA4 or CD152), expressed on activated T cells, is an inhibitory regulator of T cell activation [1–3]. It is part of the complex CTLA4/CD28/CD80/CD86 pathway, one of the most important co-stimulatory (‘second signal’) pathways essential for T cell activation, next to the major histocompatibility complex (MHC)–T cell receptor interaction. CD28 up-regulates activation when binding to B7 on an antigen-presenting cell (APC), whereas CTLA4 is described to inhibit T cell activation [3]. It is conceivable that competition with CD28 as well as intracellular signalling play a role in the immunomodulatory process of CTLA4 [4].
The therapeutic potential of modulating CTLA4-related immunity has been proved extensively in several animal transplant models [5–8]. Treatment with CTLA4Ig, a recombinant fusion protein of the extracellular domain of CTLA4 and a humanized Ig-tail could, at least in part, inhibit allograft rejection [2, 9, 10]. The CTLA4Ig construct (abatacept) and its more potent modification LEA29Y (belatacept), which dissociates more slowly from CD80/CD86 than CTLA4Ig due to two amino acid substitutions [11], are tested for use in clinical transplantation [11–14].
Next to transplantation, a role has been suggested for CTLA4 in autoimmune disease. Treatment with CTLA4Ig in experimental models was able to reverse autoimmunity [15–17]. In the non-obese diabetic (NOD) mouse model, CTLA4Ig administered early in life inhibited the development of autoimmune diabetes, but it did not when administered in a later stage of the disease [17].Various genetic polymorphisms of CTLA4 have been shown to be associated with type 1 diabetes and other autoimmune diseases [4, 18, 19]. A genetic correlate has been suggested between type 1 diabetes and decreased mRNA transcription of the non-membrane-bound, soluble form of CTLA4 (sCTLA4) [18]. In contrast, increased serum levels of sCTLA4 have been found in patients suffering from autoimmune thyroiditis and systemic lupus erythematosus [20, 21]. A recent report showed a minor, age-related increase in sCTLA4 levels in type 1 diabetes patients [22].
CTLA4Ig therapy has also shown potential in clinical autoimmune disease. Psoriasis patients were treated with CTLA4Ig with considerable success [23]. Furthermore, a pilot study showed a beneficial effect of CTLA4Ig treatment in rheumatoid arthritis patients [24]. However, CTLA4Ig has not been tested in human T cell-mediated disease type 1 diabetes.
To investigate the functionality of naturally occurring and recombinant soluble CTLA4 proteins in T cell-mediated autoimmunity, their function was tested in diabetes-associated activated T cell clones that are established in vitro tools to mimic the autoimmune process in type 1 diabetes [25–27]. The immune modulatory potential of these proteins in chronic autoreactivity was compared with their functionality in primary (allogeneic) responses, tested by mixed lymphocyte reaction (MLR).
Methods
Antigens and proteins
CTLA4Ig and LEA29Y were a gift from Dr R. Peach (Bristol Myers Squibb, Princeton, NJ, USA). Recombinant IA-2 was a gift from Dr J. Elliot (University of Alberta, Edmonton, Canada). Glutamic acid decarboxylase (GAD) was obtained from Diamyd (Stockholm, Sweden). Monoclonal fluorescently labelled anti-CD3, anti-CD80 and anti-CD86 were obtained from Pharmingen (Los Angeles, CA, USA) and anti-CTLA4 from Ancell (Bayport, MN, USA).
Production of soluble CTLA4
Soluble CTLA4 (sCTLA4) was expressed from the pIRES1neo vector (Clontech, Mountain View, CA, USA). Briefly, the sCTLA4 insert was subcloned bidirectionally from the pCR3 vector described in Oaks et al. [28] into the pIRES1neo vector via EcoRI sites. Inserts in the alpha orientation relative to the cytomegalovirus (CMV) promoter were identified through conventional restriction digest mapping, and individual plasmids were transfected with Lipofectamine 2000 (Invitrogen Corporation, Carlsbad, CA, USA) into Chinese hamster ovary (CHO) cells to verify expression. After selection of G418-resistant CHO lines, cell culture supernatants were tested for immunoreactive rat sCTLA4 using enzyme-linked immunosorbent assay (ELISA) as described previously [20]. A single plasmid that gave the highest expression of CTLA4 in the CHO system was chosen for all further expression studies. For production, cell culture supernatants were collected in protein-free CHO media (HyClone, Logan, UT, USA), purified by column chromatography and quantified by spectrophotometric analysis.
CTLA4 binding assay
Binding studies were performed to assess the ability of the CTLA4Ig construct to bind to APCs in vitro. Human leucocyte antigen (HLA) DR1 matched peripheral blood mononuclear cells (PBMCs) that were used as APCs in the rat insulinoma (RIN)-specific proliferation assay were blocked with 10% human serum in RPMI-1640 for 1 h to avoid non-specific Fc receptor binding. Subsequently, the cells were incubated for 1 h with CTLA4Ig, LEA29Y or sCTLA4 (0·9 nmol/l) at 37°C. The cells were then washed and stained for 20 min at 4°C with fluorescent-labelled monoclonal antibodies against CD3, CD80, CD86 and CTLA4 (Fig. 1a). Surface expression of markers was measured on a fluorescence activated cell sorter (FACS)Calibur (Becton Dickinson, Breda, the Netherlands) using Cellquest Pro software.
Fig. 1.
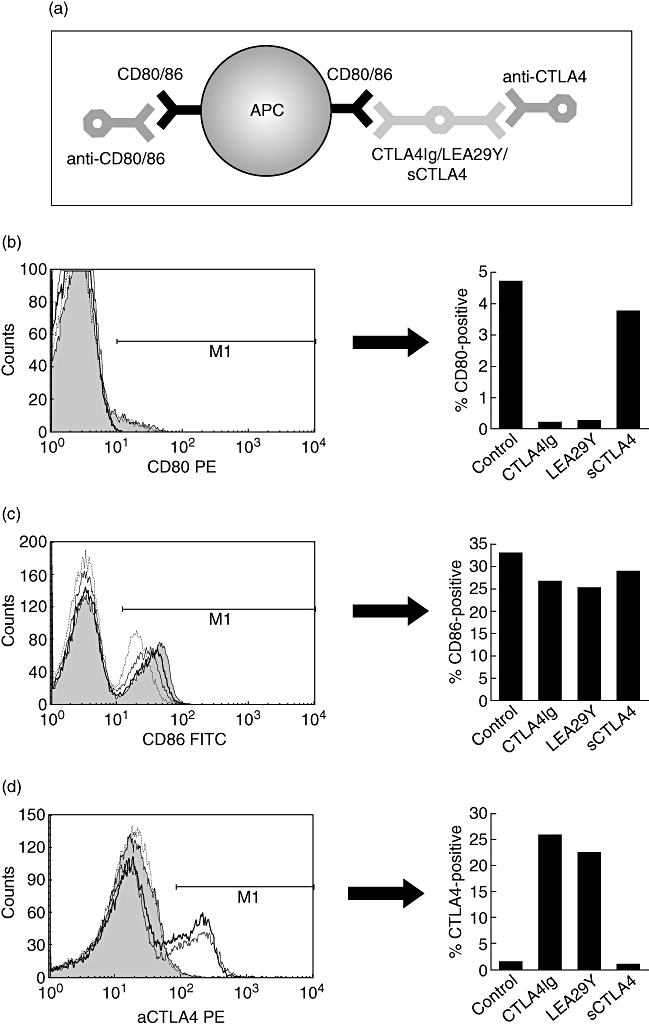
Schematic representation of cytotoxic T lymphocyte antigen 4 (CTLA4) binding and competition assay (a). Antigen-presenting cells (APCs) were incubated with CTLA4Ig, LEA29Y or sCTLA4. Subsequently, the cells were incubated with antibodies to CD3, CTLA4 (bound to the APC) and to its ligands CD80 and CD86 and expression was measured by fluorescence-activated cell sorter. Histograms show a representative example (of three independent experiments) of the expression of CD80 (b), CD86 (c) and CTLA4 (d) on APCs (CD3-negative peripheral blood mononuclear cells). Untreated APCs are represented by the grey area, APCs incubated with CTLA4Ig by the thick line, with LEA29Y by the thin line and with sCTLA4 by the dotted line. The bar graphs on the right represent the expression of the respective markers.
T cell proliferation assay
The isolation of T cell clones 1C6 (HLA-DR1-restricted, reactive to insulin secretory granules and RIN) and PM1#11 (HLA-DR4-restricted, reactive to GAD) has been described previously [25, 27]. An HLA-DR3-restricted, polyclonal autoreactive T cell line against the islet autoantigen IA-2 was derived from peripheral blood mononuclear cells of new-onset type 1 diabetes patients [29].
T cells (2 × 104/well) were incubated at 37°C with HLA-class II matched, irradiated APCs (5 × 104/well) derived from healthy blood donors and the appropriate antigens. Each condition was set up in triplicate in 96-well flat-bottomed plates in 200 μl Iscove's modified Dulbecco's medium (IMDM) with 2 mmol/l glutamine (Gibco, Paisley, Scotland, UK) and 10% pooled human serum. Different concentrations of CTLA4Ig, LEA29Y or sCTLA4 as well as monoclonal antibodies to CTLA4, CD80 and CD86 were added to the cells. Viability of cells was assessed by proliferation of T cells and APCs in response to 5% interleukin (IL)-2-containing T cell growth factor (Lymfocult, Biotest, Frankfurt, Germany) and to 1% phytohaemagglutinin, respectively. Possible toxicity of the proteins was assessed by APC-independent stimulation with T cell growth factor. After 3 days of incubation, [3H]-thymidine (0·5 μCi per well) was added and thymidine incorporation was measured 16 h later on a beta plate counter.
For calculation of the inhibitory capacity of added proteins, normalization of the data by taking the natural logarithm (ln) was required in view of the exponential nature of T cell proliferation. After normalization, inhibition was calculated by comparing the stimulation index of the test sample with the stimulation index of the positive control response (with the appropriate antigen but without addition of CTLA4 protein) using the following formula: % inhibition = 100 − (100 × [(ln test sample/ln neg.control) − 1]/[(ln pos.control/ln neg.control) − 1)].
Cytokine production assay
Cytokine release during incubation was assessed by Luminex analysis. For this procedure, culture supernatants were incubated for 45 min with antibody-coated beads specific for various human cytokines. Subsequently, plates were washed and incubated with biotinylated anti-cytokine detection antibody for 30 min. After washing, streptavidin phycoerythrin (PE) was added for 10 min to bind to the detection antibody. Specificity of the bead for a certain cytokine and the amount of binding of cytokines were analysed by a double-lasered Bio-plex reader (Bio-Rad, Veenendaal, the Netherlands).
MLR
In vitro alloreactivity was assessed as follows: 105 responder cells (PBMCs) were incubated at 37°C with 105 HLA-mismatched stimulator cells in triplicate in 96-well round-bottomed plates in 200 μl IMDM with 2 mmol/l glutamine (Gibco) and 10% pooled human serum. Different concentrations of CTLA4Ig, LEA29Y or sCTLA4 were added to the cells. After 5 days of incubation, [3H]-thymidine (0·5 μCi per well) was added for 16 h and thymidine incorporation was measured on a beta plate counter.
Results
CTLA4 binding assay
The capacity of the proteins to bind to APCs was assessed by a flow cytometry-based binding assay (Fig. 1). CTLA4Ig and LEA29Y bound to the cell surface of APCs, as indicated by the decreased availability of CD80 and CD86 to bind antibodies directed against these on CD3-negative PBMC, indicating that a proportion of CD80/86 was engaged in binding to CTLA4Ig/LEA29Y (Fig. 1b–c). Binding of CTLA4Ig/LEA29Y to the cell surface was confirmed by an increased staining intensity of anti-CTLA4 (Fig. 1d). sCTLA4 inhibited binding of anti-CD80/86 slightly, implying that sCTLA4 is less able to inhibit recognition of its ligands than CTLA4Ig/LEA29Y. We could not confirm binding of sCTLA4 to APCs with an anti-CTLA4 monoclonal antibody, due presumably to the fact that sCTLA4 is monomeric, or as a result of unavailability of the anti-CTLA4 epitope in sCTLA4, which is a splice variant.
Autoreactive T cell proliferation
High concentrations of soluble CTLA4 were able to inhibit proliferation of the IA-2-specific line and the GAD- and RIN-specific autoreactive T cell clones in a dose-dependent manner (Fig. 2a–c). In lower concentrations, similar to those reported in human serum (typically 20–70 ng/ml, 1–3 nmol/l), no inhibition of proliferation was noticed.
Fig. 2.
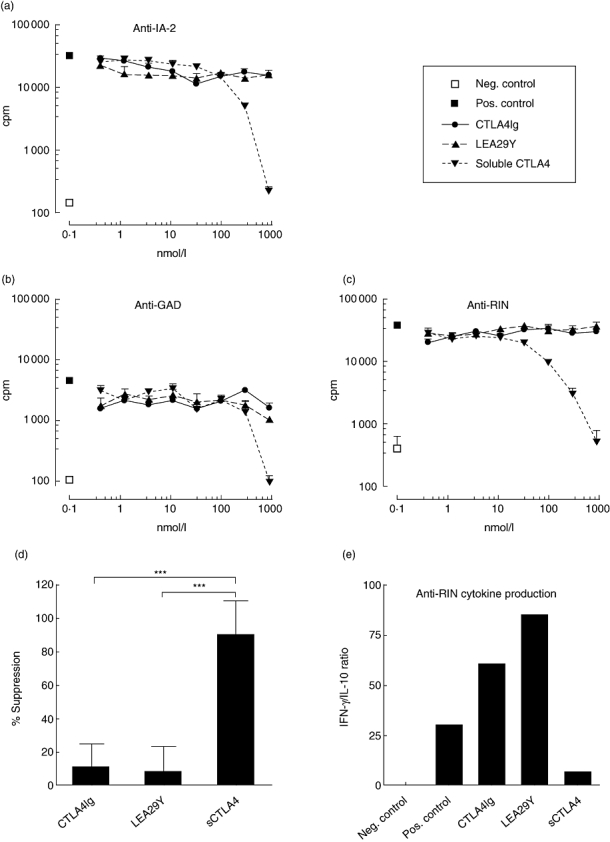
Proliferation of autoreactive T cell line specific for IA-2 (a) and T cell clones specific for glutamic acid decarboxylase (b) and rat insulinoma (RIN) (c). The x-axis indicates the added concentration of protein, the y-axis indicates proliferation in counts per minute (cpm). □ = without peptide (negative control), ▪ = with peptide (positive control), • = cytotoxic T lymphocyte antigen 4 (CTLA4)Ig, ▴ = LEA29Y, ▾ = soluble CTLA4. Shown are mean ± standard error. All data were reproduced once. (d) Average suppression of the highest concentration per protein in all six experiments performed. (e) Cytokine production (interferon-γ/interleukin-10 ratio) of the anti-RIN clone treated with the highest concentrations. ***P-value < 0·001.
Surprisingly, CTLA4Ig and LEA29Y were unable to inhibit proliferation of any of the three autoreactive T cell populations (Fig. 2a–c), even in very high concentrations (300–900 nmol/l comparable to trough levels found in the serum of transplanted animals [11]). These results were reproducible for each T cell clone/line.
Taking into account all six autoreactive proliferation experiments, the capacity of sCTLA4 to inhibit proliferation was significantly higher than that of CTLA4Ig and of LEA29Y (P < 0·001 for both by Kruskal–Wallis test corrected for multiple comparisons, Fig. 2d). Furthermore, the inhibition of proliferation was accompanied by a shift in cytokine production: the interferon (IFN)-γ/IL-10 ratio decreased in the cells treated with high-dose sCTLA4, whereas it increased with CTLA4Ig and LEA29Y (Fig. 2e).
Possible toxicity as a cause of the sCTLA4-mediated inhibition was assessed by proliferation of T cells with tissue cell growth factor only. Addition of sCTLA4 reduced this APC-independent proliferation by 17%, indicating only a small role for toxicity in the inhibition of T cell responses by sCTLA4. Furthermore, flow cytometry staining using 7-amino-actinomycin D (7-AAD) showed comparable viability of cells incubated with and without sCTLA4 (data not shown).
Alloreactive T cell proliferation
The functionality of the proteins was assessed further in a primary alloreactive setting to determine the potential to prevent, rather than intervene in, T cell activation. For this purpose, one- and two-way MLRs were performed with both single and double HLA class II mismatches. Figure 3 depicts one of these experiments, using HLA-DR3/3 responder and irradiated DR4/4 stimulator cells. Proliferation could be marginally reduced only marginally by sCTLA4 in high concentrations, whereas CTLA4Ig and LEA29Y were able to inhibit proliferation almost completely. These data were similar when using single mismatch combinations and/or two-way MLRs, confirming the inhibitory capacity of CTLA4Ig and LEA29Y in an in vitro alloreactive setting that mimics primary reactivity, a possible target for preventive immunotherapy.
Fig. 3.
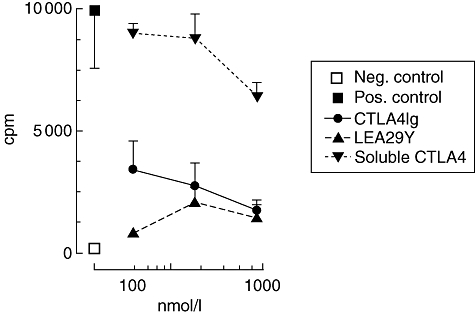
One-way mixed lymphocyte reaction (MLR), assessing the inhibitory capacity of soluble cytotoxic T lymphocyte antigen 4 (CTLA4) proteins in an unprimed alloreactive setting. Stimulator cells were human leucocyte antigen (HLA) DR/DR 4/4, responder cells were D-related 3/3. The x-axis indicates the added concentration of protein, the y-axis indicates proliferation in counts per minute (cpm). ▪ = MLR without addition (positive control), □ = medium without cells (negative control), • = CTLA4Ig, ▴ = LEA29Y, ▾ = soluble CTLA4. Shown are mean ± standard error. These results were reproduced twice.
Discussion
CTLA4 has been recognized increasingly as a key regulator molecule of T cell activation. The use of CTLA4 fusion proteins has shown considerable potential in inhibiting allograft rejection in animal transplant models, conceivably by prevention of primary immune reactivity. Experiments in autoimmune models have suggested that blockade of CTLA4 may also prevent, delay or intervene in autoimmunity [15–17], but the functional role of CTLA4 in autoimmunity is still subject to debate.
The relation between CTLA4 gene polymorphisms and autoimmunity has been proved repeatedly [4, 18, 19]. However, the suggested relation of the CT60 A–G polymorphism with lower mRNA expression of its soluble isoform seems counterintuitive, and could not be confirmed in other studies [30–32]. In our own genetic studies, we could not find a relation in 25 type 1 diabetes patients between the CT60 A–G polymorphism and sCTLA4/full-length CTLA4 mRNA ratio either (unpublished observation). Furthermore, in a recent report no correlation between the sCTLA4 polymorphism and sCTLA4 serum levels in type 1 diabetes patients was found [22]. In the same report, only minor differences in sCTLA4 serum levels between type 1 diabetes patients and controls were observed.
From our present study, however, it can be concluded that high concentrations of soluble CTLA4 are able to inhibit committed (autoreactive) T cell proliferation in vitro. Although these levels are much higher than those seen in human serum samples, sCTLA4 levels may increase locally at sites of inflammation. A first conclusion should therefore be that the relation between T cell inhibition by sCTLA4 and development of or intervention in type 1 diabetes can neither be confirmed nor rejected. Further experiments, in vitro as well as in vivo in experimental models, will be needed to elucidate the functional mechanisms explaining the association between autoimmunity and CTLA4 gene polymorphisms leading to production of splice variants of CTLA4.
In contrast to its inhibitory potential on activated T cells, sCTLA4 seems to have only a limited effect on primary responses. This may be a result of differences in primary and memory T cell reactivity, in which the interaction between membrane-bound CTLA4, CD28 and B7 is influenced. However, sCTLA4 was able to inhibit B7 binding only to a minor extent. This could be due to its monovalency or epitope binding affinity, but it should be noted that these findings obtained using monomeric, CHO-derived sCTLA4 differ from earlier results using baculovirus-derived sCTLA4 [28], where B7 binding was superior and alloreactivity could be inhibited.
The second important conclusion from our data is that the known inhibitory fusion proteins CTLA4Ig and LEA29Y were able to bind to APCs and to inhibit primary alloreactive responses, but failed to inhibit memory T cell autoreactivity. These data appear in conflict with data from several experimental in vivo models as well as a study in human rheumatoid arthritis (RA) patients [24]. This may be explained in several ways.
First, the secondary or co-stimulatory signal is of less importance in preprimed T cell responses. Antigen presentation by the MHC may suffice for activation of memory responses, rendering co-stimulation blockade less important. Secondly, the CTLA4/CD28/B7 pathway is not the only pathway inducing T cell activation. Other co-stimulatory pathways such as CD40–CD40L and OX40–OX40L may overcome the inhibitory capacity of CTLA4Ig. A third explanation would be that the function of CTLA4Ig is related to other types of cells than APCs alone. This would explain why in vitro proliferation of an autoreactive T cell population cannot be inhibited, whereas a mixed lymphocyte reaction from unprimed individuals can [8]. In this respect it is important to note that CTLA4Ig treatment can result in a reduction of the number of regulatory T cells [13]. Lastly, in a clonal T cell population CTLA4Ig might suppress the ‘normal’ CTLA4/B7 interaction which causes inhibition of the response, possibly through activity of indoleamine 2,3-dioxygenase (IDO) in the APC. This may be less the case in a heterogeneous mixed lymphocyte reaction. A recent report confirms that the inhibitory capacity of CTLA4Ig is dependent upon the activation state of the T cells: when added after 48 h of incubation, CTLA4Ig activates rather than inhibits T cells in MLR [33].
The differences observed between soluble CTLA4 and CTLA4Ig cannot be explained easily. sCTLA4 may be less able to inhibit alloreactive (primary) T cell responses as a result of the apparently lower binding rate to APC. It is more difficult to reconcile why sCTLA4 effectively inhibits activated autoreactive T cells. We favour the interpretation that sCTLA4 competes with CD28 in the primed T cell/APC interface, while this is not possible for CTLA4Ig due to steric hindrance. Alternatively, sCTLA4 leads to the activation of other or more potent signalling pathways (reviewed by Gough et al. [4]) in a primed autoreactive response.
In conclusion, our data show that sCTLA4 can inhibit autoimmune T cell proliferation in vitro. At present the physiological relevance for type 1 diabetes patients remains undetermined, and calls for further study both in vitro and in vivo. The role of type 1 diabetes-related CTLA4 polymorphisms in this process cannot be confirmed. Furthermore, our results imply that inhibition of the complex CTLA4/CD28/CD80/CD86 pathway by CTLA4Ig acts differently in primary (naive) versus recall (memory) T cell responses. Possible treatment success of CTLA4Ig in autoimmunity therefore depends largely upon the underlying pathogenesis of the disease and its effectiveness in the inhibition of autoreactive T cells. Intervening in a primed, autoreactive, T cell-driven process that has existed for years may prove more difficult than preventing the occurrence of a primary T cell reaction after transplantation. This notion is important when interpreting the results of clinical trials that are already under way.
Acknowledgments
We wish to thank Gaby Duinkerken, Kees Franken and Karen Kozinski for technical assistance. This study was supported by the Dutch Diabetes Research Foundation and the Juvenile Diabetes Research Foundation International.
References
- 1.Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–13. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 2.Sayegh MH, Akalin E, Hancock WW, et al. CD28-B7 blockade after alloantigenic challenge in vivo inhibits Th1 cytokines but spares Th2. J Exp Med. 1995;181:1869–74. doi: 10.1084/jem.181.5.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001;19:225–52. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- 4.Gough SC, Walker LS, Sansom DM. CTLA4 gene polymorphism and autoimmunity. Immunol Rev. 2005;204:102–15. doi: 10.1111/j.0105-2896.2005.00249.x. [DOI] [PubMed] [Google Scholar]
- 5.Chandraker A, Huurman V, Hallett K, et al. CTLA-4 is important in maintaining long-term survival of cardiac allografts. Transplantation. 2005;79:897–903. doi: 10.1097/01.tp.0000158275.56248.f8. [DOI] [PubMed] [Google Scholar]
- 6.Bharat A, Fields RC, Trulock EP, Patterson GA, Mohanakumar T. Induction of IL-10 suppressors in lung transplant patients by CD4+25+ regulatory T cells through CTLA-4 signaling. J Immunol. 2006;177:5631–8. doi: 10.4049/jimmunol.177.8.5631. [DOI] [PubMed] [Google Scholar]
- 7.Salvalaggio PR, Camirand G, Ariyan CE, et al. Antigen exposure during enhanced CTLA-4 expression promotes allograft tolerance in vivo. J Immunol. 2006;176:2292–8. doi: 10.4049/jimmunol.176.4.2292. [DOI] [PubMed] [Google Scholar]
- 8.Truong W, Hancock WW, Anderson CC, Merani S, Shapiro AM. Coinhibitory T-cell signaling in islet allograft rejection and tolerance. Cell Transplant. 2006;15:105–19. doi: 10.3727/000000006783982160. [DOI] [PubMed] [Google Scholar]
- 9.Lin H, Bolling SF, Linsley PS, et al. Long-term acceptance of major histocompatibility complex mismatched cardiac allografts induced by CTLA4Ig plus donor-specific transfusion. J Exp Med. 1993;178:1801–6. doi: 10.1084/jem.178.5.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levisetti MG, Padrid PA, Szot GL, et al. Immunosuppressive effects of human CTLA4Ig in a non-human primate model of allogeneic pancreatic islet transplantation. J Immunol. 1997;159:5187–91. [PubMed] [Google Scholar]
- 11.Larsen CP, Pearson TC, Adams AB, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant. 2005;5:443–53. doi: 10.1111/j.1600-6143.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- 12.Vincenti F, Larsen C, Durrbach A, et al. Costimulation blockade with belatacept in renal transplantation. N Engl J Med. 2005;353:770–81. doi: 10.1056/NEJMoa050085. [DOI] [PubMed] [Google Scholar]
- 13.Bluestone JA, St Clair EW, Turka LA. CTLA4Ig: bridging the basic immunology with clinical application. Immunity. 2006;24:233–8. doi: 10.1016/j.immuni.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Larsen CP, Knechtle SJ, Adams A, Pearson T, Kirk AD. A new look at blockade of T-cell costimulation: a therapeutic strategy for long-term maintenance immunosuppression. Am J Transplant. 2006;6:876–83. doi: 10.1111/j.1600-6143.2006.01259.x. [DOI] [PubMed] [Google Scholar]
- 15.Finck BK, Linsley PS, Wofsy D. Treatment of murine lupus with CTLA4Ig. Science. 1994;265:1225–7. doi: 10.1126/science.7520604. [DOI] [PubMed] [Google Scholar]
- 16.Khoury SJ, Akalin E, Chandraker A, et al. CD28-B7 costimulatory blockade by CTLA4Ig prevents actively induced experimental autoimmune encephalomyelitis and inhibits Th1 but spares Th2 cytokines in the central nervous system. J Immunol. 1995;155:4521–4. [PubMed] [Google Scholar]
- 17.Lenschow DJ, Ho SC, Sattar H, et al. Differential effects of anti-B7-1 and anti-B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J Exp Med. 1995;181:1145–55. doi: 10.1084/jem.181.3.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueda H, Howson JM, Esposito L, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–11. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 19.Kavvoura FK, Ioannidis JP. CTLA-4 gene polymorphisms and susceptibility to type 1 diabetes mellitus: a HuGE Review and meta-analysis. Am J Epidemiol. 2005;162:3–16. doi: 10.1093/aje/kwi165. [DOI] [PubMed] [Google Scholar]
- 20.Oaks MK, Hallett KM. Cutting edge: a soluble form of CTLA-4 in patients with autoimmune thyroid disease. J Immunol. 2000;164:5015–18. doi: 10.4049/jimmunol.164.10.5015. [DOI] [PubMed] [Google Scholar]
- 21.Liu MF, Wang CR, Chen PC, Fung LL. Increased expression of soluble cytotoxic T-lymphocyte-associated antigen-4 molecule in patients with systemic lupus erythematosus. Scand J Immunol. 2003;57:568–72. doi: 10.1046/j.1365-3083.2003.01232.x. [DOI] [PubMed] [Google Scholar]
- 22.Purohit S, Podolsky R, Collins C, et al. Lack of correlation between the levels of soluble cytotoxic T-lymphocyte associated antigen-4 (CTLA-4) and the CT-60 genotypes. J Autoimmune Dis. 2005;2:8. doi: 10.1186/1740-2557-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abrams JR, Lebwohl MG, Guzzo CA, et al. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest. 1999;103:1243–52. doi: 10.1172/JCI5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kremer JM, Westhovens R, Leon M, et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med. 2003;349:1907–15. doi: 10.1056/NEJMoa035075. [DOI] [PubMed] [Google Scholar]
- 25.Roep BO, Arden SD, de Vries RR, Hutton JC. T-cell clones from a type-1 diabetes patient respond to insulin secretory granule proteins. Nature. 1990;345:632–4. doi: 10.1038/345632a0. [DOI] [PubMed] [Google Scholar]
- 26.Roep BO, Heidenthal E, de Vries RR, Kolb H, Martin S. Soluble forms of intercellular adhesion molecule-1 in insulin-dependent diabetes mellitus. Lancet. 1994;343:1590–3. doi: 10.1016/s0140-6736(94)93055-4. [DOI] [PubMed] [Google Scholar]
- 27.Schloot NC, Batstra MC, Duinkerken G, et al. GAD65-Reactive T cells in a non-diabetic stiff-man syndrome patient. J Autoimmun. 1999;12:289–96. doi: 10.1006/jaut.1999.0280. [DOI] [PubMed] [Google Scholar]
- 28.Oaks MK, Hallett KM, Penwell RT, Stauber EC, Warren SJ, Tector AJ. A native soluble form of CTLA-4. Cell Immunol. 2000;201:144–53. doi: 10.1006/cimm.2000.1649. [DOI] [PubMed] [Google Scholar]
- 29.Hawkes CJ, Schloot NC, Marks J, et al. T-cell lines reactive to an immunodominant epitope of the tyrosine phosphatase-like autoantigen IA-2 in type 1 diabetes. Diabetes. 2000;49:356–66. doi: 10.2337/diabetes.49.3.356. [DOI] [PubMed] [Google Scholar]
- 30.Anjos SM, Shao W, Marchand L, Polychronakos C. Allelic effects on gene regulation at the autoimmunity-predisposing CTLA4 locus: a re-evaluation of the 3′+6230G>A polymorphism. Genes Immun. 2005;6:305–11. doi: 10.1038/sj.gene.6364211. [DOI] [PubMed] [Google Scholar]
- 31.Kaartinen T, Lappalainen J, Haimila K, Autero M, Partanen J. Genetic variation in ICOS regulates mRNA levels of ICOS and splicing isoforms of CTLA4. Mol Immunol. 2007;44:1644–51. doi: 10.1016/j.molimm.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 32.Mayans S, Lackovic K, Nyholm C, et al. CT60 genotype does not affect CTLA-4 isoform expression despite association to T1D and AITD in northern Sweden. BMC Med Genet. 2007;8:3. doi: 10.1186/1471-2350-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saverino D, Brizzolara R, Simone R, et al. Soluble CTLA-4 in autoimmune thyroid diseases: relationship with clinical status and possible role in the immune response dysregulation. Clin Immunol. 2007;123:190–8. doi: 10.1016/j.clim.2007.01.003. [DOI] [PubMed] [Google Scholar]