

Abstract
The success of genomic selection depends on the potential to predict genome-assisted breeding values (GEBVs) with high accuracy over several generations without additional phenotyping after estimating marker effects. Results from both simulations and practical applications have to be evaluated for this potential, which requires linkage disequilibrium (LD) between markers and QTL. This study shows that markers can capture genetic relationships among genotyped animals, thereby affecting accuracies of GEBVs. Strategies to validate the accuracy of GEBVs due to LD are given. Simulations were used to show that accuracies of GEBVs obtained by fixed regression–least squares (FR–LS), random regression–best linear unbiased prediction (RR–BLUP), and Bayes-B are nonzero even without LD. When LD was present, accuracies decrease rapidly in generations after estimation due to the decay of genetic relationships. However, there is a persistent accuracy due to LD, which can be estimated by modeling the decay of genetic relationships and the decay of LD. The impact of genetic relationships was greatest for RR–BLUP. The accuracy of GEBVs can result entirely from genetic relationships captured by markers, and to validate the potential of genomic selection, several generations have to be analyzed to estimate the accuracy due to LD. The method of choice was Bayes-B; FR–LS should be investigated further, whereas RR–BLUP cannot be recommended.
DUE to advances in molecular genetics, genomewide dense marker data are becoming available for livestock species. These can be used to estimate genome-assisted breeding values (GEBVs) as proposed by Meuwissen et al. (2001). First, marker effects are estimated with a training data set containing individuals with marker genotypes and trait phenotypes. Then, GEBVs of any genotyped individual in the population can be calculated using the estimated marker effects. The greatest advantage of this approach is the potential to predict GEBVs with high accuracy over several generations without repeated phenotyping, which results in lower costs and shorter generation intervals. This approach requires linkage disequilibrium (LD) between marker loci and quantitative trait loci (QTL), otherwise the accuracy is expected to decline fast in the generations following the estimation of marker effects. In simulation studies, Meuwissen et al. (2001) and Solberg et al. (2006) predicted the true breeding values of offspring of individuals in the training data to validate the potential advantage of GEBVs. In practical applications, cross-validation with individuals from the same population is used, and either breeding values estimated from trait phenotypes and pedigree data or progeny means corrected for environmental effects and EBVs of mates are used to validate the potential advantage of GEBVs. Thus, both in simulation and in practical applications, individuals in the validation group are related to individuals in the training data. However, markers used in the statistical models to estimate marker effects can also capture additive genetic relationships between individuals (Fernando 1998), defined here as twice the coefficient of coancestry given by Malécot (1948). This will affect the accuracy of GEBVs and thus, even if markers are not in LD with QTL, the accuracy of GEBVs will be nonzero. Furthermore, if markers are in LD with QTL, the accuracy of GEBVs is expected to be higher than accuracy due to LD alone. Legarra et al. (2007) analyzed accuracies of GEBVs for individuals related to the training data and those for individuals that were unrelated in a mouse population. They concluded that markers were able to recover family information to some extent.
The objective of this study was to show how genetic relationships between individuals are captured by markers in the statistical models used by Meuwissen et al. (2001) to estimate marker effects for prediction of GEBVs. Simulated data were used to analyze how this affects the accuracy of GEBVs over generations. On the basis of these results, strategies to validate the advantage of GEBVs due to LD in practical applications were derived.
THEORY
Statistical models:
Three statistical models were used in this study to estimate genomewide SNP-marker effects for use in computing GEBVs: fixed regression–least squares (FR–LS), random regression–BLUP (RR–BLUP), and Bayes-B as described by Meuwissen et al. (2001). The basic model underlying these methods can be written as
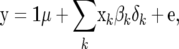 |
(1) |
where 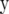 is the vector of trait phenotypes, μ is the overall mean,
is the vector of trait phenotypes, μ is the overall mean, 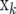 is a column vector of marker genotypes at locus k, βk is the marker effect, δk is a 0/1-indicator variable, and
is a column vector of marker genotypes at locus k, βk is the marker effect, δk is a 0/1-indicator variable, and 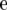 is the vector of random residual effects. In
is the vector of random residual effects. In 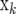 , the marker genotype of an individual is coded as the number of copies of one SNP allele it carries, i.e., 0, 1, or 2; βk is treated as fixed in FR–LS and as random in RR–BLUP and Bayes-B. The indicator variable δk = 1 for all marker loci in RR–BLUP, whereas δk can be 0 or 1 in FR–LS and Bayes-B.
, the marker genotype of an individual is coded as the number of copies of one SNP allele it carries, i.e., 0, 1, or 2; βk is treated as fixed in FR–LS and as random in RR–BLUP and Bayes-B. The indicator variable δk = 1 for all marker loci in RR–BLUP, whereas δk can be 0 or 1 in FR–LS and Bayes-B.
Let X be a matrix containing the vectors 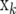 and
and 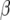 be a vector containing the elements βk for all marker loci for which δk = 1. Then, the expected value of
be a vector containing the elements βk for all marker loci for which δk = 1. Then, the expected value of 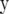 is
is 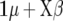 for FR–LS and
for FR–LS and 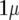 for RR–BLUP and Bayes-B. Furthermore, the variance of
for RR–BLUP and Bayes-B. Furthermore, the variance of 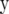 is I
is I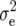 for FR-LS, XX′
for FR-LS, XX′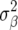 + I
+ I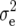 for RR–BLUP (assuming equal variance for each SNP), and
for RR–BLUP (assuming equal variance for each SNP), and 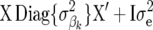 for Bayes-B, i.e., allowing SNP-specific variances. Note that the dimensions of X and
for Bayes-B, i.e., allowing SNP-specific variances. Note that the dimensions of X and 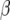 can be different for the three methods.
can be different for the three methods.
In Bayes-B, the prior probability of 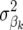 to be nonzero was defined by Meuwissen et al. (2001) as the expected proportion of segregating QTL to the total number of QTL on the genome. Note that when
to be nonzero was defined by Meuwissen et al. (2001) as the expected proportion of segregating QTL to the total number of QTL on the genome. Note that when 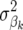 = 0 in an iteration of the Bayes approach, then δk = 0 and marker locus k is not included in the model in that iteration.
= 0 in an iteration of the Bayes approach, then δk = 0 and marker locus k is not included in the model in that iteration.
As in Meuwissen et al. (2001), FR–LS was implemented as a two-step procedure. In the first step, markers to be included in the model were selected, and in the second step, effects of these markers were estimated to predict GEBVs. In contrast to the study of Meuwissen et al. (2001), FR–LS was implemented as a forward stepwise regression as described in Kutner et al. (2005). First, simple linear regressions were fitted and t-statistics were calculated for all marker loci as
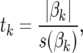 |
where tk is the t-statistic for marker locus k and s(βk) is the standard error of βk. Then, the marker locus with the lowest P-value was included in the model, if its P-value was lower than a predefined threshold α. If a marker was included in the model in the first step, the remaining marker loci were individually fitted together with the previously included marker. Another marker was added to the model, if its P-value was the lowest of the remaining markers and was also lower than α. If the model contained at least two marker loci, t-statistics for markers included earlier were obtained and the marker locus with the highest P-value greater than α was dropped from the model. The algorithm proceeded until no marker locus could be added to the model and no marker locus in the model could be dropped. Marker effects estimated from the final model were used to predict GEBVs.
Another difference compared to Meuwissen et al. (2001) is that 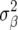 used in RR–BLUP was
used in RR–BLUP was 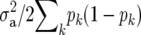 , where
, where 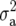 is the additive genetic variance, and pk is the allele frequency at marker locus k. The reason for doing so will be clear in the following section. In Meuwissen et al. (2001), in contrast,
is the additive genetic variance, and pk is the allele frequency at marker locus k. The reason for doing so will be clear in the following section. In Meuwissen et al. (2001), in contrast, 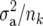 was used, where nk is the number of marker loci.
was used, where nk is the number of marker loci.
Genetic relationships captured by markers:
Denote the ith row of X by 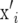 containing the marker genotypes of individual i. Thus, element i, j of XX′ is calculated by
containing the marker genotypes of individual i. Thus, element i, j of XX′ is calculated by 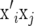 , where j denotes another individual. Treating
, where j denotes another individual. Treating 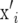 and
and 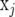 as random, the expected value of
as random, the expected value of 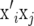 is
is
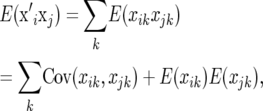 |
(2) |
where k denotes marker locus k. The covariance term in (2) is aij2pk(1 − pk), where aij is the genetic relationship coefficient between individuals i and j. Also, the expected value of xik is 2pk. Consequently, 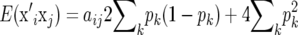 and thus
and thus
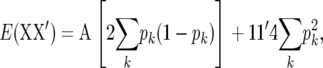 |
(3) |
which is proportional to A, apart from a constant. Note that, as the number of independent marker loci goes to infinity, XX′ converges to (3). Thus, the extent to which XX′ approximates A depends on the number of loci.
To see how genetic relationships in XX′ enter into RR–BLUP, consider the standard animal model
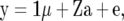 |
(4) |
where Z is an incidence matrix and 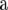 is the vector of additive genetic effects of individuals with data in
is the vector of additive genetic effects of individuals with data in 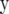 . This model used the same trait phenotypes as the other models, and further information from genetic relationships only, and is referred to as trait-pedigree–BLUP (TP–BLUP). For this model, the variance of
. This model used the same trait phenotypes as the other models, and further information from genetic relationships only, and is referred to as trait-pedigree–BLUP (TP–BLUP). For this model, the variance of 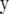 is
is
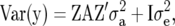 |
(5) |
where 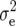 is the residual variance.
is the residual variance.
Suppose we replace A in (5) by 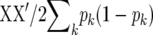 . Then, (5) is identical to the variance of
. Then, (5) is identical to the variance of 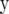 for RR–BLUP, and TP–BLUP of
for RR–BLUP, and TP–BLUP of  is identical to
is identical to 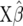 of RR–BLUP (Fernando 1998; Vanraden 2007). Note that the second term in (3) is constant for all individuals in the population and therefore its square root will be captured by the mean in the statistical model. If the number of marker loci goes to infinity and given a fixed number of trait phenotypes, RR–BLUP is equivalent to the animal model in (4) that uses the well-known numerator relationship matrix.
of RR–BLUP (Fernando 1998; Vanraden 2007). Note that the second term in (3) is constant for all individuals in the population and therefore its square root will be captured by the mean in the statistical model. If the number of marker loci goes to infinity and given a fixed number of trait phenotypes, RR–BLUP is equivalent to the animal model in (4) that uses the well-known numerator relationship matrix.
The above derivation did not require LD. Genetic relationships can enter into the analysis regardless of the amount of LD between markers and QTL. Thus, the accuracy of GEBVs is nonzero even without LD. This is also true for FR–LS and Bayes-B, which are related to RR–BLUP through modification of the marker variances. In addition, the number of markers fitted can be different for these methods. When markers are the QTL or are in LD with QTL, however, 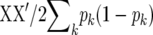 provides more information about the covariance between relatives than the numerator relationships matrix A in (5), because variation in relationships, e.g., between full sibs, is taken into account (Nejati-Javaremi et al. 1997; Vanraden and Tooker 2007).
provides more information about the covariance between relatives than the numerator relationships matrix A in (5), because variation in relationships, e.g., between full sibs, is taken into account (Nejati-Javaremi et al. 1997; Vanraden and Tooker 2007).
SIMULATION
Two main scenarios were considered in this study. The first was to demonstrate that the accuracy of GEBVs can be nonzero even without LD between markers and QTL, and the second was to analyze a more realistic situation in which markers are in LD with QTL. These two scenarios were (1) no LD between markers and QTL and (2) a population with LD based on mutation–drift equilibrium. The first scenario can also be considered as the worst case for genomic selection. The following description applies generally to both scenarios.
All simulations started with a base population of 100 individuals. Biallelic QTL effects were sampled from a standard normal distribution and alleles were sampled from a Bernoulli distribution with frequency 0.5. This differs from similar simulations conducted by, e.g., Meuwissen et al. (2001) who started with a population that was fixed for all loci and simulated multiallelic QTL with effects sampled from a gamma distribution. Starting with a segregating population, however, allowed mutation–drift equilibrium to be reached after 1000 compared to 100,000 generations of random mating. This was checked deterministically and by Monte Carlo simulation. Although the gamma distribution has slightly thicker tails than the normal distribution, use of the normal distribution for QTL effects was shown not to affect results. A mutation rate of 2.5 × 10−5 per generation was applied in the following generations, where mutations switched the allele state from 1 to 2 or from 2 to 1. Recombinations on a chromosome were modeled according to a binomial map function, where the maximum number of uniformly and independently distributed crossovers on a chromosome of 1 M was 4 (Karlin 1984), i.e., assuming interference. After a period of random mating, which was different for the two scenarios considered, the population was divided into two lines (Figure 1).
Figure 1.—

Simulated population.
Then, each line was increased in size over five generations to obtain a population size of 500 males and 500 females. In the following generations, pedigree data were recorded and 50 sires were randomly selected and mated to 500 dams in each generation, which are discrete. Each female had one male and one female offspring and thus each sire had 10 sons and 10 daughters. Line 1 provided the phenotypic training data to develop models to estimate GEBVs, whereas line 2 was used only to validate the accuracy of GEBVs independent of the impact of genetic relationships, i.e., accuracy due to LD.
Heritability of the quantitative trait was set to 0.5 by rescaling QTL effects in the generation in which pedigree recording was initiated. Phenotypes were calculated as the sum of the QTL-genotype effects of an individual plus a residual effect sampled from a standard normal distribution. The composition of training and validation data sets as well as the methods used to estimate marker effects are described separately for each scenario in the following two sections. The criterion to compare methods was the correlation between true and estimated breeding values, also referred to as the accuracy of estimated breeding values.
To evaluate the effect of genetic relationships captured by markers, TP–BLUP in (4) was used to estimate accuracies of EBVs. The additive genetic variance and the residual variance were assumed known in both TP–BLUP and RR–BLUP.
Genetic relationships captured by markers:
To show that the accuracy of GEBVs is nonzero even if there is no LD in the population, 100, 1000, and 2000 markers in linkage equilibrium (LE) with 10 QTL were simulated. To ensure linkage equilibrium between markers and QTL, each locus (markers and QTL) was located on a different chromosome. Thus, the recombination rate between any pair of loci was 0.5. Pedigree and marker data were recorded for five generations, but only the 500 males in generation 4 had trait phenotypes and thus were included in the training data. The validation data contained individuals from all 5 pedigree generations in line 1. LE markers were used to estimate GEBVs as if they were LD markers.
FR–LS, RR–BLUP, and Bayes-B were used to estimate marker effects. In FR–LS, a threshold of α = 0.2 was used. In Bayes-B, the prior probability of 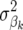 to be nonzero was set to the number of QTL divided by the number of LE markers.
to be nonzero was set to the number of QTL divided by the number of LE markers.
Accuracy of GEBVs due to linkage disequilibrium:
To analyze the accuracy of GEBVs due to LD, the population was randomly mated for 1000 generations to reach mutation–drift equilibrium before it was increased in size as described above (Figure 1). To find enough segregating markers and QTL after 1000 generations, 10,000 loci on each of 10 chromosomes, where every 100th locus was a QTL, were simulated. Loci were equally spaced and each chromosome had a length of 1 M. Marker loci were selected after 1000 generations by first dividing each chromosome in 100 bins of 1 cM and then choosing the marker with frequency closest to 0.5 in each bin. Thus, 1000 SNP markers were used in the estimations. The average marker spacing was 1 cM and thus the average distance between flanking markers and QTL was 0.5 cM. After 1000 generations, ∼50 QTL were segregating and these were randomly distributed on the genome. The frequency distribution of the minor allele of the selected markers was almost uniform from 0 to 0.5 with mean 0.27.
Pedigree and marker data were recorded for 20 generations in both lines, but only the 500 males and 500 females in generation 10 of line 1 had trait phenotypes and thus were included in the training data. The validation data consisted of individuals in lines 1 and 2 from generation 1 to 20 (Figure 1).
FR–LS with α = 0.2, RR–BLUP, and Bayes-B with two different prior probabilities for 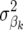 to be nonzero were used in this analysis. Bayes-B1 had a prior probability of 0.05, which corresponds to the expected proportion of segregating QTL after 1000 generations, whereas Bayes-B2 had a much smaller prior probability of 0.005. Following Meuwissen et al. (2001), 10,000 MCMC cycles were conducted for Bayes-B, where the first 1000 were discarded as burn in.
to be nonzero were used in this analysis. Bayes-B1 had a prior probability of 0.05, which corresponds to the expected proportion of segregating QTL after 1000 generations, whereas Bayes-B2 had a much smaller prior probability of 0.005. Following Meuwissen et al. (2001), 10,000 MCMC cycles were conducted for Bayes-B, where the first 1000 were discarded as burn in.
RESULTS
Genetic relationships captured by markers:
Figure 2 shows the accuracy of GEBVs for 100, 1000, and 2000 LE markers, for all males, for the 50 males in each generation that were used as parents (male parents), and for all females. These results are based on 96 replicates.
Figure 2.—

Accuracies of GEBVs obtained by fixed regression–least squares (FR–LS), random regression–BLUP (RR–BLUP), and Bayes-B using 100 (top), 1000 (center), and 2000 (bottom) LE markers in comparison to the accuracies for trait-pedigree–BLUP (TP–BLUP). Five hundred trait phenotypes in generation 4 were used as the training data for all methods (96 replicates).
The accuracy of GEBVs obtained with LE markers was always positive in the five generations considered. The maximum accuracy was obtained for fathers of individuals in the training data (generation 3), because each sire had 10 sons with trait phenotypes in the training data. As expected, the accuracy of the offspring of individuals in the training data (generation 5) is lower than that for individuals in the training data, because these individuals have no phenotypes. Furthermore, the accuracy increased and approached TP–BLUP with an increasing number of LE markers. RR–BLUP was always the closest to TP–BLUP, followed by Bayes-B, whereas FR–LS had considerably lower accuracies. The difference between Bayes-B and RR–BLUP increased with the number of LE markers, whereas the difference between Bayes-B and FR–LS decreased.
All this was observed most clearly for male parents (Figure 2, male parents). For example, the accuracy of GEBVs obtained with RR–BLUP for the fathers of individuals in the training data was 0.5 with 100 LE markers and 0.78 with 2000 LE markers. The latter was only marginally smaller than the accuracy of EBVs from TP–BLUP, which was 0.79. Even the accuracy of GEBVs from RR–BLUP for grandfathers was close to the accuracy of breeding values estimated with TP–BLUP (0.57 vs. 0.61). The accuracy of GEBVs for the offspring of individuals in the training data (generation 5) was only 0.1 using 100 LE markers, but increased to up to 0.4 using 2000 LE markers and RR–BLUP.
As the number of LE markers was increased to 2000, the accuracies of GEBVs for all males and for females also approached the accuracies of TP–BLUP, but to a lesser degree than in male parents (Figure 2). For all males, TP–BLUP had 0.07, 0.18, and 0.28% higher accuracy than RR–BLUP, Bayes-B, and FR–LS, respectively.
Accuracy of GEBVs due to linkage disequilibrium:
Figure 3 shows the accuracy of GEBVs for lines 1 and 2 using 1000 individuals in generation 10 of line 1 each with a trait phenotype and 1000 SNP markers. Furthermore, Table 1 depicts the accuracy of EBVs for individuals in the training data, for their fathers and offspring, and for generation 20. These results are based on 160 replicates.
Figure 3.—

Accuracies of GEBVs obtained by fixed regression–least squares (FR–LS), random regression–BLUP (RR–BLUP), Bayes-B1, and Bayes-B2 in lines 1 and 2 in comparison to the accuracies of EBVs obtained by trait-pedigree–BLUP (TP–BLUP) using 1000 individuals in generation 10 each with a trait phenotype and 1000 SNP markers (160 replicates).
TABLE 1.
Accuracy of GEBVs (±SE, based on 160 replicates) obtained by TP–BLUP, FR–LS, RR–BLUP, Bayes-B1, and Bayes-B2 for individuals in the training data (generation 10), their fathers (generation 9), and their offspring (generation 11) and for generation 20 (based on 1000 SNP markers and 1000 trait phenotypes in generation 10)
| Generation
|
||||
|---|---|---|---|---|
| Method | 9 | 10 | 11 | 20 |
| TP–BLUP | 0.85 ± 0.003 | 0.75 ± 0.004 | 0.53 ± 0.007 | 0.02 ± 0.010 |
| FR–LS | 0.79 ± 0.005 | 0.68 ± 0.005 | 0.61 ± 0.007 | 0.49 ± 0.009 |
| RR–BLUP | 0.87 ± 0.003 | 0.78 ± 0.002 | 0.64 ± 0.004 | 0.42 ± 0.007 |
| Bayes-B1 | 0.88 ± 0.003 | 0.78 ± 0.003 | 0.69 ± 0.005 | 0.55 ± 0.009 |
| Bayes-B2 | 0.83 ± 0.004 | 0.73 ± 0.004 | 0.66 ± 0.006 | 0.54 ± 0.009 |
The fathers of individuals in the training data (Figure 3, male parents, generation 9, line 1) generally had the highest accuracy among all pedigree individuals. The method that obtained the highest accuracy for these individuals was Bayes-B1 with 0.88 (Table 1). The individuals with the next highest accuracy were those in the training data, where RR–BLUP and Bayes-B1 resulted in the highest accuracy, which was 0.78.
The decline in accuracy between generations 10 (the generation with trait data) and 11 of line 1 for RR–BLUP was almost parallel to the decline for TP–BLUP, whereas the accuracy of GEBVs obtained with FR–LS and both Bayes-B methods decreased less. Starting in generation 11 (the offspring generation), Bayes-B1 outperformed RR–BLUP (0.69 vs. 0.64) and starting in generation 12, FR–LS outperformed RR–BLUP (Figure 3). The accuracy declined further in the following generations, but of a decreasing rate in each generation, in particular for the marker-based methods. RR–BLUP and FR–LS decreased faster in the first generations after training than the two Bayes-B methods, but finally the accuracies of GEBVs of all marker-based methods decreased at almost the same rate as the accuracy of GEBVs in line 2. Going backward in time from the father's generation to earlier generations, the decline of accuracy was similar to going forward in time.
The difference in accuracies between Bayes-B1 and Bayes-B2 was greatest in generations 9 and 10, but reduced in the following generations. In generation 20, the accuracies of both Bayes-B methods were not significantly different (Table 1).
The accuracies in generations 2 and 20 in line 1 were affected by genetic relationships to a very small extent and thus these accuracies can be used for comparisons with accuracies in line 2, in which the accuracies were only due to LD. The accuracies of both these generations were lower in line 2 than in line 1, but the difference was greater in generation 20 than in generation 2. The latter can be explained as follows. The LD pattern of individuals in generation 10 of line 1 was utilized to estimate marker effects. This LD pattern, however, changed due to recombinations between markers and QTL over generations. Thus, the longer lines 1 and 2 were separated, the more different was the LD pattern in comparison to that in the training data.
In line 2, Bayes-B1 and Bayes-B2 were not significantly different and both resulted in a higher accuracy than FR–LS and especially RR–BLUP. The accuracies of FR–LS and RR–BLUP were 0.04 and 0.13%, respectively, lower than those of the Bayes-B methods.
The decline in the accuracy over generations in line 2 for FR–LS, RR–BLUP, Bayes-B1, and Bayes-B2 was 0.0031, 0.0042, 0.0037, and 0.0034 units, respectively, per generation. Because this decline is expected to be proportional to the recombination frequency between markers and QTL, this indicates that the average recombination frequency between markers used in these methods was lower than the recombination rate of 0.00498, which corresponds to the average distance of 0.5 cM between QTL and the flanking markers. For example, the accuracies of GEBVs in line 2 from Bayes-B1 resulted from markers that were on average 0.37 cM from the QTL. The explanation is that the number of trait phenotypes in the training data was not sufficient to utilize LD between markers and QTL that were on average >0.37 cM apart. When the number of trait phenotypes was increased, markers on average >0.37 cM from the QTL also contributed to accuracy (results not shown).
The average LD between markers used and QTL can be derived approximately using the formula given by Sved (1969), being 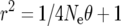 , where r2 is the measure of LD, Ne is the effective population size, and θ is the recombination frequency. Assuming Ne = 100 and using the decay shown above as average recombination frequencies, r2 ranges between 0.37 and 0.44.
, where r2 is the measure of LD, Ne is the effective population size, and θ is the recombination frequency. Assuming Ne = 100 and using the decay shown above as average recombination frequencies, r2 ranges between 0.37 and 0.44.
DISCUSSION
Genetic relationships captured by markers:
The differences in accuracy of GEBVs between FR–LS, RR–BLUP, and Bayes-B (Figure 2) can be explained mainly by the number of LE markers fitted in the model as shown in Table 2. FR–LS fitted the smallest number of LE markers and thus captured genetic relationships the least. RR–BLUP, on the other hand, fitted all LE markers and thus captured more genetic relationships than FR–LS and Bayes-B. In Bayes-B, the number of markers fitted in each round of the MCMC approach depends on the prior probability of 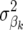 to be nonzero, which was decreased as the number of LE markers increased. Thus, the number of markers fitted in Bayes-B increased only slightly (Table 2). Despite this, the accuracy of Bayes-B increased with the number of LE markers, because in each round of the MCMC approach different markers can be fitted in the model. The differences between models are expected to reduce when the number of LE markers is increased.
to be nonzero, which was decreased as the number of LE markers increased. Thus, the number of markers fitted in Bayes-B increased only slightly (Table 2). Despite this, the accuracy of Bayes-B increased with the number of LE markers, because in each round of the MCMC approach different markers can be fitted in the model. The differences between models are expected to reduce when the number of LE markers is increased.
TABLE 2.
Average number of LE markers fitted (±SE, based on 96 replicates) in FR–LS, RR–BLUP, and Bayes-B using 100, 1000, and 2000 LE markers
| No. of LE markers used
|
|||
|---|---|---|---|
| Method | 100 | 1000 | 2000 |
| FR–LS | 1.9 ± 1.0 | 7.4 ± 2.8 | 11.0 ± 3.0 |
| RR–BLUP | 100 | 1000 | 2000 |
| Bayes-Ba | 12.6 ± 2.0 | 20.3 ± 3.1 | 21.4 ± 2.3 |
Different loci can be fitted in each MCMC round.
The reason why the accuracies of GEBVs were closer to those of TP–BLUP for male parents than for female parents is that the accuracy of GEBV for a parent depends on the genetic relationships captured by markers averaged over the relatives in the training data. The deviation of this averaged genetic relationship from the relationship in A is inversely related to the number of markers and the number of relatives in the training data. Thus, the GEBV accuracy for fathers, which had 10 times as many offspring in the training data as mothers, was closer to the accuracy of TP–BLUP.
Accuracy of GEBVs due to linkage disequilibrium:
Results of this study confirm previous simulation studies that show that genomic selection can result in sizeable accuracies of GEBVs. Simulation parameters used and the results found in the scenario with LD are comparable to those in the study of Solberg et al. (2006). They simulated 1000 phenotypes with a heritability of 0.5 to estimate 1010 SNP effects on 10 chromosomes using Bayes-B1. The accuracy of GEBVs for the offspring of individuals in the training data was 0.66 in their study, which is close to the value of 0.69 found here. Meuwissen et al. (2001), in contrast, used 1000 microsatellite markers and estimated ∼50,000 haplotype effects with 1000 trait phenotypes. They found a higher accuracy for Bayes-B1 of 0.79 as well as a higher difference between Bayes-B1 and RR–BLUP of ∼0.13%. Solberg et al. (2006) showed that microsatellites result in a higher accuracy than SNPs for a given marker density, which explains the higher accuracies found by Meuwissen et al. (2001) in comparison to this study. The greater difference between Bayes-B1 and RR–BLUP in their study, in which they estimated 50,000 haplotype effects, is likely due to the higher number of effects estimated.
The accuracies of GEBVs from FR–LS found here were considerably higher than those in Meuwissen et al. (2001). Using the simulation design explained in the previous section, they found an accuracy of 0.204 for FR–LS in the offspring of individuals in the training data, which is 0.58% lower than their accuracy for Bayes-B1. Here, however, the accuracy for FR–LS was 0.61, which is only 0.08% lower than the accuracy for Bayes-B1 (Table 1). The differences in both studies for FR–LS might be due to the different thresholds used to include markers in the model. Meuwissen et al. (2001) used a more stringent threshold than we used in this study. We observed that the accuracies of GEBVs for FR–LS were lower and more comparable to the results of Meuwissen et al. (2001), when we used a more stringent threshold of α = 0.1 (results not shown here). This is in agreement with a study by Piyasatian et al. (2006), in which higher thresholds resulted in higher breeding progress.
Scenarios with and without LD were used to demonstrate that markers capture not only effects of QTL that are in LD with markers, but also genetic relationships, and that the accuracy of GEBVs is nonzero even without LD. In reality, of course, markers on the same chromosome are not independent and thus the effect of genetic relationships on the accuracy of GEBVs is expected to be lower than that seen here with a large number of independent LE markers. Nevertheless, this effect was also demonstrated under more realistic situations using LD based on a population in mutation–drift equilibrium. Thus, the methods to estimate marker effects utilize both information from genetic relationships among individuals as well as information from LD. However, FR–LS, RR–BLUP, and Bayes-B utilize both types of information differently. Genetic relationships, on the one hand, affect the results of RR–BLUP more than those of FR–LS and Bayes-B, because in FR–LS and Bayes-B only a small proportion of the total number of markers is fitted (Table 3).
TABLE 3.
Average number of markers fitted (±SE, based on 160 replicates) in FR–LS, RR–BLUP, Bayes-B1, and Bayes-B2 using 1000 SNP markers in LD with QTL
Different loci can be fitted in each MCMC round.
Bayes-B, on the other hand, utilizes information from LD better than FR–LS as implemented here and better than RR–BLUP. Furthermore, the ranking of these methods can change over generations, because especially the contribution of genetic relationships to the prediction of GEBVs is different in each generation. This contribution can be high for the parents of individuals in the training data, but for descendant generations the information from genetic relationships is halved each generation. LD information, in contrast, is more persistent, which makes it of particular importance.
To validate the potential advantage of GEBVs, it is necessary to estimate the contribution from LD to the accuracy of GEBVs. The accuracy of GEBVs due to LD in generation 11 of line 1 (offspring of individuals in the training data) was estimated using the accuracy of GEBVs in generation 20 of line 1 and the rate of decline in line 2. The accuracy of GEBVs in generation 20 of line 1 is expected to be mostly due to LD (Table 1). Further, the rate of decline in accuracy in line 2 is entirely due to the decay of LD as depicted in Table 4. The accuracy due to LD in generation 11 was predicted as the accuracy of GEBVs in generation 20, plus nine times the decay of accuracy due to LD (Table 4).
TABLE 4.
Accuracy of GEBVs due to LD in generation 11 of line 1 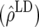 for FR–LS, RR–BLUP, Bayes-B1, and Bayes-B2 estimated by the decay of accuracy per generation in line 2 (b) × 9, plus the accuracy of GEBVs of generation 20 in line 1
for FR–LS, RR–BLUP, Bayes-B1, and Bayes-B2 estimated by the decay of accuracy per generation in line 2 (b) × 9, plus the accuracy of GEBVs of generation 20 in line 1
| FR–LS | RR–BLUP | Bayes-B1 | Bayes-B2 | |
|---|---|---|---|---|
| b | 0.0031 | 0.0042 | 0.0037 | 0.0034 |
 |
0.518 | 0.457 | 0.583 | 0.570 |
The difference between the accuracy of GEBVs (Table 1) and the accuracy of GEBVs due to LD (Table 4) in generation 11 of line 1 was 0.09% for FR–LS and Bayes-B2, 0.11% for Bayes-B1, and 0.18% for RR–BLUP. Thus, this shows again that the impact of genetic relationships was greatest for RR–BLUP and that RR–BLUP was less able to use LD between markers and QTL than the other methods. These results, however, should be specific to this simulation study, because heritability, population structure, LD, and the number of individuals in the training data affect the information from LD and from genetic relationships used to predict GEBVs. The accuracy due to LD cannot be derived from a single generation, especially not by taking the difference between the accuracy of GEBVs and the accuracy obtained by TP–BLUP. For example, in generation 11 the accuracy for Bayes-B1 was 0.69 and that for TP–BLUP was 0.53, giving a difference in accuracy of 0.16.
As seen in Figure 3, the decline of accuracies in line 1 is steep, which is due to the decay of genetic relationships. In line 2, on the other hand, the decline in accuracy is gradual, because it does not capture genetic relationship information, but only persisting LD information. Thus even without information from line 2, modeling the decay of both causes would enable prediction of the accuracy of GEBVs due to LD for any generation in line 1. The linear model
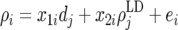 |
(6) |
was used to estimate the accuracy due to LD for generation j,  , and the difference between the accuracy of GEBVs and the accuracy due to LD in generation j, dj, where ρi is the accuracy of GEBVs for generation i and x1i is the accuracy from TP–BLUP in generation i divided by the accuracy from TP–BLUP for generation j. Thus, in doing so, x1i models the slope of accuracies from TP–BLUP from generation j to generation i as seen in Figure 3. x2i is
, and the difference between the accuracy of GEBVs and the accuracy due to LD in generation j, dj, where ρi is the accuracy of GEBVs for generation i and x1i is the accuracy from TP–BLUP in generation i divided by the accuracy from TP–BLUP for generation j. Thus, in doing so, x1i models the slope of accuracies from TP–BLUP from generation j to generation i as seen in Figure 3. x2i is 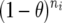 , where θ is the average recombination frequency between markers and QTL (here 0.005) and ni is the number of generations between generations i and j. Thus, x2i models the decay in accuracy due to recombinations from generation j to generation i. Note that this decay occurs not only in generations following the generation of training, but also in earlier generations, because the LD pattern of individuals in the training data is used to estimate marker effects. Finally, ei is the residual term. Generation j can be any generation for which the accuracy of GEBVs and the accuracy of TP–BLUP were observed.
, where θ is the average recombination frequency between markers and QTL (here 0.005) and ni is the number of generations between generations i and j. Thus, x2i models the decay in accuracy due to recombinations from generation j to generation i. Note that this decay occurs not only in generations following the generation of training, but also in earlier generations, because the LD pattern of individuals in the training data is used to estimate marker effects. Finally, ei is the residual term. Generation j can be any generation for which the accuracy of GEBVs and the accuracy of TP–BLUP were observed.
To demonstrate the regression model in (6), x1i and x2i are given in Table 5 for generations 6–10 of line 1. The accuracies of GEBVs from Bayes-B1 were first used to estimate both the accuracy due to LD for generation 10, 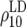 , and the difference between the accuracy of GEBVs and the accuracy due to LD, d10. These estimates were used to predict accuracies of GEBVs, accuracies due to LD, and the difference between both for generations 6–9 (Table 5). The R2-value of the fitted model was >0.99.
, and the difference between the accuracy of GEBVs and the accuracy due to LD, d10. These estimates were used to predict accuracies of GEBVs, accuracies due to LD, and the difference between both for generations 6–9 (Table 5). The R2-value of the fitted model was >0.99.
TABLE 5.
Predicted accuracy of GEBVs (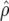 ), predicted accuracy due to LD (
), predicted accuracy due to LD (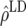 ), and predicted difference (
), and predicted difference (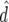 ) between accuracy of GEBVs and accuracy due to LD in generations 6–10 using the accuracies of generations 6–10 from Bayes-B1 (ρ), the accuracies obtained by TP–BLUP divided by the accuracy of TP–BLUP in generation 10 (x1), and the decay of LD at a recombination rate of 0.005 (x2)
) between accuracy of GEBVs and accuracy due to LD in generations 6–10 using the accuracies of generations 6–10 from Bayes-B1 (ρ), the accuracies obtained by TP–BLUP divided by the accuracy of TP–BLUP in generation 10 (x1), and the decay of LD at a recombination rate of 0.005 (x2)
| Generation | 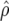 |
ρ | x1 | x2 | 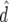 |
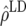 |
|---|---|---|---|---|---|---|
| 6 | 0.603 | 0.610 | 0.232 | 0.980 | 0.049 | 0.553 |
| 7 | 0.627 | 0.625 | 0.330 | 0.985 | 0.071 | 0.556 |
| 8 | 0.657 | 0.653 | 0.458 | 0.990 | 0.098 | 0.559 |
| 9 | 0.692 | 0.688 | 0.611 | 0.995 | 0.131 | 0.561 |
| 10 | 0.779 | 0.782 | 1.000 | 1.000 | 0.214 | 0.564 |
The same regression model was also applied to FR–LS, RR–BLUP, and Bayes-B2. The estimated accuracy due to LD in generation 10 (generation with trait data) was then multiplied by 1 − θ = 0.995 to predict the accuracy due to LD in the offspring generation (Table 6).
TABLE 6.
Predicted accuracy of GEBVs (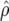 ), predicted accuracy due to LD (
), predicted accuracy due to LD (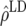 ), and predicted difference (
), and predicted difference (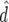 ) between accuracy of GEBVs and accuracy due to LD in generation 11 of line 1 for FR–LS, RR–BLUP, Bayes-B1, and Bayes-B2 using the accuracies of generations 6–10, the accuracies obtained by TP–BLUP divided by the accuracy of TP–BLUP in generation 10, and the decay of LD at a recombination rate of 0.005
) between accuracy of GEBVs and accuracy due to LD in generation 11 of line 1 for FR–LS, RR–BLUP, Bayes-B1, and Bayes-B2 using the accuracies of generations 6–10, the accuracies obtained by TP–BLUP divided by the accuracy of TP–BLUP in generation 10, and the decay of LD at a recombination rate of 0.005
| FR–LS | RR–BLUP | Bayes-B1 | Bayes-B2 | |
|---|---|---|---|---|
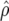 |
0.624 | 0.657 | 0.711 | 0.675 |
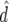 |
0.121 | 0.254 | 0.149 | 0.120 |
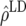 |
0.503 | 0.403 | 0.561 | 0.556 |
The predicted accuracies due to LD are only slightly lower than the accuracies due to LD obtained earlier (Table 4). A part of the difference might be caused by genetic relationships, which are still present to a small extent in generation 20 of line 1. A better approximation can be achieved by using more ancestor generations in the model (results not shown here).
The accuracies of GEBVs due to LD in both Tables 4 and 6 can be used to validate methods to predict GEBVs. Clearly, Bayes-B outperformed FR–LS and especially RR–BLUP. FR–LS was implemented here as a simple forward stepwise selection. Optimization of the model selection in FR–LS may further improve the accuracy of this method.
To show how genetic relationships affect accuracies of GEBVs and to derive the accuracy due to LD, no selection on GEBVs was applied here. In reality, however, GEBVs will be used for selection and thus accuracies in generations following the training generation will be different from those shown here. The effect of selection on the accuracy of GEBVs will be analyzed in further studies.
In practical applications, an offspring generation might not be available when marker effects are estimated. Thus, either genotyped individuals from previous generations or cross-validation will be used. As shown in the results, individuals that are most distant to individuals in the training data best approximate the accuracy due to LD. Furthermore, when using cross-validation, one has to be aware that GEBVs of individuals with progeny in the training data can have a high accuracy only due to genetic relationships. In contrast, the accuracies for individuals without direct descendants in the training data or with a small number of progeny in the training data are much less affected by genetic relationships when the number of markers is not sufficient to approximate genetic relationships accurately. In the future, however, more markers will be fitted and thus the genetic relationships of those individuals might be approximated better. As seen with Bayes-B2, it is possible to obtain a better estimate of the accuracy due to LD by decreasing the probability of a nonzero variance. Another possibility is to fit a polygenic effect, which will be analyzed in further studies.
In poultry and swine breeding, lines may be available that originate from the same population only a few generations ago. In such cases, it is possible to estimate marker effects in one line and to validate the accuracy of GEBVs due to LD in another line. This accuracy is a lower limit of the accuracy in the line used to estimate marker effects, because the LD patterns are expected to differ between both lines due to recombinations that occurred since the separation. However, the accuracy of GEBVs can also be reduced due to gene-by-gene interactions and genotype–environment interactions.
Conclusions:
The accuracy of GEBVs can result in a large part from genetic relationships captured by markers. In general, this is true for all methods that estimate marker effects for prediction of GEBVs. However, the impact of genetic relationships on the accuracy of GEBVs was greatest for RR–BLUP. As a result, to validate the potential to predict GEBVs with high accuracy for several generations following marker estimation, it is not sufficient to analyze the accuracy from only a single generation. Accuracies of GEBVs and of TP–BLUP from several generations can be used to estimate the accuracy of GEBVs due to LD as shown in the discussion.
From the accuracies due to LD, we can conclude that Bayes-B is the method of choice to estimate marker effects, whereas RR–BLUP cannot be recommended. FR–LS might be an alternative to Bayes-B and should be analyzed further.
Acknowledgments
D.H. acknowledges financial support from the Deutsche Forschungsgemeinschaft. This research was further supported by State of Iowa Hatch and Multistate Research Funds.
References
- Fernando, R. L., 1998. Genetic evaluation and selection using genotypic, phenotypic and pedigree information. Proceedings of the 6th World Congress on Genetics Applied to Livestock Production, Armidale, NSW, Australia, Vol. 26, pp. 329–336.
- Karlin, S., 1984. Theoretical aspects of genetic map functions in recombination processes, pp. 209–228 in Human Population Genetics: The Pittsburgh Symposium, edited by A. Chakravarti. Van Nostrand Reinhold, New York.
- Kutner, M. H., C. J. Nachtsheim, J. Neter and W. Li, 2005. Applied Linear Statistical Models, Ed. 5. McGraw-Hill, New York.
- Legarra, A., C. Robert-Granie, E. Manfredi and J. M. Elsen, 2007. Does genomic selection work in a mice population? Papers and Abstracts from the Workshop on QTL and Marker-Assisted Selection, edited by A. Legarra. March 22–23, 2007, Toulouse, France.
- Malécot, G., 1948. Les Mathématiques de l'Hérédité. Masson, Paris.
- Meuwissen, T. H. E., B. J. Hayes and M. E. Goddard, 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157: 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejati-Javaremi, A., C. Smith and J. P. Gibson, 1997. Effects of total allelic relationship on accuracy of evaluation and response to selection. J. Anim. Sci. 75: 1738–1745. [DOI] [PubMed] [Google Scholar]
- Piyasatian, N., L. R. Totir, R. L. Fernando and J. C. M. Dekkers, 2006. QTL detection and marker-assisted composite line development. J. Anim. Sci. 84(Suppl. 2): 134. [Google Scholar]
- Solberg, T. R., A. Sonesson, J. Wooliams and T. H. E. Meuwissen, 2006. Genomic selection using different marker types and density. 8th World Congress on Genetics Applied to Livestock Production, August 13–18, Belo Horizonte, Brazil.
- Sved, J. A., 1969. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 2: 125–141. [DOI] [PubMed] [Google Scholar]
- Vanraden, P. M., 2007. Efficient estimation of breeding values from dense genomic data. J. Dairy Sci. 90(Suppl. 1): 374–375. [Google Scholar]
- Vanraden, P. M., and M. E. Tooker, 2007. Methods to explain genomic estimates of breeding value. J. Dairy Sci. 90(Suppl. 1): 374. [Google Scholar]