

Abstract
To explore the role of pore-lining amino acids in Na+ channel ion-selectivity, pore residues were replaced serially with cysteine in cloned rat skeletal muscle Na+ channels. Ionic selectivity was determined by measuring permeability and ionic current ratios of whole-cell currents in Xenopus oocytes. The rSkM1 channels displayed an ionic selectivity sequence Na+>Li+>NH4 +>>K+>>Cs+ and were impermeable to divalent cations. Replacement of residues in domain IV showed significantly enhanced current and permeability ratios of NH4 + and K+, and negative shifts in the reversal potentials recorded in the presence of external Na+ solutions when compared to cysteine mutants in domains I, II, and III (except K1237C). Mutants in domain IV showed altered selectivity sequences: W1531C (NH4 +>K+>Na+≥Li+≈Cs+), D1532C, and G1533C (Na+>Li+≥NH4 +>K+>Cs+). Conservative replacement of the aromatic residue in domain IV (W1531) with phenylalanine or tyrosine retained Na+ selectivity of the channel while the alanine mutant (W1531A) reduced ion selectivity. A single mutation within the third pore forming region (K1237C) dramatically altered the selectivity sequence of the rSkM1 channel (NH4 +>K+>Na+≥Li+≈Cs+) and was permeable to divalent cations having the selectivity sequence Ca2+≥Sr2+>Mg2+>Ba2+. Sulfhydryl modification of K1237C, W1531C or D1532C with methanethiosulfonate derivatives that introduce a positively charged ammonium group, large trimethylammonium moiety, or a negatively charged sulfonate group within the pore was ineffective in restoring Na+ selectivity to these channels. Selectivity of D1532C mutants could be largely restored by increasing extracellular pH suggesting altering the ionized state at this position influences selectivity. These data suggest that K1237 in domain III and W1531, D1532, and G1533 in domain IV play a critical role in determining the ionic selectivity of the Na+ channel.
Keywords: mutagenesis, skeletal muscle, sulfhydryl modification, permeation
introduction
Voltage-gated Na+ channels are critical in the excitability of muscle and nerves. They conduct Na+ ions across membrane bilayers at a rate of over 106 ions per second while still being able to selectively discriminate Na+ ions against other physiological cations such as K+ and Ca2+ (Hille, 1992). This property is crucial for generating the electromotive forces required for electrical signaling. Even though several regions of the channel protein that control ion current have been identified, the underlying molecular basis for selectivity remains obscure.
The cloning of various ion channels and the development of site-directed mutagenesis has allowed the examination of the molecular structure of channel proteins. Many voltage-gated ion channels are comprised of four homologous domains, each consisting of six transmembrane spanning regions (Catterall, 1995). The four domains merge to form a barrel-like structure with a pore-forming region extending into the membrane (Catterall, 1995). The pore region was first shown to be located between the fifth and sixth transmembrane regions in K+ channels (Hartmann et al., 1991; Yellen et al., 1991; Yool and Schwartz, 1991). Further studies revealed that the conduction pathway of the Na+ channel was located in the same region (Backx et al., 1992; Heinemann et al., 1992). Regardless of this similarity, Na+ channels differ from other voltage-gated ion channels in their high selectivity for Na+ (Hille, 1992). Systematic mutations of residues within the pore region have revealed residues important for selectivity and permeation of voltage-gated K+ channels (Yool and Schwartz, 1991; Heginbotham et al., 1992; Taglialatela et al., 1993) and the L-type Ca2+ channel (Kim et al., 1993; Mikala et al., 1993; Yang et al., 1993). Similar studies in the rat brain II Na+ channel have suggested four amino acids (aspartate, glutamate, lysine, and alanine), one in each of the four pore segments, form the putative selectivity filter (Terlau et al., 1991; Heinemann et al., 1992). Neutralization of the negatively charged aspartate and glutamate residues in the first and second repeat, respectively, drastically reduced Na+ flux (Terlau et al., 1991), while conversion of the lysine and alanine residues in the third and fourth domain, respectively, to glutamate resulted in a channel that was more selective for Ca2+ over Na+ (Heinemann et al., 1992). However, the involvement of other residues within the Na+ channel pore to Na+ selectivity and permeation has not been extensively examined.
In the present study, we have examined the contribution of amino acids within the pore region of the rat skeletal muscle Na+ channel (rSkM1)1 on the properties of ionic selectivity using cysteine scanning mutagenesis. We have identified three other residues (W1531, D1532, and G1533) in the fourth domain that significantly alter Na+ selectivity but are not part of the proposed selectivity filter (D400, E755, K1237, A1529). These results suggest other residues, especially in domain IV, are important in determining the ionic selectivity of the Na+ channel.
materials and methods
Mutagenesis and Heterologous Expression
A 1.9-kb BamHI-SphI or 2.5-kb SphI-KpnI fragment of the rSkM1 Na+ channel were subcloned into pGEM-11f+ or pGEM-7f+ (Promega Corp., Madison, WI), respectively. Site-directed mutagenesis was performed using uracil-enriched single-stranded DNA according to the methods by Kunkel (1985). The mutation was confirmed by dideoxy nucleotide sequencing (Sanger et al., 1977) before and after subcloning into the expression vector pGW1-CMV (British Biotechnology, Oxford, UK) containing the full-length Na+ channel clone. The pore residues substituted by cysteine in the present study are shown in Fig 1.
Figure 1.

Structural topology of the Na+ channel α subunit. The shaded area within the pore-forming domains denotes the region mutated in the present study. The putative amino acid alignment sequence for the four pore regions is shown by the single letter amino acid code. The residues in capital letters were mutated to cysteine. The location of each residue within the primary sequence of the rSkM1 Na+ channel is shown below each amino acid letter.
Oocytes were removed from adult female Xenopus laevis (Nasco, Ft. Atkinson, WI) anesthetized in 0.2% tricaine (Sigma Chemical Co., St. Louis, MO), and digested with 2 mg/ml collagenase (Type 1A; Sigma Chemical Co.) in OR-2 containing (in mM): 88 NaCl, 2 KCl, 1 MgCl2, and 5 HEPES, pH 7.6. Oocytes were stored at room temperature in ND96 containing (in mM): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES, pH 7.6, supplemented with 50 μg/ml gentamicin, 5 mM pyruvate, and 0.5 mM theophylline. The addition of 2.5% FBS promoted the removal of the follicular layer. Nuclear injections with 50 nl of cDNA (10–100 ng/μl) consisting of the wild-type (rSkM1) or cysteine mutant, and the rat brain β1 subunit (Isom et al., 1992) (ratio of α:β1 subunit was 1:5) were performed on healthy, stage V-VI oocytes.
Electrophysiology
Whole-cell current recordings were done at room temperature (20–22°C) using a two-electrode voltage-clamp amplifier (OC-725A; Warner Instruments, Hamden, CT) 1–4 d after injection. Agarose-plugged microelectrodes (TW120F-6; World Precision Instruments, Sarasota, FL) were filled with 3 M KCl and had a final resistance of 1–3 MΩ. Whole-cell currents were evoked by step depolarizations from −60 to +50 mV from a holding potential of −120 mV. The currents were digitized at 10 kHz and low-passed filtered at 1–2 kHz (−3 dB). A P/4 protocol was used for leak and capacitance subtraction. Analysis of current records was performed using custom-written software. The recording solution (ND96) contained (in mM): 96 NaCl, 1 BaCl2, 1 MgCl2, and 5 HEPES, pH 7.6. For examining the ionic selectivity of the rSkM1 and mutant channels, NaCl was replaced with equimolar monovalent (Cs+, K+, Li+, NH4 +) or divalent cation (Ba2+, Ca2+, Mg2+, Sr2+) (as the Cl− salt) adjusted to pH 7.6 with the corresponding hydroxide salt or Tris. Substitution of NaCl with equimolar divalent cation salts results in an increase in hypertonicity of the solution. Adjustments were not made to account for this change. To prevent endogenous Ca2+-activated Cl− currents activity, either niflumic acid (1 mM) was included in the extracellular divalent solutions to block the Cl− currents or the oocytes were injected with 1 mM BAPTA. All recordings were made within the first 10 min after initially voltage clamping the oocyte to the holding potential of −120 mV.
Sulfhydryl modification by the methanethiosulfonate (MTS) derivatives (Toronto Research Chemical Co., Toronto, Canada), MTSEA (MTS-ethylammonium), MTSES (MTS-ethylsulfonate), MTSET (MTS-ethyltrimethylammonium), or MTSEB (MTS-ethylbenzoate; gift from Dr. A. Wooley, University of Toronto, Toronto, Canada) was performed by exposing the cysteine mutants to 1 mM MTS-X for 3 min followed by a 5-min washout. Modification of the cysteinyl sulfhydryl side-chain was irreversible (Akabas et al., 1992) and verified by examining the altered Cd2+ sensitivity. The MTS derivatives were prepared daily and dissolved in the recording solution.
To examine the effects of extracellular pH, the external bath solution consisted of (in mM) 96 NaCl, 1 BaCl2, 1 MgCl2, and 5 HEPES, Tris or MES (adjusted with NaOH or HCl to the desired pH).
Determination of Current and Permeability Ratios
Current ratios were determined by the ratio of peak inward current in the presence of extracellularly applied tested cations to the peak inward current in the presence of Na+. Permeability ratios (PX/PNa) for a given cation were calculated using a modified Goldman-Hodgkin-Katz equation (Hille, 1992):
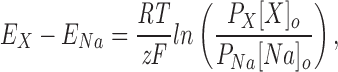 |
1 |
where E X and E Na are the reversal potentials for the tested cation (X) and Na+, respectively, z is the valence of the tested cation, and R, T, and F have their usual meanings. Both current and permeability ratios can provide equivalent interpretations of ion selectivity (Eisenman and Horn, 1983). Reversal potentials and slope conductance were calculated by fitting the current-voltage relationship to a Boltzmann distribution function:
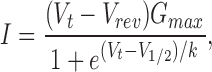 |
2 |
where I is the peak INa at the given test potential V t, V rev is the reversal potential, G max is the maximal slope conductance, V 1/2 is the half-point of the relationship and k (=RT/zF ) is the slope factor.
Data presented are the means ± SEM. Statistical significance was determined using an unpaired Student's t test with P < 0.05 representing significance.
results
Although Na+ channels are highly selective for Na+, they will permit the permeation of other cations (Hille, 1992). We examined the relative ionic selectivity of the wild-type rat skeletal muscle Na+ channel (rSkM1) expressed in Xenopus oocytes to varying monovalent and divalent cations. Fig. 2 A shows whole-cell recordings of rSkM1 in the presence of equimolar Na+, Li+, NH4 +, and K+ containing solutions with the corresponding current-voltage relationship shown in Fig. 2 B. The rSkM1 Na+ channel was highly permeable to Na+ and Li+, exhibiting substantial inward currents (Fig. 2 A) compared with NH4 + and K+. Since we are unable to control the intracellular ionic milieu of the oocyte, we are uncertain of the ionic species responsible for the outward current. No detectable inward currents were observed in the presence of Cs+ or the divalent cations (data not shown). These data give a selectivity sequence of Na+>Li+>NH4 +>>K+>>Cs+ based on current ratios. These findings are very similar to those reported for native Na+ channels (for review, see Hille, 1992) and heterologously expressed Na+ channels (Heinemann et al., 1992; Chiamvimonvat et al., 1996a , b ) using either current ratio or permeability ratio measurements. Current and permeability ratio measurements for rSkM1 gave comparable values and the same relationship regarding ion selectivity of rSkM1 (Table I).
Figure 2.

Ion selectivity of the rSkM1 Na+ channel. (A) Whole-cell recordings of the rSkM1 Na+ channel in the presence of 96 mM NaCl, LiCl, NH4Cl, and KCl solutions. Oocytes were depolarized between −60 and +50 mV from a holding potential of −120 mV. Currents traces displayed are from −50 to +40 mV in 10-mV increments. Currents were corrected for leak and cell capacity. (B) Current-voltage relationship of the same oocyte shown in A for Na+ (▪), Li+ (○), NH4 + (♦), and K+ (▵). The data were fit to a Boltzmann distribution as described in materials and methods.
Table I.
Ionic Current and Permeability Ratios of rSkM1
| Current Ratio | Permeability Ratio | |||
|---|---|---|---|---|
| PLi/PNa | 0.79 ± 0.07 | 0.97 ± 0.06 | ||
| PNH4/PNa | 0.16 ± 0.07 | 0.17 ± 0.02 | ||
| PK/PNa | 0.03 ± 0.01 | 0.05 ± 0.01 | ||
| PCs/PNa | <0.01 | 0.02 ± 0.01 |
Ionic current and permeability ratios of rSkM1 for Li+, NH4 +, K+, and Cs+. Current ratios were calculated from peak inward currents in the presence of tested cation normalized to the peak current in Na+ solution. Permeability ratios were calculated from reversal potentials using a modified Goldman-Hodgkin-Katz equation as described in materials and methods. Values represent the mean ± SEM from five oocytes.
Ionic Selectivity of Cysteine Mutants
We next examined the contribution of the pore-forming residues to ionic selectivity. The amino acids at the carboxyl end (SS2) of the four pore regions were mutated to cysteine (Fig. 1). We chose to mutate the amino acids to cysteine because of the small size and intermediate polarity (Creighton, 1993), but more importantly, it allows the determination of the spatial orientation of the residues within the pore using Cd2+ or sulfhydryl modifying agents as probes (Akabas et al., 1992; Kürz et al., 1995; Pascual et al., 1995; Li et al., 1996; Pérez-García et al., 1996). All cysteine mutants expressed functional channels with the exception of G1238C in domain III, suggesting no major structural changes to the channel protein. We implicitly assumed that only those residues which face into the aqueous pore region, and thereby are able to interact directly with the permeating ion, will be able to influence ion selectivity. It is possible that the carbonyl backbone of the protein or side-chains not facing into the aqueous pore environment may also influence selectivity. However, we are unable to distinguish these changes from side-chain effects per se. Initially, we measured the current ratios of the cysteine mutants to determine possible residues which influence ion selectivity. Fig. 3 illustrates the current ratios for Li+, NH4 +, and K+ for rSkM1 and the cysteine mutants. All cysteine mutants were equally permeable to Li+ as rSkM1 with the exception of W402C, E758C, and A1529C which had Li+ current ratios significantly larger than rSkM1 channels (Fig. 3 A).
Figure 3.

Current ratios for (A) Li+, (B) NH4 +, and (C) K+ of rSkM1 and cysteine mutant channels. Current ratios were determined from the ratio of the peak inward current in the presence of the test cation to the peak inward current in the presence of Na+. Data represent the mean ± SEM of three to five oocytes. n.e. denotes no expression. The P values are * < 0.05 and ** < 0.01.
Mutants in domains I, II and III (except E755C and K1237C) showed statistically identical selectivity to NH4 + and K+ to wild-type rSkM1 channels (Fig. 3). Similarly residues in these domains did not show significant alterations in reversal potentials (Table II). On the other hand W1531C, D1532C, and G1533C mutants in domain IV showed statistically significant enhanced selectivity towards NH4 + and K+ as measured using current ratios (Fig. 3). In each of these domain IV mutants (W1531C, D1532C, and G1533C), there was a corresponding significant shift in the reversal potential of the peak current versus voltage curve compared to rSkM1 channels. In addition, G1530C channels also showed significant shifts in reversal potential (Table II) despite a lack of change in current ratio to K+ compared to rSkM1. These results establish that residues in domain IV more profoundly influence ion selectivity with respect to NH4 + and K+ than residues in the other three domains (except E755C and K1237C).
Table II.
Na+ Reversal Potentials of rSkM1 and Cysteine Mutants
| ENa (mV) | n | |||
|---|---|---|---|---|
| rSkM1 | 54.4 ± 2.7 | 10 | ||
| Y401C | 50.7 ± 2.1 | 6 | ||
| W402C | 52.1 ± 2.3 | 6 | ||
| E403C | 51.0 ± 2.7 | 7 | ||
| E755C | 28.8 ± 2.2‡ | 7 | ||
| W756C | 51.6 ± 1.9 | 6 | ||
| I757C | 53.4 ± 1.0 | 7 | ||
| E758C | 54.0 ± 3.9 | 7 | ||
| K1237C | −1.3 ± 1.2‡ | 9 | ||
| W1239C | 49.7 ± 2.1 | 6 | ||
| M1240C | 50.2 ± 3.9 | 5 | ||
| D1241C | 54.4 ± 2.0 | 6 | ||
| A1529C | 50.0 ± 1.1 | 7 | ||
| G1530C | 41.3 ± 2.9* | 5 | ||
| W1531C | 1.6 ± 1.7‡ | 12 | ||
| D1532C | 36.1 ± 2.4‡ | 9 | ||
| G1533C | 35.9 ± 3.6‡ | 4 |
Reversal potentials of rSkM1 and cysteine mutants were calculated as described in Fig 2. Values represent the mean ± SEM.
P < 0.05,
P < 0.01.
Loss of Na+ Selectivity by K1237C, W1531C, and D1532C
Relative to other cysteine mutants, K1237C, W1531C, D1532C, and G1533C most profoundly affected the ionic selectivity compared to rSkM1 channels. Since two of these mutants (K1237C and D1532C) were substitutions for strongly hydrophilic charged residues and W1531C involved the replacement of a large aromatic residue, we therefore further characterized the nature of the changes in selectivity in these three mutants. Fig. 4 shows the whole-cell Na+ current recordings of K1237C, W1531C, and D1532C in the presence of equimolar Na+ and K+, and their corresponding current-voltage relationship. K1237C and W1531C channels displayed marked alterations in ionic selectivity towards the monovalent cations tested (Fig. 4, A and B). The ionic selectivity sequences of both K1237C and W1531C was NH4 +>K+>Na+≥Li+≈Cs+. Neither K1237C nor W1531C supported permeation by the quaternary ammonium compound, tetramethylammonium (data not shown). The marked alteration of channel selectivity by W1531C, which is not shared by the other tryptophan mutants in domain I-III (Fig. 3), suggests that this residue and K1237 are important in regulating the selectivity of the rSkM1 Na+ channel.
Figure 4.

Altered ion selectivity of K1237C, W1531C, and D1532C. Whole-cell currents of (A) K1237C, (B) W1531C, and (C) D1532C recorded in a 96 mM NaCl or 96 mM KCl solution. Current recordings were measured from oocytes held at −120 mV and depolarized from −60 to +50 mV. Current traces shown are from −50 to +50 mV in 10-mV increments. Records were corrected for leak and capacitance current. The corresponding current-voltage relationship from the same oocytes shown below, recorded in Na+ (▪), NH4 + (▵), and K+ (○) solutions. Values were normalized to the maximal peak inward current for Na+. Data were fit to a Boltzmann distribution as described in Fig. 2.
The present findings with W1531C suggest that the presence of an aromatic group in domain IV may be critical for Na+ selectivity. To address this, we constructed two conservative mutants, W1531Y and W1531F, as well as the nonconservative mutant W1531A. Fig. 5 illustrates the current ratio of rSkM1 and the tryptophan mutants. W1531C, W1531A, W1531F, and W1531Y were equally permeable to Li+ as rSkM1. A larger difference was observed with NH4 + and K+ current ratios. W1531A channels had NH4 + and K+ current ratios greater than rSkM1 channels but surprisingly much less than W1531C channels (Fig. 5). Alterations in ion selectivity with W1531A were as also reflected in a less positive reversal potential (+39.1 ± 2.9 mV, n = 6; P < 0.05) measured in Na+ solutions. The conservative mutant, W1531Y, displayed NH4 + and K+ current ratios similar to rSkM1. We were unable to detect any measurable inward currents in the presence of NH4 + or K+ solutions with W1531F. Preservation of an aromatic residue in domain IV retained Na+ selectivity, and the results with W1531A suggests that the hydrophobic character is an important determinant of Na+ selectivity in rSkM1 channels.
Figure 5.

Current ratios for tryptophan mutants. Current ratios were calculated as described in Fig. 3. Values represent the mean ± SEM of five to seven oocytes. The P values are *< 0.05, **< 0.01.
A less dramatic but significant alteration in the selectivity of the rSkM1 Na+ channel was observed with the aspartate to cysteine mutation in domain IV (D1532C) (Fig. 4 C). This mutation resulted in a channel that was significantly more permeable to NH4 + and K+ in comparison to the wild-type channel. Surprisingly, other negatively charged amino acids in the other pore regions (D400C, E403C, E758C, and D1241C) had very little effect on selectivity (Fig. 3) with the exception of E755C which was significantly more permeable to NH4 +, but interestingly not to K+.
Divalent Permeation of K1237C
Heinemann and colleagues (1992) demonstrated that replacing the lysine with glutamate in domain III (K1422E in the rat brain II Na+ channel) supported Ca2+ and Ba2+ permeation. We further examined this finding by replacing the equivalent residue with cysteine (K1237C). Exposure of K1237C to Ca2+ or Sr2+ solutions resulted in inward currents comparable to when Na+ was the permeant charge carrier, and rightward shifts in the current-voltage relationship (Fig. 6, A and B). Interestingly, replacement of Na+ in the external bath solution with Mg2+ also resulted in inward currents with a similar voltage-dependent shift in activation while replacement with Ba2+ resulted in barely detectable inward currents (Fig. 6, A and B). These results establish that replacement of K1237 with a residue not identical to that found at the homologous location in Ca2+ channels also support currents by divalents. K1237C channels were equally permeable to Ca2+ and Sr2+ compared to Na+ as measured using maximum conductance measurements (Fig. 6 C).
Figure 6.

Divalent permeation of K1237C. (A) Current recordings of K1237C in the presence of 96 mM Na+, Ca2+, Sr2+, Mg2+, and Ba2+ solutions. Currents were recorded as described in Fig. 4. (B) Corresponding current-voltage relationship from the same oocyte shown in A. Data were fit as described in Fig. 4. (C) Conductance-voltage relationship of K1237C in the presence of Na+ and the divalent cations. Symbols are the same as denoted in B. Data were normalized to the maximal Na+ conductance and fit to a Boltzmann distribution function. Values represent the mean ± SEM of five oocytes.
Since Cd2+ binds with high affinities to free sulfhydryls, we and others have used this cation as a biophysical probe to determine the spatial orientation of the amino acid side-chains by examining the Cd2+ sensitivity of the cysteine mutated channels (Chiamvimonvat et al., 1996a ; Li et al., 1996; Pérez-García et al., 1996). rSkM1 is relatively insensitive to Cd2+ block (K d = 1.8 ± 0.4 mM, n = 7), and whole-cell currents are unaffected by methanethiosulfonate (MTS) derivatives. Furthermore, there is no change in the Cd2+ sensitivity of rSkM1 after MTSEA exposure (K d = 1.9 ± 0.3 mM, n = 3). K1237C, W1531C, and D1532C all appear to have their side-chain residues accessible to the aqueous pore environment as revealed by the modification by the MTS derivatives which modify free sulfhydryl groups, and the enhanced sensitivity of these cysteine mutants to Cd2+ block compared to rSkM1 (Fig. 7). This enhanced Cd2+ sensitivity could be reversed by MTS modification (Fig. 7). These findings suggest that the alteration in selectivity result from changes in the interaction between the permeating ion and channel and not due to structural transformations of the channel protein.
Figure 7.

Effect of MTS modification on the current-voltage relationship (left) and Cd2+ sensitivity (right) of (A) K1237C, (B) W1531C, and (C) D1532C. Currents were recorded as described in Fig. 4 in the absence and presence of 1 mM MTS derivative denoted. Fractional whole-cell currents (I/Io) measured at −10 mV were plotted as a function of Cd2+ concentration and fit to the Hill equation I/I o = 1/(1 + [Cd2+]/K d) assuming a single binding site. The Kd for Cd2+ block for rSkM1 was 1,865 ± 386 μM for rSkM1 (n = 7). The Kd for Cd2+ block prior to and after modification by the MTS derivative was (A) K1237C 124 ± 22 μM (▪, n = 3) and 2,154 ± 182 μM (□, n = 3); (B) W1531C 55 ± 7 μM (○, n = 6) and 1,980 ± 176 μM (•, n = 3); (C) D1532C 583 ± 70 μM (▴, n = 6) and 2,325 ± 287 μM (▵, n = 3), respectively. Values represent the mean ± SEM.
Can Sulfhydryl Modification Reconstitute Na+ Selectivity?
A simple interpretation of the above data suggests that removal of the positively charged lysine side-chain, the aromatic moiety of tryptophan, or the negatively charged carboxylate residue may be responsible for the alterations in ionic selectivity. We hypothesized that chemically reintroducing similar groups into the pore region would restore Na+ selectivity to the channel. We used the membrane-impermeant MTS derivatives to examine this question. To restore ionic selectivity to K1237C, W1531C, and D1532C, we used the positively charged (MTSEA, MTSET) or negatively charged (MTSES) compounds, respectively.
The effects of sulfhydryl modification of K1237C with 1 mM MTSEA are illustrated in Fig. 8 A. MTSEA resulted in a reduction in peak inward current which may result from an obstruction of the conduction pathway (Fig. 7 A). Sulfhydryl modification by MTSEA was irreversible since the reduction in peak current persisted even after washout of the agent, as has been shown by other investigators (Akabas et al., 1992; Kirsch et al., 1994; Kürz et al., 1995). Modification was also confirmed by the reduced sensitivity of the channel to Cd2+ block (Fig. 7.; Li et al., 1996). Both the reduced peak inward current and the reduced sensitivity to Cd2+ could be restored after the application of dithiothreitol (data not shown). MTSEA modification did not reconstitute Na+ selectivity to the K1237C mutant since this mutant remained nonselective towards the mono-valent cations as demonstrated by the high degree of permeation by NH4 + and K+ (Fig. 8 A) and by the lack of change in the permeability ratios of K1237C after sulfhydryl modification (Table III). MTS-modified K1237C channels remained permeable to divalent cations. These data demonstrate that sulfhydryl modification of K1237C by MTSEA could not restore rSkM1-like monovalent or divalent cation selectivity to these channels. Similar studies were performed with W1531C (Fig. 8 B) and D1532C (Fig. 8 C). Modification of W1531C with 1 mM MTSET, which introduces a bulky trimethyl-ammonium group into the pore, reduced peak inward current but had no effect on selectivity as perceived by the lack of change in the NH4 + and K+ current and the permeability ratios (see Fig. 9 B, Table III). We also examined the effects of the aromatic MTS derivative, methanethiosulfonate-ethylbenzoate (MTSEB), which would introduce the more appropriate phenolic group into the pore. However, attempts to chemically modify the cysteine residue with MTSEB failed since there was no change in the Cd2+ sensitivity of W1531C after MTSEB exposure. Failure of MTSEB to modifiy this channel mutant may be due to the inaccessibility of this reagent to the site within the pore. Lastly, replacement of a negative charge into D1532C channels using MTSES modification resulted in an enhancement of current (Fig. 7 C) but again did not restore channel selectivity (Fig. 8 C).
Figure 8.

Effect of sulfhydryl modification of (A) K1237C, (B) W1531C, and (C) D1532C on ionic selectivity. Whole-cell currents were recorded in the presence of 96 mM NaCl or KCl solutions as described in Fig. 4. The corresponding current-voltage relationships of the cysteine mutants in the presence of Na+ (▪), K+ (○), and NH4 + (▴) are shown below. K1237C, W1531C, and D1532C were modified with 1 mM of MTSEA, MTSET, and MTSES, respectively.
Table III.
Permeability Ratios of rSkM1, K1237C, W1531C, and D1532C: Effect of Sulfhydryl Modification
| PLi/PNa | PNH4/PNa | PK/PNa | PCs/PNa | |||||
|---|---|---|---|---|---|---|---|---|
| rSkM1 | 0.97 ± 0.06 | 0.17 ± 0.02 | 0.05 ± 0.01 | 0.02 ± 0.01 | ||||
| K1237C | 1.00 ± 0.03 | 1.19 ± 0.04 | 1.01 ± 0.06 | 0.87 ± 0.03 | ||||
| K1237C + MTSEA | 1.01 ± 0.02 | 1.15 ± 0.05 | 1.02 ± 0.03 | 0.86 ± 0.02 | ||||
| W1531C | 1.01 ± 0.01 | 1.24 ± 0.01 | 1.03 ± 0.02 | 0.89 ± 0.05 | ||||
| W1531C + MTSET | 1.03 ± 0.02 | 1.23 ± 0.02 | 0.99 ± 0.01 | 0.90 ± 0.01 | ||||
| D1532C | 0.88 ± 0.07 | 0.56 ± 0.04 | 0.20 ± 0.03 | 0.11 ± 0.01 | ||||
| D1532C + MTSES | 0.96 ± 0.04 | 0.54 ± 0.06 | 0.22 ± 0.04 | 0.07 ± 0.03 |
Permeability ratios (PX/PNa) were calculated from reversal potentials using a modified Goldman-Hodgkin-Katz equation as described in materials and methods. Channels were modified with 1 mM of the corresponding MTS derivative. Values represent the mean ± SEM of three to five experiments.
Figure 9.

Effect of extracellular pH on rSkM1 and D1532C. (A) Effect of pH 5.6 (○), 7.6 (▪), and 9.6 (▵) on the current-voltage relationship of rSkM1 and D1532C. Currents were recorded as described in Fig. 4. (B) Effect of extracellular pH on the reversal potential (ENa) of rSkM1 (▪) and D1532C (○). Data represent the mean ± SEM of four to five oocytes. P values as in Fig. 3.
These data suggest that a simple reintroduction of a positively charged group, bulky hydrophobic moiety, or negatively charged group into the pore of K1237C, W1531C, or D1532C, respectively, is not sufficient to reestablish selectivity. However, these findings do not negate the importance of the side-chains of these residues for selectivity since not only the charge or size of the side-chain, but the localization of these groups within the pore may influence selectivity. Modification of the cysteine mutants with the MTS derivatives most likely do not localize the replaced group within the same vicinity because of their attachment to the ethyl alkyl chain.
Effects of Extracellular pH on the Selectivity of D1532C
To determine whether the localization of the negatively charged group is critical for determining ionic selectivity, we examined the effects of extracellular pH on D1532C mutant channels. We reasoned that alterations in extracellular pH will affect the ionized state of the cysteinyl sulfhydryl side-chain (pKa cysteine 8.5) thereby reintroducing a negative charge. Fig. 9 illustrates the effects of changing extracellular pH on the current-voltage relationship of the rSkM1 and D1532C Na+ channel. Decreasing the extracellular pH from 7.6 to 5.6 resulted in a reduction in peak inward current and a rightward shift in the current-voltage relationship of the rSkM1 Na+ channel (Fig. 9 A). These effects have been previously attributed to proton block of the channel and screening of negative surface charges (Woodhull, 1973). Elevating the pH from 7.6 to 9.6 produced a modest increase in peak current of rSkM1. Increasing the extracellular pH had two effects on D1532C. There was a large increase in peak inward current at pH 9.6 (Fig. 9 A). Secondly, there was a positive shift in the reversal potential as extracellular pH was made more basic (Fig. 9 A). This effect is more clearly illustrated in Fig. 9 B where the reversal potential (ENa) is plotted as a function of the extracellular pH. There was a shift in the reversal potential from +33.2 ± 2.2 mV (pH 5.6) to +41.3 ± 1.6 mV (pH 9.6) (n = 5) which tended to converge towards the rSkM1 curve. Changes in pH did not influence ENa of rSkM1 (ENa: +47.1 ± 2.6 mV, pH 5.6; +46.9 ± 3.1 mV, pH 9.6; n = 4). As observed with rSkM1, inward currents of D1532C were reduced and there was a depolarizing shift in activation at pH 5.6 (Fig. 9 A). These data suggest that not only the charge but the location of the negatively-charged group is critical for influencing ionic selectivity.
discussion
Mutagenesis studies have revealed critical residues important for a number of intrinsic properties of the Na+ channel including activation and inactivation (Stühmer et al., 1989; West et al., 1992) and ionic selectivity (Heinemann et al., 1992). We have further used mutagenic strategies to probe the spatial orientation of the pore-forming residues of the rat skeletal muscle Na+ channel (Li et al., 1996). Most of the residues mutated were exposed to the aqueous pore environment. We have furthered these studies and examined the contribution of these residues to ion selectivity. Cysteine mutations within the pore appear to have localized effects since all but one cysteine mutant (G1238C) expressed functional channels with relatively little effect on channel gating.
Alterations in Selectivity Revealed by Cysteine Mutations
The present study demonstrated that four residues markedly influence ion selectivity of the rSkM1 Na+ channel; K1237, W1531, D1532, and G1533. Three of these residues (W1531, D1532, and G1533) have not been previously implicated in forming the selectivity filter of the Na+ channel. Studies with the rat brain II Na+ channel (Terlau et al., 1991; Heinemann et al., 1992) have suggested four amino acids (aspartate, glutamate, lysine, alanine), one in each of the four pore regions form the selectivity filter. More notably, the lysine and alanine residues in domain III and IV had the most influence on Na+ selectivity. Mutating both these residues to glutamate, which are found at the equivalent positions in the Ca2+ channel, conferred Ca2+ channel characteristics to the Na+ channel. The single lysine to glutamate mutation alone had a large effect on ion selectivity of the channel such that the channel was relatively nonselective towards the monovalent cations tested, similar to the loss of selectivity we observed with K1237C. Furthermore, as observed with the lysine to glutamate mutant, our K1237C mutant showed appreciable inward currents in the presence of divalent cations. The selectivity sequence of K1237C for divalent cations based on conductance and reversal potential measurements was Ca2+≥Sr2+>Mg2+>Ba2+. However, the selectivity sequence of K1237C for the alkaline earth cations cannot be inferred from measurements of atomic radii (Ba2+>Sr2+>Ca2+>Mg2+) or hydration energies (Mg2+>Ca2+>Sr2+>Ba2+) (Hille, 1992), as it can be for the L-type Ca2+ channel using single-channel conductance (Ba2+>Sr2+∼Ca2+) or reversal potentials (Ca2+>Sr2+>Ba2+) (Hess et al., 1986). L-type Ca2+ channels were impermeable to Mg2+. The permeation of Mg2+ through the K1237C channel is quite surprising since Mg2+ does not permeate most ion channels due to the slow rate of dehydration (Diebler et al., 1969). Therefore, dehydration of the ion may not play a significant role in Mg2+ permeation through this channel. The finding that Ba2+ is weakly permeable through the K1237C channel is also unexpected since Ba2+ has a higher dehydration rate than the other divalent cations tested (Diebler et al., 1969) and a similar atomic radius as the permeant K+ ion (Hille, 1992). The presence of a positively charged residue at position 1237 in domain III appears to be critical in preventing divalent permeation since the mutation K1237R abolished Ca2+ conductance, however, still rendered the channel nonselective towards monovalent cations (Favre et al., 1996).
Negatively charged residues have a strong influence on ion selectivity and permeation of Na+ channels (Terlau et al., 1991; Heinemann et al., 1992; Chiamvimonvat et al., 1996a , b ) and Ca2+ channels (Kim et al., 1993; Mikala et al., 1993; Yang et al., 1993; Ellinor et al., 1995), whereas for voltage-gated K+ channels, less hydrophilic residues control K+ selectivity (Yool and Schwarz, 1991; Taglialatela et al., 1993; Heginbotham et al., 1994). It has been speculated that the conserved tyrosine residue within the pore of the voltage-gated K+ channels is important for coordinating cation-π interactions between the permeating K+ ion and the channel protein (Heginbotham and MacKinnon, 1992; Kumpf and Dougherty, 1993; Lü and Miller, 1995) and furthermore, may form part of the selectivity filter of the channel (Durell and Guy, 1992). It is intriguing that mutating the hydrophobic tryptophan residue in domain IV to cysteine (W1531C) but not the other tryptophan residues in the domains I-III rendered the channel unable to discriminate against the monovalent cations tested. Furthermore, maintaining an aromatic group at this position (W1531F and W1531Y) retained selectivity similar to that of the wild-type channel. The alterations observed with W1531C strongly suggest that size and chemical nature of the residue at this position is a critical determinant in conferring Na+ selectivity to rSkM1.
D1532C showed prominent inward current with both NH4 + and K+. Mutating the negatively charged residues in the domains I-III (D400C, E403C, E755C, E758C, and D1241C) did not have as a dramatic effect on selectivity as D1532C, as has been previously observed (Heinemann et al., 1992; Kontis and Goldin, 1993; Chiamvimonvat et al., 1996a , b ; Favre et al., 1996). The importance of negatively charged residues in the pore have been attributed to electrostatic focusing or binding of the permeant ions and determinants of ion translocation for the voltage-gated Na+ channel (Hille, 1972; Terlau et al., 1991; Chiamvimonvat et al., 1996b ). Our data further support this notion and that the negatively charged residue in domain IV plays a critical role in Na+ permeation. These data and the results with W1531C also suggest that each domain does not contribute equally to the property of ion selectivity. Such asymmetry in the conduction pathway has been recently described for the Na+ (Chiamvimonvat et al., 1996a , b ) and Ca2+ channel (Kim et al., 1993; Mikala et al., 1993; Yang et al., 1993; Ellinor et al., 1995).
Both glycine residues in domain IV (G1530C and G1533C) disrupted selectivity. Glycines allow for high degree of protein backbone flexibility and, furthermore, are major constituents of β hairpin turns within proteins (Creighton, 1993). It may be possible that the two glycine residues are involved in allowing for the coordination of a Na+ ion with the channel protein, through cation-π interactions with W1531 (Dougherty, 1996) or electrostatic interactions with D1532 (Chiamvimonvat et al., 1996), as has been suggested for the glycine residues located in the “signature sequence” of the voltage-gated K+ channel (Heginbotham et al., 1994) and in the Ca2+ channel (Kim et al., 1993).
Heinemann and colleagues (1992) demonstrated the importance of the lysine residue in domain III in conferring Na+ selectivity to the Na+ channel and suggested that it comprised part of the selectivity filter. A more recent study further examined the role of this residue on selectivity and demonstrated the importance of the chemical nature at this locus in domain III for determining Na+ selectivity over K+ and Ca2+ (Favre et al., 1996). The present study supports these findings and furthermore demonstrates that the residues in domain IV influence ion selectivity more so than the residues in the other domains. The unique characteristic of this region may be associated with the high degree of flexibility within the pore region of domain IV, as we have recently demonstrated (Li et al., 1996). This property of the channel may be important for ion coordination, selectivity and permeation.
Effect of Sulfhydryl Modification on Selectivity
The development of the methanethiosulfonate compounds has provided a unique probe to examine the tertiary nature of the pore-forming residues in ion channels when combined with cysteine scanning mutagenesis (Akabas et al., 1992). We (Li et al., 1996) and others (Akabas et al., 1992; Kürz et al., 1995; Pascual et al., 1995; Pérez-García et al., 1996) have used these compounds to study the topology of pore residues in a number of ion channels. We have further employed these probes to determine whether ion selectivity can be restored by reintroducing specific groups into the pore to mimic the mutated residue. Modification of K1237C, W1531C, and D1532C with specific methanethiosulfonate derivatives which reproduce the positively charged, bulky moiety, or negatively charged side-chains did not restore selectivity suggesting that a simple replacement is not sufficient for reconstituting selectivity of the channel. Furthermore, the presence of the positively charged ammonium group within the pore of K1237C did not prevent divalent permeation. This is unlike the finding with K1237R which exhibited no Ca2+ permeation (Favre et al., 1996). Recently, Marban and colleagues demonstrated that the loss in selectivity of rSkM1 with D1532C could be partially restored by 10 mM MTSES as measured by whole-cell flux and single-channel permeability ratios (Chiamvimonvat et al., 1996b ). Although we used 1 mM MTSES to modify D1532C channels, we observed a similar 50% increase in peak current (Fig. 8 C) as observed with 10 mM MTSES (Chiamvimonvat et al., 1996b ) suggesting that differences in the degree of channel modification cannot explain the discrepancy in results.
Extracellular pH Influences Selectivity
It should be noted that conservative replacements of pore-forming amino acids do not ensure conservation of selectivity and vice versa, as has been demonstrated for the L-type Ca2+ channel (Ellinor et al., 1995) and voltage-gated K+ channels (Taglialatela et al., 1993; Heginbotham et al., 1994). Side-chain length, charge, and polarity may all influence selectivity of the channel. In this regard, although we have tried to preserve charge and polarity using the MTS compounds, the localization of the desired groups is confounded by their attachment to an ethyl alkyl chain. To overcome this problem, we changed the extracellular pH to alter the ionized state of the sulfhydryl side-chain of D1532C to maintain side-chain length, charge, and polarity. D1532C became more selective as we changed the cysteinyl sulfhydryl side-chain to a thiolate derivative as extracellular pH increased. This further supports our findings that this negatively charged residue is important for ion selectivity.
Summary
We have demonstrated that four residues (K1237, W1531, D1532, and G1533) in the pore of the rat skeletal muscle Na+ channel play an important role in ion selectivity. Three of these residues have not been shown to influence ion selectivity, and one in particular (W1531) appears to strongly regulate selectivity. It is reasonable to suggest that W1531 also forms part of the selectivity filter given the degree of nonselectivity that is observed when this residue is mutated to cysteine. We speculate that this residue may directly interact with the positively charged lysine group in domain III or with the permeating Na+ ion through cation-π interactions which have been recently ascribed to be important for a number of biological interactions (Dougherty, 1996).
Acknowledgments
We thank Dr. Andrew Wooley of the Department of Chemistry for synthesizing the methanethiosulfonate-ethylbenzoate derivative.
This work was supported by the Medical Research Council of Canada. P.H. Backx is a Medical Research Council of Canada Scholar. R.G. Tsushima was supported by a fellowship from the Department of Medicine, University of Toronto.
Footnotes
Abbreviations used in this paper: MTSEA, methanethiosulfonate-ethyl-ammonium; MTSEB, methanethiosulfonate-ethylbenzoate; MTSES, methanethiosulfonate-ethylsulfonate; MTSET, methanethiosulfonate-ethyltrimethylammonium; rSkM1, rat skeletal muscle Na+ channel.
references
- Akabas MH, Stauffer DA, Xu M, Karlin A. Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science (Wash DC) 1992;258:307–310. doi: 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
- Backx PH, Yue DT, Lawrence JH, Marban E, Tomaselli GF. Molecular localization of an ion-binding site within the pore of mammalian sodium channels. Science (Wash DC) 1992;257:248–251. doi: 10.1126/science.1321496. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and function of voltage-gated ion channels. Annu Rev Biochem. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- Chiamvimonvat N, Pérez-García MT, Fanjan R, Marban E, Tomaselli GF. Depth asymmetries of the pore-lining segments of the Na+channel revealed by cysteine mutagenesis. Neuron. 1996a;16:1037–1047. doi: 10.1016/s0896-6273(00)80127-0. [DOI] [PubMed] [Google Scholar]
- Chiamvimonvat N, Pérez-García MT, Tomaselli GF, Marban E. Control of ion flux and selectivity by negatively charged residues in the outer mouth of rat sodium channels. J Physiol (Lond) 1996b;491:51–59. doi: 10.1113/jphysiol.1996.sp021195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton, T.E. 1993. Proteins. Structures and Molecular Properties. W. H. Freeman and Company, New York.
- Diebler H, Eigen M, Ilgenfritz G, Maas G, Winkler R. Kinetics and mechanism of reactions of main group metal ions with biological carriers. Pure Appl Chem. 1969;20:93–115. [Google Scholar]
- Dougherty DA. Cation-π interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science (Wash DC) 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- Durell SR, Guy HR. Atomic scale structure and functional models of voltage-gated potassium channels. Biophys J. 1992;62:238–250. doi: 10.1016/S0006-3495(92)81809-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman G, Horn R. Ionic selectivity revisited: the role of kinetic and equilibrium processes in ion permeation through channels. J Membr Biol. 1983;76:197–225. doi: 10.1007/BF01870364. [DOI] [PubMed] [Google Scholar]
- Ellinor PT, Yang J, Sather WA, Zhang J-F, Tsien RW. Ca2+ channel selectivity at a single locus for high-affinity Ca2+interactions. Neuron. 1995;15:1121–1132. doi: 10.1016/0896-6273(95)90100-0. [DOI] [PubMed] [Google Scholar]
- Favre I, Moczydlowski E, Schild L. On the structural basis for ionic selectivity among Na+, K+, and Ca2+in the voltage-gated sodium channel. Biophys J. 1996;71:3110–3125. doi: 10.1016/S0006-3495(96)79505-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann HA, Kirsch GE, Drewe JA, Taglialatela M, Joho RH, Brown AM. Exchange of conduction pathways between two related K+channels. Science (Wash DC) 1991;251:942–944. doi: 10.1126/science.2000495. [DOI] [PubMed] [Google Scholar]
- Heginbotham L, MacKinnon R. The aromatic binding site for tetraethylammonium ion on potassium channels. Neuron. 1992;8:483–491. doi: 10.1016/0896-6273(92)90276-j. [DOI] [PubMed] [Google Scholar]
- Heginbotham L, Lu Z, Abramson T, MacKinnon R. Mutations in the K+channel signature sequence. Biophys J. 1994;66:1061–1067. doi: 10.1016/S0006-3495(94)80887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann SH, Terlau H, Stühmer W, Imoto K, Numa S. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature (Lond) 1992;356:441–443. doi: 10.1038/356441a0. [DOI] [PubMed] [Google Scholar]
- Hess P, Lansman JB, Tsien RW. Calcium channel selectivity for divalent and monovalent cations. Voltage and concentration dependence of single channel current in ventricular heart cells. J Gen Physiol. 1986;88:293–319. doi: 10.1085/jgp.88.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. The permeability of the sodium channel to metal cations in myelinated nerve. J Gen Physiol. 1972;59:637–658. doi: 10.1085/jgp.59.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille, B. 1992. Ionic Channels of Excitable Membranes. 2nd Ed. Sinauer Associates, Inc. Sunderland, MA.
- Isom LL, DeJongh KS, Patton DE, Reber BFX, Offord J, Carbonneau H, Walsh K, Goldin AL, Catterall WA. Primary structure and functional expression of the β1-subunit of the rat brain sodium channel. Science (Wash DC) 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- Kim MS, Morii T, Sun LX, Imoto K, Mori Y. Structural determinants of ion selectivity in brain calcium channel. FEBS Lett. 1993;318:145–148. doi: 10.1016/0014-5793(93)80009-j. [DOI] [PubMed] [Google Scholar]
- Kirsch GE, Alam M, Hartmann HA. Differential effects of sulfhydryl reagents on saxitoxin and tetrodotoxin block of voltage-dependent Na channels. Biophys J. 1994;67:2305–2315. doi: 10.1016/S0006-3495(94)80716-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontis KJ, Goldin AL. Site-directed mutagenesis of the putative pore region of the rat IIA sodium channel. Mol Pharmacol. 1993;43:635–644. [PubMed] [Google Scholar]
- Kumpf RA, Dougherty DA. A mechanism for ion selectivity in potassium channels: computational studies of cation-π interactions. Science (Wash DC) 1993;261:1708–1710. doi: 10.1126/science.8378771. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kürz LL, Zühlke RD, Zhang H-J, Joho RH. Side-chain accessibilities in the pore of a K+channel probed by sulfhydryl-specific reagents after cysteine-scanning mutagenesis. Biophys J. 1995;68:900–905. doi: 10.1016/S0006-3495(95)80266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R., R. Tsushima, and P. Backx. 1996. Determination of Na+ channel pore structure using single and multiple cysteine substitutions. Biophys. J. 70:A24 (Abstr.)
- Lü Q, Miller C. Silver as a probe of pore-forming residues in a potassium channel. Science (Wash DC) 1995;268:304–307. doi: 10.1126/science.7716526. [DOI] [PubMed] [Google Scholar]
- Mikala G, Bahinski A, Yatani A, Tang S, Schwartz A. Differential contribution by conserved glutamate residues to an ion-selectivity site in the L-type Ca2+channel pore. FEBS Lett. 1993;335:265–269. doi: 10.1016/0014-5793(93)80743-e. [DOI] [PubMed] [Google Scholar]
- Pascual JM, Shieh C-C, Kirsch GE, Brown AM. K+pore structure revealed by reporter cysteines at inner and outer surfaces. Neuron. 1995;14:1055–1063. doi: 10.1016/0896-6273(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Pérez-García MT, Chiamvimonvat N, Marban E, Tomaselli GF. Structure of the sodium channel pore revealed by serial cysteine mutagenesis. Proc Natl Acad Sci USA. 1996;93:300–304. doi: 10.1073/pnas.93.1.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stühmer W, Conti F, Suzuki H, Wang X, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature (Lond) 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Taglialatela M, Drewe JA, Kirsch GE, De Biasi M, Hartmann HA, Brown AM. Regulation of K+/Rb+ selectivity and internal TEA blockade by mutations at a single site in K+pores. Pflüg Arch. 1993;423:104–112. doi: 10.1007/BF00374967. [DOI] [PubMed] [Google Scholar]
- Terlau H, Heinemann SH, Stühmer W, Pusch M, Conti F, Imoto K, Numa S. Mapping the site of block by tetrodo-toxin and saxitoxin of sodium channel II. FEBS Lett. 1991;293:93–96. doi: 10.1016/0014-5793(91)81159-6. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhull AM. Ionic blockade of sodium channels in nerve. J Gen Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ellinor PT, Sather WA, Zhang J-F, Tsien RW. Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+channels. Nature (Lond) 1993;366:158–161. doi: 10.1038/366158a0. [DOI] [PubMed] [Google Scholar]
- Yellen G, Jurman ME, Abramson T, MacKinnon R. Mutations affecting internal TEA blockade identify the probable pore-forming region. Science (Wash DC) 1991;251:939–942. doi: 10.1126/science.2000494. [DOI] [PubMed] [Google Scholar]
- Yool AJ, Schwartz TL. Alteration of ionic selectivity of a K+channel by mutation of the H5 region. Nature (Lond) 1991;349:700–704. doi: 10.1038/349700a0. [DOI] [PubMed] [Google Scholar]