

Abstract
Endocannabinoids, including anandamide (arachidonoyl ethanolamide) have been implicated in the regulation of a growing number of physiological and pathological processes. Anandamide can be generated from its membrane phospholipid precursor N-arachidonoyl phosphatidylethanolamine (NAPE) through hydrolysis by a phospholipase D (NAPE-PLD). Recent evidence indicates, however, the existence of two additional, parallel pathways. One involves the sequential deacylation of NAPE by α,β-hydrolase 4 (Abhd4) and the subsequent cleavage of glycerophosphate to yield anandamide, and the other one proceeds through phospholipase C-mediated hydrolysis of NAPE to yield phosphoanandamide, which is then dephosphorylated by phosphatases, including the tyrosine phosphatase PTPN22 and the inositol 5′ phosphatase SHIP1. Conversion of synthetic NAPE to AEA by brain homogenates from wild-type and NAPE-PLD−/− mice can proceed through both the PLC/phosphatase and Abdh4 pathways, with the former being dominant at shorter (<10 min) and the latter at longer incubations (60 min). In macrophages, the endotoxin-induced synthesis of anandamide proceeds uniquely through the phospholipase C/phosphatase pathway.
1. Introduction
N-acylethanolamines were first identified as endogenous lipids in the hypoxic myocardium (Epps et al., 1979) where their biosynthesis was found to occur in two steps. First, a transacylase N-acylates membrane phosphatidyl ethanolamine (PE) (Natarajan et al., 1981), which is then hydrolyzed by a phospholipase D (PLD) to yield N-acylethanolamines and phosphatidic acid (Schmid et al., 1983). A low abundance member of this class of lipids, N-arachidonoyl ethanolamide (AEA, also called anandamide) was later identified as the first endogenous cannabinoid ligand (Devane et al., 1992). Since then, endocannabinoids (ECs) acting at specific cannabinoid receptors in the brain and periphery have been implicated in the regulation of a growing number of physiological functions and pathological processes (Pacher et al., 2006). Compounds that selectively inhibit EC actions at cannabinoid receptors or enhance them by blocking EC degradation show great therapeutic potential (Pacher et al., 2006). Inhibitors of enzymes that metabolize ECs were found to potentiate some but not other EC actions (Kathuria et al., 2003), which suggests that blocking EC biosynthesis may have a pharmacological profile different from that achieved by blocking cannabinoid receptors, and thus may be used therapeutically to avoid some of the unwanted side effects of the latter.
Anandamide is generated from the membrane phospholipid precursor N-arachidonoyl PE (NAPE) through a two-step process similar to that described above (Di Marzo et al., 1994). The calcium-dependent transacylase that catalyzes the transfer of arachidonic acid (AA) from the sn-1 position of phosphatidyl choline to the nitrogen atom of PE to generate NAPE has not yet been identified. However, a NAPE-specific PLD that hydrolyzes NAPE to yield anandamide has been cloned (Okamoto et al., 2004) and the purified protein characterized (Wang et al., 2006). When overexpressed in cells, NAPE-PLD selectively reduces NAPE and increases anandamide levels (Okamoto et al., 2005). Unexpectedly, no significant reduction in anandamide levels was found in the brain of NAPE-PLD knockout mice (Leung et al., 2006), or in macrophages following siRNA knockdown of NAPE-PLD (Liu et al., 2006). Furthermore, tissues from NAPE-PLD−/− mice contained an enzymatic activity capable of converting NAPE to AEA in a calcium-independent manner (Leung et al., 2006), suggesting the existence of additional, parallel biosynthetic pathways. There has been evidence since the 1980’s for enzymatic activities that can generate N-acylethanolamines from NAPE through sequential deacylations yielding lyso-NAPE and glycerophospho-ethanolamines, respectively (Natarajan et al., 1984). Accordingly, a recently identified group IB secretory phospholipase A2 can convert NAPE to 2-lyso-NAPE, which is then metabolized to anandamide through a calcium-independent mechanism sensitive to inhibition by methyl arachidonoyl fluorophosphate (MAFP) (Sun et al., 2004). However, the restricted tissue expression of this PLA2 suggested the existence of additional enzymes involved in generating lyso-NAPE. One such enzyme is the recently identified αβ-hydrolase 4 (Abhd4), which can act on either NAPE or lyso-NAPE to generate the glycerophospho-arachidonoyl ethanolamide (GpAEA), which is then acted on by a metal-dependent, phosphodiesterase to yield AEA (Simon and Cravatt, 2006). Another alternative pathway identified in the RAW264.7 mouse macrophage cell line involves the hydrolysis of NAPE by a phospholipase C to yield phosphoanandamide (pAEA) which is then dephosphorylated by phosphatases, including the putative tyrosine phosphatase PTPN22 (Liu et al., 2006). This latter pathway was found to be responsible for the endotoxin (LPS)-induced increase in AEA biosynthesis in macrophages (Liu et al., 2003; 2006). In the present study we have analyzed the relative importance of the NAPE-PLD, Abhd4 and PLC/phosphatase pathways in AEA biosynthesis in RAW264.7 cells and in mouse brain.
2. Methods and Materials
2.1. NAPE-PLD, PTPN22 and SHIP1 knockout mice
NAPE-PLD knockout mice were generated by R.D. Palmiter and S. Luquet. A 4.4 kb EcoRI/NaeI fragment containing the exon 2 (left arm) of the NAPE-PLD and the 6.5 kb NaeI/NheI fragment (right arm) containing the exon 3 were subcloned in the 4517D plasmid to generate the targeting construct. LoxP sites flank exon 3. The Sv-Neo (SV40 promoter-neomycin-phosphotransferase) gene is flanked by frt sites from removal by the action of FLPase. The targeting construct contains a Pgk-diphtheria toxin A gene at the 5′ end of the left arm and an HSV thymidine kinase gene at the 3′ end of the right arm for negative selection. Linearized plasmid (20 μg) was electroporated into ~ 107 AK18.1 embryonic stem cells (129S4/SvJaeSor; provided by P. Soriano) and plated on mitomycin C-treated SNL feeders and selected in G418 (300 μg/ml) and gancyclovir (2 μM). Individual colonies were picked, expanded for analysis, correctly targeted clones (5/80) were identified by Southern blot, and injected into blastocysts. After removal of the Sv-Neo cassette by breeding with FLPer mice, exon 3 was removed in all cells by breeding with Mox2-Cre mice (Tallquist & Soriano, 2000) to generate heterozygous animals with one null NAPE-PLD allele. These mice were bred with C57BL/6 mice to remove the Mox2-Cre gene, and then bred together to generate mice homozygous for the NAPE-PLD-null allele. Mice are on a mixed 129S4 x C57Bl/6 genetic background.
PTPN22 knockout mice were generated as described (Hasegawa et al., 2004). The generation of SHIP1 (inpp5d) knockout mice was reported earlier (Helgason et al., 1998).
2.2. Cell culture
RAW264.7 mouse macrophages were obtained from ATCC (Manassas, VA) and maintained under standard culturing conditions as described (Liu et al., 2003). To test the effect of bacterial endotoxin (lipopolysaccharide [LPS], Escherichia coli, 0127:B8), preconfluent cultures of cells were incubated for 90 min with 10 ng/ml LPS or vehicle, and then washed, harvested and extracted for measurement of AEA content as described (Liu et al., 2003). The extraction protocol used (2 volumes of chloroform:methanol 2:1 added to one volume of aqueous tissue or cell homogenate, repeated 2 more times) yielded >99% recovery of synthetic AEA and > 87% recovery of synthetic pAEA.
2.3. siRNA knockdown and real-time PCR
siRNA knockdown of PTPN22 and NAPE-PLD expression in RAW264.7 cells and verification of the degree of knockdown by real-time PCR was done as described previously (Liu et al., 2006). For siRNA knockdown of Abhd4, predesigned siRNAs and primer mix for real-time PCR were purchased from Qiagen (Valencia, CA).
2.4. Enzymatic conversion of synthetic NAPE, GpAEA or pAEA to AEA
To measure the enzymatic conversion of synthetic NAPE, GpAEA or pAEA to AEA, RAW264.7 cell or mouse brain homogenates (10 μg protein) were incubated for the indicated times with 10 nmol of the given synthetic precursor in 50 mM Tris buffer, with or without the indicated enzyme inhibitors. The reaction was stopped by the addition of 2 volumes of chloroform:methanol 2:1 containing 5 nmol of [2H4]AEA, and AEA in the extracted sample was measured by LC/MS as described below.
2.5. Synthesis of NAPE, GpAEA and pAEA
The synthesis of NAPE (N-arachidonoyl-1,2-dioleoyl-phosphatidyl ethanolamine, Sugiura et al., 1996) and pAEA (Liu et al, 2006) were as described earlier. A synthetic sequence was developed for the preparation of GpAEA and deposited as Supplementary Material. Briefly, GpAEA was synthesized from commercially available solketal, which was transformed to acetic acid 2-acetoxy-1-hydroxymethyl-ethyl ester [(via (i) protection of the hydroxyl as benzyl, (ii) opening of the acetal (acetic acid), (iii) protection of the hydroxyls as acetates and finally (iv) debenzylation (H2, Pd/C, methanol)]. The primary alcohol was then converted to the corresponding phosphatidylethanolamine using an established protocol (Eibl, 1978; Li & Pascal, 1993). The amine was then coupled to arachidonic ester in the presence of NaHCO3 in THF/H2O (10:12) mixture to give the arachidonoyl derivative. Treatment of this intermediate with LiOH.H2O in methanol/H2O (5:1) mixture gave the target compound Gp-AEA, which was purified (Combiflash) by reverse phase column chromatography using water/metyhanol as the gradient. All compounds were characterized by TLC and mass spectroscopy (Agilent 1100 Series HPLC-MS) and 1HNMR (300 MHz, Jeol Eclipse).
2.6. Measurement of AEA and pAEA
Measurement of tissue or cellular levels of AEA by stable isotope dilution HPLC/MS and simultaneous measurement of AEA by HPLC/ESI-MS/MS were done as described before (Liu et al., 2006).
3. Results
3.1. Effect of siRNA knockdown of NAPE-PLD or Abhd4 on basal and LPS-stimulated AEA levels in RAW264.7 macrophages
In order to explore the enzymatic pathways involved in the LPS-induced synthesis of AEA in RAW264.7 cells, we first examined the effect of LPS on NAPE-PLD, Abhd4 and PTPN22 gene expression, using real-time PCR. Incubation of the cells with 10 ng/ml LPS for 90 min resulted in a > 50% reduction in NAPE-PLD mRNA, a modest 27% increase in Abhd4 mRNA and a 2-fold increase in PTPN22 mRNA (Fig. 1).
Fig. 1.

Effect of LPS on NAPE-PLD, PTPN22 and Abhd4 mRNA levels in RAW264.7 cells. Cells were treated with vehicle or LPS (10 ng/ml) for 90 min. mRNA was quantified by real-time PCR as described in Methods and Materials. Means ± SE from three to four experiments are shown. * indicate significant difference (P < 0.05) from values in vehicle-treated control cells, as determined by using the paired t test.
Next we analyzed the effect of siRNA knockdown of these enzymes on basal and LPS-stimulated AEA levels in RAW264.7 cells. As illustrated in Fig. 2, siRNA knockdown of NAPE-PLD resulted in a 42% decrease in the cellular level of NAPE-PLD mRNA, but did not affect basal AEA levels and the LPS-induced increase in AEA was actually greater in these cells than in mock-transfected controls. siRNA knockdown of Abhd4 resulted in a 51 ± 4 % decrease in Abhd4 mRNA. Again, basal levels of AEA remained unchanged and the LPS-induced increase in AEA was also unaffected by the knockdown. siRNA knockdown of PTPN22 reduced PTPN22 mRNA levels by 72 % and whereas it did not affect baseline levels of AEA, it caused a 36 % reduction in LPS-induced increase in AEA levels.
Fig. 2.

The effect of siRNA knockdown of NAPE-PLD, Abhd4 or PTPN22 on LPS-induced AEA synthesis in RAW264.7 cells. The degree of knockdown was verified by real-time PCR in mock-transfected (white columns) vs siRNA-transfected cells (shaded columns, left side). The right two pairs of columns indicate the cellular levels of AEA following vehicle (open columns) or LPS treatment (hatched columns) of mock- or siRNA transfected cells. Means ± SE from three experiments are shown. Significant difference from corresponding vehicle-treated group (*) or from corresponding mock-transfected group (#) is indicated, P < 0.05. Comparison of relative mRNA levels was analyzed by the paired t test, whereas the effect of LPS on cellular AEA levels was analyzed using the unpaired t test.
3.2. Role of the PLC/phosphatase pathway in LPS-induced AEA synthesis in macrophages
The findings described above suggest that the PLC/phosphatase pathway, but not the NAPE-PLD or Abhd4 pathways, is involved in the LPS-induced synthesis of AEA in macrophages. Indeed, preincubation of RAW264.7 cells with 3 mM neomycin, a PLC inhibitor, or 1 mM of the tyrosine phosphatase inhibitor NaVO3 nearly completely prevented the LPS-induced increase in cellular AEA levels (Fig. 3). Earlier studies have identified PTPN22 as one of the genes induced by LPS in RAW264.7 macrophages, overexpression of which resulted in elevated cellular AEA levels (Liu et al., 2006). The finding that siRNA knockdown of PTPN22 caused only a partial reduction in the effect of LPS suggested the possible involvement of additional phosphatases in the dephosphorylation of pAEA. One such phosphatase may be the inositol 5′ phosphatase SHIP1, whose expression in RAW264.7 macrophages is also induced by LPS (An et al., 2005). Although the expression of PTPN22 is low in the brain (Liu et al., 2006) and SHIP1 may only be expressed in microglia, we analyzed the conversion of synthetic pAEA to AEA in brain extracts from PTPN22 and SHIP1 knockout mice and their wild-type controls, under conditions described in Methods. For SHIP1, the amount of AEA generated was 3.23 ± 0.32 nmol/mg/min in controls vs 2.46 ± 0.15 nmol/mg/min in knockouts (P < 0.05), and for PTPN22 the respective values were 3.04 ± 0.19 vs 2.06 ± 0.12 nmol/mg/min (P < 0.01). These findings suggest that both PTPN22 and SHIP1 can contribute to the generation of AEA through the PLC/phosphatase pathway.
Fig. 3.
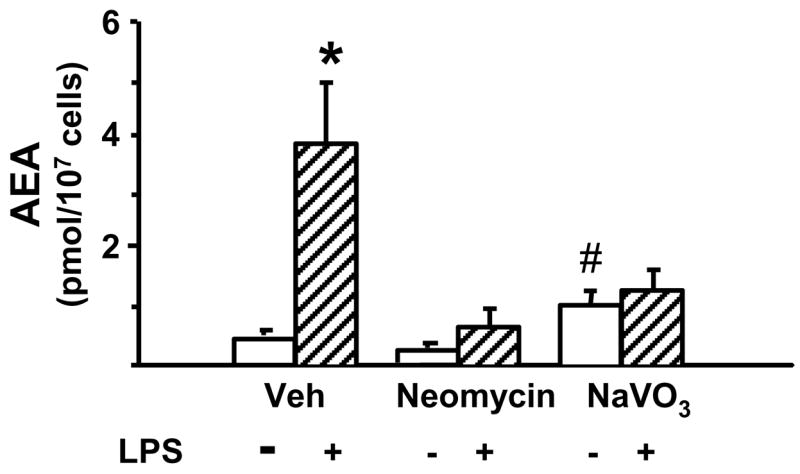
Blocking PLC or tyrosine phosphatases prevents LPS-induced AEA synthesis in RAW264.7 macrophages. The cells were preincubated for 30 min with 3 mM neomycin or with 1 mM NaVO3 before the addition of vehicle or 10 ng/ml LPS for 90 min. AEA was then quantified in lipid extracts of vehicle- (open columns) or LPS-treated (hatched columns) cells as described in Methods and Materials. Values represent means ± SE from 3–4 experiments. * indicate significant difference (P < 0.05) from values in corresponding control cells.
3.3. Mechanisms of enzymatic conversion of NAPE to AEA in brain tissue
To analyze the enzymatic pathways involved in the generation of AEA from NAPE, aliquots of brain homogenate (10 μg protein) were incubated with 10 nmol of synthetic NAPE in the absence or presence of various inhibitors, and the amount of AEA generated was measured by LC/MS. The accumulation of AEA was linear for up to an hour, and it could be nearly abolished by boiling the samples before incubation. The genetic ablation of NAPE-PLD resulted in a ~75% reduction in the amount of AEA generated from exogenous NAPE, from 56.0 ± 5.0 pmol/mg/min in wild-type controls to 13.5 ± 2.5 pmol/mg/min in NAPE-PLD knockouts (P < 0.01), as illustrated in Fig. 4 (note the difference in scale for wildtype vs knockout). In samples incubated for 1 hour, blocking the PLC/phosphatase pathway with neomycin, NaVO3 or their combination did not affect AEA synthesis (Fig. 4B). Activity of the Abhd4 pathway can be inhibited by MAFP or EDTA (Sun et al., 2004; Simon & Cravatt, 2006). Although EDTA caused a ~ 30% reduction in the amount of AEA generated, MAFP or MAFP + EDTA were ineffective (Fig. 4B). With much shorter, 1 min incubations the situation was reversed, now neomycin and/or NaVO3 were effective in reducing AEA accumulation, whereas MAFP and EDTA were ineffective (Fig. 4A).
Fig. 4.
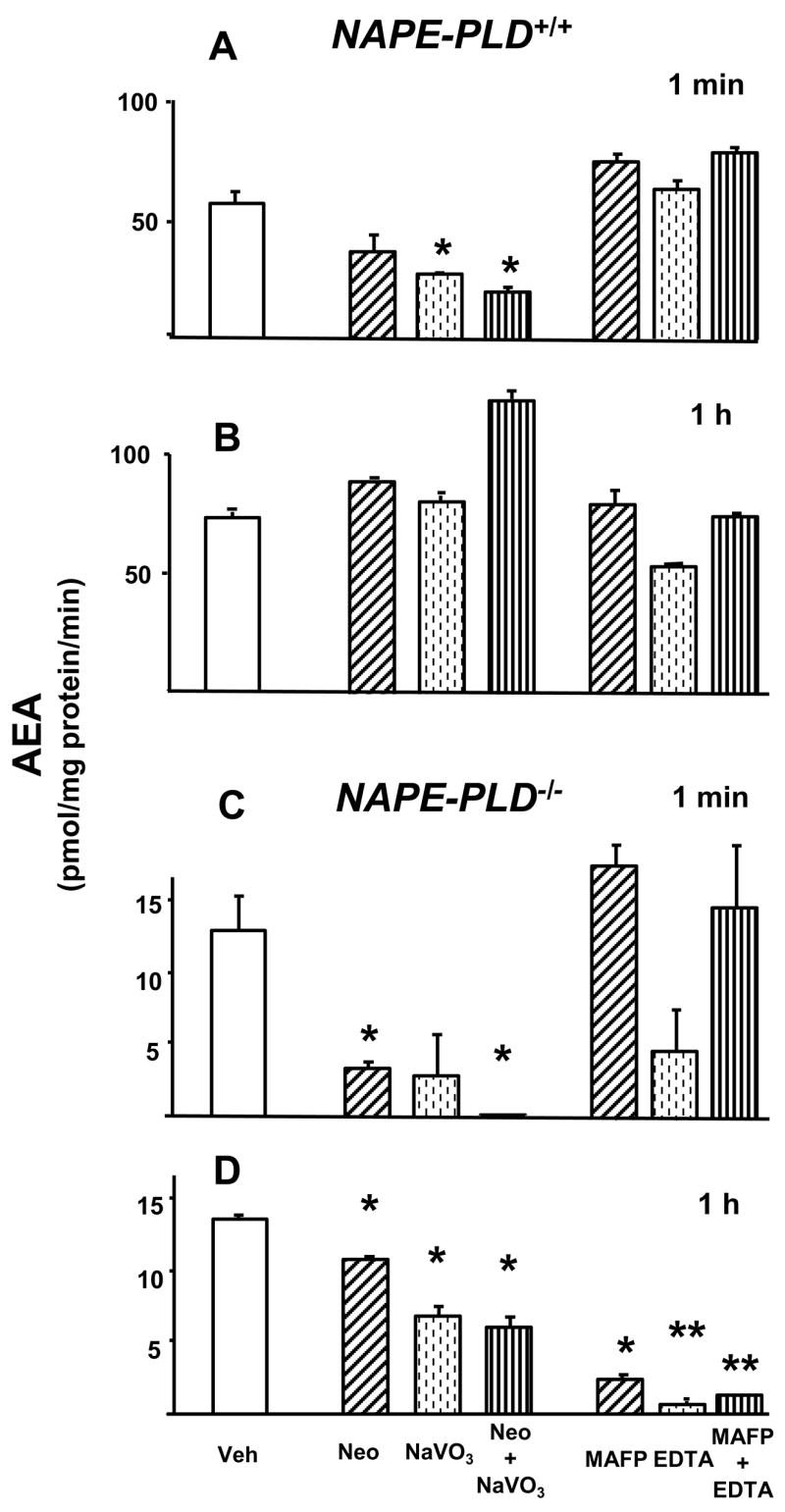
Conversion of NAPE to AEA in brain extracts from NAPE-PLD+/+ and NAPE-PLD−/− mice. Brain homogenates (10 μg protein) were pretreated with 3mM neomycin (hatched columns), 1 mM NaVO3 (dotted columns), neomycin + NaVO3 (vertical hatched columns); or 5 mM MAFP (hatched columns), 2 mM EDTA (dotted columns) or MAFP + EDTA (vertical hatched columns) for 30 min at room temperature before incubating with 10 nmol NAPE for the indicated time period at 37°C. The reaction was then terminated and AEA assayed as described in Methods and Materials. Means ± SE from three experiments are shown. Asterisks indicate significant difference (* P < 0.05; ** P < 0.005) from values in corresponding vehicle-treated controls (open columns), as analyzed by the unpaired t test.
In the absence of NAPE-PLD, the contribution of the other two pathways was increased. When brain extracts from NAPE-PLD−/− mice were incubated for 1 hour, MAFP and/or EDTA nearly completely blocked AEA generation and neomycin and/or NaVO3 caused partial inhibition (Fig. 4D), whereas in samples incubated for 1 min, neomycin plus NaVO3 caused complete inhibition, whereas EDTA (but not MAFP) was partially effective (Fig. 4C). The time course of this change was tested using brain homogenates from PLD−/− mice. In the presence of neomycin + NaVO3, AEA generated from synthetic NAPE was reduced by 98%, 84% or 20% at 1, 10 or 60 min, respectively, whereas in the presence of MAFP + EDTA the corresponding values were 0%, 48% and 91%. This indicates that the PLC/phosphatase pathway still plays a major role at 10 min.
3.4. pAEA is an intermediate of AEA biosynthesis in mouse brain
We have earlier identified pAEA by HPLC/ESI-MS/MS as an intermediate of AEA synthesis in RAW264.7 cells (Liu et al., 2006). pAEA could also be detected in mouse brain, where its levels were markedly increased in the presence of the tyrosine phosphatase inhibitor NaVO3, and NaVO3 also inhibited the conversion of synthetic pAEA to AEA by brain extracts (Liu et al., 2006). The putative role of PTPN22 in this conversion is further indicated by the increase in pAEA levels in brain tissue from PTPN22−/− compared to wild-type mice, as illustrated in Fig. 5.
Fig. 5.

Determination of pAEA and AEA by HPLC/ESI-MS/MS in brain samples. The transitions of m/z 428/330, 348/287 and 352/287 were selected for pAEA, AEA and 2H4AEA (internal standard), respectively. Note the higher pAEA and lower AEA level in the PTPN22−/− vs control brain extract.
GpAEA has been proposed to be converted to AEA through the action of a metal-dependent phosphodiesterase (Simon & Cravatt, 2006). We tested whether this conversion may also proceed through an alternative 2-step pathway where a putative lyso-PLC first converts GpAEA to pAEA, which is then dephosphorylated to AEA. As indicated by the data in Table 1, incubation of aliquots of brain homogenate from a control mouse with 10 nmol GpAEA resulted in the generation of 663 pmol/mg/min AEA and detectable levels of pAEA. In agreement with the results of Simon & Cravatt (2006), the generation of AEA was nearly completely blocked in the presence of 2 mM EDTA. EDTA also caused a parallel 4.5-fold increase in the amount of pAEA, suggesting that by blocking the predominant metal-sensitive pathway, metabolism of GpAEA is shifted toward the alternative 2-step pathway. NaVO3 alone caused a modest, ~8% reduction in the amount of AEA generated from GpAEA, which was accompanied by a >3-fold increase in pAEA, suggesting that this pathway is active even in the absence of EDTA. The addition of neomycin to NaVO3 caused a slight further reduction in the amount of AEA generated, but substantially increased the amount of pAEA (Table 1). This could be due to blockade of the reacylation of pAEA to NAPE, which may occur when levels of pAEA are elevated due both to its increased generation from an exogenous precursor and lack of dephosphorylation in the presence of NaVO3.
Table 1.
Conversion of GpAEA to pAEA and AEA in brain extract from a control mouse.
| AEA (pmol/mg/min) | pAEA/d4AEA ×100 (%) | |
|---|---|---|
| Vehicle | 663 | 0.16 |
| Neo | 648 | 0.27 |
| NaVO3 | 610 | 0.51 |
| Neo-NaVO3 | 562 | 2.3 |
| EDTA | 13 | 0.72 |
4. Discussion
As signaling molecules, endocannabinoids are unique in that they are not stored but synthesized ‘on demand’ in post-synaptic neurons or non-neuronal cells. This means that mechanisms that regulate their biosynthesis are directly coupled to and modulate their biological actions, and may be targeted for the purpose of therapeutic drug development. Such efforts may be complicated by emerging evidence that the biosynthesis of endocannabinoids occurs via multiple parallel pathways.
The evidence reviewed in the introduction and additional findings presented here clearly indicate that there may be at least three different pathways through which the membrane phospholipid precursor NAPE can be converted to AEA, as illustrated schematically in Figure 6. This may explain why genetic ablation/knockdown or pharmacological inhibition of any one of these pathways does not eliminate or cause a substantial reduction in the basal cellular levels of AEA (Fig. 2). However, the ‘on demand’ synthesis of endocannabinoids may involve mechanisms different from those operating to maintain basal tissue levels. Bacterial endotoxin (LPS) causes a major increase in the biosynthesis of AEA in RAW264.7 macrophages, as a result of increases in the rate of both the generation of NAPE and its subsequent conversion to AEA (Liu et al., 2003). This LPS-induced AEA synthesis is unaffected by knockdown of NAPE-PLD or Abhd4, but is nearly completely inhibited by blocking PLC and/or tyrosine phosphatase activities (Liu et al., 2006; see also Fig. 3). This suggests activation of this pathway during the ‘on demand’ synthesis of AEA. An analogous situation for the other endocannabinoid, 2-arachidonoylglycerol (2-AG), may exist in the hippocampus where Gq/G11-mediated signaling was found not to be involved in basal 2-AG production, but to be indispensable for the kainate-induced formation of 2-AG (Wettschureck et al., 2006). Whether this dichotomy reflects distinct metabolic and signaling-related subcellular EC pools remains to be seen.
Fig. 6.

Cellular pathways of anandamide biosynthesis. For explanation, see text.
The different time-course of activation of the various pathways involved in the conversion of NAPE to AEA is also compatible with a unique role of the PLC/phosphatase pathway in rapid, ‘on demand’ signaling. As illustrated by the data in Fig. 4, the conversion of exogenous NAPE to AEA is reduced by ~75% in brain tissue form NAPE-PLD−/− compared to control mice, which is very similar to the degree of reduction reported by Leung et al. (2006) under similar conditions. This illustrates the dominant role of NAPE-PLD in this process, which is also indicated by the finding that blocking the other two pathways fails to impact on AEA generation over a 1 hour incubation period. However, the initial rate of AEA synthesis in the first minute is substantially reduced by blocking PLC and/or phosphatases, suggesting a role for this pathway in the rapid initial synthesis of AEA. This is even more striking in preparations from NAPE-PLD−/− mice, where the synthesis of AEA is fully dependent on alternative pathways. Whereas Abhd4 appears to be dominant in the long-term synthesis of AEA (Fig. 4D), the rapid initial synthesis of AEA appears to be predominantly through the PLC/phosphatase pathway (Fig. 4C), as tested in tissue homogenates. Further studies are needed to test whether this also applies to the intact brain under in vivo conditions.
The role of PTPN22 in LPS-induced AEA synthesis in macrophages was indicated by its upregulation in LPS-treated RAW264.7 cells, and by the reduced conversion of pAEA to AEA following siRNA knockdown of its expression (Liu et al., 2006; also Fig. 1). The findings of reduced AEA (Liu et al., 2006) and increased pAEA levels in the brain of PTPN22−/− compared to wild-type mice (Fig. 5) suggests a similar role in brain. The partial nature of these changes implicates additional phosphatases. The present findings suggest that SHIP1, an inositol 5′ phosphatase which is similarly induced by LPS in RAW264.7 cells (An et al., 2005), can also dephosphorylate pAEA, as suggested by the reduced rate of this reaction in brain extracts from SHIP1−/− mice. Alternatively, the increase in pAEA levels in the absence of phosphatases or following their blockade by NaVO3 could be due to lack of dephosphorylation of the PLC involved in generating pAEA from NAPE.
The activity of biosynthetic pathways under conditions of a large excess of exogenous substrate is not necessarily indicative of their importance in the metabolism of physiological levels of endogenous substrate. The findings in RAW264.7 macrophages clearly indicate the unique role of a neomycin/NaVO3-sensitive pathway in the LPS-stimulated synthesis of endogenous AEA (Fig. 3). Further studies of the relative role of these pathways in the basal versus stimulated synthesis of anandamide in the brain will benefit from an experimental model in which the biosynthesis of AEA can be robustly increased by a well defined physiological or pathological stimulus. A further goal of these studies will be the identification of the PLC isozyme and any additional phosphatases involved. The existence of parallel pathways of AEA synthesis makes it unlikely that targeting a single enzyme will allow the depletion of tissue AEA. However, the dominant role of a single pathway in the stimulated synthesis of AEA could make this pathway a target for drug development, in view of emerging evidence that overactivity of the endocannabinoid system may underlie certain diseases, such as obesity/metabolic syndrome (Osei-Hyiaman et al., 2005; Engeli et al., 2005).
Supplementary Material
Acknowledgments
This work was supported by the Division of Intramural Clinical and Biological Research, NIAAA, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- An H, Xu H, Zhang M, Zhou J, Feng T, Qian C, Qi R, Cao X. Src homology 2 domain-containing inositol-5-phosphatase 1 (SHIP1) negatively regulates TLR4-mediated LPS reponse primarily through a phosphatase activity- and PI-3K-independent mechanism. Blood. 2005;105:4685–4692. doi: 10.1182/blood-2005-01-0191. [DOI] [PubMed] [Google Scholar]
- Devane Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- Eibl H. Phospholipid synthesis: oxazaphospholanes and dioxaphospholanes as intermediates. Proc Natl Acad Sci USA. 1978;75:4074–4077. doi: 10.1073/pnas.75.9.4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engeli S, Bohnke J, Feldpausch M, Gorzelniak K, Janke J, Batkai S, Pacher P, Harvey-White J, Luft FC, Sharma AM, Jordan J. Activation of the peripheral endocannabinoid system in human obesity. Diabetes. 2005;54:2838–2843. doi: 10.2337/diabetes.54.10.2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epps DE, Schmid PC, Natarajan V, Schmid HHO. N-Acylethanolamine accumulation in infarcted myocardium. Biochem Biophys Res Commun. 1979;90:628–633. doi: 10.1016/0006-291x(79)91281-6. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Martin F, Huang G, Tumans D, Diehl L, Chan AC. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science. 2004;303:685–689. doi: 10.1126/science.1092138. [DOI] [PubMed] [Google Scholar]
- Helgason CD, Damen JE, Rosten P, Grewal R, Sorensen P, Chappel SM, Borowski A, Jirik F, Krystal G, Humphries RK. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. doi: 10.1101/gad.12.11.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Leung D, Saghatelian A, Simon GM, Cravatt BF. Inactivation of N-Acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry. 2006;45:4720–4726. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Pascal RA. Sulfur-substituted phosphatidylethanolamines. J Org Chem. 1993;58:1952–1954. [Google Scholar]
- Liu J, Bátkai S, Pacher P, Harvey-White J, Wagner JA, Cravatt BF, Kunos G. Activation of anandamide synthesis in mouse macrophages and its role in endotoxin-induced hypotension. J Biol Chem. 2003;278:45034–45039. doi: 10.1074/jbc.M306062200. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim H-Y, Kunos G. A biosynthetic pathway for anandamide. Proc Natl Acad Sci USA. 2006;103:13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan V, Reddy pV, Schmid PC, Schmid HH. On the biosynthesis and metabolism of N-acylethanolamine phospholipids in infarcted dog heart. Biochim Biophys Acta. 1981;664:445–448. doi: 10.1016/0005-2760(81)90067-9. [DOI] [PubMed] [Google Scholar]
- Okamoto Y, Morishita J, Wang J, Schmid PC, Krebsbach RJ, Schmid HHO, Ueda N. Mammalian cells stably overexpressing N-acylphosphatidylethanolamine-hydrolysing phospholipase D exhibit significantly decreased levels of N-acylphosphatidylethanolamines. Biochem J. 2005;389:241–247. doi: 10.1042/BJ20041790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. Molecular characterization of phospholipase D generating anandamide and its congeners. J Biol Chem. 2004;279:5298–5305. doi: 10.1074/jbc.M306642200. [DOI] [PubMed] [Google Scholar]
- Osei-Hyiaman D, DePetrillo M, Pacher P, Liu J, Radaeva S, Batkai S, Harvey-White J, Mackie K, Offertaler L, Wang L, Kunos G. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Invest. 2005;115:1298–1305. doi: 10.1172/JCI23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid PC, Reddy PV, Natarajan V, Schmid HHO. Metabolism of N-acylethanolamine phospholipids by a mammalian phosphodiesterase of the phospholipase D type. J Biol Chem. 1983;258:9302–9306. [PubMed] [Google Scholar]
- Simon GM, Cravatt BF. Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for α/β-hydrolase 4 in this pathway. J Biol Chem. 2006;281:26465–26472. doi: 10.1074/jbc.M604660200. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Tonegawa T, Nakane S, Yamashita A, Ishima Y, Waku K. Transacylase-mediated and phosphodiesterase-mediated synthesis of N-arachidonoylethanolamine, an endogenous cannabinoid-receptor ligand, in rat brain microsomes. Comparison with synthesis from free arachidonic acid and ethanolamine. Eur J Biochem. 1996;240:53–62. doi: 10.1111/j.1432-1033.1996.0053h.x. [DOI] [PubMed] [Google Scholar]
- Sun YX, Tsuboi K, Okamoto Y, Tonai T, Murakami M, Kudo I, Ueda N. Biosynthesis of anandamide and N-palmitoylethanolamine by sequential actions of phospholipase A2 and lysophospholipase D. Biochem J. 2004;380:749–756. doi: 10.1042/BJ20040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallquist MD, Soriano P. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis. 2000;26:113–115. doi: 10.1002/(sici)1526-968x(200002)26:2<113::aid-gene3>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Wang J, Okamoto Y, Morishita J, Tsuboi K, Miyatake A, Ueda N. Functional analysis of the purified anandamide-generating phospholipase D as a member of the metallo-β-lactamase family. J Biol Chem. 2006;281:12325–12335. doi: 10.1074/jbc.M512359200. [DOI] [PubMed] [Google Scholar]
- Wettschureck N, van der Stelt M, Tsubokawa H, Krestel H, Moers A, Petrosino S, Schutz G, Di Marzo V, Offermanns S. Forebrain-specific inactivation of Gq/G11 family G proteins results in age-dependent epilepsy and impaired endocannabinoid formation. Mol Cell Biolformation Mol Cell Biol. 2006;26:5888–5894. doi: 10.1128/MCB.00397-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.