

Abstract
Background and purpose:
Selective cannabinoid CB2 receptor agonists have demonstrated analgesic activity across multiple preclinical pain models. AM1241 is an indole derivative that exhibits high affinity and selectivity for the CB2 binding site and broad spectrum analgesic activity in rodent models, but is not an antagonist of CB2 in vitro functional assays. Additionally, its analgesic effects are μ-opioid receptor-dependent. Herein, we describe the in vitro and in vivo pharmacological properties of A-796260, a novel CB2 agonist.
Experimental approach:
A-796260 was characterized in radioligand binding and in vitro functional assays at rat and human CB1 and CB2 receptors. The behavioural profile of A-796260 was assessed in models of inflammatory, post-operative, neuropathic, and osteoarthritic (OA) pain, as well as its effects on motor activity. The receptor specificity was confirmed using selective CB1, CB2 and μ-opioid receptor antagonists.
Key results:
A-796260 exhibited high affinity and agonist efficacy at human and rat CB2 receptors, and was selective for the CB2 vs CB1 subtype. Efficacy in models of inflammatory, post-operative, neuropathic and OA pain was demonstrated, and these activities were selectively blocked by CB2, but not CB1 or μ-opioid receptor-selective antagonists. Efficacy was achieved at doses that had no significant effects on motor activity.
Conclusions and implications:
These results further confirm the therapeutic potential of CB2 receptor-selective agonists for the treatment of pain. In addition, they demonstrate that A-796260 may be a useful new pharmacological compound for further studying CB2 receptor pharmacology and for evaluating its role in the modulation of pain.
Keywords: cannabinoid, CB2, A-796260, inflammatory pain, neuropathic pain
Introduction
Although the analgesic properties of cannabinoid ligands have been recognized for many years, the therapeutic potential of cannabis-based medicines is severely restricted by the undesired psychotropic side-effects associated with this class of drugs (Pertwee, 2001; Fox and Bevan, 2005; Ibrahim et al., 2006; Mackie, 2006; Whiteside et al., 2007). The pharmacology of natural and synthetic cannabinoid ligands is derived from their interaction with two cannabinoid receptor subtypes, CB1 and CB2. It is well established that the psychotropic effects of cannabis are derived largely, if not exclusively, by activity at the CB1 subtype; and based on rodent studies, it is becoming apparent that the CB2 receptor plays an important role in the mediation of pain processing (Malan et al., 2002, 2003; Ibrahim et al., 2005; Fox and Bevan, 2005). There is an emerging body of evidence to suggest that expression of the CB2 receptor is upregulated as a consequence of tissue or nerve injury, supporting a potential role for CB2-selective ligands in the treatment of inflammatory, postoperative and neuropathic pain (Zhang et al., 2003; Wotherspoon et al., 2005; Romero-Sandoval and Eisenach, 2007).
Over the years, a number of CB2-selective ligands have emerged and have been shown to exhibit analgesic, antihyperalgesic and antiallodynic activity in various rodent pain models. JWH-133 and HU-308 are structurally related to Δ9-tetrahydrocannabinol, and display high radioligand-binding affinity and selectivity towards the human CB2 receptor (Hanus et al., 1999; Huffman et al., 1999). JWH-133 has been characterized as a full agonist in a GTPγS-binding assay at the human CB2 receptor (Huffman et al., 2004), and HU-308 has been characterized as a selective full agonist in human CB2 cyclic GMP assays (Hanus et al., 1999). However, their activities at rodent CB1 and CB2 receptors have not been studied. In preclinical studies, JWH-133 has demonstrated efficacy in models of both inflammatory and neuropathic pain, and antagonist blockade studies have indicated that these activities are mediated through activation of the CB2 subtype (Elmes et al., 2004, 2005; Sagar et al., 2005). HU-308 exhibited activity in a formalin-induced peripheral pain model (Hanus et al., 1999) and a skin-incision model of postoperative pain (LaBuda et al., 2005). In both these models, the effects of HU-308 were blocked by the CB2 antagonist SR144528.
Several structurally related indole derivatives, including AM1241 and L-768,242 (GW405833) have been identified as CB2-selective ligands exhibiting activity in several rodent pain models. AM1241 has been found to exhibit high affinity at human, rat and mouse CB2 receptors (Makriyannis and Deng, 2002; Ibrahim et al., 2003a; Nackley et al., 2003a; Yao et al., 2006; Bingham et al., 2007). However, the agonist efficacy profile of AM1241 (as the racemate) is complex and condition-dependent, ranging from inverse agonist to partial agonist at the human CB2 receptor (Yao et al., 2006; Bingham et al., 2007) and is an apparent inverse agonist at the rat CB2 receptor (Bingham et al., 2007). Nevertheless, the behavioural profile of AM1241 across multiple pain models is consistent with CB2 agonist activity. The efficacy of AM1241 in models of inflammatory (Malan et al., 2001), neuropathic (Ibrahim et al., 2003a) and postoperative pain (LaBuda et al., 2005) has been shown to be mediated by the CB2 receptor, as the effects are blocked by CB2- but not CB1-selective antagonists. Despite an apparent lack of affinity for the μ-opioid receptor, the in vivo analgesic activity of AM1241 in a model of acute thermal pain is antagonized by both naloxone and β-endorphin antiserum, and AM1241 is inactive in the μ-opioid receptor knockout mouse (Ibrahim et al., 2005). L-768,242 is characterized by high affinity for human and rat CB2 receptors and high selectivity vs the CB1-binding sites (Gallant et al., 1996). Functionally, it displays partial agonist activity in human CB2 receptor-mediated inhibition of forskolin-stimulated cAMP release assays (Valenzano et al., 2005). Like AM1241, L-768,242 is effective in models of inflammatory, postoperative and neuropathic pain (Valenzano et al., 2005), but unlike AM1241, the effects of L-768,242 are not antagonized by a μ-opioid receptor antagonist (Whiteside et al., 2005).
Finally, GW-842166X, a pyrimidine derivative structurally distinct from other known CB2 agonists, has been shown to exhibit agonist activity at human and rat CB2 receptors in a yeast reporter gene assay, and is effective in a complete Freund's adjuvant (CFA) model of chronic inflammatory pain (Giblin et al., 2007). This compound has been selected as a clinical candidate for the treatment of pain associated with osteoarthritis and rheumatoid arthritis.
Despite supporting evidence provided by multiple CB2-selective ligands for the therapeutic potential of this pharmacological class as novel analgesic agents, inconsistencies exist with respect to both their in vitro and in vivo profiles. The compounds described above either fail to exhibit potent or robust agonist efficacy or have not been fully characterized in functional assays at rodent CB2 receptors. Differences exist with respect to potential interactions with the opioid system, suggesting that different mechanisms may be contributing to the in vivo properties of these compounds. These shortcomings and inconsistencies highlight the need for additional compounds to help assess the potential that this pharmacological class may hold for the development of new drugs for the treatment of pain.
Methods
Compounds
Racemic AM1241, SR141716A and SR144528 were prepared at Abbott Laboratories as previously described (Yao et al., 2006). CP 55,940, WIN 55,212-2 and AM630 were purchased from Tocris (Ellisville, MO, USA). L-768,242 was synthesized as described by Valenzano et al. (2005). GW-842166X was synthesized as described by Giblin et al. (2007). The para-toluenesulphonate salt of A-796260 was synthesized as depicted in the scheme and described herein. Briefly, acylation of indole with 2,2,3,3-tetramethylcyclopropanecarbonyl chloride afforded a 1.6:1 mixture of the desired C(3)- and undesired N-acylated regioisomers, which were separated by silica gel chromatography. Alkylation of the indole with the methanesulphonate ester of 2-morpholinoethanol gave the free base of A-796260 that was subsequently transformed into its toluenesulphonate salt, which was utilized in all biological experiments in the current study. Doses of A-796260 are presented as mg kg−1 of the free base (mol wt=354.50). Characterization of the free base and toluenesulphonate salt of A-796260 are as described below: Free base of A-796260, 1H NMR (CD3OD, 300 MHz) δ p.p.m. 1.33 (s, 12H), 2.13 (s, 1H), 2.46–2.54 (m, 4H), 2.79 (t, J=6.4 Hz, 2H), 3.61–3.71 (m, 4H), 4.37 (t, J=6.4 Hz, 2H), 7.16–7.30 (m, 2H), 7.45–7.53 (m, 1H), 8.11 (s, 1H), 8.20–8.30 (m, 1H), MS (DCI/NH3) m/z 355 (M+H)+; Tosylate salt of A-796260: [1-(2-morpholin-4-yl-ethyl)-1H-indol-3-yl]-(2,2,3,3-tetramethylcyclopropyl)-methanone p-toluenesulphonic acid, 1H NMR (MeOH-d4, 300 MHz) δ 1.33 (s, 6H), 1.34 (s, 6H), 2.15 (s, 1H), 2.36 (s, 3H), 3.40 (m, 4H), 3.68 (dd, J=7.1, 7.1 Hz, 2H), 3.90 (m, 4H), 4.73 (dd, J=7.1, 7.1 Hz, 2H), 7.23 (br d, J=7.8 Hz, 2H), 7.26 (ddd, J=8.1, 8.1, 1.4 Hz, 1H), 7.33 (ddd, J=7.1, 7.1, 1.0 Hz, 1H), 7.56 (br d, J=8.1 Hz, 1H), 7.72 (br d, J=8.5 Hz, 2H), 8.15 (s, 1H), 8.29 (dt, J=7.8, 1.0 Hz, 1H); MS (DCI/NH3) m/z 355 (M+H)+; anal (C22H30N2O2·C7H8O3S) C,H,N. 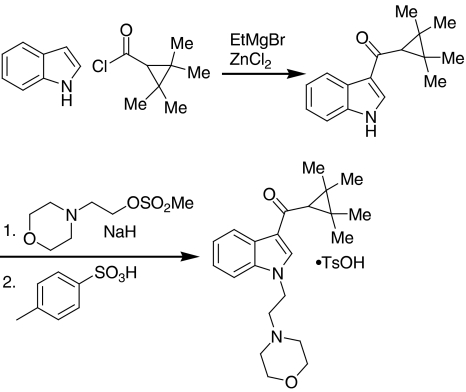
Cell culture
Reagents for cell culture were purchased from Invitrogen (Carlsbad, CA, USA) unless indicated otherwise. Human embryonic kidney (HEK; American Type Culture Collection, Rockville, MD, USA) cells stably expressing the human CB2, rat CB2 or rat CB1 receptor were grown in Dulbecco's modified Eagle's medium containing high glucose supplemented with 10% fetal bovine serum and 25 μg ml−1 zeocin in a 37 °C incubator in the presence of 5% CO2. HEK cells stably co-expressing the chimeric Gαq/o5 protein with either the human or rat CB2 receptor were grown under similar conditions, except that in addition to the components described above, the growth medium was further supplemented with 200 μg ml−1 hygromycin. The Chinese hamster ovary cell lines stably expressing the human CB1 receptor were purchased from Euroscreen (Brussels, Belgium), and the cells were grown under the conditions recommended by the vendor.
Radioligand-binding assays
Membrane samples prepared from HEK cells stably expressing the human CB2, rat CB2 or rat CB1 receptor, and Chinese hamster ovary cells stably expressing the human CB1 receptor were used to perform radioligand-binding assays using [3H]-CP 55,940 ligand as previously described (Mukherjee et al., 2004). Briefly, competition experiments were conducted using 0.5 nM [3H]-CP 55,940 in the presence of variable concentrations of test compounds in an assay buffer containing 50 mM Tris-HCl, pH 7.4, 2.5 mM EDTA, 5 mM MgCl2 and 0.05% fatty acid free BSA. After 90 min incubation at 30 °C, the reactions were terminated by rapid vacuum filtration through UniFilter-96 GF/C filter plates (Perkin Elmer Boston, MA, USA) and four washes with cold assay buffer. Nonspecific binding was defined by 10 μM unlabelled CP 55,940. Ki values from competition binding assays were determined with one-site competition curve fitting using the Prism software (GraphPad, San Diego, CA, USA).
Cyclase functional assays
The cyclase functional assays were performed using the HitHunter cAMP assay kit from DiscoveRx (Fremont, CA, USA) in suspension forms according to the manufacturer's protocol and as described previously (Yao et al., 2006). Briefly, cell suspensions were incubated at 37 °C for 20 min with variable concentrations of test ligands or 10 μM CP 55,940 as a positive control in the presence of a fixed concentration of forskolin (18 μM for the rat CB2 line; and 37 μM for human CB1 and CB2, and rat CB1 lines) in D-phosphate-buffered saline (PBS) buffer (Invitrogen) supplemented with BSA (0.01% final concentration). The reactions were terminated by the addition of lysis buffer and the luminescence was detected following the procedure according to the manufacturer's instructions. The positive control, CP 55,940 10 μM, produced significant inhibition of cAMP levels induced by forskolin in the cell lines expressing the human CB1 (84% inhibition; n=10), human CB2 (71% inhibition; n=10), rat CB1 (90% inhibition; n=10) and rat CB2 receptors (63% inhibition; n=10). Receptor activation by ligands is expressed as % response compared to that of 10 μM CP 55,940. EC50 values were calculated by sigmoidal dose–response curve fitting using Prism (GraphPad) software.
Fluorescence imaging plate reader functional assays
Fluorescence imaging plate reader (FLIPR) assays were performed using HEK cells stably co-expressing the chimeric Gαq/o5 protein with either human or rat CB2 receptors (Mukherjee et al., 2004). Briefly, cells were seeded at 75 000 cells per well 1 day prior to the assay and the assays were performed with no-wash dye (FLIPR Calcium Assay Kit; Molecular Device, Sunnyvale, CA, USA) following the manufacturer's instructions. Variable concentrations of test compounds (0.3 nM to 10 μM), CP 55,940 (at 10 μM final concentration) positive control or vehicle negative control were added to cells in the presence of assay buffer (10 mM HEPES, pH 7.4, 130 mM NaCl, 1 mM MgCl2, 5 mM KCl, 2 mM CaCl2 and 0.05% BSA), and fluorescence responses were measured immediately with a FLIPR machine. Net peak responses were compared with that of 10 μM CP 55,940 and expressed as percentages of the CP 55,940-evoked response. EC50 values were analysed with sigmoidal dose–response curve fitting using Prism.
Animals
Adult Sprague–Dawley rats (male, 250–300 g body weight; obtained from Charles River, Portage, MI, USA) were used for all experiments. All animals were housed in Association for Assessment and Accredidation of Laboratory Animal Care (AALAC) approved facilities at Abbott Laboratories in a temperature-regulated environment under a controlled 12 h:12 h light–dark cycle, with lights on at 0600 h. Food and water were available ad libitum at all times except during testing. All testing was carried out following procedures outlined in protocols approved by Abbott's Institutional Animal Care and Use Committee.
In vivo pain models
Chronic inflammatory pain model
Chronic inflammatory thermal hyperalgesia was induced by injection of 150 μl of CFA solution (50%) in PBS into the plantar surface of the right hind paw of the rats. Thermal hyperalgesia was assessed 48 h post-CFA injection.
Thermal hyperalgesia was determined using a commercially available thermal paw stimulator (UARDG; University of California, San Diego, CA, USA) described by Hargreaves et al. (1988). Rats were placed in individual plastic cubicles mounted on a glass surface maintained at 30 °C, and allowed a 20 min habituation period. A thermal stimulus, in the form of radiant heat emitted from a focused projection bulb, was then applied to the plantar surface of each hind paw. The stimulus current was maintained at 4.50±0.05 A, and the maximum time of exposure was set at 20.48 s to limit possible tissue damage. The elapsed time until a brisk withdrawal of the hind paw from the thermal stimulus was recorded automatically using photodiode motion sensors. The right and left hind paws of each rat were tested in three sequential trials at approximately 5-min intervals. Paw withdrawal latency (PWL) was calculated as the mean of the two shortest latencies. PWL was measured 30 min post-A-796260 administration in both the CFA-treated and uninjected paws.
Neuropathic pain model
A model of chronic constriction injury (CCI)-induced neuropathic pain was produced by following the method of Bennett and Xie (1988). The right common sciatic nerve was isolated at mid-thigh level, and loosely ligated by four chromic gut (5-0) ties separated by an interval of 1 mm. All animals were left to recover for at least 2 weeks and no more than 5 weeks prior to testing tactile allodynia.
A-796260 was injected i.p. 30 min before the test for tactile allodynia. Tactile allodynia was measured using calibrated von Frey filaments (Stoelting, Wood Dale, IL, USA) as previously described (Chaplan et al., 1994). Rats were placed into inverted individual plastic containers (20 cm × 12.5 cm × 20 cm) on top of a suspended wire mesh grid, and acclimatized to the test chambers for 20 min. von Frey filaments were presented perpendicularly to the plantar surface of the selected hind paw, and then held in this position for approximately 8 s with enough force to cause a slight bend in the filament. Positive responses included an abrupt withdrawal of the hind paw from the stimulus, or flinching behaviour immediately following removal of the stimulus. A 50% withdrawal threshold was determined using an up–down procedure (Dixon, 1980). Only rats with a baseline threshold score of less than 5 g were used in this study, and animals demonstrating motor deficit were excluded. A 15 g threshold was used as the maximal possible effect in this assay. Drug effects in this model were conducted in a randomized, blind manner.
Postoperative pain model
A model of postoperative pain was performed as described by Brennan et al. (1996). The plantar aspect of the rat left hind paw was exposed through a hole in a sterile plastic drape, and a 1 cm longitudinal incision was made through the skin and fascia, starting 0.5 cm from the proximal edge of the heel and extending towards the toes. The plantaris muscle was elevated and incised longitudinally, leaving the muscle origin and insertion points intact. After homoeostasis by application of gentle pressure, the skin was apposed with two mattress sutures using 5-0 nylon. Animals were then allowed to recover for 2 h after surgery, at which time tactile allodynia was assessed as described above. Drug effects in this model were conducted in a randomized, blind manner.
Knee joint osteoarthritic pain model
Unilateral knee joint osteoarthritis was induced in the rats by a single intra-articular (i.a.). injection of sodium monoiodoacetate (MIA) (Sigma-Aldrich, St Louis, MO, USA) (3 mg in 0.05 ml sterile isotonic saline) into the joint cavity under light (1–3%) isoflurane anaesthesia using a 26 G needle (Guzman et al., 2003; Pomonis et al., 2005). Following the injection, the animals were allowed to recover from anaesthesia (usually 5–10 min) before being returned to their home cages. To maintain uniformity across the study, the right knee joint of each animal was injected with MIA. Hindlimb grip force assessment was carried out 20 days following MIA injection as discussed below. The dose of the MIA (3 mg per i.a. injection) was selected based on results obtained from preliminary studies wherein optimal pain behaviour was observed at this dose.
Pain behavioural assessment of hindlimb grip force following unilateral injection of MIA was used as a behavioural measure of activity-induced pain in adult osteoarthritic (OA) rats (body weight 20 days following MIA injection: 325–350 g). Measurements of peak hindlimb grip force were conducted by recording the maximum compressive force exerted on the hindlimb strain gauge set up, in a commercially available grip force measurement system (Columbus Instruments, Columbus, OH, USA). During testing, each rat was gently restrained by grasping it around its rib cage and then allowed to grasp the wire mesh frame attached to the strain gauge. The experimenter then moved the animal in a rostral-to-caudal direction until the grip was broken. Each rat was sequentially tested twice at approximately 2–3 min interval to obtain a raw mean grip force (CFmax). This raw mean grip force data were converted to a maximum hindlimb cumulative compressive force (CFmax) (g force) per kg body weight for each animal. For assessing the effects of A-796260, the evaluation of hindlimb grip force was conducted 20 days following the i.a. injection of MIA. A group of age-matched naive animals were added to each experiment and the data obtained from the different dose groups for the compound being tested were compared to the naive group. Experiments were conducted in a randomized, blind manner, and drug effects are expressed as raw mean grip force (CFmax)/body weight (kg).
Locomotor activity
To determine potential CNS side-effects of A-796260, spontaneous exploratory behaviour was examined in naive rats (30 min post-compound injection). Rats were individually placed into test chambers, and horizontal (locomotion) activity was recorded by a photo beam detector system for 30 min (AccuScan Instruments, Columbus, OH, USA).
Data analysis
Statistical analyses were carried out using Graph Pad Prism (version 4.03; Graph Pad Software Inc., San Diego, CA, USA). The results are presented as mean±s.e.mean. Statistical significance between group means was measured by one-way ANOVA, followed by Bonferroni post hoc analysis. In all cases, P<0.05 was assumed as the level for statistical significance. ED50 values were also calculated by linear regression analysis and presented with CI 95% (Graph Pad).
Results
Ligand affinities and selectivity at CB2 receptors in radioligand-binding assays
The receptor-binding potencies of A-796260, WIN 55,212-2, CP 55,940, AM1241, SR144528, SR141716A, GW-842166X and L-768,242 (GW405833) were determined in [3H]-CP 55,940 competition binding assays performed with membrane preparations generated from recombinant cell lines expressing the human CB1, human CB2, rat CB1 or rat CB2 receptor, respectively. The results are summarized in Table 1. A-796260 exhibited high binding potencies at both human and rat CB2 receptors with Ki values of 4.37 and 13.0 nM, respectively. Selectivity for the CB2- over the CB1-binding site was 193-fold in human and 30-fold in rat samples. This is comparable to the potencies and selectivity exhibited by AM1241. GW-842166X did not demonstrate detectable [3H]-CP 55,940 displacement at either human or rat CB1 (up to 10 μM) and had weak affinities at the human and rat CB2 receptors (2000 and 2580 nM, respectively) in the current assays, whereas L-768,242 showed good binding affinity at the human and rat CB2 receptors. The binding potencies of the non-selective agonists WIN 55,212-2 and CP 55,940, CB1-selective antagonist SR141716A and CB2-selective antagonist SR144528 displayed binding properties similar to those published previously (Howlett et al., 2002).
Table 1.
Radioligand competition binding assays using [3H]CP 55,940
| Ligands |
Mean Ki, nM (n) |
|||||
|---|---|---|---|---|---|---|
|
95% CI of Ki values, nM |
||||||
| hCB1 | hCB2 | hCB2/hCB1 selectivity | rCB1 | rCB2 | rCB2/rCB1 selectivity | |
| A-796260 | 845 (13) | 4.37 (14) | 193 | 395 (5) | 13.0 (5) | 30 |
| 410–1740 | 1.87–10.2 | 173–899 | 6.35–26.6 | |||
| WIN 55,212-2 | 13.3 (27) | 1.30 (44) | 10 | 4.47 (11) | 1.64 (19) | 2.7 |
| 7.5–23.6 | 0.971–1.74 | 3.02–6.65 | 0.871–3.11 | |||
| CP 55,940 | 2.11 (71) | 1.08 (99) | 2.0 | 1.54 (29) | 0.987 (56) | 1.6 |
| 1.62–2.77 | 0.925–1.27 | 1.15–2.07 | 0.764–1.28 | |||
| AM1241 | 1270 (10) | 11.5 (21) | 110 | 115 (4) | 3.37 (15) | 34 |
| 354–4545 | 6.99–18.8 | 7.75–1710 | 1.24–9.15 | |||
| SR144528 | 264 (12) | 5.66 (17) | 47 | 428 (6) | 1.80 (7) | 238 |
| 175–398 | 3.86–8.31 | 159–1150 | 0.477–6.81 | |||
| SR141716A | 2.36 (29) | 429 (13) | 182 × CB1 selective | 2.80 (23) | 127 (4) | 45 × CB1 selective |
| 1.41–3.95 | 270–682 | 1.94–4.03 | 35.4–452 | |||
| GW-842166X | >10 000 (2) | 2000 (7) | NA | >10 000 (2) | 2580 (8) | NA |
| 561–7140 | 993–6700 | |||||
| L-768,242 (GW405833) | 282 (2) | 7.62 (4) | 37 | ND | 11.1 (4) | NA |
| 1370–5830 | 2.23–26.1 | 4.74–25.9 | ||||
Abbreviation: NA, not available.
In a recent publication (Ibrahim et al., 2005), AM1241 has been shown to stimulate β-endorphin release via a μ-opioid receptor-dependent pathway. To investigate if CB2-selective ligands directly interact with the μ-opioid receptor, [3H]-DAMGO-binding assays were performed according to the method described by Spetea et al. (1998). Neither A-796260, AM1241 nor SR144528 exhibited affinity at the μ-opioid receptor, up to ligand concentrations of 10 μM, in this assay, whereas μ-opioid receptor ligand naloxone had a Ki value of 17 nM in these experiments.
In vitro ligand characterization in cyclase and FLIPR functional assays
To assess agonist properties of these ligands, receptor-coupled adenylyl cyclase activity and calcium influx response were measured, respectively, in cyclase and FLIPR functional assays, employing cell lines that express the recombinant cannabinoid receptors. In cyclase assays, A-796260 behaved as a potent and efficacious agonist at human and rat CB2 receptors with EC50 values of 0.710 nM (Emax of 78%) and 1.63 nM (Emax of 69%), respectively (Table 2). A-796260 is highly selective at CB2 receptors, exhibiting 1380- and 175-fold selectivity at human and rat CB2 receptors compared to CB1 receptors of the respective species. In contrast, AM1241 did not exhibit either agonist or inverse agonist activity at human and rat CB2 receptors in these assays, but it was shown to produce a full agonist efficacy at both human and rat CB1 receptors at high concentrations (human CB1 EC50=2650 nM, Emax=101%; and rat CB1 EC50=1970 nM, Emax=81%). L-768,242 was a potent inverse agonist at both human (EC50=44.4 nM) and rat (EC50=3.26 nM) CB2 receptors, producing maximum (–30% (human) and –39% (rat)) negative efficacy relative to that of CP 55,940. In contrast, it was a weak, but efficacious, agonist at the rat CB1 receptor. GW-842166X was inactive at human and weakly efficacious at rat CB1 receptors; however, it exhibited full agonist activity at human and rat CB2 receptors. Reference standards, WIN 55,212-2, CP 55,940, SR144528 and SR141716A, exhibited properties similar to previously published results (Felder et al., 1995; Bonhaus et al., 1998; Tao and Abood, 1998; Lunn et al., 2006).
Table 2.
Cyclase and FLIPR functional assays
| Ligands |
Mean agonist EC50, nM (n), efficacy % relative to CP 55,940 (%) |
|||||
|---|---|---|---|---|---|---|
|
95% CI of EC50 values, nM |
||||||
|
Cyclase assays |
FLIPR assays |
|||||
| hCB1 | hCB2 | rCB1 | rCB2 | hCB2 | rCB2 | |
| A-796260 | 983 (7), 118 | 0.710 (6), 78 | 286 (8), 97 | 1.63 (7), 69 | 17.3 (8), 71 | 31.0 (4), 39 |
| 618–1560 | 0.315–1.60 | 206–397 | 0.505–5.28 | 10.64–28.2 | 7.87–125 | |
| WIN 55,212-2 | 47.2 (5), 113 | 0.650 (5), 98 | 16.5 (5), 114 | 1.16 (6), 46 | 119 (10), 74 | >10 000 (13) |
| 23.5–94.9 | 0.156–2.72 | 3.74–72.5 | 0.367–3.65 | 57.94–245 | ||
| CP 55,940 | 1.81 (12), 103 | 0.864 (41), 106 | 4.12 (17), 103 | 0.682 (42), 99 | 31.3 (212), 99 | 13.0 (29), 106 |
| 0.777–4.21 | 0.626–1.19 | 2.51–6.76 | 0.444–1.05 | 28.3–34.5 | 9.44–17.8 | |
| AM1241 | 2650 (5), 101 | >1000 (9) | 1970 (6), 81 | >1000 (8) | >10 000 (18) | >10 000 (9) |
| 1730–4050 | 842–4610 | |||||
| SR144528 | 9420 (4), 52 | 73.4 (20), −106 | 5960 (3), 39 | 34.3 (17), −75 | >10 000 (12) | >10 000 (6) |
| 4340–20 400 | 48.4–111 | 1780–20 000 | 24.9–47.3 | |||
| SR141716A | 21.3 (4), −67 | >3000 (4) | 31.6 (7), −61 | >3000 (2) | >10 000 (3) | >10 000 (6) |
| 5.73–79.1 | 16.2–61.8 | |||||
| GW-842166X | >25 000 (2) | 133 (4), 101 | 26 000 (4), 33 | 96.2 (6), 95 | 7780 (5), 84 | ND |
| 94–187 | 24 200–27 900 | 65.5–141 | 2700–22 400 | |||
| L-768,242 (GW405833) | ND | 44.4 (2), −30 | 916 (2), 70 | 3.26 (4), −39 | >10 000 (4) | ND |
| 22.4–88.1 | 839–1000 | 1.09–9.78 | ||||
Abbreviation: ND, not determined.
In FLIPR assays, where a chimeric Gαq/o protein was employed to facilitate redirection of the Gαi/o signalling to intracellular calcium release responses (Yao et al., 2006), receptor activation was readily measured by the use of a FLIPR machine. To characterize A-796260 and other cannabinoid receptor ligands further, FLIPR functional assays were performed. A-796260 displayed high potency and full efficacy at the human CB2 receptor. In contrast, AM1241 and L-768,242 failed to exhibit agonist efficacy, and GW-842166X exhibited weak agonist potency with an EC50 of 7780 nM and Emax of 84%. Non-selective agonists, WIN 55,212-2 and CP 55,940, exhibited high potencies and full efficacy, whereas SR144528 and SR141716A were inactive. WIN 55,212-2, a partial agonist at the rat CB2 receptor in cyclase assays, was inactive in this assay. Similar to the result with the human CB2 receptor, AM1241 was also inactive at the rat CB2 receptor in FLIPR assays (Figure 1).
Figure 1.

Characterization of cannabinoid ligands in recombinant rat CB1 and CB2 cyclase assay systems. A-796260 (a), WIN 55,212-2 (b) and CP 55,940 (c) were tested in rat CB1 and CB2 cyclase assays. Levels of receptor activation are calculated as the reduction of cAMP levels and are expressed as a percentage relative to the response of 10 μM CP 55,940. Typically, 10 μM CP 55,940 inhibits forskolin-induced cAMP levels in rat CB1 and CB2 cyclase assays by 90 and 63%, respectively; these responses were normalized to 100% activation of receptor activity. (d) The effects of A-796260, AM1241, GW-842166X and L-768,242 (GW405833) at the rat CB2 receptor.
Effects of A-796260 on CFA-induced chronic inflammatory thermal hyperalgesia
Two days following CFA injection into the hind paw, a significant decrease in PWL to thermal stimulation was observed (PWL uninflamed: 10.7±0.3 s vs PWL inflamed: 5.3±0.1 s, P<0.01), demonstrating the development of thermal hyperalgesia (Figure 2a). A-796260 dose-dependently attenuated CFA-induced thermal hyperalgesia (Figure 2a; F7,88=68.2, P<0.0001, ED50=2.8 mg kg−1 i.p., CI 95%=2.1–3.4 mg kg−1), demonstrating full efficacy (94±7%) at 11 mg kg−1. No effect was observed in the uninflamed hind paw.
Figure 2.

Effects of A-796260 (i.p.) on complete Freund's adjuvant (CFA)-induced chronic inflammatory thermal hyperalgesia. (a) At 48 h following CFA injection, A-796260 was injected 30 min before testing. Data represent mean±s.e.mean. *P<0.05, **P<0.01 as compared to vehicle-treated rats (n=12 per group). (b) Reversal of A-796260-evoked analgesia by pretreatment with CB2-selective antagonists SR144528 or AM630 (SR2, 4.8 mg kg−1 i.p.; AM630, 3.0 mg kg−1 i.p.), but the CB1-selective antagonist SR141716A (SR1, 14 mg kg−1 i.p) had no effect. The antagonists were administered 15 min before A-796260 injection. (c) Effects of the opioid antagonist naloxone (Nal) on the analgesic activity induced by A-796260 or AM1241. Naloxone (10 mg kg−1 i.p.) was administered 15 min before the injection of A-796260 (11 mg kg−1 i.p.) or AM1241 (15 mg kg−1 i.p.). In panels b and c data are expressed as mean±s.e.mean (n=6). **P<0.01 as compared to vehicle-treated rats, ++P<0.01 as compared to A-796260 control.
Using this in vivo inflammatory pain model, the receptor specificity was investigated using receptor-selective antagonists (Figure 2b). Pretreatment with SR144528 (4.8 mg kg−1 i.p.), a CB2 receptor-selective antagonist (Rinaldi-Carmona et al., 1998), significantly reversed the analgesic efficacy induced by treatment with A-796260 at 11 mg kg−1 i.p. (F7,40=112.2, P<0.0001). AM630, another CB2 receptor-selective antagonist (Hosohata et al., 1997; Ross et al., 1999), when pre-administered at 3 mg kg−1 i.p., also significantly blocked the antihyperalgesic activity induced by A-796260 (F7,38=77.7, P<0.0001). In contrast, pretreatment with SR141716A (14 mg kg−1 i.p.), a selective CB1 receptor antagonist (Rinaldi-Carmona et al., 1994), had no effect on the analgesia induced by A-796260 (F7,40=13.2, P<0.0001). The rats treated with CB2 receptor antagonists alone at the doses used in the present studies did not exhibit any change in PWL as compared with the vehicle-treated animals.
Release of endogenous opioids has been proposed as a mechanism of action for the analgesic activities of CB2 agonists, and the effects of the CB2 ligand AM1241 have been shown to be dependent on β-endorphin release and require a functional μ-opioid receptor (Ibrahim et al., 2005). To investigate whether the analgesic effects of A-796260 are mediated via μ-opioid receptor-dependent activity, CFA-treated rats were injected with the opioid receptor antagonist naloxone (10 mg kg−1 i.p.) prior to the administration of A-796260 (11 mg kg−1 i.p.). Naloxone pretreatment had no effect on the antihyperalgesic activity of A-796260 (Figure 2c; F7,52=14.1, P<0.0001). However, under similar conditions, pretreatment with naloxone significantly reversed the analgesic effects induced by AM1241 at 15 mg kg−1 i.p. (Figure 2c; F7,52=51.5, P<0.0001).
Effects of A-796260 in the skin incision model of acute postoperative pain
Two hours following skin incision surgery, tactile allodynia was observed on the injured paw (PWTvonfrey=1.91±0.17 g). Skin incision did not affect tactile PWT in the uninjured paw. A-796260, administered i.p. 1.5 h post-surgery, dose-dependently attenuated tactile allodynia (Figure 3a; F4,48=39.8, P<0.0001, ED50 18 mg kg−1, CI 95%=13–25 mg kg−1), with a maximal efficacy of 68±7% (P<0.01 vs vehicle) at the highest dose tested (Figure 3a). Intraperitoneal administration of morphine (6 mg kg−1) also produced a statistically significant reversal of allodynia 30 min post-dosing.
Figure 3.

Effects of A-796260 on skin incision-induced acute postoperative pain. (a) A-796260, administered 1.5 h post-surgery, dose-dependently attenuated tactile allodynia (ED50=18 mg kg−1 i.p.). Morphine (6 mg kg−1 i.p.) was included in the same study. Data represent mean±s.e.mean. **P<0.01 as compared to vehicle-treated animals (n=12 per group). (b) Analgesic effect of A-796260 (35 mg kg−1 i.p.) and morphine (3 mg kg−1 s.c.) in rats after repeated administration for 5 days. Data are expressed as mean±s.e.mean (n=6). **P<0.01 as compared to vehicle-treated animals; ++P<0.01 as compared to the acute single dosing group. (c) Reversal of A-796260 analgesic effects by CB2-selective antagonist. SR144528 (SR2, 4.8 mg kg−1 i.p.) was administered 15 min before A-796260 injection (35 mg kg−1 i.p.). Data are expressed as mean±s.e.mean (n=6). **P<0.01 as compared to vehicle-treated animals; ++P<0.01 as compared to A-796260 control.
A separate study was conducted to determine whether the analgesic effect of A-796260 was maintained after repeat administration (twice daily for 5 days). The results, shown in Figure 3b, indicate that tolerance to the analgesic effect of A-796260, 35 mg kg−1 i.p., did not develop following repeated administration for 5 days (64±4%, P<0.01 vs vehicle for acute dosing vs 60±10%, P<0.01 vs vehicle for repeated dosing). The plasma levels of A-796260 were measured after the behavioural testing, and no difference was seen between the two treatment groups (data not shown). In contrast to the results with A-796260, the analgesic effects of morphine were almost completely abolished after the repeated administration with the same dosing regime (Figure 3b; F5,41=29.4, P<0.0001).
To examine whether the analgesic effects of A-796260 in the skin incision model of postoperative pain are mediated via CB2 receptors, studies were conducted in conscious rats treated with A-796260, 35 mg kg−1 i.p., before and after pretreatment with the CB2 antagonist SR144528. A-796260 produced 75±11% analgesia (P<0.01 vs vehicle) when given alone, whereas the same dose of A-796260 had no significant effect (26±6%) in rats pretreated with SR144528 4.8 mg kg−1 i.p. (Figure 3c; F3,20=29.9, P<0.0001).
Effects of A-796260 on tactile allodynia in a neuropathic pain model
In rats, CCI of the sciatic nerve (Bennett and Xie, 1988) produced a decrease in PWT to mechanical stimulation 2 weeks following surgery (PWTvonfrey=3.7 ±0.4 g), demonstrating the development of tactile allodynia. A-796260 treatment attenuated CCI-induced tactile allodynia in a dose-related manner with an ED50 of 15 mg kg−1 i.p. (CI 95%=5.3–26 mg kg−1) and an efficacy of 66±9% (P<0.01 vs vehicle) at the highest dose tested (Figure 4; F4,54=18.0, P<0.0001). Under the same conditions, morphine (6 mg kg−1 i.p.) also produced a statistically significant reversal of allodynia 30 min post-administration.
Figure 4.

Effects of A-796260 on tactile allodynia in a rat neuropathic pain model, sciatic nerve chronic constriction injury (CCI). A-796260 dose-dependently attenuated neuropathic pain in this model (ED50=12 mg kg−1 i.p.), which is seen as an increase in the withdrawal threshold of the nerve-injured paw. Data are expressed as mean±s.e.mean. **P<0.01 vs vehicle-treated group, n=12 per group.
Effects of A-796260 in an OA pain model
The analgesic effects of A-796260 on activity-induced pain behaviour were evaluated in a rat model of MIA-induced OA joint pain (Guzman et al., 2003; Pomonis et al., 2005), observed 20 days following the i.a. injection of MIA. Previously we showed that i.a. administration of MIA produces a long lasting decrease in hindlimb grip force when compared to saline-injected animals (Chandran et al., 2006). A-796260 (i.p.) reversed, in a dose-related manner, MIA-induced decrease in the grip force with 56±7% effect at 35 mg kg−1 (P<0.01 vs the vehicle CFmax 370±87 g). The effects were comparable to the 62% efficacy elicited by celecoxib (38 mg kg−1 i.p.), a clinically effective analgesic for OA pain (Figure 5; F5,30=31.6, P<0.0001).
Figure 5.

Effects of A-796260 on hindlimb grip force in rats 20 days following injection of monoiodoacetate (MIA). A-796260 demonstrated dose-related reversal of activity-induced pain behaviour in osteoarthritic (OA) rats with effects comparable to celecoxib (38 mg kg−1 i.p.), a clinically relevant analgesic for OA pain. Data are expressed as mean±s.e.mean. **P<0.01 vs vehicle-treated group, n=6 per group.
Effects of A-796260 on locomotion
Locomotor activity in rats was evaluated by automatically recording distance travelled in an open field. Systemic administration of A-796260 (1–35 mg kg−1 i.p.) did not significantly affect locomotor activity (Figure 6). The highest dose tested in this study has been shown to produce full or nearly full efficacy across models of chronic inflammatory, postoperative, neuropathic and OA pain. In this study, haloperidol (1 mg kg−1 i.p.) significantly decreased locomotion in rats (Figure 6; F5,42=7.10, P<0.0001).
Figure 6.

Effects of A-796260 treatment on spontaneous horizontal locomotor activity. Average (±s.e.mean; n=8 rats per group) levels of horizontal locomotor activity exhibited by rats 30 min following compound injection (i.p.) are shown. Haloperidol (1 mg kg−1 i.p.) was included as a positive control. **P<0.01, vs vehicle control.
Discussion and conclusions
Selective activation of the cannabinoid CB2 receptor has been proposed as a new mechanism for the treatment of pain, with potential for the development of broad-spectrum analgesics devoid of the CNS-mediated side-effects associated with non-selective cannabinoid ligands. The evidence supporting this hypothesis has been largely derived from the study of a handful of CB2-selective ligands that share efficacy across multiple rodent pain models, but have significantly different in vitro pharmacological profiles at the CB2 receptor and also differ with respect to the potential involvement of the μ-opioid receptor in their mechanism of action. In the present study, we describe the in vitro and in vivo properties of a new CB2-selective ligand, A-796260.
Similar to the structurally related indole derivatives AM1241 and L-768,242 (GW405833), A-796260 is characterized by high affinity for human and rat CB2 receptors and moderate to excellent selectivity vs the CB1 receptor subtypes. Previous studies on AM1241 have shown it has affinities for both the human and mouse CB2 receptors in the range from 1.6 (Makriyannis and Deng, 2002) to 28.7 nM (Bingham et al., 2007) for the human receptor, and from 2 (Nackley et al., 2003b) to 28.8 nM (Bingham et al., 2007) for the mouse receptor, and 26.7 nM for the rat CB2 receptor (Bingham et al., 2007). In this study, we observed comparable affinities and selectivity ratios to these values for AM1241 (see Table 1). However, in previous studies discrepancies in binding affinity data for L-768,242 have been obtained; Valenzano et al. (2005) reported a Ki of 3.92 nM at the human CB2-binding site and >1200-fold selectivity vs the human CB1 site, whereas Gallant et al. (1996) reported an affinity of 12 nM at the human CB2 receptor and 160-fold selectivity vs the human CB1 site. The values obtained in this study are intermediate between these two results (Table 1). Based on radioligand-binding experiments, A-796260 exhibits a comparable level of affinity and selectivity for the human and rat CB2 receptors relative to previously described CB2-selective ligands.
In in vitro functional assays, A-796260 exhibits a profile distinct from other available CB2 ligands, characterized by high potencies and full or near full agonist efficacy at human and rat CB2 receptors, and a high degree of selectivity vs CB1 receptors (Table 2). This is in contrast to structurally related ligands that, in this study, either exhibit no agonist efficacy (AM1241) at either the human or rat CB2 receptor or display inverse agonist activity (L-768,242) at the rat CB2 receptor. Bingham et al. (2007) showed that AM1241 exhibits partial agonist efficacy when a low (1 μM) concentration of forskolin was used to stimulate the cAMP release, a finding consistent with that of Yao et al. (2006), where it was demonstrated that at the human CB2 receptor, AM1241 exhibits no antagonistic activity or very low efficacy as an inverse agonist at high forskolin concentrations (37 μM), but is a partial agonist at lower (8 μM) forskolin concentrations. Further, the effects of AM1241 are species-dependent; it is a partial agonist at human CB2 receptor and an inverse agonist at the rat CB2 receptor (Bingham et al., 2007). L-768,242 has been shown to exhibit partial agonist efficacy at the recombinant human CB2 receptor (Valenzano et al., 2005), but its effects on rat CB1 and CB2 receptors had not been characterized previously. In this study, L-768,242 exhibited inverse agonist activities at both human and rat CB2 receptors. The pharmacological properties of AM1241 and L-768,242 are consistent with those of protean agonists. In contrast, the fully efficacious agonist CP 55,940 and the inverse agonist SR144528 are unaffected by the assay conditions used and the level of receptor constitutive receptor activity (Yao et al., 2006). GW-842166X, despite its low potency for displacement of [3H]-CP 55,940 from the human and rat CB2 receptors (2000 and 2580 nM, respectively), did exhibit full agonist efficacy in cyclase assays in the present study (Table 2), exhibiting potencies similar to those found by Giblin et al. (2007) in a yeast reporter gene assay (human CB2 EC50=63 nM, Emax=95%; rat CB2 EC50=91 nM, Emax=100%). It is possible that GW-842166X may activate signalling through another site in addition to the CP 55,940-binding site on the CB2 receptor. Compared with the CB2 ligands studied previously, the consistent pharmacology, robust and fully efficacious agonist activities, as well as selectivity at CB2 receptors displayed by A-796260 demonstrate that this compound may be a useful tool for studying CB2 receptor pharmacology and for evaluating the therapeutic potential of CB2 agonists in rodent models.
In this study, A-796260 exhibited robust analgesic activity in rat models of inflammatory, postoperative, neuropathic and OA pain. In the CFA model of chronic inflammatory pain (Figure 2a), A-796260 produced a dose-dependent antihyperalgesic effect, but did not increase the response time to thermal stimulus in the uninjured paw. The antihyperalgesic effects of A-796260 were reversed by the CB2-selective antagonists AM630 and SR144528 but not by the CB1-selective antagonist SR141716A (Figure 2b), supporting a CB2-mediated mechanism of action. However, unlike AM1241, whose analgesic activity requires a functional μ-opioid receptor, A-796260-mediated analgesic effects were not dependent on μ-opioid receptors. Extending the previous studies by Ibrahim et al. (2005), we demonstrated that the effects of AM1241 in the CFA model are blocked by the μ-opioid antagonist naloxone (Figure 2c), whereas the antihyperalgesic effects of A-796260 were unaffected by naloxone under identical conditions, suggesting that the mechanisms of action of these two CB2-selective indole derivatives are clearly distinct. However, although the mechanism whereby μ-opioid receptors are involved in AM1241-evoked analgesic activity is not clearly understood, it does not appear to involve a direct receptor interaction as AM1241 has no affinity for the μ-opioid receptor.
Similarly, in a skin-incision model of postoperative pain (Brennan et al., 1996), A-796260 was efficacious in reversing allodynia (2 h post-incision), with a maximal efficacy comparable to that observed with morphine (Figure 3a). This effect of A-796260 was completely inhibited by the CB2-selective antagonist SR144528, indicating that CB2 receptors are involved in this model as well (Figure 3c). The efficacy of cannabinoid agonists has previously been demonstrated in a postoperative pain model (Romero-Sandoval and Eisenach, 2007); it was demonstrated that both CB1 and CB2 activation can contribute to reversal of incisional pain and the antiallodynic effects of the non-selective ligand CP 55,940 were partially reversed by both CB1 and CB2 antagonists, whereas the effects of the CB2-selective agonist JWH-015 were antagonized by the CB2 antagonist and not the CB1 antagonist. Consistent with this work, we also observed that systemic administration of the CB1 full agonist and CB2 partial agonist WIN 55,212 (see Table 2) produced a reversal of allodynia in the skin incision model, and that these effects were preferentially blocked by the CB1 antagonist SR141716A (data not shown). In addition, L-768,242 has also been shown to be active in this model at a 24 h time point post-surgical incision (Valenzano et al., 2005). These results provide further support for the potential of CB2 agonists in the management of post-surgical pain.
Tolerance develops rapidly to the analgesic effects of morphine, particularly in animal models. In the present study, tolerance to the effects of A-796260 did not develop at a dosing protocol shown to induce tolerance to the analgesic effects of morphine. Although chronic dosing studies of a substantially longer duration are required before it can be concluded that tolerance to the analgesic effects of A-796260 does not occur, these preliminary data suggest that this compound has less propensity for the development of tolerance than opioid agonists.
In the CCI of the sciatic nerve model of neuropathic pain (Bennett and Xie, 1988), A-796260 produced a dose-dependent reversal of tactile allodynia, suggesting that CB2 receptor agonists have potential as a treatment of neuropathic pain (Figure 4). These findings are consistent with the effects observed of other CB2-selective ligands, such as AM1241 (Ibrahim et al., 2003a) and L-768,242 (Valenzano et al., 2005) in models of neuropathic pain.
I.a. injection of sodium monoiodoacetate into the knee joint of rats has been used to induce morphological and behavioural changes that mimic OA in humans (Guingamp et al., 1997; Guzman et al., 2003; Combe et al., 2004). A behavioural end point that measures grip force in rats has been characterized for several classes of drugs with demonstrated clinical efficacy in the management of pain associated with OA (Chandran et al., 2006). In this model, A-796260 exhibited dose-dependent restoration of performance comparable to the clinically effective COX-2 inhibitor celecoxib. To our knowledge, this is the first time a CB2 agonist has been demonstrated to be an effective analgesic in a model of chronic OA pain. Compounds, such as morphine and non-selective agonists, which produce sedation or decreased motor coordination, also have a negative effect on the grip force task that the animals are required to perform even in uninjured animals (data not shown). In contrast, the present findings indicate that, within the dose range used, A-796260 has minimal effects that could be attributed to activation of central CB1 receptors.
To address the behavioural consequences of the modest selectivity exhibited by A-796260 in rat radioligand-binding studies (30-fold selective rat CB2 vs rat CB1) directly, its effects on spontaneous horizontal motor activity were assessed across a behaviourally effective range. No significant effects on motor activity were observed, indicating a separation of the analgesic effect mediated by the activation of CB2 receptor in vivo from the locomotor deficit attributable to CB1 receptor activation.
The pain models described in this study are all models of tissue or nerve injury, and thus compound-mediated effects in these models represent reversal of hyperalgesia or allodynia, and not antinociceptive activity. Indeed, in the CFA model, the inability of A-796260 to increase PWL to a thermal stimulus in the contralateral paw within the behaviourally effective range for reversal of hyperalgesia (as measured in the ipsilateral paw; Figure 3a) suggests that A-796260 does not possess significant antinociceptive activity, at least within this dose range. In a separate experiment in a naive rat model of acute thermal nociception (data not shown), A-796260 exhibited no effect on PWL at 3.5 and 11 mg kg−1, and only a modest increase in PWL (45% of a maximum possible effect) at a dose of 35 mg kg−1 i.p. These data contrast with the antinociceptive activity of AM1241 observed in a model of acute thermal pain (Malan et al., 2001). Also, within the same dose range AM1241 has been shown to reverse capsaicin-induced hyperalgesia (Hohmann et al., 2004) and tactile allodynia in a model of neuropathic pain (Ibrahim et al., 2003b). These different effects of AM1241 compared to A-796260 may be due to the involvement of an opioid mechanism in the effects of AM1241 but not A-796260.
In this study, we describe the properties of a new ligand, A-796260, with attributes suggesting it could be useful compound for the further exploration of CB2-selective receptor agonists in the treatment of pain. A-796260 was found to be a potent and selective CB2 agonist with broad-spectrum antihyperalgesic and antiallodynic activity across models of chronic inflammatory, postoperative, neuropathic and OA pain, and these effects were shown to be mediated through the CB2 receptor and independent of an interaction with the μ-opioid receptor. These properties differentiate A-796260 from many of the existing CB2 agonists both with respect to its agonist efficacy profile in vitro and lack of opioid receptor dependence in vivo. The studies described here also add additional support for the hypothesis that selective CB2 receptor agonists may be an attractive approach for the development of new drugs for the treatment of chronic pain; they are less likely to cause side-effects compared to the currently available treatments.
Abbreviations
- A-796260
[1-(2-morpholin-4-yl-ethyl)-1H-indol-3-yl]-(2,2,3,3-tetramethylcyclopropyl)-methanone
- AM1241
(2-iodo-5-nitrophenyl)-[1-(1-methylpiperidin-2-ylmethyl)-1H-indol-3-yl-methanone
- CP 55,940
5-(1,1-dimethyl-heptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxy-propyl)-cyclohexyl]-phenol
- GW-842166X
2-(2,4-dichloro-phenylamino)-4-trifluoromethyl-pyrimidine-5-carboxylic acid (tetrahydro-pyran-4-ylmethyl)-amide
- L-768,242 (GW405833)
(2,3-dichloro-phenyl)-[5-methoxy-2-methyl-3-(2-morpholin-4-yl-ethyl)-indol-1-yl]-methanone
- SR141716A (SR1)
5-(4-chloro-phenyl)-1-(2,4-dichloro-phenyl)-4-methyl-1H-pyrazole-3-carboxylic acid piperidin-1-ylamide
- SR144528 (SR2)
5-(4-chloro-3-methyl-phenyl)-1-(4-methyl-benzyl)-1H-pyrazole-3-carboxylic acid ((1S,2S,4R)-1,3,3-trimethyl-bicyclo[2.2.1]hept-2-yl)-amide
- WIN 55,212-2
(R)-(+)-[2,3-Dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de)-1,4-benzoxazin-6-yl]-1-napthalenylmethanone
Conflict of interest
All authors are employees of Abbott Laboratories, which funded the research.
References
- Bennett GJ, Xie Y-K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Bingham B, Jones PG, Uveges AJ, Kotnis S, Lu P, Smith VA, et al. Species-specific in vitro pharmacological effects of the cannabinoid receptor 2 (CB2) selective ligand AM1241 and its resolved enantiomers. Br J Pharmacol. 2007;151:1061–1070. doi: 10.1038/sj.bjp.0707303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhaus DW, Chang LK, Kwan J, Martin GR. Dual activation and inhibition of adenylyl cyclase by cannabinoid receptor agonists: evidence for agonist-specific trafficking of intracellular responses. J Pharmacol Exp Ther. 1998;287:884–888. [PubMed] [Google Scholar]
- Brennan TJ, Vandermeulen EP, Gebhart GF. Characterization of a rat model of incisional pain. Pain. 1996;64:493–501. doi: 10.1016/0304-3959(95)01441-1. [DOI] [PubMed] [Google Scholar]
- Chandran P, Hsieh GC, Pai M, Lee L, Faltynek C, Decker MW, et al. TRPV1 receptor mediates the modulation of a novel activity-induced pain behavior in a rat model of osteoarthritis. Soc Neurosci Abstr. 2006;32:642.12. [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh L. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Combe R, Bramwell S, Field MJ. The monosodium iodoacetate model of osteoarthritis: a model of chronic nociceptive pain in rats? Neurosci Lett. 2004;370:236–240. doi: 10.1016/j.neulet.2004.08.023. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Elmes SJR, Jhaveri MD, Smart D, Kendall DA, Chapman V. Cannabinoid CB2 receptor activation inhibits mechanically evoked responses of wide dynamic range dorsal horn neurons in naive rats and in rat models of inflammatory and neuropathic pain. Eur J Neurosci. 2004;20:2311–2320. doi: 10.1111/j.1460-9568.2004.03690.x. [DOI] [PubMed] [Google Scholar]
- Elmes SJR, Winyard LA, Medhurst SJ, Clayton NM, Wilson AW, Kendall DA, et al. Activation of CB1 and CB2 receptors attenuates the induction and maintenance of inflammatory pain in the rat. Pain. 2005;118:327–335. doi: 10.1016/j.pain.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, et al. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1995;48:443–450. [PubMed] [Google Scholar]
- Fox A, Bevan S. Therapeutic potential of cannabinoid receptor agonists as analgesic agents. Exp Opin Investig Drugs. 2005;14:695–703. doi: 10.1517/13543784.14.6.695. [DOI] [PubMed] [Google Scholar]
- Gallant M, Dufresne C, Gareau Y, Guay D, Leblanc Y, Prasit P, et al. New class of potent ligands for the human peripheral cannabinoid receptor. Bioorg Med Chem Lett. 1996;6:2263–2268. [Google Scholar]
- Giblin GMP, O'shaughnessy CT, Naylor A, Mitchell WL, Eatherton AJ, Slingsby BP, et al. Discovery of 2-[(2,4-dichlorophenyl)amino]-N-[(tetrahydro-2H-pyran-4-yl)methyl]-4-(trifluoromethyl)-5-pyrimidinecarboxamide, a selective CB2 receptor agonist for the treatment of inflammatory pain. J Med Chem. 2007;50:2597–2600. doi: 10.1021/jm061195+. [DOI] [PubMed] [Google Scholar]
- Guingamp C, Gegout-Pottie P, Philippe L, Terlain B, Netter P, Gillet P. Mono-iodoacetate-induced experimental osteoarthritis: a dose–response study of loss of mobility, morphology, and biochemistry. Arthritis Rheum. 1997;40:1670–1679. doi: 10.1002/art.1780400917. [DOI] [PubMed] [Google Scholar]
- Guzman RE, Evans MG, Bove S, Morenko B, Kilgore K. Mono-iodoacetate-induced histologic changes in subchondral bone and articular cartilage of rat femorotibial joints: an animal model of osteoarthritis. Toxicol Pathol. 2003;31:619–624. doi: 10.1080/01926230390241800. [DOI] [PubMed] [Google Scholar]
- Hanus L, Breuer A, Tchilibon S, Shiloah S, Goldenberg D, Horowitz M, et al. HU-308: a specific agonist for CB2, a peripheral cannabinoid receptor. Proc Natl Acad Sci US A. 1999;96:14228–14233. doi: 10.1073/pnas.96.25.14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Farthing JN, Zvonok AM, Makriyannis A. Selective activation of cannabinoid CB2 receptors suppresses hyperalgesia evoked by intradermal capsaicin. J Pharmacol Exp Ther. 2004;308:446–453. doi: 10.1124/jpet.103.060079. [DOI] [PubMed] [Google Scholar]
- Hosohata Y, Quock RM, Hosohata K, Makriyannis A, Consroe P, Roeske WR, et al. AM630 antagonism of cannabinoid-stimulated [35S]GTPgS binding in the mouse brain. Eur J Pharmacol. 1997;321:R1–R3. doi: 10.1016/s0014-2999(97)00047-2. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Huffman JW, Liddle J, Yu S, Aung MM, Abood ME, Wiley JL, et al. 3-(1′,1′-Dimethylbutyl)-1-deoxy-D8-THC and related compounds: synthesis of selective ligands for the CB2 receptor. Bioorg Med Chem. 1999;7:2905–2914. doi: 10.1016/s0968-0896(99)00219-9. [DOI] [PubMed] [Google Scholar]
- Huffman JW, Zengin G, Wu M-J, Lu J, Hynd G, Bushell K, et al. Structure–activity relationships for 1-alkyl-3-(1-naphthoyl)indoles at the cannabinoid CB1 and CB2 receptors: steric and electronic effects of naphthoyl substituents. New highly selective CB2 receptor agonists. Bioorg Med Chem. 2004;13:89–112. doi: 10.1016/j.bmc.2004.09.050. [DOI] [PubMed] [Google Scholar]
- Ibrahim MM, Deng H, Zvonok A, Cockayne DA, Kwan J, Mata HP, et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci USA. 2003a;100:10529–10533. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Deng H, Zvonok A, Cockayne DA, Kwan J, Mata HP, et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci USA. 2003b;100:10529–10533. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Porreca F, Lai J, Albrecht PJ, Rice FL, Khodorova A, et al. CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc Natl Acad Sci USA. 2005;102:3093–3098. doi: 10.1073/pnas.0409888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Rude ML, Stagg NJ, Mata HP, Lai J, Vanderah TW, et al. CB2 cannabinoid receptor mediation of antinociception. Pain. 2006;122:36–42. doi: 10.1016/j.pain.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Labuda CJ, Koblish M, Little PJ. Cannabinoid CB2 receptor agonist activity in the hindpaw incision. Eur J Pharmacol. 2005;527:172–174. doi: 10.1016/j.ejphar.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Lunn CA, Fine JS, Rojas-Triana A, Jackson JV, Fan X, Kung TT, et al. A novel cannabinoid peripheral cannabinoid receptor-selective inverse agonist blocks leukocyte recruitment in vivo. J Pharmacol Exp Ther. 2006;316:780–788. doi: 10.1124/jpet.105.093500. [DOI] [PubMed] [Google Scholar]
- Mackie K. Cannabinoid receptors as therapeutic targets. Annu Rev Pharmacol Toxicol. 2006;46:101–122. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- Makriyannis A, Deng H.Preparation of CB2 cannabinoid receptor-selective cannabimimetic aminoalkylindoles, particularly (piperidinylmethyl)- and (morpholinylmethyl)indoles, and their use PCT Int Appl 2002University of Connecticut, USA; pp 31; pp WO20020129 [Google Scholar]
- Malan TP, Ibrahim MM, Vanderah TW, Makriyannis A, Porreca F. Inhibition of pain responses by activation of CB2 cannabinoid receptors. Chem Phys Lipids. 2002;121:191–200. doi: 10.1016/s0009-3084(02)00155-x. [DOI] [PubMed] [Google Scholar]
- Malan TP, Jr, Ibrahim MM, Deng H, Liu Q, Mata HP, Vanderah T, et al. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. 2001;93:239–245. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- Malan TP, Jr, Ibrahim Mohab M, Lai J, Vanderah Todd W, Makriyannis A, Porreca F. CB2 cannabinoid receptor agonists: pain relief without psychoactive effects? Curr Opin Pharmacol. 2003;3:62–67. doi: 10.1016/s1471-4892(02)00004-8. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Adams M, Whiteaker K, Daza A, Kage K, Cassar S, et al. Species comparison and pharmacological characterization of rat and human CB2 cannabinoid receptors. Eur J Pharmacol. 2004;505:1–9. doi: 10.1016/j.ejphar.2004.09.058. [DOI] [PubMed] [Google Scholar]
- Nackley AG, Makriyannis A, Hohmann AG. Selective activation of cannabinoid CB2 receptors suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience. 2003a;119:747–757. doi: 10.1016/s0306-4522(03)00126-x. [DOI] [PubMed] [Google Scholar]
- Nackley AG, Makriyannis A, Hohmann AG. A peripheral cannabinoid mechanism suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience. 2003b;117:659–670. doi: 10.1016/s0306-4522(02)00870-9. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Cannabinoid receptors and pain. Prog Neurobiol. 2001;63:569–611. doi: 10.1016/s0301-0082(00)00031-9. [DOI] [PubMed] [Google Scholar]
- Pomonis JD, Boulet JM, Gottshall SL, Phillips S, Sellers R, Bunton T, et al. Development and pharmacological characterization of a rat model of osteoarthritis pain. Pain. 2005;114:339–346. doi: 10.1016/j.pain.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Héaulme M, Shire D, Calandra B, Congy C, et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Millan J, Derocq J-M, Casellas P, Congy C, et al. SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther. 1998;284:644–650. [PubMed] [Google Scholar]
- Romero-Sandoval A, Eisenach JC. Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision. Anesthesiology. 2007;106:787–794. doi: 10.1097/01.anes.0000264765.33673.6c. [DOI] [PubMed] [Google Scholar]
- Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, et al. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656 and AM630. Br J Pharmacol. 1999;126:665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar DR, Kelly S, Millns PJ, O'shaughnessey CT, Kendall DA, Chapman V. Inhibitory effects of CB1 and CB2 receptor agonists on responses of DRG neurons and dorsal horn neurons in neuropathic rats. Eur J Neurosci. 2005;22:371–379. doi: 10.1111/j.1460-9568.2005.04206.x. [DOI] [PubMed] [Google Scholar]
- Spetea M, Nevin ST, Hosztafi SN, Ronai ASZ, Toth G, Borsodi A. Affinity profiles of novel Î-receptor selective benzofuran derivatives of non-peptide opioids. Neurochem Res. 1998;23:1211–1216. doi: 10.1023/a:1020738304036. [DOI] [PubMed] [Google Scholar]
- Tao Q, Abood ME. Mutation of a highly conserved aspartate residue in the second transmembrane domain of the cannabinoid receptors, CB1 and CB2, disrupts G-protein coupling. J Pharmacol Exp Ther. 1998;285:651–658. [PubMed] [Google Scholar]
- Valenzano KJ, Tafesse L, Lee G, Harrison JE, Boulet JM, Gottshall SL, et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology. 2005;48:658–672. doi: 10.1016/j.neuropharm.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Whiteside GT, Gottshall SL, Boulet JM, Chaffer SM, Harrison JE, Pearson MS, et al. A role for cannabinoid receptors, but not endogenous opioids, in the antinociceptive activity of the CB2-selective agonist, GW405833. Eur J Pharmacol. 2005;528:65–72. doi: 10.1016/j.ejphar.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Whiteside GT, Lee GP, Valenzano KJ. The role of the cannabinoid CB2 receptor in pain transmission and therapeutic potential of small molecule CB2 receptor agonists. Curr Med Chem. 2007;14:917–936. doi: 10.2174/092986707780363023. [DOI] [PubMed] [Google Scholar]
- Wotherspoon G, Fox A, Mcintyre P, Colley S, Bevan S, Winter J. Peripheral nerve injury induces cannabinoid receptor 2 protein expression in rat sensory neurons. Neuroscience. 2005;135:235–245. doi: 10.1016/j.neuroscience.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Yao BB, Mukherjee S, Fan Y, Garrison TR, Daza AV, Grayson GK, et al. In vitro pharmacological characterization of AM1241: a protean agonist at the cannabinoid CB2 receptor? Br J Pharmacol. 2006;149:145–154. doi: 10.1038/sj.bjp.0706838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hoffert C, Vu Huy K, Groblewski T, Ahmad S, O'donnell D. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur J Neurosci. 2003;17:2750–2754. doi: 10.1046/j.1460-9568.2003.02704.x. [DOI] [PubMed] [Google Scholar]