

Abstract
The genetic basis of variation in recombination in higher plants is polygenic and poorly understood, despite its theoretical and practical importance. Here a method of detecting quantitative trait loci (QTL) influencing recombination in recombinant inbred lines (RILs) is proposed that relies upon the fact that genotype data within RILs carry the signature of past recombination. Behavior of the segregational genetic variance in numbers of chromosomal crossovers (recombination) over generations is described for self-, full-sib-, and half-sib-generated RILs with no dominance in true crossovers. This genetic variance, which as a fraction of the total phenotypic variance contributes to the statistical power of the method, was asymptotically greatest with half sibbing, less with sibbing, and least with selfing. The statistical power to detect a recombination QTL declined with diminishing QTL effect, genome target size, and marker density. For reasonably tight marker linkage power was greater with less intense inbreeding for later generations and vice versa for early generations. Generational optima for segregation variance and statistical power were found, whose onset and narrowness varied with marker density and mating design, being more pronounced for looser marker linkage. Application of this method to a maize RIL population derived from inbred lines Mo17 and B73 and developed by selfing suggested two putative QTL (LOD > 2.4) affecting certain chromosomes, and using a canonical transformation another putative QTL was detected. However, permutation tests failed to support their presence (experimentwise α = 0.05). Other populations with more statistical power and chosen specifically for recombination QTL segregation would be more effective.
GENETIC recombination is central to the evolutionary process, the study of genome organization, and plant and animal breeding. It pertains to the evolution of sex (Crow and Kimura 1965; Maynard Smith 1978) and the evolution of genome organization and the units of selection (Lewontin 1964, 1971; Felsenstein 1974; Felsenstein and Yokoyama 1976; Charlesworth et al. 1979) and is a fundamental consideration for the effect of selection acting on multiple loci (Hedrick et al. 1978). Further, the modification of recombination in plant breeding and thus its genetic basis are of current practical interest (Sall 1990; Busso et al. 1995).
The best understanding of recombination mechanisms arises from genetic and molecular studies in bacteria and yeast (reviewed in Petes et al. 1991). Much less is known about mechanisms of recombination in plants and animals. Meiotic recombination in plants and animals is influenced by sex and genetic background, in particular cis effects of chromosomal rearrangements, centromeres, and heterochromatin, and supernumary chromosomes that affect recombination in trans (Grant 1975; Korol et al. 1994). At the molecular level, somatic recombination rates in repeated sequence transgenes in plants have revealed the influence of homology (Gal et al. 1991), repeat length (Peterhans et al. 1990), and genome position (Swoboda et al. 1993). Effects of homology upon meiotic recombination in maize have also been studied by Dooner and Martinez-Ferez (1997) and Okagaki and Weil (1997). Trans-acting polygenic control of general or specific genomic recombination is suggested by variation in recombination frequencies among RFLP and morphological markers in a variety of segregating maize populations (Beavis and Grant 1991; Tulsieram et al. 1992; Fatmi et al. 1993; Timmermans et al. 1997). As with most quantitative traits, however, polygenic control of recombination is probably most common and thus more difficult to study than discrete mutations.
In this article we note that since recombinant inbred lines (RILs) provide information about their number of crossovers and hence segregating quantitative trait loci (QTL) influencing recombination (hereafter referred to as recombination QTL), data on existing RILs may allow the detection of such QTL in animals and plants. We sketch the basic theory behind this idea and examine the effects of QTL effect, mating design, recombination target size, and map density upon the variance in numbers of crossovers among inbred lines caused by recombination QTL. Because the fraction of phenotypic variance caused by genetic segregation is key to predicting the power of detection (Lander and Botstein 1989; Lynch and Walsh 1998), we use the expected fraction of phenotypic variance caused by segregational variance to compare the relative power of designs under different parameters of the model. We further apply this method to a maize RIL population as an illustration of the approach.
GENETIC MODEL
RIL populations are produced by crossing two genetically distinct inbreds, selfing or sibbing the F1's to produce F2's, and selfing or sibbing individual F2's and their subsequent progeny for several generations (six or more) to produce a series of highly homozygous inbred lines. Each RIL contains a mixture of genomic segments from the two original inbred parents.
If original parents differ at loci influencing recombination, alleles will segregate and be partitioned into various inbred lines, in the case of selfing, or equivalently the variance of allelic frequencies among lines increases due to genetic drift within inbreeding lines (but randomly mating within). This generates variation in recombination frequency among lines that should be associated with the number of detectable crossovers among RIL genotypes.
While the greatest variance in observed crossovers would occur when recombination loci are fixed among different lines and recombining segments are heterozygous, since all neutral loci become homozygous at the same rate with inbreeding, the merit of this approach seems dubious. Further, detection of recombination relies upon linkage disequilibria among marker loci that go to zero as inbreeding and recombination proceed. An additional problem is that loci that increase recombination decrease linkage disequilibria between marker loci, thus decreasing the number of detected crossovers and vice versa. Our motivation is therefore to develop a simple quantitative model to evaluate the statistical power of this method by determining the number of inbred lines scored necessary to detect a QTL with a certain effect under different allelic effects, mating systems, sizes of the genomic region(s) (hereafter referred to as the target) affected by the recombination QTL, and marker densities. We begin by examining the genetic expectations.
Consider a single QTL with effects upon the total number of those detectable occurring in a RIL in a certain generation. Define 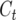 as the number of detected crossovers expected per chromosome within a certain genomic region (the target) of a diploid individual within a certain inbred line following t generations of inbreeding. As crossovers accumulate, the number of crossovers following T generations of inbreeding is given by
as the number of detected crossovers expected per chromosome within a certain genomic region (the target) of a diploid individual within a certain inbred line following t generations of inbreeding. As crossovers accumulate, the number of crossovers following T generations of inbreeding is given by
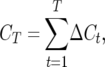 |
(1) |
where 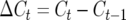 is the change in total number of crossovers between adjacent generations.
is the change in total number of crossovers between adjacent generations.
For any system of mating with a Poisson distribution of crossovers in a target with n heterozygous loci and no interference, the Haldane (1919) map function applies. Thus,
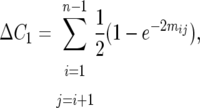 |
(2) |
where 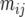 is the mean number of actual crossovers occurring between adjacent loci i and j. Recombination between loci i and j in only double heterozygotes affects
is the mean number of actual crossovers occurring between adjacent loci i and j. Recombination between loci i and j in only double heterozygotes affects 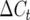 , adding numbers of observable crossovers to double heterozygotes in coupling and subtracting them in repulsion double heterozygotes. Because the difference in coupling and repulsion heterozygotes in an inbred (bottlenecked) segregating line with random mating within is
, adding numbers of observable crossovers to double heterozygotes in coupling and subtracting them in repulsion double heterozygotes. Because the difference in coupling and repulsion heterozygotes in an inbred (bottlenecked) segregating line with random mating within is 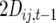 , where
, where 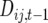 is the within line linkage disequilibrium between loci i and j in generation t − 1,
is the within line linkage disequilibrium between loci i and j in generation t − 1,
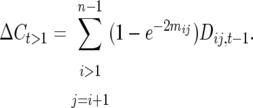 |
(3) |
The disequilibria may be determined for a system of inbreeding by using the digenic descent measures of Cockerham and Weir (1968, 1977). Consider the inbreeding coefficient (Wright 1922) in generation t, 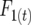 to represent the probability that each gamete uniting to form a zygote in this generation carries an allele at a certain locus that is descended from the same initial gamete at the start of a pedigree. Similarly, the recombinant coefficient
to represent the probability that each gamete uniting to form a zygote in this generation carries an allele at a certain locus that is descended from the same initial gamete at the start of a pedigree. Similarly, the recombinant coefficient 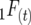 is the probability that alleles at two different loci inherited from both uniting gametes are descended from the same initial gamete, and the parental coefficient
is the probability that alleles at two different loci inherited from both uniting gametes are descended from the same initial gamete, and the parental coefficient 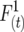 is the probability that two alleles at different loci inherited from one gamete are descended from the same initial gamete. Both
is the probability that two alleles at different loci inherited from one gamete are descended from the same initial gamete. Both 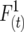 and
and 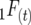 are digenic measures since they refer to two alleles at separate loci. Note that if two loci are completely linked,
are digenic measures since they refer to two alleles at separate loci. Note that if two loci are completely linked, 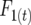 =
= 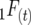 and
and 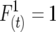 , and in general the linkage disequilibrium within lines equals
, and in general the linkage disequilibrium within lines equals 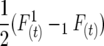 with initial gametes coming from two inbred parents (cf. Cockerham and Weir 1977). Formulas for calculating these coefficients for RILs generated by selfing, full sibbing, and half sibbing are given in appendix a.
with initial gametes coming from two inbred parents (cf. Cockerham and Weir 1977). Formulas for calculating these coefficients for RILs generated by selfing, full sibbing, and half sibbing are given in appendix a.
We consider crossover rate to differ among genotypes at a locus influencing recombination such that the mean true number occurring any generation between the adjacent loci i and j is 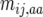 in one homozygote,
in one homozygote, 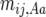 in the heterozygote, and
in the heterozygote, and 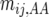 in the other homozygote, with
in the other homozygote, with 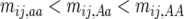 . Similarly,
. Similarly, 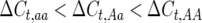 at least in early generations (see below). Expected values of
at least in early generations (see below). Expected values of 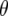 for the three genotypes at the recombination locus in generation t are accordingly
for the three genotypes at the recombination locus in generation t are accordingly 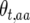 ,
, 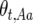 , and
, and 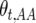 . Define the difference in change in observable crossovers between a line and its expectation at generation t as
. Define the difference in change in observable crossovers between a line and its expectation at generation t as 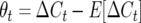 , with
, with 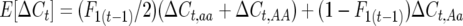 . Using Equation 1, the segregation variance among lines in total crossovers at generation T is
. Using Equation 1, the segregation variance among lines in total crossovers at generation T is
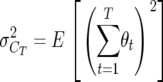 |
(4) |
Taking into account that the segregation variance is due to drift in inbred lines at the recombination locus, and expanding, Equation 4 may be rewritten as
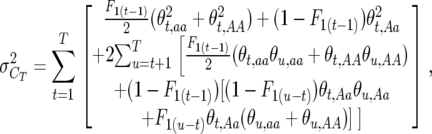 |
(5) |
which illustrates the contribution of both the segregation variance within generations and the covariance among them due to fixation of recombination alleles. The (u − t) subscripts refer to the probability of identity by descent (Wright 1922) t generations after heterozygosity.
To evaluate the statistical power of this method, a computer program was written to compute the segregation variance under various mating systems and other parameters of the model such as allelic effects (defined as the difference plus or minus a percentage of the heterozygote m that additively influences the true recombination fraction m), target size, and map densities for a single QTL. We assumed, for simplicity, a uniform distribution of n genetic markers, giving n − 1 recombination intervals. Thus, n − 1 replaced the summation over marker intervals in Equations 1 and 2. The number of inbred lines necessary to detect a recombination QTL was computed using a type I error rate of α = 0.01 and a type II error rate of β = 0.10. The low value of α was intended to accommodate the practical problem of multiple genomic region comparisons in QTL mapping (cf. Lander and Botstein 1989).
Lynch and Walsh (1998) have given expressions for the number of required measured individuals in segregating F2 and backcross populations to detect a QTL with probability 1 − β at significance level α and note that these are quite similar for related designs. We extended their formula for F2 populations to allow for t generations of inbreeding to compute the number of inbred lines required as approximately
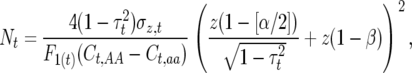 |
(6) |
where 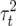 is the proportion of phenotypic variance due to segregation,
is the proportion of phenotypic variance due to segregation, 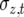 is the phenotypic standard deviation, and
is the phenotypic standard deviation, and 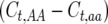 is the difference between the homozygote with more observed crossovers minus that with fewer. This expression reflects a t-test between different homozygote categories. The
is the difference between the homozygote with more observed crossovers minus that with fewer. This expression reflects a t-test between different homozygote categories. The 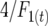 factor compensates for the increase in homozygotes as inbreeding proceeds. The nongenetic component of variance in crossovers was conservatively approximated as the binomial variance (assuming no interference, i.e., a number of Bernoulli trials) for n − 1 regions
factor compensates for the increase in homozygotes as inbreeding proceeds. The nongenetic component of variance in crossovers was conservatively approximated as the binomial variance (assuming no interference, i.e., a number of Bernoulli trials) for n − 1 regions 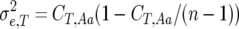 . This value is conservative because it does not take into account environmental or epistatic variation in recombination due to, e.g., temperature and gender. Thus, N may be biased downward for some conditions.
. This value is conservative because it does not take into account environmental or epistatic variation in recombination due to, e.g., temperature and gender. Thus, N may be biased downward for some conditions.
Figure 1a shows how varying levels of allelic effects influence required numbers of full-sibbed lines to detect the recombination QTL where its effect is upon a 2000-cM region (a typical genome size for many plants) with markers spaced c = 0.10 (10 cM) apart. Homozygote effects were given as a percentage, plus or minus, of the number of true crossovers in the QTL heterozygote; e.g., for 20% effect, 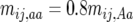 , and
, and 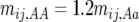 . Many more lines are needed for weaker effects, e.g., 1%, as expected. Figure 1b illustrates N to detect for the alleles with stronger effects, e.g., 20, 10, and 5%. To put it into perspective, consider that if all loci have equal and independent effects in a segregating cross, with the total segregation variance equal to 30% of the phenotypic variance, if individual QTL have effects of 20, 10, 5, and 1%, there are respectively ∼7, 29, 121, and 2330 QTL influencing recombination segregating within the cross. Clearly major genes are more easily detected.
. Many more lines are needed for weaker effects, e.g., 1%, as expected. Figure 1b illustrates N to detect for the alleles with stronger effects, e.g., 20, 10, and 5%. To put it into perspective, consider that if all loci have equal and independent effects in a segregating cross, with the total segregation variance equal to 30% of the phenotypic variance, if individual QTL have effects of 20, 10, 5, and 1%, there are respectively ∼7, 29, 121, and 2330 QTL influencing recombination segregating within the cross. Clearly major genes are more easily detected.
Figure 1.—


For Figures 1–6, generation 1 is the F1 generation. The number of recombinant inbred lines (RILs) N generated by full sibbing needed to detect a recombination QTL as a function of allelic (homozygous) effect is shown. Type I and II errors have been set to 0.01 and 0.10, respectively, for Figures 1–6. A recombination QTL acts upon a 2000-cM target with mean map distance among markers (marker density) of 10 cM (e.g., for individuals in which the recombination QTL is heterozygous). (a) Results are shown for 20, 10, 5, and 1% plus or minus the additive homozygote effect upon true numbers of true crossovers  relative to QTL heterozygotes. (b) The same as a, except the 1% homozygote effect has been deleted to provide greater detail for the larger allelic effects.
relative to QTL heterozygotes. (b) The same as a, except the 1% homozygote effect has been deleted to provide greater detail for the larger allelic effects.
The number of RILs needed to detect recombination QTL depends on the size of the target genome. Figure 2 illustrates N required under conditions of fullsibbing and target size ranging from 2000 to 100 cM, with mean marker density of 10 cM and 10% homozygote effect. For a given marker density our method can most easily detect QTL that influence larger genomic regions because of the greater opportunity for segregation variance in regions containing more genetic markers. One must be careful, therefore, to not interpret different frequencies of detection as evidence for a different number or magnitude effect of recombination QTL when target regions differ in size.
Figure 2.—

The number of RILs N generated by full sibbing needed to detect a recombination QTL as a function of recombination target sizes of 2000, 1000, 400, 200, and 100 cM, with homozygote allelic effect of 10%, as in Figure 1.
Marker density and mating design have striking effects upon statistical power (Figure 3, a–c). In general, tighter marker linkage yields fewer N required to detect recombination QTL. This pattern is related to the finding that larger recombination targets require fewer N, as in both cases there are more markers in the target, which increases segregation variance. Regardless of marker density, less intense inbreeding yields the least N required, over the 15 generations evaluated. This effect is more pronounced for tighter linkage (cf. Figure 3, a and c). However, for early generations of inbreeding (up to the 5th generation), more intense inbreeding, e.g., selfing, leads to a lower N required. Surprisingly, especially for more gradual inbreeding, e.g., half sibbing, and relatively sparse marker density (mean = 15 cM), there are minimum values of N required for detection following ∼6 generations of inbreeding (Figure 3a). This pattern is not seen (at least within 15 generations) for tighter marker linkage (mean = 10 or 5 cM; Figure 3, b and c).
Figure 3.—



Effect of marker density and mating system upon N required for recombination QTL detection. The QTL acts upon a 2000-cM region with homozygote effect of 10%. (a) Effect of mating system with mean marker density of 15 cM. (b) Effect of mating system with mean marker density of 10 cM. (c) Effect of mating system with mean marker density of 5 cM.
These unexpected patterns are due to the effects of marker density on the segregation variance. Figure 4a shows a maximum for the variance near six generations of inbreeding, at loose marker linkage (mean =15 cM), which parallels the expectation from Figure 3a. This maximum is not apparent (or barely so) for tighter linkage (mean = 5 cM; Figure 4b), again paralleling the pattern expected from Figure 3c.
Figure 4.—


Effect of marker density and mating design upon segregation variance among RILs for number of crossovers, using the Haldane mapping function (no interference). The recombination QTL acts upon a 2000-cM region with homozygote effect of 10%. (a) Effect of mating design with mean marker density of 15 cM. (b) Effect of mating design with mean marker density of 5 cM.
To explain these results it is informative to investigate the behavior of 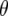 -values, which represent the difference between the per generational change in crossover number of each QTL genotype and its expectation (shown for QTL homozygotes in Figure 5). Under the same parameter values as in Figures 3a and 4a (e.g., mean marker density = 15 cM), under half sibbing
-values, which represent the difference between the per generational change in crossover number of each QTL genotype and its expectation (shown for QTL homozygotes in Figure 5). Under the same parameter values as in Figures 3a and 4a (e.g., mean marker density = 15 cM), under half sibbing 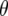 switches sign about generation 7 (Figure 5a). For tighter linkage (mean = 5 cM), and the same parameter values as in Figure 3c, for half sibbing
switches sign about generation 7 (Figure 5a). For tighter linkage (mean = 5 cM), and the same parameter values as in Figure 3c, for half sibbing 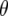 -values do not switch sign and become only gradually reduced in magnitude (Figure 5b). The switch in sign produces negative covariances of crossover number among earlier and later generations (Equation 5). Surprisingly, this means that the homozygote with greater recombination accumulates fewer new crossovers than the contrasting homozygote. Thus, one would be wrong to infer gene action from a limited observation of per generational changes in later generations with such loose linkage.
-values do not switch sign and become only gradually reduced in magnitude (Figure 5b). The switch in sign produces negative covariances of crossover number among earlier and later generations (Equation 5). Surprisingly, this means that the homozygote with greater recombination accumulates fewer new crossovers than the contrasting homozygote. Thus, one would be wrong to infer gene action from a limited observation of per generational changes in later generations with such loose linkage.
Figure 5.—


Effect of marker density upon the between-generational changes in observed crossovers and the expected value over all genotypes; e.g., 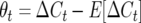 for generation t in QTL homozygotes under half sibbing, using the Haldane mapping function (no interference). The recombination QTL acts upon a 2000-cM region with homozygote effect of 10%. (a) Effect of mating design with mean marker density of 15 cM. (b) Effect of mating design with mean marker density of 5 cM.
for generation t in QTL homozygotes under half sibbing, using the Haldane mapping function (no interference). The recombination QTL acts upon a 2000-cM region with homozygote effect of 10%. (a) Effect of mating design with mean marker density of 15 cM. (b) Effect of mating design with mean marker density of 5 cM.
The behavior of 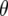 -values reflects the fact that lines that have an excess of alleles enhancing recombination enhance the number of crossovers for earlier generations and so decrease the disequillibria among markers. The accumulation of new observed crossovers then ultimately becomes less than that of lines that have an excess of alleles decreasing recombination. The switch occurs more rapidly and with larger effect for loose linkage because the accumulation of new observed crossovers depends on
-values reflects the fact that lines that have an excess of alleles enhancing recombination enhance the number of crossovers for earlier generations and so decrease the disequillibria among markers. The accumulation of new observed crossovers then ultimately becomes less than that of lines that have an excess of alleles decreasing recombination. The switch occurs more rapidly and with larger effect for loose linkage because the accumulation of new observed crossovers depends on 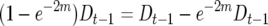 (cf. Equation 3). As m increases the second term on the right side approaches zero, leaving the contrast in
(cf. Equation 3). As m increases the second term on the right side approaches zero, leaving the contrast in 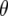 between different QTL homozygotes as predominantly depending upon their difference in
between different QTL homozygotes as predominantly depending upon their difference in 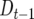 .
.
Figures 3 and 4 demonstrate the efficiency of different mating designs on detecting recombination QTL as a function of marker density. Figure 3a shows that the effect of loose linkage (= 15 cM) in causing a generational minimum in N required for QTL detection is exacerbated by a lesser rate of inbreeding, e.g., half sibbing. This pattern is also evident by inspection of the segregational variance (Figure 4a). With tighter linkage, e.g., mean = 10 or 5 cM (Figure 3, b and c), this effect is not seen. Moreover, tighter linkage enhances the segregation variance for more gradual inbreeding, e.g., full- and half-sib designs, thus decreasing N necessary for detection (cf. Figures 3 and 4). In summary, looser marker linkage reduces statistical power for detection of recombination QTL because (1) fewer crossovers are possible, reducing the segregation variance, and (2) the segregation variance declines more quickly due to the effect of recombination alleles on marker disequilibria.
To evaluate effects of interference of recombination, which has been heretofore assumed absent, we modified Equation 2 to utilize the Kosambi mapping function (Kosambi 1944), which presumes that positive interference decreases linearly as m increases (Ott 1985). As expected, segregation variance results were very similar to those using the Haldane mapping function (cf. Figures 4 and 6). Interference has little effect upon qualitative patterns of response as functions of marker density and mating design. However, with looser linkage the asymptote in segregation variance is more sharply defined (cf. Figures 4a and 6a). It is difficult to compare results for N required with interference to those without, as we assumed independence (no interference) of crossovers to estimate the nongenetic component of variance in observed crossovers. However, as both genetic and nongenetic components of total variance in observed crossovers are expected to behave similarly with interference, it seems that no qualitative changes in our conclusions would follow from using a more biologically realistic model that includes interference.
Figure 6.—


Effect of marker density and mating design upon segregation variance among RILs for numbers of crossovers, using the Kosambi mapping function (interference allowed). The recombination QTL acts upon a 2000-cM region with homozygote effect of 10%. (a) Effect of mating design with mean marker density of 15 cM. (b) Effect of mating design with mean marker density of 5 cM.
APPLICATION TO A MAIZE RECOMBINANT INBRED POPULATION
We applied this method to marker data derived from 208 RILs arising from a cross between maize inbreds Mo 17 and B73, which was kindly provided by L. Senior (North Carolina State University). This cross was previously described by Stuber et al. (1992), who used it to generate segregating populations used to map QTL involved in heterosis. Following seven generations of selfing, loci were scored for a total of 202 RFLP, microsatellite, allozyme, and morphological markers with a mean density of 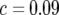 , for a total genome size of ∼1818 cM, represented by 10 chromosomes. The number of crossovers was scored by counting the number of crossovers of homozygote genotypes. For example, the ordered sequence of A and B parent homozygote genotypes over the four adjacent loci AABB reflects one crossover between the second and the third locus. For heterozygotes, three possibilities exist, and for these we extrapolated to the expected number of crossovers at complete homozygosity. Thus, AHA or BHB yields
, for a total genome size of ∼1818 cM, represented by 10 chromosomes. The number of crossovers was scored by counting the number of crossovers of homozygote genotypes. For example, the ordered sequence of A and B parent homozygote genotypes over the four adjacent loci AABB reflects one crossover between the second and the third locus. For heterozygotes, three possibilities exist, and for these we extrapolated to the expected number of crossovers at complete homozygosity. Thus, AHA or BHB yields 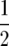 crossover on average, AHB or BHA yields one, and a sequence where H is terminal, e.g., AH terminus or BH terminus, yields
crossover on average, AHB or BHA yields one, and a sequence where H is terminal, e.g., AH terminus or BH terminus, yields 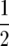 . Very rarely 2 or 3 adjacent markers were heterozygous, e.g., AHHA or BHHB; these were treated as one heterozygous locus, which slightly underestimates the expected number of crossovers by neglecting multiple crossovers.
. Very rarely 2 or 3 adjacent markers were heterozygous, e.g., AHHA or BHHB; these were treated as one heterozygous locus, which slightly underestimates the expected number of crossovers by neglecting multiple crossovers.
Since missing data preclude observation of certain crossovers and thus cause less informative individuals to have fewer, we corrected by assigning them the empirical expected number of crossovers derived from the informative subset (appendix b). We excluded data from individuals in which more than three adjacent internal chromosomal loci or two terminal loci had missing data, causing the exclusion of 25 of 208 RILs for a final sample size of 183. Correction may yield a conservative test of QTL presence since it will reduce segregation variance . Because missing data are expected to be unrelated to genotype, tests of the presence of QTL should not be biased by using uncorrected data; however, estimates of the distribution of crossovers will. We therefore used both corrected and uncorrected data in tests for presence of QTL. These were done using qGene 2.20 (provided courtesy of Clare Nelson, Cornell University). qGene uses a flanking-marker regression technique (Haley and Knott 1992) that gives results similar to those of maximum-likelihood analysis but is computationally faster. A regression was performed at every interval of 1 cM and the F or LOD statistic was found (see Doerge 1995 for their relationship). In addition to LOD scores we used the better supported method of permutation tests (Churchill and Doerge 1994), using 1000 genomewide permutations of the relevant F statistic to evaluate the experimentwise significance of putative recombination QTL with qGene 2.20.
The distribution of total crossovers was slightly skewed upward with a mean of 23.230 and variance of 34.239, but was approximately normally distributed on the basis of P-P and Q-Q normality plots for both the total number of crossovers and subsets of these data (SPSS 1994). Transformation to natural logs improved normality but had little effect on the analyses; hence results are described only for the untransformed data.
QTL influencing recombination could act genomewide (globally) or upon certain sections of the genome (locally). If they act globally, we expect a positive correlation among all pairs of chromosomes within lines in number of crossovers, whereas if they act upon certain chromosomes, only some pairs of chromosomes would be positively correlated. Environmental influences upon recombination could also act globally or locally. Using a Bonferroni correction for multiple comparisons, chromosomes 8 and 10 were significantly correlated (r = 0.282; P = 0.000). Six other of the 45 pairwise correlations showed positive correlations with P < 0.05 but these were not significant because of the multiple comparisons (results not shown).
Although the numbers of targets within certain regions could be partitioned in many different nonindependent ways, we a priori defined the following traits: (1) total genomic crossovers, (2) individual chromosome crossovers, (3) combinations of crossovers derived by summing the numbers of positively correlated chromosomes (with P < 0.05), and (4) the numbers of proximal (middle 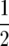 ) vs. distal (outer
) vs. distal (outer 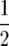 ) crossovers within the quartiles of marker locations of individual chromosomes. The motivation for trait 3 was that QTL effects upon two chromosomes jointly could be measured by their total number of crossovers, and that for trait 4 by previous results indicating a differential impact of B chromosomes and the K10 chromosome upon recombination in centromeric proximal and distal regions (Hanson 1969; Nel 1973; Rhoades 1978).
) crossovers within the quartiles of marker locations of individual chromosomes. The motivation for trait 3 was that QTL effects upon two chromosomes jointly could be measured by their total number of crossovers, and that for trait 4 by previous results indicating a differential impact of B chromosomes and the K10 chromosome upon recombination in centromeric proximal and distal regions (Hanson 1969; Nel 1973; Rhoades 1978).
In sum, we constructed a total of 20 different more- or less-independent quantitative traits to test for recombination QTL. With our uncertainty of the method of action (especially global or local) of recombination QTL and the infinite number of ways in which the total data may be restructured, multiple comparisons of these different data sets greatly increase the probability of type I errors.
Churchill and Doerge (1994) suggested that the problem of multiple comparisons among different genetic markers is best treated by permutation tests using experimentwise error levels. The problem of multiple quantitative trait comparisons has been addressed more recently (e.g., Korol et al. 1995; Spelman et al. 1996; Weller et al. 1996; Zhang et al. 1998). To increase the otherwise reduced statistical power by assuming that different quantitative traits are independent, Weller et al. (1996) suggested canonical transformation of the trait data set and analysis of the derived canonical variables. Threshold values for the test statistic are derived from the number of independent canonical variables. If QTL affecting the canonical variables are judged significant, the effects on the original variables could be derived by the reverse transformation, i.e., the inverse of the eigenvector matrix multiplied by the vector of effects. Alternatively, threshold values for canonical or principal-component variables may be used to test for effects upon the untransformed traits (cf. Spelman et al. 1996; Zhang et al. 1998). We applied these methods by first standardizing the traits (crossovers values) to have mean zero and unit variance and then multiplying the trait matrix by the matrix of eigenvectors of the (co)variance (correlation) matrix, using SAS (1997). These resulted in canonical variables equal to the number of traits (20), of which 8 had eigenvalues, representing coefficients of determination of the canonical variables, which accounted for >95% of the overall variance. Therefore, we concluded that the data set represented 8 independent traits. Thus, the experimentwise type I error level was 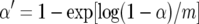 = 0.006 with α = 0.05 and m = 8. We verified that the same procedures may equivalently utilize the principal-component coefficients, using PROC PRINCOMP (SAS Institute 1997), which gave the same results.
= 0.006 with α = 0.05 and m = 8. We verified that the same procedures may equivalently utilize the principal-component coefficients, using PROC PRINCOMP (SAS Institute 1997), which gave the same results.
Due to the low fraction of missing data, the corrected data set gave similar results to the uncorrected data set and hence results are given only for the corrected data set. The QTL analysis for corrected data found two regions with LOD > 2.4 (Table 1), which seemed to have effects on crossovers on other different chromosomes. None of the other sets of genomic regions (combinations of two chromosomes, or proximal vs. distal comparisons) suggested the presence of QTL (LOD < 2.4). However, experimentwise permutation tests showed significance levels in excess of P = 0.05 (experimentwise α′ = 0.006) for both QTL with LOD > 2.4; thus they were clearly not significant. The comparison of canonical variables also found a putative QTL on chromosome 2 with LOD = 2.55 (Table 1), affecting canonical variable 6, but this too was judged insignificant by the permutation tests with P = 0.06, far above the experimentwise α′ = 0.006.
TABLE 1.
Putative QTL influencing recombination
| QTL location (chromosome) | Effect upon | Location (cM; closest marker) | LOD | Contributor |
|---|---|---|---|---|
| 3 | Chromosome 2 | 113; umc60 | 2.65 | Mo17 |
| 1 | Chromosome 9 | 161; umc128, phi002 | 2.45 | B73 |
| 2 | Canonical variable 6 | 19; umc36b | 2.55 | B73 |
Contributor is the parent whose allele increases observed recombination.
DISCUSSION
There are several reasons why our failure to detect recombination QTL at the experimentwise level in this first empirical test is not surprising. First, consider genome target size effects. With a genome size of ∼2000 cM and chromosome sizes of ∼200 cM, as in this study, only 1 of the 20 traits we considered had a target size of ∼2000 cM (total genome target), which would be most powerful (Figure 2), and the concomitant restructuring of the data (per genomic fragment or combination thereof) presents statistical problems. Of the others, 7 were ∼400 cM, as the sum of two correlated chromosomes, another 10 were ∼200 cM for each of the individual chromosomes, and 2 others were the total genomic sum of distal or proximal regions, ∼1000 cM.
Second, by devising new composite traits, less statistical power is available. We illustrated the canonical or principal-component transformations as a solution to this problem but their biological interpretations are less obvious. The most effective approach would seem to use a series of sequential tests, choosing to detect effects of a recombination QTL upon total genomic crossovers first, which is most powerful, or use an a priori most likely specific method based upon the question in mind. Sequential tests based upon a priori knowledge of biology or power would minimize the statistical problems of multitrait comparisons. Third, the selfing design used here is less superior in general to the full or half sibbing designs, especially when marker density is reasonably high, as in this case (Figures 1–4). Fourth, statistical power would have been increased by using a more saturated map, (e.g., mean marker recombination fraction c < 0.09; Figures 3 and 4). Fifth, especially with smaller QTL effects, a sample size much larger than 183 inbred lines would be necessary to detect QTL (Figures 1–4), and finally, this cross was not chosen to maximize the possible segregation of recombination QTL, as may be the case when parents are suspected to differ in recombination rate. Also, it should be noted that segregation of recombination QTL would increase the error in estimates of recombination rates in target areas; however, these are unlikely to affect the ordering of loci, which is the only error (aside from scoring errors) that would influence this approach.
Since many other RIL data sets already exist, a search for recombination QTL may easily be accomplished as we have illustrated here. It is also possible that doubled haploids could also be used in a similar fashion to detect recombination QTL, but such data sets are far less available than data from RILs. These recombination QTL could be further examined by fine-scale mapping, development of BAC contigs, and further isolation to small genetic fragments to examine and validate their function and utility (e.g., Deng et al. 2001).
In addition to identifying loci modifying recombination to improve transfer of useful traits in plant and animal breeding, our approach suggests that RILs extracted from natural populations, while perhaps less variable for most markers, could indicate the basis of genetic variation at QTL for recombination in nature. This information, in conjunction with assessment of overall polygenic variation in recombination, could be used to estimate naturally occurring levels of genetic variation in recombination as well as the degree to which it is under major or minor gene control.
Our approach also suggests a means for testing the hypothesis that a balance between plant breeding systems and intragenomic recombination evolves. For example, Stebbins (1950) suggested that inbreeders might have greater recombination rates than outbreeders with the same chromosome number due to stabilizing selection on the overall genetic system (cf. Mather 1943; Darlington 1958). While this notion has been supported by comparing chiasma frequency in a number of related outbreeders and inbreeders, the relationship between chiasma frequency and genetic recombination is frequently unclear (Grant 1958, 1975; Brown 1961). In organisms with nearly perfect cytological behavior, it may be possible to relate chiasma frequency with recombination (e.g., King et al. 2002). In these cases chiasma frequency may serve as an alternative to our molecular marker approach. However, for example, in maize, relating chismata counts to actual recombination is very impractical (D. Weber, unpublished data). One would predict under Mather's, Stebbins', and Darlington's hypothesis that alleles increasing recombination would more frequently map to the inbreeding parent in crosses among closely related species differing in mating system (e.g., Dole and Jain 1992; Dole and Kesseli 1997; Lin and Ritland 1997; Fishman et al. 2002), especially when the evolution of recombination is due to QTL with large effects. In any case, the genetic architecture of the coevolution of recombination and mating systems can be studied with this approach.
Acknowledgments
We thank L. Senior for providing the RIL data set; M. Schneerman for helpful advice, encouragement, and comments on the manuscript; B. Beavis for helpful insights; J. Crow for helpful suggestions; and an anonymous reviewer for recommendations.
APPENDIX A
Formulas for computation of the monogametic and digametic descent measures 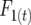 ,
, 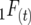 , and
, and 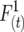 are shown. These coefficients depend upon the linkage parameter
are shown. These coefficients depend upon the linkage parameter 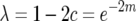 for this model (Cockerham and Weir 1977), where c is the observed recombination fraction. Generation t = 0 is the F1 generation, in contrast to the figures where generation t = 1 is the F1 generation. The half sibbing model follows Li (1976).
for this model (Cockerham and Weir 1977), where c is the observed recombination fraction. Generation t = 0 is the F1 generation, in contrast to the figures where generation t = 1 is the F1 generation. The half sibbing model follows Li (1976).
Selfing:
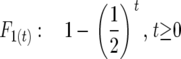 |
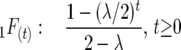 |
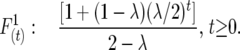 |
Full sibbing:
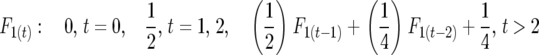 |
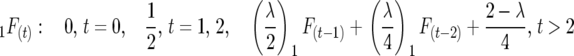 |
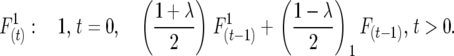 |
Half sibbing:
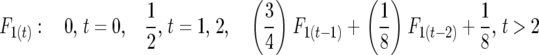 |
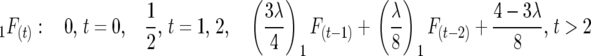 |
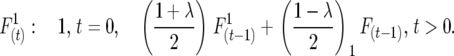 |
APPENDIX B
To correct for missing data, we used the following approach. For the following sequence of missing data, indicated by “-,” there are n + 1 relevant crossovers. For example, the sequence A–A has three relevant crossover frequencies, x1, x2, and x3, the first and the last one at the border of the missing sequence and the flanking informative markers. Empirical estimates of the x's were derived from the portion of the data set that was informative for the region in question. Then we applied the following corrections, adding the expected number of crossovers in a sequence 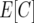 to the observed numbers of crossovers in an individual genotype:
to the observed numbers of crossovers in an individual genotype:
| Case | 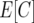 |
|---|---|
| A-B | 0 |
| A-A | 2x1x2 |
| A–B | 2x1x2x3 |
| A–A | 2[(1 − x1)x2x3 + x1(1 − x2)x3 + x1x2(1 − x3)] |
| A—B | 2[(1 − x1)x2x3x4 + x1(1 − x2)x3x4 + x1x2(1 − x3)x4 + x1x2x3(1 − x4)] |
| A—A | 2[(1 − x1)(1 − x2)x3x4 + x1(1 − x2)(1 − x3)x4 + x1x2(1 − x3)(1 − x4) + (1 − x1)x2(1 − x3)x4 + x1(1 − x2)x3(1 − x4)] |
| A-terminal | x1 |
| A–terminal | 2x1x2 + x1(1 − x2) + (1 − x1)x2 |
References
- Beavis, W. D., and D. Grant, 1991. A linkage map based on information from four F2 populations of maize (Zea mays L.). Theor. Appl. Genet. 82: 636–644. [DOI] [PubMed] [Google Scholar]
- Brown, H.S., 1961. Differential chiasma frequencies in self-pollinating and cross- pollinating species of the genus Gilia. Aliso 5: 67–81. [Google Scholar]
- Busso, C. F., C. J. Liu, C. T. Hash, J. R. Witcombe, K. M. Devos et al., 1995. Analysis of recombination rate in female and male gametogenesis in pearl millet (Pennisetum glaucum), using RFLP markers. Theor. Appl. Genet. 90: 242–246. [DOI] [PubMed] [Google Scholar]
- Charlesworth, D., B. Charlesworth and C. Strobeck, 1979. Selection for recombination in partially self-fertilizing populations. Genetics 93: 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138: 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerham, C. C., and B. S. Weir, 1968. Sib mating with two linked loci. Genetics 60: 629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockerham, C. C, and B. S. Weir, 1977. Digenic measures for finite populations. Genet. Res. 30: 121–147. [Google Scholar]
- Crow, J. F., and M. Kimura, 1965. Evolution in sexual and asexual populations. Am. Nat. 99: 439–450. [Google Scholar]
- Darlington, C. D., 1958. The Evolution of Genetic Systems. Basic Books, New York.
- Deng, Z., S. Huang, P. Ling, C. Yu, Q Tao et al., 2001. Fine genetic mapping and BAC contig development for the citrus tristeza virus resistance gene locus in Poncirus trifoliata (Raf.) Mol. Gen. Genomics 265: 739–747. [DOI] [PubMed] [Google Scholar]
- Doerge, R. W., 1995. The relationship between the LOD score and the analysis of variance F statistic when detecting QTL using single markers. Theor. Appl. Genet. 90: 980–981. [Google Scholar]
- Dole, J. A., and S. K. Jain, 1992. Genetics of new crop genus Limnanthes. I. Five morphological marker loci in L. alba x L. gracilis progenies. Plant Breed. 109: 198–202. [Google Scholar]
- Dole, J., and R. Kesseli, 1997. Inheritance of pale flower, calyx spotting, and glandular pubescence in Mimulus guttatus x M. platycalyx progenies. J. Hered. 88: 42–46. [Google Scholar]
- Dooner, H. K., and I. M. Martinez-Ferez, 1997. Recombination occurs uniformly within the bronze gene, a meiotic recombination hotspot in the maize genome. Plant Cell 9: 1633–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatmi, A., C. G. Poneleit and T. W. Pfeiffer, 1993. Variability of recombination frequencies in the Iowa Stiff Stalk Synthetic (Zea mays L.). Theor. Appl. Genet. 86: 859–866. [DOI] [PubMed] [Google Scholar]
- Fishman, L., A. J. Kelly and J. H. Willis, 2002. Minor quantitative trait loci underlie floral traits associated with mating system divergence in Mimulus. Evolution 56: 2138–2155. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J., 1974. The evolutionary advantage of recombination. Genetics 78: 737–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein, J., and S. Yokoyama, 1976. The evolutionary advantage of recombination II. Individual selection for recombination. Genetics 83: 845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal, S., B. Pisan, T. Hohn, N. Grimsle and B. Hohn, 1991. Genomic homologous recombination in planta. EMBO J. 10: 1571–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant, V., 1958. The regulation of recombination in plants. Cold Spring Harbor Symp. Quant. Biol. 23: 337–363. [DOI] [PubMed] [Google Scholar]
- Grant, V., 1975. Genetics of Flowering Plants. Columbia University Press, New York.
- Haldane, J. B. S., 1919. The combination of linkage values and the calculation of distances between the loci of linked factors. J. Genet. 2: 3–19. [Google Scholar]
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantititative trait loci in line crossess using flanking markers. Heredity 69: 315–324. [DOI] [PubMed] [Google Scholar]
- Hanson, G. P., 1969. B-chromosome-stimulated crossing over in maize. Genetics 63: 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick, P. W., S. Jain and L. Holden, 1978. Multilocus systems in evolution. Evol. Biol. 11: 101–182. [Google Scholar]
- King, J., L. A. Roberts, M. J. Kearsey, H. M. Thomas, R. M. Jones et al., 2002. A demonstration of a 1:1 correspondence between chiasma frequency and recombination using a Lolium perrene/Festuca pratensis substitution. Genetics 161: 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korol, A. B., I. A. Preygel and S. I. Preygel, 1994. Recombination Variability and Evolution. Chapman & Hall, London.
- Korol, A. B, Y. I. Ronin and V. M. Kirzhner, 1995. Interval mapping of quantitative trait loci employing correlated trait matrices. Genetics 140: 1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosambi, D. D., 1944. The estimation of map distances from recombination values. Ann. Eugen. 12: 172–175. [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin, R. C., 1964. The interaction of selection and linkage. I. General considerations; heterotic models. Genetics 49: 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin, R. C., 1971. The effect of linkage on the mean fitness of a population. Proc. Natl. Acad. Sci. USA 68: 984–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. C., 1976. First Course in Population Genetics. Boxwood Press, Pacific Grove, CA.
- Lin, J., and K. Ritland, 1997. Quantitative trait loci differentiating the outbreeding Mimulus guttatus from the inbreeding M. platycalyx. Genetics 146: 1115–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, MA.
- Mather, K., 1943. Variation and selection of polygenic characters. Biol. Rev. 18: 32–64. [Google Scholar]
- Maynard-Smith, J., 1978. The Evolution of Sex. Cambridge University Press, London.
- Nel, P. M., 1973. The modification of crossing over in maize by extraneous chromosomal elements. Theor. Appl. Genet. 43: 196–202. [DOI] [PubMed] [Google Scholar]
- Okagaki, R. J., and C. F. Weil, 1997. Analysis of recombination sites within the maize waxy locus. Genetics 147: 815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott, J., 1985. Analysis of Human Genetic Linkage. John Hopkins University Press, Baltimore.
- Peterhans, A., H. Schlupmann, C. Basse and J. Paszkowski, 1990. Intrachromosomal recombination in plants. EMBO J. 9: 3437–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petes, T. D., R. E. Malone and L. S. Symington, 1991. Recombination in yeast, pp. 407–521 in The Molecular and Cellular Biology of the Yeast Saccharomyces, Vol. I, edited by J. R. Broach, J. R. Pringle and E. W. Jones. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Rhoades, M. M., 1978. Genetic effects of heterochromatin in maize, pp. 641–671 in Maize Breeding and Genetics, edited by D. B. Walden. Wiley & Sons, New York.
- Sall, T., 1990. Genetic control of recombination in barley. Hereditas 112: 171–178. [Google Scholar]
- SAS Institute, 1997. Users Guide Version. 6.12. SAS Institute, Carey, NC.
- Spelman, R. J., W. Doppieters, L. Karim, J. A. M. Van Arendonk and H. Bovenhuis, 1996. Quantitative trait locus analysis for five milk production traits on chromosome six in the Dutch Holstein-Friesian population. Genetics 144: 1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPSS, 1994. SPSS 6.12 Base System Users Guide. SPSS, Chicago.
- Stebbins, G. L., 1950. Variation and Evolution in Plants. Columbia University Press, New York.
- Stuber, C. W., S. E. Lincoln, D. W. Wolff, T. Helantjaris and E. S. Lander, 1992. Identification of genetic factors contributing to heterosis in a hybrid from two elite maize inbred lines using molecular markers. Genetics 132: 823–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swoboda, P., B. Hohn and S. Gal, 1993. Somatic homologous recombination in planta: the recombination frequency is dependent on the alleleic state of recombining sequences and may be influenced by genomic position effects. Mol. Gen. Genet. 237: 33–40. [DOI] [PubMed] [Google Scholar]
- Timmermans, M. C., O. P. Das, J. M. Bradeen and J. Messing, 1997. Region specific cis- and trans-acting factors contribute to genetic variability in meiotic recombination in maize. Genetics 146: 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulsieram, L., W. A. Compton, R. Morris, M. Thomas-Compton and K. Eskridge, 1992. Analysis of recombination in maize populations using molecular markers. Theor. Appl. Genet. 84: 65–72. [DOI] [PubMed] [Google Scholar]
- Weller, J. I., G. R. Wiggans, P. M. Vanradan and M. Ron, 1996. Application of a canonical transformation to detection of quantitative trait loci with the aid of genetic markers in a multi-trait experiment. Theor. Appl. Genet. 92: 998–1002. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1922. Coefficients of inbreeding and relationship. Am. Nat. 56: 330–338. [Google Scholar]
- Zhang, Q., D. Boichard, I. Hoeschele, C. Ernst, A. Eggen et al., 1998. Mapping quantitative trait loci for milk production and health of dairy cattle in a large outbred pedigree. Genetics 149: 1959–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]