

Abstract
The distribution of fitness effects of new mutations (DFE) is important for addressing several questions in genetics, including the nature of quantitative variation and the evolutionary fate of small populations. Properties of the DFE can be inferred by comparing the distributions of the frequencies of segregating nucleotide polymorphisms at selected and neutral sites in a population sample, but demographic changes alter the spectrum of allele frequencies at both neutral and selected sites, so can bias estimates of the DFE if not accounted for. We have developed a maximum-likelihood approach, based on the expected allele-frequency distribution generated by transition matrix methods, to estimate parameters of the DFE while simultaneously estimating parameters of a demographic model that allows a population size change at some time in the past. We tested the method using simulations and found that it accurately recovers simulated parameter values, even if the simulated demography differs substantially from that assumed in our analysis. We use our method to estimate parameters of the DFE for amino acid-changing mutations in humans and Drosophila melanogaster. For a model of unconditionally deleterious mutations, with effects sampled from a gamma distribution, the mean estimate for the distribution shape parameter is ∼0.2 for human populations, which implies that the DFE is strongly leptokurtic. For Drosophila populations, we estimate that the shape parameter is ∼0.35. Differences in the shape of the distribution and the mean selection coefficient between humans and Drosophila result in significantly more strongly deleterious mutations in Drosophila than in humans, and, conversely, nearly neutral mutations are significantly less frequent.
THE distribution of fitness effects of new mutations (DFE) specifies the probability of a new mutation having a given fitness effect. This distribution is therefore of interest for several questions in genetics. The DFE is an important determinant of the amount and nature of genetic variation for fitness and other quantitative traits (Eyre-Walker et al. 2006; Eyre-Walker and Keightley 2007). For example, if quantitative genetic variation is maintained by a balance between mutation and selection and there is substantial variation in the effects of mutations, most of the variance is likely to be contributed by mutations segregating at low frequencies. This is relevant to the nature of complex genetic disease variation in humans, since the genetic mapping of rare alleles subject to strong negative selection is expected to be difficult (Reich and Lander 2001). The DFE is also of critical importance in determining how quickly fitness is expected to change due to an accumulation of new deleterious mutations (Bataillon 2000). This may be important for designing optimal strategies to conserve species or populations that have small effective population sizes (Lande 1995). The maximum rate at which fitness can decline is determined by the product of the mutation rate per genome and the mean mutational effect. If, however, there is substantial variation in the fitness effects of mutations, and some mutations are very strongly deleterious, then selection may be effective against the most damaging mutations, even in quite small populations (Schultz and Lynch 1997). Finally, the DFE is potentially important for several evolutionary questions, including the evolution of sex and recombination (Butcher 1995; Peck et al. 1997), the stability of the molecular clock (Ohta 1977; Eyre-Walker et al. 2006), and the extent to which linkage retards selection response (Keightley and Hill 1987).
A number of different methods have been developed to estimate the DFE from DNA sequence data (Nielsen and Yang 2003; Piganeau and Eyre-Walker 2003; Yampolsky et al. 2005; Eyre-Walker et al. 2006; Loewe et al. 2006). For the most part, these use summaries of the data to infer the distribution and thus discard potentially valuable information. One exception is the method proposed by Eyre-Walker et al. (2006), which attempts to infer the DFE from the distribution of allele frequencies in a sample of sequences, a distribution that is usually referred to as the site-frequency spectrum (SFS). However, any method that uses the SFS for inference about selection needs to consider population demography, because this can affect the SFS in similar ways to selection. For example, population expansion leads to a skew toward rare alleles, whereas population contraction has the opposite effect. Eyre-Walker et al. (2006) suggest an approximate method by which many of the effects of demography can be accounted for. However, their method tends to overestimate the mean strength of selection if there has been population size expansion. It is clearly desirable to account for demographic change by estimating parameters of a demographic model along with the DFE. Williamson et al. (2005) have made a step in this direction by developing a method by which the “mean” strength of selection can be inferred within a model in which the population is allowed to go through an instantaneous population size increase or decrease. A. Boyko (personal communication) has recently extended this approach to simultaneously infer the DFE under the demographic model used by Williamson et al. (2005). Here, we develop a method that is similar to that of A. Boyko; we infer both the DFE and the demography of the population from the DNA sequence data. However, our method potentially has some advantages. The inference of demography is simultaneous, whereas A. Boyko infers the demography and then the distribution of effects under the assumption that the demographic model is correct. By inferring all parameters simultaneously using all the available information in the data we expect to derive more realistic confidence intervals, since the error in the demographic model is incorporated into the estimate of the error in the DFE. We also infer confidence limits on our parameter estimates by bootstrapping in an attempt to account for nonindependence between linked sites. We apply our new method to polymorphism data from humans and Drosophila melanogaster.
MATERIALS AND METHODS
Model:
The frequency distribution of segregating alleles at nucleotide sites in the genome of a diploid population of size N1 is assumed to be at an equilibrium between mutation, selection, and drift. All sites are assumed to be unlinked and have the same mutation rate, and mutations are assumed to be sufficiently rare that no more than two alleles segregate at a given site. A class of selected sites is subject to new deleterious mutations, and the fitnesses of the wild-type, heterozygote, and mutant homozygote genotypes are 1, 1 − s/2, and 1 − s, respectively. Different mutations have independent s, which are assumed to be drawn from a gamma distribution with shape parameter β and scale α. We assume a gamma distribution, since it can take a wide variety of shapes and only has two parameters. These include distributions ranging from highly leptokurtic if β → 0 to a platykurtic form that becomes a spike at the distribution's mean (β/α) if β → ∞ and includes the exponential distribution (β = 1). We assume that there is a class of neutral sites at which mutant alleles have no effect on fitness. The equilibrium population of size N1 experiences a step change in size (upward or downward) to size N2, and the population remains at this size for t2 generations, at which point the frequencies of alleles at a number of selected and neutral sites in the genome are surveyed in a sample of individuals.
Generation of the expected allele frequency vector:
The computation of the likelihood of the polymorphism SFS is based on an allele-frequency vector (AFV), v(s), which is a scaled, weighted sum of two vectors, w(s) and x(s), containing the expected numbers of mutations segregating at the time of sampling in different frequency classes attributable to mutations that occur before and after the change of population size, respectively. These vectors were generated by transition matrix methods using a Fisher–Wright transition matrix, M, which specifies stochastic change in the allele-frequency distribution. The elements of M are:
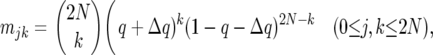 |
(1) |
where q = j/2N, and
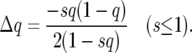 |
(2) |
The elements of M specify the probability that a mutation present in j of the 2N chromosomes is present in k of 2N chromosomes in the next generation. Using this transition matrix it is possible to derive the AFV in the following manner.
We start by considering the contribution of mutations to the AFV that occur after the change in population size. Let f(t2) be a row vector of dimension 2N2 whose elements f(t)i (0 ≤ i ≤ 2N2) are the probabilities that the population has an allele frequency i/2N2 t2 generations after the occurrence of a new mutation. For example, at t2 = 0 f(0)1 = 1, and all the other elements are zero. The vector f(t2) at generation t2 (t2 > 0) is obtained by iterating
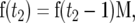 |
(3) |
where M has dimension 2N2 × 2N2. f(t2) gives the AFV for a single mutation t2 generations after it occurred. However, the SFS for a real population would contain a sample of mutations that had occurred at times in the past up to and including generation t2. The cumulative frequency vector x(s), containing the sum of contributions from mutations that occurred t2, t2 − 1, … , 0 generations ago, is
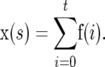 |
(4) |
The gamma distribution of mutational effects can potentially have a long tail with a substantial part of the density >1. As Ns increases the contribution of a new mutation is expected to decrease. We therefore modeled the contributions of mutations for s > 1 by setting x(s)1 to 2/s and all other elements to zero, so that the mean allele frequency was proportional to the expectation at mutation–selection balance.
To compute w(s) (the vector containing the contribution from mutations that occur before the change in population size), we first computed a vector, u(s), containing the relative numbers of mutations in different frequency classes at mutation–selection–drift equilibrium in a population of size N1. This could be obtained by transition matrix iteration using Equation 4 for large t2 (i.e., t2 ≫ N1), but a faster method is to obtain this sum from
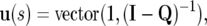 |
(5) |
(Kemeny and Snell 1960), where vector(i, matrix) is the operation that extracts the vector corresponding to column i of the matrix, Q is the square submatrix of M of dimension 2N1 − 1, defined by qij = mij for 1 ≤ i, j ≤ 2N1 − 1, and I is the identity matrix. To generate the vector w(s), specifying the numbers of alleles segregating at different frequencies subsequent to the operation of selection and drift (but not mutation) in a population of size N2, we apply a transition matrix with dimensions 2N1 × 2N2 to u(s) for one generation followed by iteration with a square transition matrix of dimension 2N2 for t − 1 generations.
Let the vector v′(s) be the sum of w(s) and x(s), weighted by N1 and N2, respectively, which are proportional to the numbers of mutations occurring per generation in the populations before and after the change of population size; i.e.,
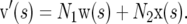 |
(6) |
Note that v′(s) specifies relative numbers of mutations that segregate in the population, whereas we require a vector of allele frequencies that includes the frequency of sites at which mutations have been eliminated by selection or that have not experienced a mutation. Elements of this probability vector v(s) were therefore obtained by scaling v′(s) as
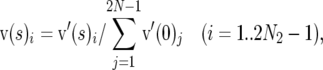 |
(7) |
where the subscripts refer to the element of a vector. This scaling implies that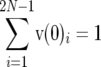 , whereas
, whereas 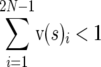 for s < 0. The difference between the scaled density for s = 0 and the scaled density for s < 0 is due to mutations that have become selectively eliminated, so
for s < 0. The difference between the scaled density for s = 0 and the scaled density for s < 0 is due to mutations that have become selectively eliminated, so
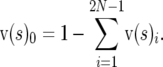 |
(8) |
To account for sites that are invariant due to never having experienced a mutation, we introduced an additional parameter in the model, f0. The frequency of this nonsegregating class was estimated by dividing elements of v(s)i by 1 − f0 (for i = 0 to 2N2 − 1) and incrementing v(s)0 by f0.
Computation of likelihood:
The SFS data for nonsynonymous and silent sites consist of vectors p(N) and p(S) of numbers of sites p(N)i and p(S)i containing i (0 ≤ i < nT) derived alleles in a sample of nT alleles from the population. For simulated data the derived (i.e., mutant) allele is known. However, it is not possible to know this with certainty for real data. One possibility is to infer the derived allele by parsimony using an outgroup species, but this introduces bias because parsimony assignments can be inaccurate even when two species are quite closely related and this will tend to lead to an excess of common variants. We therefore folded the SFS data vectors and v(s) as follows:
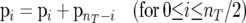 |
(9) |
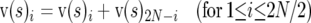 |
(10) |
Simulation results suggest there is relatively little information lost by using the folded vector.
For a given selection coefficient, s, the probability of observing i derived alleles is obtained from the sum of probabilities, weighted by the elements of the AFV v(s)j, over all possible frequencies of mutant alleles (j/2N2) in the population (0 ≤ j < 2N2), under the assumption that i is binomially distributed. For the sites assumed to be under selection, this sum was integrated numerically over the distribution of mutation selection coefficients f(s), which in our case is a gamma distribution. For unfolded distributions, the log likelihood of the data corresponding to the sites assumed to be under selection was
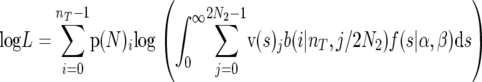 |
(11) |
where b(i | n, p) is the binomial probability function for i derived alleles in a sample of n alleles with the probability of occurrence p. Similarly, the log likelihood with folded distributions (assuming odd numbers of alleles) was
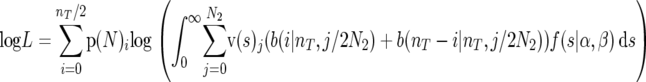 |
(12) |
The log likelihood for the silent-site data (p(S)) was computed from Equation 11 or 12 for a point value s = 0, omitting the integration. The overall log likelihood was the sum of log likelihoods for the selected and neutrally evolving sites.
Algorithm for maximization of likelihood:
Likelihood was evaluated for fixed population sizes N1. The variable parameters estimated in the model were N2, t2, f0, α, and β. To speed up the likelihood calculations, the expected gene frequency vectors (EGFs) w(s) and x(s) were precomputed. The generation of these vectors can be expensive in computing resources, the time required being approximately proportional to 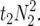 Computing time for maximization of likelihood is not a serious issue for N2 up to 1000. Most evaluations were done for N1 = 20 or N1 = 100. EGFs for values of N2 were generated from 2 to 2000 in steps increasing by 5% or 1, whichever was the higher. Values of t2 went from 1 to 5000, again in steps increasing by 5%. The numerical integration procedure used 250 s points in four ranges with an increasing density of points close to s = 0; results of likelihood evaluations that used 125 of these points were almost identical to those that used the full 250 points (data not shown). For a fixed value of N2, the downhill simplex method (Nelder and Mead 1965; Press et al. 1992) was used to find a local maximum likelihood (ML), and a search over N2 was carried out to find the value closest to the global ML. The variable t2 is discrete, whereas the simplex algorithm requires continuous values, so the likelihood calculations used EGFs for noninteger values of t2 that were generated by linear interpolation for each element. To compute confidence intervals, we ranked parameter estimates obtained from 200 bootstrap data sets, resampled with replacement over loci. Confidence limits obtained from profile likelihoods using values corresponding to drops in natural log L of 2 units from ML estimates were in good agreement with these (data not shown).
Computing time for maximization of likelihood is not a serious issue for N2 up to 1000. Most evaluations were done for N1 = 20 or N1 = 100. EGFs for values of N2 were generated from 2 to 2000 in steps increasing by 5% or 1, whichever was the higher. Values of t2 went from 1 to 5000, again in steps increasing by 5%. The numerical integration procedure used 250 s points in four ranges with an increasing density of points close to s = 0; results of likelihood evaluations that used 125 of these points were almost identical to those that used the full 250 points (data not shown). For a fixed value of N2, the downhill simplex method (Nelder and Mead 1965; Press et al. 1992) was used to find a local maximum likelihood (ML), and a search over N2 was carried out to find the value closest to the global ML. The variable t2 is discrete, whereas the simplex algorithm requires continuous values, so the likelihood calculations used EGFs for noninteger values of t2 that were generated by linear interpolation for each element. To compute confidence intervals, we ranked parameter estimates obtained from 200 bootstrap data sets, resampled with replacement over loci. Confidence limits obtained from profile likelihoods using values corresponding to drops in natural log L of 2 units from ML estimates were in good agreement with these (data not shown).
Simulations:
The performance of the method was checked by analyzing simulated data sets, generated by transition matrix methods, as described above. All variable parameters described above were estimated. The simulated data were either generated assuming a two-epoch model, as assumed by the analysis, or a three-epoch model, which violates the assumptions of the analysis. Both scenarios involved a steady-state population of size N1, followed by a change in population size to N2 for t2 generations. In the three-epoch model, there was a further step change in population size to N3 individuals for t3 generations. Allele frequencies were sampled in proportion to their probabilities in the AFV, and then numbers of individuals containing the mutant allele were sampled from a binomial distribution. These analyses were carried out using unfolded distributions (Equation 11); results are similar if folded distributions are used in the analysis (data not shown). Checks were also carried out using a full Monte Carlo simulation, in which the fates of freely recombining mutations were tracked in populations of initial size N1 parents, which changed to N2 parents for t2 generations. Results from these simulations agreed closely with simulated parameter values (data not shown).
Data:
Human nucleotide sequences were downloaded from the Environmental Genome Project (EGP) website (University of Washington, Seattle; http://egp.gs.washington.edu; January 2007; Livingston et al. 2004) and from the Program for Genomic Applications (PGA) website (NHLBI SeattleSNPs, Seattle; http://pga.gs.washington.edu; February 2007). Alleles of African and European origin were analyzed separately. For these data sets, intronic bases, with the exception of bases corresponding to sites known to be involved in splicing (the first 6 and last 16 bases of each intron) served as the neutrally evolving standard. The frequency of CpG dinucleotides varies dramatically between coding and nocoding DNA in mammals, leading to differences in mutation rates due to the hypermutable nature of these sites (Kondrashov et al. 2006). We therefore restricted our analysis to those sites that are not part of a CpG dinucleotide. The two data sets are not random collections of genes, and there are substantial differences between them in diversity at intronic and especially nonsynonymous sites (Table 1). This could reflect different mean strengths of selection on the two sets of genes.
TABLE 1.
Polymorphism data sets analyzed
| Species | Population | Data set | No. loci | No. alleles analyzed | Intronic/ fourfold sites | Proportion segregating | Zerofold sites | Proportion segregating |
|---|---|---|---|---|---|---|---|---|
| H. sapiens | Africa | PGA | 288 | 38 | 3,675,233 | 0.0035 | 241,794 | 0.0014 |
| Europe | 38 | 0.0021 | 0.0092 | |||||
| Africa | EGP | 221 | 42 | 3,326,733 | 0.0033 | 196,133 | 0.0011 | |
| Europe | 34 | 0.0018 | 0.0067 | |||||
| D. melanogaster | Africa | Shapiro et al. (2007) | 418 | 12 | 32,837 | 0.040 | 137,796 | 0.0036 |
| Zimbabwe | 8 | 34,209 | 0.035 | 143,139 | 0.0029 | |||
| Non-Africa | 6 | 37,234 | 0.027 | 154,136 | 0.0024 |
D. melanogaster nucleotide sequences described in Shapiro et al. (2007) were kindly provided by Joshua Shapiro. The African data set consists of nucleotide sequences that originated in Zimbabwe (10 alleles) and Botswana and Zambia (2 alleles each). The alleles originating in Zimbabwe were analyzed separately; this data set is therefore a subset of the African data set. Non-African alleles (6) are from diverse regions worldwide, but not from Africa. For the Drosophila data sets, fourfold degenerate sites served as the neutral standard. In cases where >2 alleles segregate at a site, the derived allele frequency was taken as the sum of the frequencies of the rarest alleles.
In both humans and Drosophila, the DFE was fitted to zerofold degenerate sites. In all data sets, the numbers of alleles sampled at a given nucleotide site vary, but the likelihood calculations are speeded up considerably if each site has the same number of alleles. We therefore disregarded sites with less than a minimum acceptable number of alleles (nmin), so that we disregarded ∼5% of segregating sites. If the number of alleles at a site exceeded nmin, we sampled nmin alleles without replacement. Table 1 gives some details of each data set analyzed.
RESULTS
Simulations:
To evaluate the performance of the inference method, we checked whether estimated parameter values matched simulations under a range of scenarios. The transition matrix approach makes it possible to infer the AFV for a population of a particular size that has been subject to past expansion or contraction. It is important to note that the true population size from which the data were sampled is not known and cannot be inferred without additional information about the mutation rate (u). However, population genetic theory suggests that the dynamics should scale with Neu and Nes. Hence, if we arbitrarily choose a value of N1, the inferred values of N2E(s) = N2β/α and β should be unbiased estimates of their true values. To investigate this we simulated data under one value of N1 and used different values of N1 to estimate the parameters. For cases in which the simulated data included a single phase of population expansion or contraction (so conformed to the analysis model), results suggest that the method recovers mean simulated values for the mutational distribution parameters with little bias in most cases (Table 2). Two cases in which bias arises were noted. First, under population contraction, analysis with small N1 (i.e., 20), implying even smaller N2 gives downwardly biased estimates of β and upwardly biased estimates of N2E(s), particularly if the simulated distribution is leptokurtic. This suggests that large N1 should be used if population contraction is apparent. Second, for simulations involving platykurtic distributions (β = 5), some replicates gave β-estimates considerably larger than the value simulated (i.e., implying that the ML-estimated distribution approaches equal effects, β → ∞); these outliers can therefore lead to upwardly biased mean estimates of β in these cases. However, most analyses from DNA sequence data suggest the DFE is fairly leptokurtic, so this should not arise in practice.
TABLE 2.
Simulation results: simulated data conform to the analysis model
| Mean parameter estimates (SD)
|
||||
|---|---|---|---|---|
| Simulated β | Simulated N2E(s) | N1 assumed | β | N2E(s) |
| a. Constant population | ||||
| 0.2 | 5 | 20 | 0.19 (0.054) | 5.0 (1.45) |
| 100 | 0.21 (0.057) | 4.6 (1.14) | ||
| 1 | 20 | 0.84 (0.18) | 5.6 (1.03) | |
| 100 | 0.88 (0.63) | 5.7 (0.85) | ||
| 5 | 20 | 5.5 (2.19) | 5.2 (0.72) | |
| 100 | 6.0 (2.33) | 4.6 (0.20) | ||
| b. Twofold population contraction | ||||
| 0.2 | 10 | 20 | 0.11 (0.022) | 138 (186) |
| 100 | 0.23 (0.035) | 8.4 (2.34) | ||
| 1 | 20 | 0.93 (0.47) | 18.5 (5.28) | |
| 100 | 0.93 (0.080) | 11.1 (0.98) | ||
| 5 | 20 | 3.3 (0.94) | 15.7 (2.93) | |
| 100 | 5.6 (2.01) | 12.4 (2.26) | ||
| c. Twofold population expansion | ||||
| 0.2 | 10 | 20 | 0.22 (0.066) | 8.9 (3.57) |
| 100 | 0.22 (0.076) | 11.0 (7.15) | ||
| 1 | 20 | 1.0 (0.18) | 9.7 (1.95) | |
| 100 | 1.0 (0.22) | 9.3 (1.52) | ||
| 5 | 20 | 5.3 (1.19) | 9.6 (0.98) | |
| 100 | 5.6 (1.43) | 9.0 (0.63) | ||
| d. Fourfold population expansion | ||||
| 0.2 | 20 | 20 | 0.18 (0.018) | 17.2 (1.4) |
| 100 | 0.18 (0.016) | 23.4 (4.8) | ||
| 1 | 20 | 1.1 (0.12) | 19.3 (2.0) | |
| 100 | 1.2 (0.13) | 17.0 (1.4) | ||
| 5 | 20 | 7.2 (2.35) | 21.7 (3.5) | |
| 100 | 9.4 (4.10) | 20.5 (3.0) | ||
Replicate data sets were generated by Monte Carlo simulation for three values of β and E(s) = 0.1 under four demographic models: (a) N1 = 50, N2 = 50; (b) N1 = 200, N2 = 100; (c) N1 = 50, N2 = 100; and (d) N1 = 50, N2 = 200. Other parameter values were t2 = 200 and f0 = 0.95. The data sets consisted of 40 alleles sampled for 10,000 neutral sites and 2000 selected sites. Each data set was analyzed separately assuming two different N1's (20 and 100), and estimates of N2, t, β, E(s), and f0 were obtained. There were five replicates for β = 1 and β = 5 and 10 replicates for β = 0.2.
We also simulated data that depart from the assumptions of the analysis method by incorporating two-step changes in population size. Results from simulations of mild or severe bottlenecks or accelerating population expansion suggest that the method is quite robust to such departures, and parameter values are recovered from the SFS data with little bias (Table 3).
TABLE 3.
Simulation results: simulated data violate assumptions of the analysis model
| Mean parameter estimates (SD)
|
||||
|---|---|---|---|---|
| Simulated β | Simulated N3E(s) | N1 assumed | β | N3E(s) |
| a. Mild bottleneck followed by population expansion | ||||
| 0.2 | 10 | 20 | 0.20 (0.057) | 15.2 (14.6) |
| 100 | 0.20 (0.058) | 15.0 (14.8) | ||
| 1 | 20 | 1.16 (0.31) | 9.8 (3.13) | |
| 100 | 1.17 (0.31) | 9.7 (2.97) | ||
| 5 | 20 | 5.4 (1.14) | 9.8 (0.64) | |
| 100 | 5.6 (1.56) | 11.6 (2.60) | ||
| b. Severe bottleneck followed by population expansion | ||||
| 0.2 | 10 | 20 | 0.25 (0.083) | 8.2 (3.12) |
| 100 | 0.27 (0.091) | 7.7 (3.05) | ||
| 1 | 20 | 1.08 (0.28) | 9.7 (2.71) | |
| 100 | 1.11 (0.31) | 9.7 (2.43) | ||
| 5 | 20 | 5.6 (2.40) | 9.6 (0.69) | |
| 100 | 6.4 (3.25) | 9.3 (0.67) | ||
| c. Two phases of accelerating population expansion | ||||
| 0.2 | 10 | 20 | 0.22 (0.081) | 10.6 (7.61) |
| 100 | 0.24 (0.10) | 10.3 (8.52) | ||
| 1 | 20 | 1.1 (0.22) | 10.1 (1.84) | |
| 100 | 1.2 (0.31) | 9.4 (1.83) | ||
| 5 | 20 | 5.6 (2.00) | 9.7 (0.88) | |
| 100 | 7.0 (3.09) | 9.2 (0.99) | ||
Replicate data sets were generated by Monte Carlo simulation for three values of β and E(s) = 0.1 under demographic models involving three epochs that violated the assumptions of the model simulated: (a) N1 = 100, N2 = 25, N3 = 100, t2 = 5, t3 = 100; (b) N1 = 100, N2 = 10, N3 = 100, t2 = 10, t3 = 100; and (c) N1 = 25, N2 = 50, N3 = 100, t2 = 100, t3 = 100. The parameter f0 = 0.95. The data sets consisted of 40 alleles sampled for 10,000 neutral sites and 2000 selected sites. Each data set was analyzed separately assuming two different N1's (20 and 100), and estimates of N2, t, β, E(s), and f0 were obtained. There were 5 replicates for β = 1 and β = 5 and 10 replicates for β = 0.2.
Humans:
As expected, the demographic models that best fit the African and European polymorphism data differ markedly (Table 4) (Adams and Hudson 2004; Marth et al. 2004; Garrigan and Hammer 2006). Frequencies of polymorphisms in the European data sets are consistent with a modest recent population contraction. In the case of the European EGP data set, however, the fit of this model is only marginally better than a constant population, whereas the PGA data set gives strong evidence of population contraction (Table 4). These somewhat contrasting results may reflect limitations of the simple two-epoch model that we have fitted. For example, a population bottleneck followed by an expansion is consistent with the recent history of European populations (Adams and Hudson 2004; Marth et al. 2004). Notably, the EGP genes are substantially less polymorphic than the PGA ones (Table 1), implying that their local effective population size is lower, on average. The European EGP SFS may therefore be more strongly affected by a recent population expansion. In the case of the African data sets, the best-fitting models give increasing likelihood as N2 increases for a given N1 while t/N2 remains constant at ∼2.5, and E(s) and β remain approximately constant. We found that this behavior can also be produced in simulations if there is a strong population expansion far in the past (i.e., several N generations ago; supplemental Figure 1 at http://www.genetics.org/supplemental/). In these cases, the data do not seem to contain information that makes it possible to disentangle the magnitude of the population expansion and the time at which it occurred. For the African data sets, models with populations that have changed in size fit the data better than constant-population models, and the differences in log likelihood are large (Table 4).
TABLE 4.
Estimates of demographic parameters for humans and Drosophila
| Species | Population | Data set | N2/N1 | t/N2 | Log L |
|---|---|---|---|---|---|
| Homo sapiens | Africa | PGA | a | 2.7 | 130 |
| Europe | 0.67 | 1.4 | 46 | ||
| Africa | EGP | a | 2.1 | 242 | |
| Europe | 0.89 | 1.2 | 0.8 | ||
| D. melanogaster | Africa | Shapiro et al. (2007) | 20 | 2.4 | 16 |
| Zimbabwe | 4.9 | 1.5 | 18 | ||
| Non-Africa | 7.2 | 1.7 | 9.9 |
Log L is the difference in log likelihood between the models that allow/do not allow a population size change. The initial population size, N1, was 20 for Drosophila and African humans; for European humans N1 was 100.
Likelihood increases toward a plateau as N2 increases for a given N1; the parameter values reported were for N2 = 1000.
To check the fit of the model to the data, we calculated expected SFSs for neutral and selected sites on the basis of the maximum-likelihood estimates (MLEs) of the parameters of the model. The observed and expected SFSs (plotted in Figure 1 for the neutral data) suggest that the fit is very good in all cases. For the human data sets, the proportion of variance (r2) of the observed SFS explained by the expected SFS exceeds 96% in all cases.
Figure 1.—

Site-frequency spectra for the intronic data of human populations compared to expectation generated under the assumption of MLE parameter values.
The human polymorphism data sets give fairly consistent estimates for the shape parameter of the gamma distribution, β, of ∼0.2 (Table 5), implying that the distribution of fitness effects of new mutations is strongly leptokurtic. Estimates for NeE(s), the mean selective effect of a new mutation, are quite large and consistently larger in the African than in the European populations. Presumably, this difference at least partly reflects a difference between the populations' long-term effective sizes (Tenesa et al. 2007). In general NeE(s) is rather imprecisely estimated. This may be caused by the lack of information coming from strongly deleterious mutations, since these are very rarely present in a sample of DNA sequences. However, the proportions of mutations in different effects classes are estimated with somewhat higher confidence (Table 6). Approximately 30% of amino acid-changing mutations behave as nearly neutral (i.e., Nes < 1), but there is quite a conspicuous difference between the EGP and the PGA data sets. There is also an indication that there is a higher proportion of strongly selected mutations with effects Nes >100 in Africans than in Europeans.
TABLE 5.
Estimates of mean selective effect and distribution shape parameter for deleterious mutations in humans and Drosophila
| Species | Population | Data set | NeE(s) (95% C.I.) | β (95% C.I.) |
|---|---|---|---|---|
| H. sapiens | Africa | PGA | 5,300 (160, →∞) | 0.10 (0.01, 0.19) |
| Europe | 51 (16, →∞) | 0.19 (0.04, 0.32) | ||
| Africa | EGP | 2,500 (180, →∞) | 0.15 (0.02, 0.25) | |
| Europe | 61 (16, 4.8 × 105) | 0.29 (0.08, 0.54) | ||
| D. melanogaster | Africa | Shapiro et al. (2007) | 1,800 (520, 25,000) | 0.38 (0.26, 0.49) |
| Zimbabwe | 9,800 (700, →∞) | 0.30 (0.15, 0.49) | ||
| Non-Africa | 14,000 (290, →∞) | 0.27 (0.04, 0.59) |
TABLE 6.
Proportions of mutations with effects in different Nes ranges in humans and Drosophila
|
Nes range (95% C.I.)
|
||||||
|---|---|---|---|---|---|---|
| Species | Population | Data set | 0–1 | 1–10 | 10–100 | >100 |
| H. sapiens | Africa | PGA | 0.34(0.26, 0.43) | 0.09(0.01, 0.16) | 0.12(0.01, 0.25) | 0.45(0.28, 0.59) |
| Europe | 0.37(0.29, 0.46) | 0.20(0.04, 0.31) | 0.27(0.04, 0.35) | 0.15(0.02, 0.45) | ||
| Africa | EGP | 0.24(0.18, 0.33) | 0.10(0.02, 0.16) | 0.15(0.02, 0.28) | 0.51(0.33, 0.67) | |
| Europe | 0.23(0.15, 0.33) | 0.22(0.06, 0.33) | 0.36(0.07, 0.48) | 0.19(0.01, 0.54) | ||
| D. melanogaster | Africa | Shapiro et al. (2007) | 0.05(0.03, 0.06) | 0.06(0.05, 0.08) | 0.15(0.08, 0.23) | 0.74(0.65, 0.81) |
| Zimbabwe | 0.05(0.03, 0.07) | 0.05(0.03, 0.07) | 0.10(0.04, 0.21) | 0.80(0.69, 0.87) | ||
| Non-Africa | 0.06(0.03, 0.09) | 0.05(0.01, 0.09) | 0.09(0.01, 0.30) | 0.79(0.55, 0.89) | ||
Estimates for β tend to be larger and N2E(s) smaller if CpG sites are included (data not shown). This arises because hypermutable CpGs are considerably less frequent in introns (the assumed neutrally evolving standard) than in nonsynonymous sites (the sites assumed to be under selection), so the average mutation rate per site is relatively lower in introns compared to nonsynonymous sites, if CpG sites are included. This leads to relatively fewer nonsegregating intronic sites and thus to the appearance of weaker selection under the equal mutation rates model assumed. The results for non-CpG sites are the more relevant, however, since we assume that all sites mutate at the same rate. A more powerful method for dealing with this difference in the mutation rate might be to estimate separate f0 parameters for CpG and non-CpG sites.
D. melanogaster:
The polymorphism-frequency spectra suggest that there has been a population expansion in all populations (Table 4). This is consistent with what has previously been inferred for African D. melanogaster populations (Li and Stephan 2006; Stephan and Li 2007). As with human populations, non-African D. melanogaster seems to have gone through a bottleneck followed by a population expansion (Li and Stephan 2006), and the present analysis may be picking up the population expansion signal from the SFS. Estimates of β (Table 5) tend to be higher than those seen in human populations, and several are significantly higher at the 5% level (Table 7). This implies that the distribution of selective effects may be less leptokurtic in Drosophila than in humans. Mean Nes is imprecisely estimated for all data sets, presumably for the same reasons as mentioned above for humans. However, the splitting of the distribution of mutation effects into ranges (Table 6) shows that there are far fewer nearly neutral mutations (Nes < 1) in Drosophila than in human populations (P < 0.01 for all comparisons; Table 7) and that there are many more strongly deleterious mutations (Nes > 100) (P < 0.05 for all comparisons; Table 7). As with the human data, the fit of the SFSs to their expectations is excellent (r2 > 0.97; Figure 2).
TABLE 7.
Tests of significance of differences in properties of the DFE between Drosophila and humans
| Drosophila data set
|
|||
|---|---|---|---|
| Human data set | Africa | Zimbabwe | Non-Africa |
| Africa PGA | 0, 0, 0 | 0.02, 0, 0 | 0.05, 0, 0.01 |
| Europe PGA | 0.03, 0, 0 | 0.13, 0, 0 | 0.19, 0, 0.01 |
| Africa EGP | 0.01, 0, 0.01 | 0.06, 0, 0 | 0.12, 0, 0.02 |
| Europe EGP | 0.18, 0, 0 | 0.39, 0, 0 | 0.44, 0, 0.01 |
Numbers in each cell are the proportions of pairs of human/Drosophila bootstrap samples (of 200) in which (a) β(human) − β(Drosophila) > 0, (b) the proportion of mutations with NE(s) in the range 0–1 (human) − the proportion of mutations with NeE(s) in the range 0–1 (Drosophila) < 0, and (c) the proportion of mutations with NeE(s) > 100 (human) − the proportion of mutations with NeE(s) > 100 (Drosophila) > 0. P-values in the text are obtained by doubling the values in the table, since the values above refer to one-sided tests.
Figure 2.—

Site-frequency spectra for the synonymous-site data of Drosophila populations compared to expectation generated under the assumption of MLE parameter values.
DISCUSSION
There are several interesting contrasts between the results from the different data sets. First, the distributions of effects of amino acid-changing mutations are strongly leptokurtic in humans and Drosophila. However, estimates for the gamma distribution shape parameter suggest that the distribution may be substantially less leptokurtic in Drosophila than in humans. It is unknown what biological factors could cause this difference in the shape of the distribution. Second, the mean effect of an amino acid substitution is imprecisely estimated in all data sets, in spite of the large number of genes sequenced. This lack of power probably reflects the relatively low numbers of alleles sequenced and the inability to ascertain the frequency of rare, strongly deleterious polymorphisms that have a major impact on the tail of the distribution of mutational effects. However, point estimates for NeE(s) for humans suggest substantially lower values for European than for African populations, presumably due to recent bottlenecks that affected Europeans (Marth et al. 2004; Garrigan and Hammer 2006). Third, although imprecisely estimated, point estimates for NeE(s) are similar in African humans and Drosophila. This is surprising, given that previous estimates for the effective population size, obtained by combining nucleotide diversity and between-species divergence, differ by about two orders of magnitude between these species (Eyre-Walker et al. 2002), which suggests that E(s) differs considerably between Drosophila and humans. However, the confidence intervals on our estimates do not allow us to discern whether there is a real difference between the two species. It is also possible that Ne in Drosophila has been overestimated because the mutation rate per base pair may have been underestimated (Haag-Liautard et al. 2007). Finally, there are far fewer effectively neutral amino acid-changing mutations in Drosophila than in humans. In addition, mutations of very strong effect (Ns > 100) are more frequent in Drosophila.
Our results are broadly concordant with previous analyses. Eyre-Walker et al. (2006) inferred the DFE for humans using other data from the Environmental Genome Project not used here. By assuming a gamma distribution they estimated that β = 0.23 and Ns = 850, which are similar to the values estimated here. This is remarkable given that different sets of genes were analyzed and Eyre-Walker et al. (2006) used only an approximate correction for demography. We also compared our results with those obtained by applying the method of Eyre-Walker et al. (2006) to our current data sets (supplemental Table 1 at http://www.genetics.org/supplemental/). In general, similar parameter estimates are obtained, although estimates of β tend to be slightly higher using the current method. Our results are also similar to those of Yampolsky et al. (2005) who estimated the distribution using a more heuristic approach. In Drosophila, Loewe et al. (2006) have estimated the DFE by different methods; their point estimates for β are 0.30 and 0.56, and for NeE(s) they are 2200 and 41,000, which are similar to our values even though they were obtained using a different set of genes in different species. However, their confidence intervals are even larger than ours.
Methods have been previously developed to infer the distribution of effects of mutations from DNA sequence data (Piganeau and Eyre-Walker 2003; Nielsen and Yang 2003; Sawyer et al. 2003; Eyre-Walker et al. 2006; Loewe et al. 2006), but none have attempted to estimate selection and demographic parameters together. From a statistical point of view, this is desirable because uncertainty concerning population demography should be taken into account when making inferences about the mutational parameters, and this is particularly important if the relative amount of neutral-site data is small, as is the case for synonymous sites. Furthermore, information from the SFS of the selected sites affects the demographic parameter estimates. Previous work has shown that it is important to correct for demographic changes that alter the SFS (Bustamante et al. 2003; Eyre-Walker et al. 2006). For example, if a constant-population model is fitted to the African Drosophila data, the estimate for β is 0.52 (instead of 0.38 for an expanding population, Table 5) and NeE(s) is 5 × 106 (instead of 1800). Furthermore, under the constant-population model, the proportion of mutations inferred to have effects in the range 10 < Ns < 100 is markedly higher than that under population expansion. As expected, if ML estimates of the demographic model parameters obtained from the neutral site data are treated as fixed, estimates of confidence limits on the mutational parameters become somewhat narrower (results not shown).
The method introduced here has several limitations. First, the choice of sites at which neutral evolution is assumed to occur can be problematical. In mammals, intronic sequences, excluding those sequences involved in splicing, evolve only marginally more slowly than transposable-element remnants (Gaffney and Keightley 2006), so are a reasonable choice as a neutral standard. In contrast, synonymous sites evolve more slowly than introns or transposable-element remnants and seem to be under some form of purifying selection (Chamary et al. 2006). The situation is more difficult in Drosophila, because selection on all categories of noncoding DNA seems to be common (Andolfatto 2005; Halligan and Keightley 2006). For example, weak purifying selection at synonymous sites could generate the negative Tajima's D values seen in African D. melanogaster (Shapiro et al. 2007). It is generally thought that selection is no longer operating on synonymous codon use in D. melanogaster (Akashi 1996; McVean and Vieira 2001). However, to investigate the potential effects of selection on synonymous codon use and its influence on our estimates, we fitted a constant-population model that includes a parameter for selection (of the same magnitude) on all synonymous sites. We found that this model fits only slightly worse than a population-expansion model with no selection on synonymous sites. For example, the difference in log L is 1.3 and the ML estimate for the strength of selection on synonymous sites is Ns = 0.8 for the African Drosophila data. This is similar to the strength of selection inferred, for example, in D. simulans (McVean and Vieira 2001). Although this model fits almost as well as the population size-change model, parameter estimates for nonsynonymous mutational effects are somewhat different; e.g., for Africa, β = 0.51 and NE(s) = 540, as opposed to 0.38 and 1800. It was not feasible to estimate both selection on synonymous sites and a demographic change affecting all sites, because selection and population expansion or contraction can affect the SFS in similar ways, so the model becomes overparameterized.
A second limitation concerns the simple model of selection. Additive mutational effects have been assumed, but if recessive mutations are common, then selection against the heterozygote would be overestimated if alleles reached high enough frequencies to give an appreciable chance of the homozygote appearing. Furthermore, it is possible that the SFS for recessive mutations is qualitatively different from that for semidominant mutations. Unfortunately, there is no obvious way of estimating dominance of mutations from the SFS. The high frequency of weakly deleterious mutations in the best-fitting models, particularly in humans, suggests that slightly advantageous mutations should also be considered, but this would involve the fitting of at least one additional parameter, and it is unclear if the data contain information that could be used to estimate it. Advantageous mutations of large effect have little impact on the SFS, because they spend little time segregating, so it is reasonable to ignore their contribution to the SFS. Clearly, if there are large numbers of sites linked to alleles subject to some form of balancing selection, the results would be biased because such sites contribute to intermediate frequencies of the SFS.
Finally, the method is somewhat limited by the demographic scenarios that have been modeled. The step change in population size does not, for example, model the bottleneck followed by expansion that appears to have affected European human populations (Adams and Hudson 2004; Marth et al. 2004; Garrigan and Hammer 2006). A constantly expanding population might also fit African polymorphism data better than a single-step change. Incorporating these models is possible in principle, but would require the estimation of at least one additional parameter in each case and a considerable increase in the computational complexity and computing time of the method.
Acknowledgments
We thank Josh Shapiro for providing a data set of Drosophila nucleotide sequences and John Welch, Brian Charlesworth, Dan Halligan, Ian White, and Bill Hill for helpful comments and suggestions.
References
- Adams, A. M., and R. R. Hudson, 2004. Maximum-likelihood estimation of demographic parameters using the frequency spectrum of unlinked single-nucleotide polymorphisms. Genetics 168: 1699–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, H., 1996. Molecular evolution between Drosophila melanogaster and D. simulans: reduced codon bias, faster rates of amino acid substitution, and larger proteins in D. melanogaster. Genetics 144: 1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto, P., 2005. Adaptive evolution of non-coding DNA in Drosophila. Nature 437: 1149–1152. [DOI] [PubMed] [Google Scholar]
- Bataillon, T., 2000. Estimation of spontaneous genome-wide mutation rate parameters: Whither beneficial mutations? Heredity 84: 497–501. [DOI] [PubMed] [Google Scholar]
- Bustamante, C. D., R. Nielsen and D. L. Hartl, 2003. Maximum likelihood and Bayesian methods for estimating the distribution of selective effects among classes of mutations using DNA polymorphism data. Theor. Popul. Biol. 63: 91–103. [DOI] [PubMed] [Google Scholar]
- Butcher, D., 1995. Muller's ratchet, epistasis and mutation effects. Genetics 141: 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamary, J. V., J. L. Parmley and L. D. Hurst, 2006. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 7: 98–108. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker, A., and P. D. Keightley, 2007. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8: 610–618. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker, A., P. D. Keightley, N. G. C. Smith and D. Gaffney, 2002. Quantifying the slightly deleterious model of molecular evolution. Mol. Biol. Evol. 19: 2142–2149. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker, A., M. Woolfit and T. Phlelps, 2006. The distribution of fitness of new deleterious amino acid mutations in humans. Genetics 173: 891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney, D. J., and P. D. Keightley, 2006. Genomic selective constraints in murid noncoding DNA. PLoS Genet. 2: e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrigan, D., and M. F. Hammer, 2006. Reconstructing human origins in the genomic era. Nat. Rev. Genet. 7: 669–680. [DOI] [PubMed] [Google Scholar]
- Haag-Liautard, C., M. Dorris, X. Maside, S. Macaskill, D. L. Halligan et al., 2007. Direct estimation of per nucleotide and genomic deleterious mutation rates in Drosophila. Nature 445: 82–85. [DOI] [PubMed] [Google Scholar]
- Halligan, D. L., and P. D. Keightley, 2006. Ubiquitous selective constraints in the Drosophila genome revealed by a genome-wide interspecies comparison. Genome Res. 16: 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., and W. G. Hill, 1987. Directional selection and variation in finite populations. Genetics 117: 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemeny, J. F., and J. L. Snell, 1960. Finite Markov Chains. Van Nostrand, Princeton, NJ.
- Kondrashov, F. A., A. Y. Ogurtsov and A. S. Kondrashov, 2006. Selection in favor of nucleotides G and C diversifies evolution rates and levels of polymorphism at mammalian synonymous sites. J. Theor. Biol. 240: 616–626. [DOI] [PubMed] [Google Scholar]
- Lande, R., 1995. Mutation and conservation. Conserv. Biol. 9: 782–791. [Google Scholar]
- Li, H., and W. Stephan, 2006. Inferring the demographic history and rate of adaptive substitution in Drosophila. PLoS Genet. 2: e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston, R. J., A. von Niederhausern, A. G. Jegga, D. C. Crawford, C. S. Carlson et al., 2004. Pattern of sequence variation across 213 environmental response genes. Genome Res. 14: 1821–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe, L., B. Charlesworth, C. Bartolomé and V. Noel, 2006. Estimating selection on non-synonymous mutations. Genetics 172: 1079–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth, G. T., E. Czabarka, J. Murvai and S. T. Sherry, 2004. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics 166: 351–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean, G. A. T., and J. Vieira, 2001. Inferring parameters of mutation, selection and demography from patterns of synonymous site evolution in Drosophila. Genetics 157: 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelder, J. A., and R. Mead, 1965. A simplex method for function minimization. Comput. J. 7: 308–313. [Google Scholar]
- Nielsen, R., and Z. Yang, 2003. Estimating the distribution of selection coefficients from phylogenetic data with applications to mitochondrial and viral DNA. Mol. Biol. Evol. 20: 1231–1239. [DOI] [PubMed] [Google Scholar]
- Ohta, T., 1977. Extension of the neutral mutation drift hypothesis, pp. 148–167 in Molecular Evolution and Polymorphism, edited by M. Kimura. National Institute of Genetics, Mishima, Japan.
- Peck, J. R., G. Barreau and S. C. Heath, 1997. Imperfect genes, Fisherian mutation and the evolution of sex. Genetics 145: 1171–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piganeau, G. V., and A. Eyre-Walker, 2003. Estimating the distribution of fitness effects from DNA sequence data: implications for the molecular clock. Proc. Natl. Acad. Sci. USA 100: 10335–10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press, W. H., S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, 1992. Numerical Recipes in C, Ed. 2. Cambridge University Press, Cambridge/London/New York.
- Reich, D. E., and E. S. Lander, 2001. On the allelic spectrum of human disease. Trends Genet. 17: 502–510. [DOI] [PubMed] [Google Scholar]
- Sawyer, S. A., R. J. Kulathinal, C. D. Bustamante and D. L. Hartl, 2003. Bayesian analysis suggests that most amino acid replacements in Drosophila are driven by positive selection. J. Mol. Evol. 57: S154–S164. [DOI] [PubMed] [Google Scholar]
- Schultz, S. T., and M. Lynch, 1997. Mutation and extinction: the role of variable mutational effects, synergistic epistasis, beneficial mutations, and degree of outcrossing. Evolution 51: 1363–1371. [DOI] [PubMed] [Google Scholar]
- Shapiro, J. A., W. Huang, C. Zhang, M. J. Hubisz, J. Lu et al., 2007. Adaptive genic evolution in the Drosophila genomes. Proc. Natl. Acad. Sci. USA 104: 2271–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan, W., and H. Li, 2007. The recent demographic and adaptive history of Drosophila melanogaster. Heredity 98: 65–68. [DOI] [PubMed] [Google Scholar]
- Tenesa, A., P. Navarro, B. J. Hayes, D. L. Duffy, G. M. Clarke et al., 2007. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 17: 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, S., R. Hernandez, A. Fledel-Alon, L. Zhu, R. Nielsen et al., 2005. Simultaneous inference of selection and demography from patterns of variation in the human genome. Proc. Natl. Acad. Sci. USA 102: 7882–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yampolsky, L. Y., F. A. Kondrashov and A. S. Kondrashov, 2005. Distribution of the strength of selection against amino acid replacements in human proteins. Hum. Mol.Genet. 14: 3191–3201. [DOI] [PubMed] [Google Scholar]