

Abstract
It remains unclear how misfolded membrane proteins are selected and destroyed during endoplasmic reticulum associated degradation (ERAD). For example, chaperones are thought to solubilize aggregation-prone motifs, and some data suggest that these proteins are degraded at the ER. To better define how membrane proteins are destroyed, the ERAD of Ste6p*, a twelve transmembrane protein, was reconstituted. We found that specific Hsp70/40s act before ubiquitination and facilitate Ste6p* association with an E3 ubiquitin ligase, suggesting an active role for chaperones. Furthermore, polyubiquitination was a prerequisite for retro-translocation, which required the Cdc48 complex and ATP. Surprisingly, the substrate was soluble, and extraction was independent of a ubiquitin chain extension enzyme (Ufd2p). However, Ufd2p increased the degree of ubiquitination and facilitated degradation. These data indicate that polytopic membrane proteins can be extracted from the ER, and define the point of action of chaperones and the requirement for Ufd2p during membrane protein quality control.
Keywords: molecular chaperone, Hsp70, ubiquitination, Cdc48/p97, Ufd2, proteasome
Newly synthesized soluble secretory and transmembrane proteins fold in the endoplasmic reticulum (ER), but those that fail to achieve their correct conformations may be retained in the ER and degraded by the cytoplasmic proteasome via a process referred to as ER-associated degradation (ERAD) (Kostova and Wolf, 2003; Meusser et al., 2005; Romisch, 2005; Nishikawa et al., 2005). Current evidence suggests that different ERAD pathways are employed depending on the location of the misfolded lesion, and that molecular chaperones and chaperone-like lectins help select ERAD substrates (Carvalho et al., 2006; Denic et al., 2006; Nishikawa et al., 2005; Vashist and Ng, 2004). Soluble ERAD substrates are retro-translocated, or dislocated from the ER to the cytoplasm after selection, and are easily accessed by the proteasome. In contrast, the identification and targeting of membrane proteins—particularly those that possess multiple transmembrane spans—are likely to involve more elaborate machineries given substrate complexity and difficulties during substrate solubilization and proteasome recruitment.
The vast majority of ERAD substrates are ubiquitinated by E2 conjugating enzymes and E3 ligases, which, respectively, catalyze the transfer of ubiquitin from a ubiquitin activating enzyme (E1) and are thought to aid in substrate recognition (Elsasser and Finley, 2005). In the yeast Saccharomyces cerevisiae two integral membrane E3 ligase complexes, the Hrd1p complex and the Doa10p complex, play important roles during ERAD (Carvalho et al., 2006; Vashist and Ng, 2004). Both Hrd1p and Doa10p possess a RING domain on the cytoplasmic face of the ER and function with the ER associated E2 enzymes, Ubc6p and Ubc7p (Gardner et al., 2001; Swanson et al., 2001). Although Hrd1p and Doa10p may directly recognize an unfolded domain or a specific amino acid motif (Bays et al., 2001; Gardner and Hampton, 1999; Ravid et al., 2006), molecular chaperones contribute to substrate ubiquitination. Chaperones can help prevent the aggregation of misfolded lumenal proteins and soluble domains in membrane proteins during ERAD (Nishikawa et al., 2005); therefore, the simplest view is that they augment substrate access to E3 ligases. However, recent reports suggested that an Hsp70-containing or putative lectin-containing complex in the ER helps recruit a misfolded substrate to the Hrd1p complex (Hebert et al., 2005; Denic et al., 2006; Gauss et al., 2006). Moreover, in vivo studies have demonstrated that cytoplasmic chaperones form a high-order network, which may specifically escort substrates to folding or degradation pathways (Albanese et al., 2006; McClellan et al., 2005; Meunier et al., 2002; Wang et al., 2006). Overall, the mechanism by which chaperones facilitate the degradation of a given class of substrates is not well understood.
After selection, ubiquitinated ERAD substrates are delivered to the 26S proteasome, which is composed of two 19S “caps” (PA700) that mediate substrate de-ubiquitination and ATP-dependent unfolding, and a single proteolytic 20S “core” (Voges et al., 1999). Proteasome delivery requires an ER-associated AAA ATPase, Cdc48p (in yeast) or p97 (in mammals) (Jentsch and Rumpf, 2007). During the degradation of membrane proteins, Cdc48p might actively pull a transmembrane domain and a subsequent lumenal domain from the ER (Carlson et al., 2006; Ye et al., 2003; Ravid et al., 2006). Alternatively, Cdc48p might “segregate” a polypeptide that has already been extracted or retro-translocated from the ER membrane. Cdc48p might also act after retro-translocation of the ERAD substrate by virtue of its interaction with a series of ubiquitin binding proteins, including the E4 polyubiquitin chain extension enzyme, Ufd2p (Richly et al., 2005; Rumpf and Jentsch, 2006).
In principle, the degradation of polytopic membrane substrates could start from either end of the polypeptide, as proposed for FtsH in bacteria (Akiyama and Ito, 2003), or degradation may commence from an internal site after an endoproteolytic “clip” (Liu et al., 2003; Piwko and Jentsch, 2006). In these models, it is assumed that degradation and retro-translocation are tightly coupled and occur at the ER membrane (Mayer et al., 1998; Plemper et al., 1998; Xiong et al., 1999). On the other hand, degradation intermediates of various soluble and membrane substrates have been detected in the cytosol (Carlson et al., 2006; Jarosch et al., 2002; McCracken and Brodsky, 1996; Meusser and Sommer, 2004; Wiertz et al., 1996). In addition, treatment of cells with proteasome inhibitors can result in the cytosolic deposition of aggregated Cystic Fibrosis Transmembrane conductance Regulator (CFTR) and other polytopic membrane substrates. The resulting aggresomes concentrate at a peri-centriolar locus (Johnston et al., 1998; Wigley et al., 1999), suggesting that ubiquitinated membrane proteins can be degraded in the cytoplasm after they are completely released from the ER membrane.
To define the ERAD of misfolded polytopic membrane proteins, we reconstituted each step in this reaction using components (either microsomes or cytosol) prepared from the yeast S. cerevisiae. We demonstrate that ER-membrane-associated Hsp70 and Hsp40s assist the recognition of a substrate by the E3 ligase, Doa10p, and must act “in cis”, and that Cdc48p binds to the ubiquitinated substrate and releases it from the ER membrane. Further, we show that Ufd2p elongates the polyubiquitin chain before the substrate is processed by the proteasome. Substrate ubiquitination could also be reconstituted with two other integral membrane proteins (CFTR and Sec61-2p). Thus, our system is robust and in principle may be employed to elucidate the ERAD pathway of any membrane protein that can be expressed in yeast.
RESULTS
Ste6p* ubiquitination in vitro requires the same E2s, E3s and chaperones as those that facilitate ERAD in vivo
Ste6p*, a 12 transmembrane ERAD substrate, is retained in the ER and is degraded by the proteasome in yeast. In addition, degradation is slowed when specific E2 conjugating enzymes (Ubc6p and Ubc7p), E3 ligases (Doa10p and Hrd1p), an Hsp70 (Ssa1p), and functionally redundant Hsp40s (Ydj1p and Hlj1p) are disabled (Huyer et al., 2004; Loayza and Michaelis, 1998; Vashist and Ng, 2004). Because the ERAD pathway for this substrate is relatively well-defined, and because of its structural similarity to CFTR, Ste6p* was selected for our first attempts to reconstitute the ERAD of a constitutively degraded membrane protein. ER-derived microsomes were prepared from cells expressing HA-tagged Ste6p*, and the membranes were incubated with yeast cytosol (which supplies the E1 ubiquitin activating enzyme and other factors potentially needed for ERAD), an ATP regenerating system and 125I-labeled ubiquitin (125I-Ub). We chose 125I-Ub because the subsequent isolation of the ubiquitinated substrate would provide quantitative data. After Ste6p* was immunoprecipitated with anti-HA antibody and resolved by SDS-PAGE, ubiquitinated Ste6p* was observed as a “smear” with a minimum molecular mass of ~140 kDa, which corresponds approximately to the native size of Ste6p* (Figure 1A, upper panel, lanes 2 and 3). Substrate ubiquitination depended upon cytosol, physiological temperature, and ATP (lanes 4, 5, and 7). Methylated ubiquitin (Me-Ub), an inhibitor for polyubiquitin chain formation (Hershko and Heller, 1985), reduced the extent of ubiquitination (lane 8). The addition of MG132, a proteasome inhibitor, did not alter the amount of either the polyubiquitinated protein or the unmodified protein (see below). Consistent with in vivo data, microsomes lacking Ubc6p and Ubc7p failed to support Ste6p* ubiquitination (Figure 1B, lanes 2 and 8). Other in vivo data indicated that depletion of Doa10p does not completely inhibit Ste6p* turn-over, possibly because Hrd1p partially compensates for Doa10p’s absence (Huyer et al., 2004; Figure S1). Indeed, microsomes lacking Doa10p or Hrd1p supported Ste6p* ubiquitination, whereas the simultaneous deletion of the genes encoding these enzymes inhibited ubiquitination (Figure 1B, lanes 3–6 and 9–12).
Figure 1. Ste6p* is ubiquitinated in vitro.
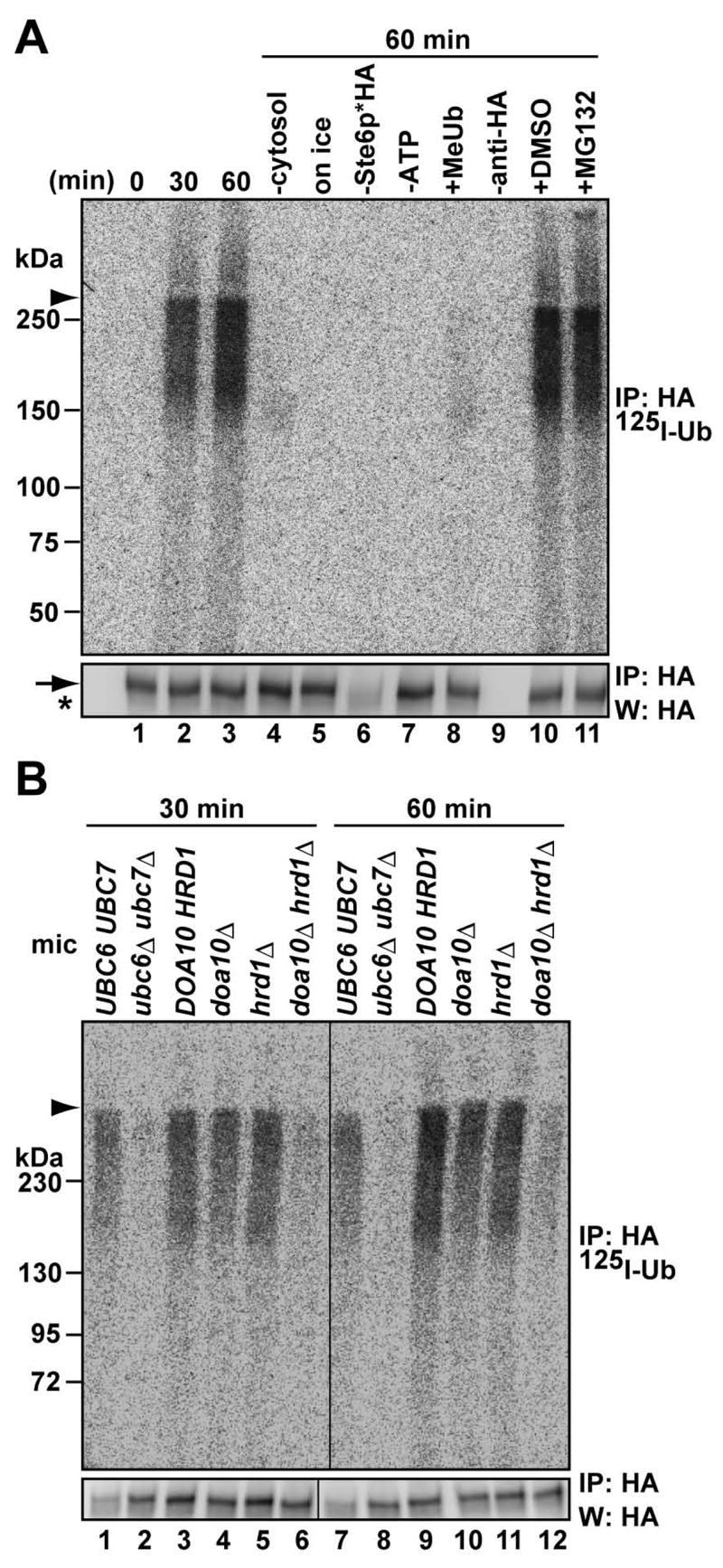
(A) Microsomes containing Ste6p* were incubated with 2 mg/ml cytosol, an ATP regenerating system and 125I-labeled ubiquitin at 23°C or on ice for the indicated times before the reaction was quenched and Ste6p* was immunoprecipitated with anti-HA antibody. Immunoprecipitates were resolved by SDS-PAGE and the products were visualized by phosphor imager (top) or western blot analysis (bottom). Where indicated, reactions were prepared without cytosol, or with 0.02 u/μl apyrase (“-ATP”), 0.5 μg/μl methylated ubiquitin (“Me-Ub”), or 100 μM of MG132; “-anti-HA” denotes that the precipitation was performed in the absence of antibody, and “-Ste6p*HA” denotes that microsomes were prepared from cells lacking Ste6p*HA.
(B) In vitro ubiquitination requires E2 and E3 enzymes. Microsomes were prepared from the indicated cells expressing Ste6p*HA, and the in vitro reaction was performed in the presence of WT cytosol (4 mg/ml) at 23°C for the indicated times. In A and B, the arrowhead indicates the 4% stacking gel-6% running gel boundary, the arrow indicates immuno-detected Ste6p*, and the asterisk indicates IgG: mic; microsomes.
The Hsp70 chaperone, Ssa1p, and the Hsp40 cochaperones, Ydj1p and Hlj1p, are required for Ste6p* ERAD in vivo, and as observed for other substrates these Hsp70 and Hsp40 homologues facilitate substrate ubiquitination (Han et al., 2007; reviewed in Nishikawa et al., 2005). However, their mechanism of action has not been investigated. We therefore prepared cytosol and Ste6p*-containing microsomes from wild-type SSA1 and temperature-sensitive ssa1-45 mutant cells. Before harvesting, cells were shifted to a nonpermissive temperature of 37°C for 45 min, which inactivates Ssa1-45p (Becker et al., 1996; Brodsky et al., 1999). Strikingly, Ste6p* was ubiquitinated in the SSA1 microsomes, whereas ssa1-45 mutant microsomes failed to support ubiquitination (Figure 2A, lanes 2 and 4). A similar result was obtained when microsomes were prepared from a strain bearing a temperature sensitive allele of YDJ1 (ydj1-151) and a deletion of HLJ1 (lanes 6 and 8). It is notable that these components must be supplied in microsomes and that cytosolic supplementation is ineffective (see Discussion). To generalize these results, we measured the in vitro ubiquitination of CFTR, whose degradation in yeast similarly depends on Ubc6p/Ubc7p, Doa10p/Hrd1p and Ssa1p (Gnann et al., 2004; Zhang et al., 2001). As anticipated, CFTR was ubiquitinated in vitro in an ATP, cytosol, Ubc6p/Ubc7p, and Doa10p/Hrd1p dependent manner, and the ssa1-45 mutant microsomes failed to support ubiquitination (Figure S2). Together, the results indicate that these chaperones facilitate substrate ubiquitination.
Figure 2. Ste6p* ubiquitination requires the Hsp70 and Hsp40 chaperones.
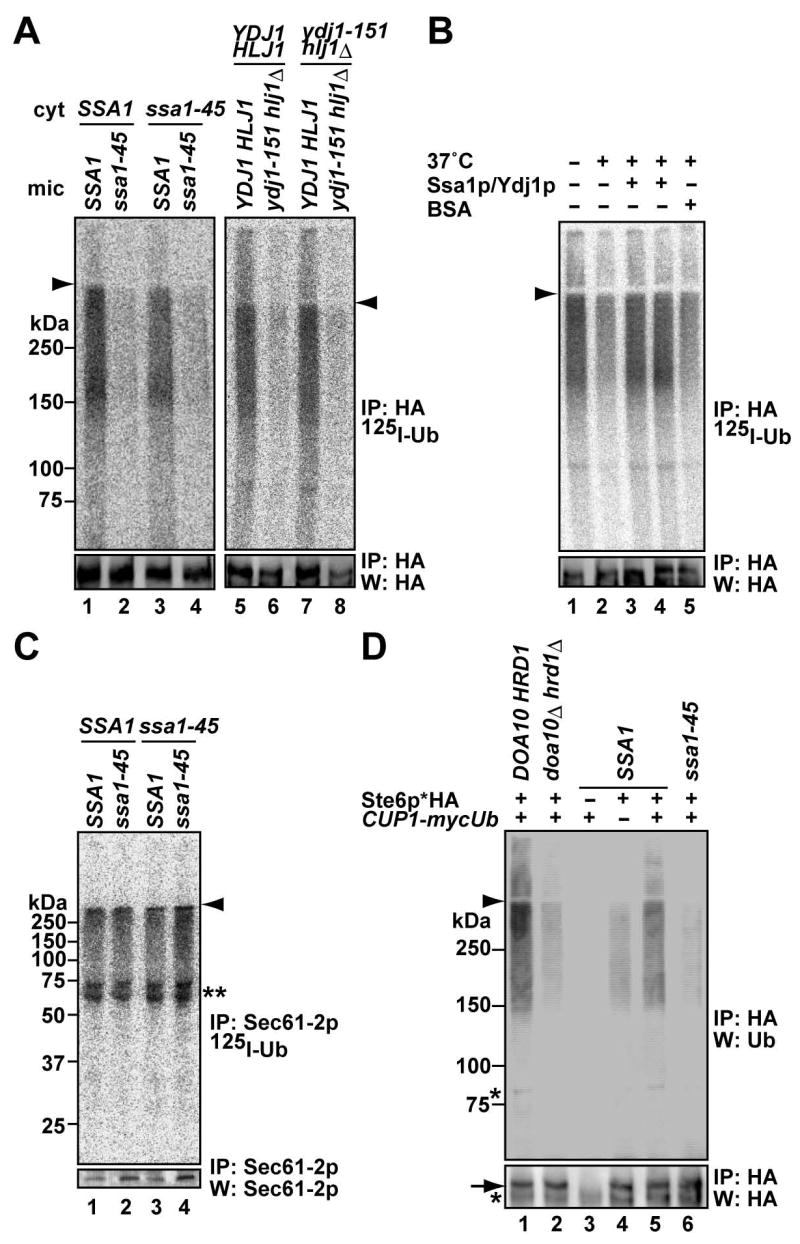
(A) Microsomes were prepared by glass bead disruption from the indicated cells expressing Ste6p*HA grown at 23°C and shifted to 37°C for 45 min. Cytosol (“cyt”) was prepared from the same strains at 23°C and shifted to 37°C for 45 min. Reactions were performed with 1 mg/ml cytosol at 23°C for 20 min (left panel) or 30°C for 40 min (right panel). Although Ste6p* is expressed in ydj1-151/hlj1Δ cells at lower levels than in the isogenic WT strain (lane 6 and 8), there is a 30–70% reduction in ubiquitination efficiency in replicated assays.
(B) Exogenously added Ssa1p and Ydj1p improve ubiquitination efficiency. Microsomes from WT cells were preincubated with 1.75 μM (lane 3) or 2.5 μM (lane 4) Ssa1p/Ydj1p and an ATP regenerating system at 23°C for 5 min and shifted to 37°C for 15 min before 125I-labeled ubiquitin and WT cytosol (1 mg/ml) were added and the reaction was incubated at 23°C for 40 min. The reaction was also performed either without chaperones at (lane 1, 2), or in the presence of 2.5 μM BSA (lane 5).
(C) Sec61-2p ubiquitination is Ssa1p-independent. Microsomes were prepared by glass bead disruption from sec61-2/SSA1 and sec61-2/ssa1-45 cells grown at 23°C and shifted to 37°C for 45 min, and cytosol was prepared from SSA1 and ssa1-45 cells as in (A). The reaction was performed with 1 mg/ml cytosol at 30°C for 50 min. The double asterisk indicates 125I-labelled contaminants in bovine ubiquitin that are immunoprecipitated with anti-Sec61p antiserum (see Figure S3).
(D) In vivo ubiquitination of Ste6p* is Ssa1p-dependent. Microsomes were prepared by glass bead disruption from cells expressing Ste6p*HA and myc-tagged ubiquitin under the control of the CUP1 promoter. DOA10/HRD1 and doa10Δ/hrd1Δ cells were grown at 30°C, and SSA1 and ssa1-45 cells were grown at 23°C and shifted to 37°C for 45 min. Ubiquitin expression was induced by 100 μM copper sulfate for 3 hr. Where indicated, cells contained a vector control (“-”). Ste6p*HA was immunoprecipitated and products were analyzed by western blot analysis with anti-ubiquitin (top panel) or anti-HA antibodies (bottom panel). A full size blot for the bottom panel is shown in Figure S4A. Further support for these data is shown in Figure S4B.
Because Ssa1p and Ydj1p/Hlj1p play multiple roles during protein translocation, folding and degradation (Nishikawa et al., 2005), we wondered if the impaired ubiquitination of Ste6p* resulted from a nonspecific secondary effect. However, we observed that the inclusion of purified Ssa1p/Ydj1p improved ubiquitination efficiency when added during a pre-incubation prior to cytosol addition (Figure 2B). Second, we assayed the in vitro ubiquitination of Sec61-2p, an ERAD substrate whose degradation is independent of Ssa1p (Nishikawa et al., 2001; Figure S3). Sec61-2p was ubiquitinated in vitro in an ATP and cytosol dependent manner (Figure S3). Moreover, Sec61-2p ubiquitination was robust in ssa1-45 mutant microsomes (Figure 2C).
We next asked whether Ste6p* ubiquitination is Ssa1p-dependent in vivo. Consistent with the results obtained from our in vitro assay, Ste6p* was poorly ubiquitinated in doa10Δ/hrd1Δ and ssa1-45 cells (Figure 2D). Given the fact that microsomes contain not only unmodified Ste6p* but also ubiquitinated Ste6p*, 125I-labeled Ub may be attached directly onto Ste6p* and/or onto pre-existing, ubiquitin chains on Ste6p*. Regardless, our results demonstrate that cytoplasmic Hsp70/Hsp40 chaperones directly facilitate Ste6p* ubiquitination in vivo and in vitro and validate the ability of the in vitro assay to recapitulate cellular phenomena.
Cytoplasmic Hsp70 (Ssa1p) facilitates Step* binding to Doa10p
Based on these data, we hypothesized that the chaperones facilitate the incorporation of Ste6p* into the Doa10p complex and performed a chemical cross-linking experiment. Microsomes were prepared from Ste6p*HA and Doa10p13myc-expressing SSA1 and ssa1-45 cells grown at 23°C and shifted to 37°C (Figure 3A). The microsomes were then treated with a membrane-permeable cross-linker (DSP) and Ste6p* was precipitated with anti-HA antibody. Although Doa10p precipitated with Ste6p*, approximately half as much of the E3 ligase precipitated with Ste6p* when Ssa1-45p function was ablated (Figure 3B). In parallel, we performed the in vitro ubiquitination assay and observed a corresponding decrease in Ste6p* ubiquitination (Figure 3C). We also noted that the Doa10p-Ste6p* co-immunoprecipitation efficiency and Ste6p* ubiquitination efficiency rose in SSA1 microsomes from cells shifted to 37°C. This may result from the induction of Ssa1p and/or the ubiquitination machinery. In addition, we employed another cross-linker, DTSSP, a water-soluble analog of DSP. As shown in one of three representative experiments, a ~2-fold reduction in Ste6p*-Doa10p interaction was observed when ssa1-45 microsomes were employed (Figure 3D, lane 6). Furthermore, we noted a decrease in Ste6p*-Doa10p association when microsomes from the ydj1-151/hlj1Δ strain were used (data not shown). These results suggest that Hsp70-Hsp40 facilitate Ste6p* ubiquitination by assisting in E3 ligase recognition.
Figure 3. Hsp70 facilitates the interaction between Ste6p* and Doa10p.

(A) Schematic of culturing conditions for microsome preparation
(B) Chemical cross-linking was performed using microsomes containing Ste6p*HA and Doa10p13myc. Microsomes were prepared as in (A) by glass bead disruption from SSA1/DOA10-13myc and ssa1-45/DOA10-13myc cells expressing Ste6p*HA. The Doa10p13myc fusion protein supports the degradation of a model substrate, Deg1-Ura3p (Kreft et al., 2006) and Ste6p* (Figure S5). After cross-linking, Ste6p*HA was immunoprecipitated and resolved by SDS-PAGE under reducing conditions (top two panels). Lysate before antibody addition was also resolved and examined by western blot analysis (bottom two panels) with anti-HA (Ste6p*) or anti-myc antibody (Doa10p), and a quantification of the co-immunoprecipitated Doa10p13myc is shown in the right panel. (Quantification takes into account the relative amounts of immunoprecipitated Ste6p*; the amount of material at t = 0 was set to “1”. Note that the crosslinking in lane 6 represents efficient Ste6p*-Doa10p interaction in the mutant at the permissive temperature.)
(C) The in vitro ubiquitination reaction was performed using microsomes prepared as in (B) and 4 mg/ml cytosol prepared from SSA1 or ssa1-45 yeast as in Figure 2A. The amount of ubiquitinated Ste6p* is shown in the right panels. The amount of material at t = 0 in the presence of WT cytosol and microsomes was set to “1”.
(D) Cross-linking with DTSSP was performed using microsomes containing Ste6p*HA and Doa10p13myc prepared from cells shifted to 37°C for 60 min as in (A). Ste6p*HA was immunoprecipitated with anti-HA affinity matrix. Based on the relative amount of immunoprecipitated Ste6p*, Doa10p-Ste6p* association from the ssa1-45 strain was ~35–50% as efficient compared to microsomes from WT cells.
The ubiquitination, degradation, and E3 interaction defects in the ssa1-45 mutant are reversible
Substrate solubility is vital for the ubiquitination and degradation of ERAD substrates (Nishikawa et al., 2005). Therefore, it was possible that Ste6p* aggregates in the ssa1 mutant and is excluded from the Doa10p complex. However, we failed to detect a significant loss of Ste6p* detergent solubility in ssa1-45 mutant microsomes as compared to WT microsomes at 18,000g, a condition at which CFTR aggresomes can be pelleted (Johnston et al., 1998) and at 30,000g (Figure S6, and data not shown). We next asked if Ste6p* degradation might be re-activated in ssa1-45 mutant cells, which would otherwise be unlikely if the substrate formed a dead-end aggregate. Ssa1-45 cells were grown at a nonpermissive temperature for 40 min and then returned to 23°C (Figure 4A). To focus on the population of Ste6p* which was in a ubiquitination and degradation-incompetent state, CHX was added to the media before the temperature was lowered. We found that the Ste6p*-Doa10p interaction (Figure 4B upper and lower panels) and that Ste6p* ubiquitination and degradation (Figure 4C–D, respectively) were restored after the temperature had been returned to 23°C. Moreover, Ssa1p was a component of a putative multi-chaperone complex, which may include Ydj1p and Hsc82p, and Ste6p* (Figure 4E).
Figure 4. Ste6p* remains a productive intermediate for degradation in ssa1 mutant yeast.

(A) Schematic of culturing condition for microsome preparation. Ssa1-45 cells expressing Ste6p*HA and Doa10p13myc were grown at 23°C, shifted to 37°C for 40 min, and CHX was added. Half of the culture was left at 37°C (a) and the other half was shifted to 23°C (b).
(B) Microsomes were prepared by glass bead disruption from cells grown as in (A). After cross-linking, Ste6p* was immunoprecipitated, and Ste6p*HA and Doa10p13myc were detected as in Figure 3B. The amount of co-immunoprecipitated Doa10p13myc was quantified as in Figure 3B and the means of two independent experiments are shown in the bottom panel.
(C) In vitro ubiquitination of Ste6p* was performed using microsomes prepared as in (A). Cytosol was prepared from SSA1 and ssa1-45 yeast as in Figure 2A.
(D) Hsp70 mutant (ssa1-45/DOA10-13myc) cells expressing Ste6p*HA were cultured as in (A) and the degradation of Ste6p* was analyzed by western blot following CHX addition. Sec61p serves as a loading control and the relative amount of Ste6p* remaining over time is indicated below the anti-HA western blot.
(E) Ssa1p is one member of an Ste6p*-associated multi-protein complex. A Triton X-100 solubilized cell lysate was prepared from cells expressing Ste6p*HA and Doa10p-13myc. Ste6p* was immunoprecipitated with anti-HA affinity matrix and the indicated proteins were detected by western blot analysis. Cells containing a vector that lacked the Ste6p* insert were used as a control. Immunoprecipitations were also performed under denaturing conditions (“den”) to confirm the identities of distinct proteins and support the efficacy of Hsp70-Hsp40 interaction with the Ste6p* complex. Note, however, that a fraction of Hsp82p was also precipitated under denaturing conditions.
Ufd2p extends the ubiquitin chain and catalyzes Ste6p* degradation
Ubiquitin chain assembly usually requires only an E1, an E2, and an E3. However, polyubiquitination sometimes requires the E4 polyubiquitin chain extension enzyme, which is encoded by UFD2 (Koegl et al., 1999). Although the degradation of two ERAD substrates in vivo is slowed when UFD2 is deleted (Richly et al., 2005), direct evidence for Ufd2p-catalyzed polyubiquitin chain extension during ERAD is lacking. Of interest, we noted that the ubiquitinated Ste6p* species converts from a low to a high molecular weight form as the concentration of cytosol is increased (Figure 5A, lanes 1–6). To test whether ubiquitin extension was Ufd2p-mediated, cytosol and Ste6p*-expressing microsomes were prepared from WT and ufd2Δ cells, and the cytosols were titrated into the in vitro assay. When Ufd2p was absent in the microsome fraction (“mic”), no change in the ubiquitinated forms was observed. In contrast, when Ufd2p was absent from cytosol (“cyt”), Ste6p* remained in the LMW form (Figure 5A, compare lanes 7–12 and 19–24). To confirm this result, a GST-Ufd2p fusion protein was purified from bacteria (Figure 5B) and added into the reaction. As little as 0.5 μg of purified Ufd2p (~1% of total protein) restored ubiquitin elongation to the HMW form when ufd2Δ mutant microsomes and cytosol were examined (Figure 5C, lane 2–4). We then examined the in vivo ubiquitination state of Ste6p* in WT and ufd2Δ yeast. When Ste6p* was immunoprecipitated from cells, the HMW form was significantly weaker than the LMW form in ufd2Δ cells (Figure 5D, compare lanes 3 to 4), but strong ubiquitin overexpression in the presence of copper partially suppressed the ubiquitin extension defect (Figure 5D, lanes 5 and 6). In addition, the ERAD of this substrate was significantly slowed in ufd2Δ cells (Figure 5E). These in vitro and in vivo results strongly suggest that Ufd2p facilitates Ste6p* degradation by increasing the extent of the polyubiquitin chain.
Figure 5. Ufd2p elaborates the ubiquitin chain and facilitates Ste6p* degradation.

(A) In vitro ubiquitination of Ste6p* was assessed using microsomes prepared from UFD2 or ufd2Δ cells expressing Ste6p*HA and cytosol prepared from UFD2 or ufd2Δ cells at the indicated concentrations. HMW; high molecular weight ubiquitinated species, LMW; low molecular weight ubiquitinated species.
(B) Purified GST-Ufd2p and GST were resolved by SDS-PAGE and stained with Coomassie Brilliant blue.
(C) In vitro ubiquitination was performed as in (A) using ufd2Δ microsomes and cytosol at a final concentration of 1 mg/ml in the presence of the indicated amounts of the GST-Ufd2p fusion protein or GST.
(D) In vivo ubiquitination of Ste6p* in UFD2 and ufd2Δ cells. Microsomes were prepared from UFD2 or ufd2Δ cells expressing Ste6p*HA and myc-tagged ubiquitin under the control of the CUP1 promoter. In lanes 3 and 4, no copper was added, and in lanes 5 and 6, the expression of ubiquitin was induced by 100 μM copper sulfate for 3 hr before the cells were harvested. Ste6p*HA was immunoprecipitated and analyzed by western blot analysis with anti-ubiquitin (top panel) or anti-HA antibodies (bottom panel). The in vivo ubiquitination of Ste6p* in DOA10/HRD1 and doa10Δ/hrd1Δ cells was analyzed in parallel (lanes 1 and 2).
(E) The degradation of Ste6p*HA in UFD2 or ufd2Δ cells was analyzed by western blot analysis of cell extracts prepared at the indicated time points after the addition of CHX. Cells were incubated at 30°C. The relative amount of Ste6p* was quantified and the means of two independent experiments are shown.
Polyubiquitinated Ste6p* solubilization is Cdc48p and ATP-dependent
Conflicting data have accumulated on whether ubiquitinated membrane substrates are degraded at the ER membrane or in the cytoplasm (see Introduction). To test if Ste6p* is released into the cytosol, we included a centrifugation step after the in vitro ubiquitination reaction. As the reaction proceeded, ~50% of the ubiquitinated Ste6p* was observed in the supernatant (Figure 6A lanes 9–12, upper panel), although most unmodified Ste6p* remained in the microsomes (bottom panel). Consistent with the in vitro result, polyubiquitinated Ste6p* was also observed in the supernatant when lysates from cim5-1 yeast were subjected to centrifugation (data not shown). Because the Cdc48p-Npl4p-Ufd1p complex is required for Ste6p* degradation (Huyer et al., 2004), and contributes to the extraction of some polypeptides from the ER membrane (Ye et al., 2003; Jentsch and Rumpf, 2007), we asked if this complex catalyzes Ste6p* release from microsomes. A ~6.5-fold difference was observed in the liberation of ubiquitinated Ste6p* when reactions were performed using cytosols from a WT strain and from a cdc48-3 mutant (Figure 6B, compare lanes 3 to 7, and 4 to 8). A defect in Ste6p* solubilization was also observed in the presence of ufd1-1 cytosol (Figure S7). In addition, we found that a TAP-tagged version of Cdc48p co-precipitated with ubiquitinated substrate (Figure 6C, lane 8). Therefore, the Cdc48p complex catalyzes Ste6p* release from the membrane.
Figure 6. In vitro polyubiquitinated Ste6p* is released from the membrane by the Cdc48p complex.

(A) In vitro ubiquitination of Ste6p* was performed for the indicated times and microsome and cytosol fractions were separated by centrifugation before Ste6p* was immunoprecipitated. Cytosol was prepared from WT cells and used at 4 mg/ml. T: total reaction; P: pellet; S: supernatant.
(B) The same assay was performed as in (A) except that cytosol was prepared from CDC48 and cdc48-3 cells grown at 23°C and shifted to 37°C for 5 hr. The reaction was performed using 4 mg/ml cytosol at 23°C for 50 min. In lanes 4 and 8, a 2.5-fold volume of supernatant was subjected to immunoprecipitation.
(C) The same assay was performed as in (A) except that 4 mg/ml cytosol prepared from CDC48 or CDC48-TAP cells grown at 30°C was used. In lanes 4 and 8, an 8.5-fold volume of supernatant was subjected to a non-denaturing immunoprecipitation with IgG Sepharose. The immunocomplex was then subjected to a second round of immunoprecipitation under denaturing conditions with anti-HA antibody.
(D) Microsomes were prepared by glass bead disruption from WT cells expressing Ste6p*HA and myc-tagged ubiquitin under the control of the CUP1 promoter at 30°C. The expression of ubiquitin was induced by 100 μM copper sulfate for 3 hr. Microsomes were incubated with 4 mg/ml of the indicated cytosol and an ATP regenerating system for 40 min at 23°C. After separating the microsomes and cytosol by centrifugation, each fraction was subjected to immunoprecipitation with anti-HA antibody and analyzed by western blot with anti-ubiquitin (top panel) or anti-HA antibodies (bottom panel). Where indicated, cytosol was omitted (“buffer”), or 0.02 u/μl of apyrase was added in place of the ATP regenerating system (“-ATP”). (E) The supernatant after the in vitro assay was enriched by filtration, and the fate of ubiquitinated Ste6p* was assessed in RRL in the presence of DMSO (lanes 1–4) or 125 μM MG132 and 90 μM epoxomicin (lanes 5–8) for the indicated times. Ste6p* was immunoprecipitated with anti-HA antibody and analyzed by phosphor imager.
The HMW Ste6p* species was preferentially released into the cytosolic fraction (Figure 6A, lanes 9–12). However, Ste6p* release was unaffected when cytosol and microsomes were prepared from ufd2Δ cells, and in these experiments even the LMW ubiquitinated species was released from the membrane over time (Figure S8). These data suggest that the Ufd2p-mediated extension of the polyubiquitin chain of Ste6p* occurs in the cytosol, downstream of Cdc48p-mediated release (Figure 6B lane 5). Consistent with this hypothesis, Ufd2p is a soluble protein (Huh et al., 2003; Richly et al., 2005). At this point, however, we cannot exclude the possibility that Ufd2p may also act on a membrane-bound population of Ste6p*.
Because the US11-dependent retro-translocation of MHC class I heavy chain requires ATP hydrolysis (Ye et al., 2003), we asked if the Cdc48p-dependent release of Ste6p* requires ATP. To focus on Cdc48p mediated-release, we took advantage of the fact that microsomes contain ubiquitinated Ste6p* as well as unmodified species at steady-state (Figure 2C, top and bottom lanels); therefore, the fate of Ste6p* which had already been ubiquitinated in vivo could be monitored. First, microsomes containing Ste6p* were prepared from cells over-expressing ubiquitin. Then, the microsomes were incubated with the WT or cdc48-3 cytosol or buffer and in the presence or absence of ATP (Figure 6D). The reaction was subsequently centrifuged to assess the amount of released, ubiquitinated Ste6p*. Compared to reactions using WT microsomes in the presence of ATP, the level of ubiquitinated Ste6p* released into the cytosol decreased 2-fold when cdc48-3 cytosol was used (lanes 3 and 6) and was almost absent when WT cytosol lacking ATP was used (lanes 3 and 12). These data provide further support that Ste6p* solubilization requires the Cdc48p complex and ATP.
Polyubiquitinated Ste6p* in the cytosol is deubiquitinated and/or degraded by the proteasome
Finally, we tested if the polyubiquitinated Ste6p* released into the cytosol was a productive intermediate in the ERAD reaction. Cytosol containing the released substrate was enriched and free 125I-labeled ubiquitin was removed using a membrane filter. Because yeast cytosol failed to support the degradation of ubiquitinated Ste6p* (Figure 1A), as reported for another ubiquitinated substrate (Deshaies et al., 1995), we used rabbit reticulocyte lysate (RRL) (Carlson et al., 2006; Xiong et al., 1999). The 125I-ubiquitinated Ste6p*-enriched cytosol was mixed with RRL and ATP and Ste6p* degradation was monitored after precipitation. As shown in Figure 6E, ubiquitinated Ste6p* disappeared over time, but degradation was slowed upon the addition of proteasome inhibitors (the disappearance of Ste6p* in the presence of the inhibitors may arise from contaminating proteases). When the results of two experiments were averaged, 20% and 40% of the substrate remained after the 40 min incubation in the absence or presence of the proteasome inhibitors, respectively. Although the disappearance of the poylubiquitinated material may also arise from de-ubiquitinating enzymes, these data suggest that the soluble form of Ste6p* is a productive intermediate for the proteasome degradation pathway.
DISCUSSION
Polytopic membrane proteins constitute an important class of macromolecules, representing ~20–30% of the proteome (Wallin and von Heijne, 1998). Because of their complexity they are prone to misfold, and indeed many proteins in this class traffic inefficiently beyond the ER (Brodsky, 2007). To better understand the pathway by which these proteins are destroyed, each step during the ERAD of Ste6p* was reconstituted. Based on in vivo studies and our findings, a model for the ERAD of Ste6p* is shown in Fig. 7. While it was previously known that the cytoplasmic Hsp70, Ssa1p, is required for ERAD, we demonstrate that the chaperone facilitates substrate-E3 ligase interaction. After ubiquitination, Ste6p* is captured by Cdc48p and released into the cytosol. Next, Ufd2p enhances the extent of ubiquitination. Finally, the soluble, species is a substrate for proteasome-mediated processing. Our results suggest that polytopic membrane proteins are ubiquitinated at the ER membrane but may be released into the cytosol for degradation.
Figure 7. A proposed pathway for the ERAD of a polytopic membrane protein.

Doa10p, in conjugation with the E2 enzymes (Ubc6p and Ubc7p), catalyzes Ste6p* ubiquitination. This step is enhanced by specific ER-associated Hsp70 and Hsp40 molecular chaperones that facilitate Ste6p*-Doa10p association. Ubiquitinated Ste6p* is then recognized by the Cdc48p complex and can be released into the cytoplasm, where Ufd2p elaborates the ubiquitin chain. Finally, polyubiquitinated Ste6p* is de-ubiquitinated and/or degraded. At present, it is not clear if cytoplasmic Ste6p* is fragmented by the proteasome. Note that Ubc7p is recruited to the ER membrane by Cue1p, and the location of the ubiquitinated residue(s) in Ste6p* are unknown. The proteasome image was adopted from Voges et al., 1999.
One view of chaperone function is that they bind misfolded substrates and prevent aggregate formation, thereby promoting recognition by the ubiquitination machinery. However, recent studies suggest that cytoplasmic chaperones form a high-order network and escort substrates to folding or degradative pathways (Albanese et al., 2006; McClellan et al., 2005; Wang et al., 2006). Indeed, we found that Ste6p* remains detergent extractable after Ssa1p inactivation, and Ste6p*’s interaction with Doa10p—and its ubiquitination and degradation competence—are resurrected upon Ssa1p reactivation. In the absence of functional Ssa1p, Ste6p* solubility may be maintained by other chaperones, such as Hsp90 (Hsp82p in yeast, Figure S9). Therefore, a multi-chaperone assembly may prevent the aggregation of ERAD substrates, and Hsp70 is most likely a component of this complex (Figure 4E, but note that distinct members of the complex may bind in a mutually exclusive manner). We also suggest that Hsp70 promotes Ste6p* recognition by Doa10p. At this time, it is not clear if Doa10p similarly recognizes a nonnative polypeptide motif in ERAD substrates, as proposed for Hrd1p (Bays et al., 2001), or instead detects a degradation signal (Gardner and Hampton, 1999; Ravid et al., 2006).
Our inability to complement the Ste6p* ubiquitination defect in Hsp70 mutant microsomes upon the addition of WT cytosol was initially surprising (Figure 2A), especially since the ubiquitination and degradation of Ste6p* could be restored by Hsp70 reactivation in vivo. Moreover, repeated attempts to rescue the ssa1-45 mutant phenotype upon the addition of purified proteins were unsuccessful. We propose three models to explain these results. First, Ste6p* that resides in the Hsp70 mutant microsomes may be associated with an inert chaperone complex, and thus externally supplied Hsp70 would be sterically restricted for substrate access. Second, Hsp70 may facilitate Ste6p*-Doa10p interaction and ubiquitination by activating membrane-integrated factors. Therefore, WT cytosol would be unable to rescue the mutant phenotype. Third, Ste6p* in ssa1-45 mutant microsomes may lack a critical component which could not be supplied from cytosol or purified chaperones. Nevertheless, we cannot completely exclude the possibility that Hsp70 modulates Doa10p by subtly altering its conformation. However, we note that the defect in Doa10p-Ste6p* association in ssa1-45 microsomes was observed using two different crosslinkers, and that Doa10p solubility is unchanged when microsomes are prepared from either WT or ssa1-45 mutant yeast (data not shown).
It is not completely clear how the ER quality control machinery surveys integrating membrane proteins to identify those that are inappropriately assembled. Because Ste6p* ERAD was recovered upon Ssa1p reactivation and after translation arrest, we suggest that Doa10p recognizes Ste6p* post-translationally. This view is consistent with the fact that Ste6p* lacks the C-terminal 42 amino acids found in the full length, WT protein; thus, the conformations of translation and translocation intermediates of Ste6p* and WT Ste6p are identical, and the nature of the Ste6p* misfolding defect should only be evident post-translationally.
Several lines of evidence indicate that Doa10p is the ligase for membrane substrates with misfolded cytoplasmic domains, such as Ste6p* (Carvalho et al., 2006; Vashist and Ng, 2004). However, degradation and ubiquitination is incompletely inhibited when Doa10p is absent, which suggests that Hrd1p contributes to the ubiquitination of this class of substrates (Huyer et al., 2004; this study). One means to explain this observation is that cytoplasmic domain misfolding might influence the assembly of the intermembrane domain, which is recognized by the Hrd1p complex. Alternatively, the Hrd1p complex may recognize a cytoplasmic misfolded region. Substrates for Hrd1p are not restricted to lumenal and membrane proteins (Arteaga et al., 2006). This notion is also supported by the fact that the degradation of a general amino acid permease requires both Hrd1p and Doa10p (Kota et al., 2007). Overall, depending on the topology and the location of the misfolded lesion, complex polytopic membrane proteins could be sorted to redundant pathways, which converge at the proteasome.
A long-standing question is whether polytopic ERAD substrates are degraded by the proteasome in situ at the membrane or whether they are extracted prior to proteolysis. Because solubilized, polyubiquitinated Ste6p* was precipitated with anti-HA antibody, which recognizes a lumenally disposed epitope in Ste6p*HA, our results indicate that the transmembrane domain of Ste6p* became solvent-exposed. More generally, our data provide the first in vitro evidence that aggresomes may form from the retro-translocation of a polytopic protein in the ER membrane.
It is unclear whether polytopic membrane proteins are intact or clipped prior to or during extraction. We note that the cytoplasmic polyubiquitinated species begin at and are higher than the molecular weight of the native species, suggesting strongly that unclipped proteins are released into the cytosol. If so, how is the substrate pulled from the ER? And, how is the solubility of the membrane domain maintained? The degradation of Ste6p* does not require proposed retro-translocation channels (Huyer et al., 2004; Kreft et al., 2006). Thus, Ste6p* could either be retro-translocated through an ill-defined channel or from the membrane. It should be noted that fusion of green fluorescent protein (GFP) to Ste6p* juxtaposed to the HA epitope tag in the ER lumen does not impair degradation (Huyer et al., 2004), suggesting that the GFP moiety can be retro-translocated or extracted. Alternatively, the reconfiguration of membrane lipids could support extraction/release, and attached lipids may maintain the solubility of transmembrane domains (Ploegh, 2007). Candidates for proteinaceous factors that could maintain the solubility of transmembrane domains include cytoplasmic chaperones, Cdc48p, proteasome-associated factors such as Rad23p/Dsk2p (Richly et al., 2005), and the 19S particle. The in vitro system reported in this study provides a means to address these questions.
MATERIALS AND METHODS
Strains and Plasmids
Yeast strains and plasmids are shown in the Supplemental Data.
In Vitro Ubiquitination Assay
Unless otherwise indicated, ER-derived microsomes were prepared by homogenization and cytosol was prepared by liquid nitrogen lysis (McCracken and Brodsky, 1996). Chaperone mutant microsomes were prepared by glass bead disruption as described in the Supplemental Data. A typical ubiquitination reaction (20 μl), including 20 μg of microsomes, an ATP regenerating system and the indicated concentration of yeast cytosol in buffer 88 (20 mM HEPES, pH 6.8, 150 mM KOAc, 250 mM sorbitol, 5 mM MgOAc), was pre-warmed at 23°C (Ste6p*) or at 30°C (Sec61-2p) for 10 min. After 2 μl of 125I-labeled ubiquitin (~1.0×106 cpm/μl) was added, the reaction was incubated for up to 60 min. The reaction was quenched with 1% SDS plus protease inhibitors and 10 mM N-ethylmaleimide. Ste6p* and Sec61-2p were immunoprecipitated with anti-HA antibody or anti-Sec61p antiserum, and were resolved by SDS-PAGE. Half of each sample was used for phosphor imager analysis and the other half was used for western blot analysis. Detailed buffer composition and protocols for the degradation assay are provided in the Supplemental Data.
Detection of the In Vivo Ubiquitinated Substrate
The extent of the polyubiquitinated substrate in yeast was detected using affinity purified anti-ubiquitin antibody (a kind gift from C. Pickart) or anti-myc antibody (a kind gift from O. Weisz and G. Apodaca) as described (Ahner et al., 2007).
Cross-linking Assay
ER-derived microsomes were prepared using buffer 88 at pH 7.5 instead of pH 6.8 from a total of 20–30 OD600 equivalents of log phase cells (~OD600=1.0), and 250 μg of microsomes were incubated with the indicated crosslinkers for 60 min on ice. The reaction was quenched with 230 mM Tris pH 7.5, the membranes were solubilized with 1% SDS, insoluble material was removed, and Ste6p* was immunoprecipitated with anti-HA antibody. The immunoprecipitated proteins were analyzed by SDS-PAGE under reducing conditions and by western blot analysis with anti-HA or anti-myc antibody. Detailed protocols on this assay and the native immunoprecipitation are provided in the Supplemental Data.
Cycloheximide Chase Degradation Assay
CHX degradation analyses were performed as described previously (Zhang et al., 2001) with minor modifications (see Supplemental Data).
Supplementary Material
Acknowledgments
We would like to thank A. Oztan, C. Rondanino and G. Apodaca for their generosity, G. Apodaca, K. Arndt, M. Hochstrasser, R. Hampton, J. Martens, S. Nishikawa, C. Pickart, H. Rao, R. Schekman, C. Stirling, and O. Weisz for reagents, strains and plasmids, and members of the Brodsky laboratory for discussions and technical assistance. K.N. was supported by a post-doctoral grant from the Uehara Memorial Foundation and from the American Heart Association. This study was supported by grants GM75061 (to J. L. B.) and GM51508 (to S. M.) from the National Institutes of Health, and by grant BRODSK05P0 to J. L. B. from the Cystic Fibrosis Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahner A, Nakatsukasa K, Zhang H, Frizzell RA, Brodsky JL. Small heat-shock proteins select deltaF508-CFTR for endoplasmic reticulum-associated degradation. Mol Biol Cell. 2007;18:806–814. doi: 10.1091/mbc.E06-05-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama Y, Ito K. Reconstitution of membrane proteolysis by FtsH. J Biol Chem. 2003;278:18146–18153. doi: 10.1074/jbc.M302152200. [DOI] [PubMed] [Google Scholar]
- Albanese V, Yam AY, Baughman J, Parnot C, Frydman J. Systems analyses reveal two chaperone networks with distinct functions in eukaryotic cells. Cell. 2006;124:75–88. doi: 10.1016/j.cell.2005.11.039. [DOI] [PubMed] [Google Scholar]
- Arteaga MF, Wang L, Ravid T, Hochstrasser M, Canessa CM. An amphipathic helix targets serum and glucocorticoid-induced kinase 1 to the endoplasmic reticulum-associated ubiquitin-conjugation machinery. Proc Natl Acad Sci U S A. 2006;103:11178–11183. doi: 10.1073/pnas.0604816103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol. 2001;3:24–29. doi: 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- Becker J, Walter W, Yan W, Craig EA. Functional interaction of cytosolic hsp70 and a DnaJ-related protein, Ydj1p, in protein translocation in vivo. Mol Cell Biol. 1996;16:4378–4386. doi: 10.1128/mcb.16.8.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL. The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated-degradation) Biochem J. 2007;404:353–363. doi: 10.1042/BJ20061890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL, Werner ED, Dubas ME, Goeckeler JL, Kruse KB, McCracken AA. The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J Biol Chem. 1999;274:3453–3460. doi: 10.1074/jbc.274.6.3453. [DOI] [PubMed] [Google Scholar]
- Carlson EJ, Pitonzo D, Skach WR. p97 functions as an auxiliary factor to facilitate TM domain extraction during CFTR ER-associated degradation. EMBO J. 2006;25:4557–4566. doi: 10.1038/sj.emboj.7601307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, Chau V, Kirschner M. Ubiquitination of the G1 cyclin Cln2p by a Cdc34p-dependent pathway. EMBO J. 1995;14:303–312. doi: 10.1002/j.1460-2075.1995.tb07004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsasser S, Finley D. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol. 2005;7:742–749. doi: 10.1038/ncb0805-742. [DOI] [PubMed] [Google Scholar]
- Gardner RG, Hampton RY. A ‘distributed degron’ allows regulated entry into the ER degradation pathway. EMBO J. 1999;18:5994–6004. doi: 10.1093/emboj/18.21.5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Shearer AG, Hampton RY. In vivo action of the HRD ubiquitin ligase complex: mechanisms of endoplasmic reticulum quality control and sterol regulation. Mol Cell Biol. 2001;21:4276–4291. doi: 10.1128/MCB.21.13.4276-4291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauss R, Jarosch E, Sommer T, Hirsch C. A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nature Cell Biol. 2006;8:849–854. doi: 10.1038/ncb1445. [DOI] [PubMed] [Google Scholar]
- Gnann A, Riordan JR, Wolf DH. Cystic fibrosis transmembrane conductance regulator degradation depends on the lectins Htm1p/EDEM and the Cdc48 protein complex in yeast. Mol Biol Cell. 2004;15:4125–4135. doi: 10.1091/mbc.E04-01-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Liu Y, Chang A. Cytoplasmic Hsp70 promotes ubiquitination for endoplasmic reticulum-associated degradation of a misfolded mutant of the yeast plasma membrane ATPase, PMA1. J Biol Chem. 2007;282:26140–26149. doi: 10.1074/jbc.M701969200. [DOI] [PubMed] [Google Scholar]
- Hebert DN, Garman SC, Molinar M. The glycan code of the endoplasmic reticulum: asparagines-linked carbohydrates as protein maturation and quality control tags. Trends Cell Biol. 2005;15:364–370. doi: 10.1016/j.tcb.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Hershko A, Heller H. Occurrence of a polyubiquitin structure in ubiquitin-protein conjugates. Biochem Biophys Res Commun. 1985;128:1079–1086. doi: 10.1016/0006-291x(85)91050-2. [DOI] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL, Michaelis S. Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J Biol Chem. 2004;279:38369–38378. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T. Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol. 2002;4:134–139. doi: 10.1038/ncb746. [DOI] [PubMed] [Google Scholar]
- Jentsch S, Rumpf S. Cdc48 (p97): a “molecular gearbox” in the ubiquitin pathway? Trends Biochem Sci. 2007;32:6–11. doi: 10.1016/j.tibs.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabani M, Kelley SS, Morrow MW, Montgomery DL, Sivendran R, Rose MD, Gierasch LM, Brodsky JL. Dependence of endoplasmic reticulum-associated degradation on the peptide binding domain and concentration of BiP. Mol Biol Cell. 2003;14:3437–3448. doi: 10.1091/mbc.E02-12-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell. 1999;96:635–644. doi: 10.1016/s0092-8674(00)80574-7. [DOI] [PubMed] [Google Scholar]
- Kostova Z, Wolf DH. For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 2003;22:2309–2317. doi: 10.1093/emboj/cdg227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota J, Gilstring CF, Ljungdahl PO. Membrane chaperone Shr3 assists in folding amino acid permeases preventing precocious ERAD. J Cell Biol. 2007;176:617–628. doi: 10.1083/jcb.200612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreft SG, Wang L, Hochstrasser M. Membrane topology of the yeast endoplasmic reticulum-localized ubiquitin ligase Doa10 and comparison with its human ortholog TEB4 (MARCH-VI) J Biol Chem. 2006;281:4646–4653. doi: 10.1074/jbc.M512215200. [DOI] [PubMed] [Google Scholar]
- Liu CW, Corboy MJ, DeMartino GN, Thomas PJ. Endoproteolytic activity of the proteasome. Science. 2003;299:408–411. doi: 10.1126/science.1079293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loayza D, Tam A, Schmidt WK, Michaelis S. Ste6p mutants defective in exit from the endoplasmic reticulum (ER) reveal aspects of an ER quality control pathway in Saccharomyces cerevisiae. Mol Biol Cell. 1998;9:2767–2784. doi: 10.1091/mbc.9.10.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer TU, Braun T, Jentsch S. Role of the proteasome in membrane extraction of a short-lived ER-transmembrane protein. EMBO J. 1998;17:3251–3257. doi: 10.1093/emboj/17.12.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan AJ, Scott MD, Frydman J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell. 2005;121:739–748. doi: 10.1016/j.cell.2005.03.024. [DOI] [PubMed] [Google Scholar]
- McCracken AA, Brodsky JL. Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J Cell Biol. 1996;132:291–298. doi: 10.1083/jcb.132.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham GC, Lu Z, King S, Sorscher E, Tousson A, Cyr DM. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999;18:1492–1505. doi: 10.1093/emboj/18.6.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier L, Usherwood YK, Chung KT, Hendershot LM. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol Biol Cell. 2002;13:4456–4469. doi: 10.1091/mbc.E02-05-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- Meusser B, Sommer T. Vpu-mediated degradation of CD4 reconstituted in yeast reveals mechanistic differences to cellular ER-associated protein degradation. Mol Cell. 2004;14:247–258. doi: 10.1016/s1097-2765(04)00212-6. [DOI] [PubMed] [Google Scholar]
- Nishikawa S, Brodsky JL, Nakatsukasa K. Roles of molecular chaperones in endoplasmic reticulum (ER) quality control and ER-associated degradation (ERAD) J Biochem (Tokyo) 2005;137:551–555. doi: 10.1093/jb/mvi068. [DOI] [PubMed] [Google Scholar]
- Nishikawa SI, Fewell SW, Kato Y, Brodsky JL, Endo T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J Cell Biol. 2001;153:1061–1070. doi: 10.1083/jcb.153.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piwko W, Jentsch S. Proteasome-mediated protein processing by bidirectional degradation initiated from an internal site. Nat Struct Mol Biol. 2006;13:691–697. doi: 10.1038/nsmb1122. [DOI] [PubMed] [Google Scholar]
- Plemper RK, Egner R, Kuchler K, Wolf DH. Endoplasmic reticulum degradation of a mutated ATP-binding cassette transporter Pdr5 proceeds in a concerted action of Sec61 and the proteasome. J Biol Chem. 1998;273:32848–32856. doi: 10.1074/jbc.273.49.32848. [DOI] [PubMed] [Google Scholar]
- Ploegh HL. A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature. 2007;448:435–438. doi: 10.1038/nature06004. [DOI] [PubMed] [Google Scholar]
- Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25:533–543. doi: 10.1038/sj.emboj.7600946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120:73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Romisch K. Endoplasmic reticulum-associated degradation. Annu Rev Cell Dev Biol. 2005;21:435–456. doi: 10.1146/annurev.cellbio.21.012704.133250. [DOI] [PubMed] [Google Scholar]
- Rumpf S, Jentsch S. Functional division of substrate processing cofactors of the ubiquitin-selective Cdc48 chaperone. Mol Cell. 2006;21:261–269. doi: 10.1016/j.molcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Swanson R, Locher M, Hochstrasser M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev. 2001;15:2660–2674. doi: 10.1101/gad.933301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, Gurkan C, Kellner W, Matteson J, Plutner H, et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell. 2006;127:803–815. doi: 10.1016/j.cell.2006.09.043. [DOI] [PubMed] [Google Scholar]
- Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- Wigley WC, Fabunmi RP, Lee MG, Marino CR, Muallem S, DeMartino GN, Thomas PJ. Dynamic association of proteasomal machinery with the centrosome. J Cell Biol. 1999;145:481–490. doi: 10.1083/jcb.145.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Chong E, Skach WR. Evidence that endoplasmic reticulum (ER)-associated degradation of cystic fibrosis transmembrane conductance regulator is linked to retrograde translocation from the ER membrane. J Biol Chem. 1999;274:2616–2624. doi: 10.1074/jbc.274.5.2616. [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol. 2003;162:71–84. doi: 10.1083/jcb.200302169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nijbroek G, Sullivan ML, McCracken AA, Watkins SC, Michaelis S, Brodsky JL. Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol Biol Cell. 2001;12:1303–1314. doi: 10.1091/mbc.12.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.