

Abstract
Cardiac amyloidosis can result from any of the systemic amyloidoses. The disease is often characterized by a restrictive cardiomyopathy although the particular signs and symptoms depend in part on the underlying cause. In addition to managing the symptoms of heart failure, treatment options vary depending on the etiology of amyloid deposition. It is therefore critical to identify the cause of cardiac amyloidosis before initiating definitive therapy. We present a patient with presumed immunoglobulin (AL) amyloidosis who had a circulating lambda monoclonal protein, but a bone marrow biopsy with kappa predominant plasma cells. This unusual finding called into question the diagnosis of AL amyloidosis and highlights the importance and difficulty of determining the cause of cardiac amyloid deposition before initiating treatment. We review the different forms of cardiac amyloidosis and propose a diagnostic algorithm to help identify the etiology of cardiac amyloid deposition before beginning therapy.
KEY WORDS: cardiac amyloidosis, AL amyloidosis, familial amyloidosis, restrictive cardiomyopathy
INTRODUCTION
Cardiac amyloidosis can result from any of the systemic amyloidoses. The disease is often characterized by a restrictive cardiomyopathy, although the particular signs and symptoms depend in part on the underlying cause. The treatment of cardiac amyloidosis depends on the underlying etiology. As therapies include conservative management, chemotherapy, autologous stem cell transplantation, and solid organ transplantation, identifying the cause of amyloid deposition is critical to determining a treatment regimen. However, the diagnosis of the type of cardiac amyloidosis is not always straightforward. We present here an unusual case of cardiac amyloidosis that highlights the potential difficulties in determining the cause of amyloid deposition before initiating therapy.
CASE DESCRIPTION
A 65-year-old Caucasian male with a history of hypertension and coronary artery disease was transferred from an outside hospital with shortness of breath, orthostatic hypotension, and lower extremity edema.
His shortness of breath began approximately 8 months earlier with dyspnea on exertion. His symptoms gradually progressed and he began to have frequent syncopal episodes. During one of these episodes he was found to be bradycardic, prompting insertion of a dual chamber pacemaker at the outside hospital. He continued to have syncopal episodes despite pacemaker placement and was readmitted to the outside hospital after a fall for further evaluation. During this admission he suffered an asystolic arrest. Spontaneous circulation returned after approximately 5 minutes of cardiopulmonary resuscitation. He was subsequently transferred to our hospital for further management.
On arrival to our hospital he complained of mild dyspnea at rest and marked dyspnea on exertion. He described stable 3 pillow orthopnea for the last 2 months. He denied any cough or chest discomfort. He had not noted any palpitations, even in the setting of his prior syncopal episodes. He did complain of mild diffuse abdominal pain that had worsened over a several-day period.
On physical examination he was ill-appearing, afebrile, with a blood pressure of 106/62 and heart rate of 68. He was breathing at a rate of 27 breaths per minute and his oxygen saturation was 94% on 3 L of oxygen by nasal cannula. His jugular venous pressure was markedly elevated at 18 cm with a rapid y descent and a positive hepatojugular reflux. Cardiac exam revealed a regular rate and rhythm with an audible third heart sound. On lung exam, he had bilateral rales at the bases. His abdomen was tense, distended, and tender diffusely to palpation. A prominent fluid wave was appreciated. He had 3+ edema in the lower extremities to the thigh as well as 3+ sacral and scrotal edema.
Laboratory values on admission are shown in Table 1. Urinalysis showed 3+ protein. Electrocardiogram revealed first-degree AV block, low voltage QRS complexes in all leads, and evidence of an inferior infarct of unknown age (see Fig. 1).
Table 1.
Admission laboratory values
| Laboratory test | Normal values | Admission laboratory values |
|---|---|---|
| Sodium (meq/L) | 135–148 | 136 |
| Potassium (meq/L) | 3.5–5.0 | 4.2 |
| Chloride (meq/L) | 96–109 | 101 |
| BUN (mg/dL) | 7–22 | 46 |
| Creatinine (mg/dL) | 0.6–1.3 | 1.7 |
| Glucose (mg/dL) | 60–109 | 115 |
| Calcium (mg/dL) | 8.4–10.5 | 9.1 |
| Total Protein (mg/dL) | 6.0–8.2 | 6.7 |
| Albumin (mg/dL) | 3.5–5.3 | 3.4 |
| Total bilirubin (mg/dL) | 0.1–1.2 | 1.0 |
| Alanine aminotransferase (units/L) | 0–40 | 19 |
| Aspartate aminotransferase (units/L) | 0–37 | 27 |
| Alkaline phosphatase (units/L) | 30–120 | 60 |
| Bicarbonate (meq/L) | 21–31 | 23 |
| Troponin I (ng/dL) | 0–0.50 | 0.16 |
| Creatine kinase (units/L) | 24–195 | 48 |
| Creatine kinase–MB fraction (ug/L) | 0–7 | 3 |
| White blood cell count (cells/mm3) | 4,500–11,500 | 3,670 |
| Hemoglobin (g/dL) | 13.9–16.3 | 10.1 |
| Packed cell volume (%) | 41–53 | 30.1 |
| Platelet count (K cells/mm3) | 150–350 | 133 |
| Mean corpuscular volume (fL) | 80–100 | 98 |
| Red cell distribution width (%) | 11.5–14.5 | 25.5 |
Figure 1.
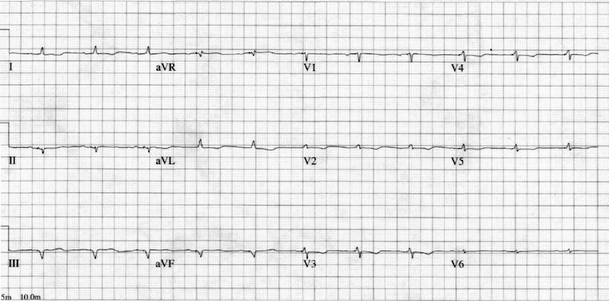
Electrocardiogram reveals low-voltage QRS complexes in all leads with evidence of an inferior myocardial infarction of unknown age.
Twenty-four-hour urine protein collection revealed nephrotic range proteinuria of 5.95 g. Serum protein electrophoresis demonstrated a spike of 1.8 g/dL in the gamma globulin region. Immunofixation revealed an IgG lambda monoclonal gammopathy. Urine protein electrophoresis showed a band of restricted mobility in the IgG lane with a corresponding band in the lambda lane. Immunofixation confirmed the presence of a monoclonal IgG lambda protein.
Two-dimensional transthoracic echocardiogram performed on hospital day 2 revealed a small left ventricular cavity with evidence of severe left and right ventricular hypertrophy. The left ventricular ejection fraction was 55%. Both the left and right atria were moderately to severely dilated. Four-chamber views of the ventricles and atria suggested the possibility of a restrictive cardiomyopathy. There were no reported valvular abnormalities. Right ventricular systolic pressure was estimated at 38 mmHg (normal <35).
A subcutaneous fat pad biopsy to look for amyloid deposition was performed on the third hospital day and was nondiagnostic.
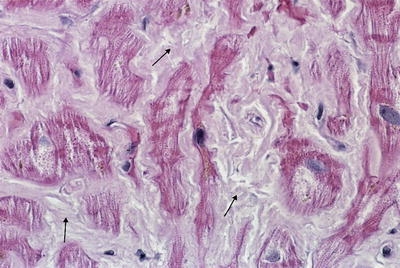
Bone marrow biopsy was performed on the fifth hospital day and was notable for an atypical plasma cell infiltrate accounting for 20–25% of the total cellularity. The plasma cells stained positive for CD138 and demonstrated kappa immunoglobulin and light chain restriction (see Fig. 2). Several of the medium-sized vessels were noted to have abnormally thickened walls with intramural deposition of a homogeneous eosinophilic material that demonstrated green birefringence when stained with Congo Red under polarized light.
Figure 2.

Bone marrow biopsy demonstrates multiple plasma cells with kappa immunoglobulin and light chain restriction (small arrows) (magnification ×160; immunoperoxidase stain for kappa light chain).
Right heart catheterization with endomyocardial biopsy was performed on hospital day number 7 (see Table 2). Endomyocardial biopsies demonstrated abundant interstitial, subendocardial, and small artery media deposits of amyloid (see Fig. 3), which were confirmed on Thioflavin T staining.
Table 2.
Right heart catheterization results
| Measure | Normal Range | Our patient |
|---|---|---|
| Cardiac index (L/min/BSA) | 2.5–3.6 | 1.8 |
| RA mean (mmHg) | 0–6 | 12 |
| Right ventricular systolic pressure (mmHg) | 15–30 | 32 |
| Right ventricular diastolic pressure (mmHg) | 0–6 | 12 |
| Pulmonary artery systolic pressure (mmHg) | 15–30 | 36 |
| Pulmonary artery diastolic pressure (mmHg) | 5–12 | 20 |
| Pulmonary artery mean pressure (mmHg) | 10–15 | 26 |
| Mean pulmonary capillary wedge pressure (mmHg) | 5–12 | 19 |
| Pulmonary vascular resistance (dyne × s)/cm5 | 120–250 | 160 |
| Systemic vascular resistance index (dyne × s)/cm5/BSA | 800–1,200 | 3,405 |
Figure 3.
Endomyocardial biopsy shows abundant interstitial, subendocardial, and small artery media deposits of an eosinophilic substance (small arrows), which was later confirmed to be amyloid by Thioflavin T staining (magnification ×160; hematoxylin and eosin stain).
The fact that the monoclonal protein in the serum was IgG lambda, but the bone marrow biopsy revealed a predominance of kappa restricted plasma cells raised the possibility that our patient had a monoclonal gammopathy of undetermined significance in addition to a nonsecretory form of AL amyloidosis. The possibility of a hereditary form of amyloidosis was also considered. However, given the advanced state of his cardiac decompensation, further diagnostic testing was deferred in accordance with the wishes of the patient and his family. He was discharged to home hospice and expired 12 weeks after discharge.
DISCUSSION
Amyloidosis refers to a number of different disease entities that share in common the extracellular deposition of insoluble fibrillar proteins in organs and tissues.1 The 4 major types of systemic amyloidosis are classified by the etiology of the underlying disease and include: Immunoglobulin (AL) amyloidosis, Familial amyloidosis (or hereditary amyloidosis), Secondary (SAA) amyloidosis (or reactive amyloidosis), and Senile systemic amyloidosis. A fifth type of systemic amyloid has been described in patients with chronic renal failure on hemodialysis.1–4 AL amyloidosis is the most common type, with an incidence of approximately 8.9 cases per million person years.5
Amyloid proteins can infiltrate a number of organ systems resulting in a variety of clinical syndromes that differ according to the type of amyloid protein and the organ system involved. Cardiac involvement is most common and most severe in AL amyloidosis, but can occur in all types of systemic amyloidosis.6 In general, patients with cardiac amyloidosis suffer from a restrictive cardiomyopathy, thought to result from replacement of normal myocardial contractile elements by infiltrative and interstitial deposits of amyloid. This process renders the myocardium firm, rubbery, and noncompliant.7 Patients commonly present with signs of right-sided heart failure, including an elevated jugular venous pressure, right-sided third heart sound, peripheral edema, and hepatomegaly.1 Amyloid deposition can also occur in the sinoatrial (SA) and atrioventricular (AV) nodes and in the bundle branches as well, leading to a variety of arrhythmias that seem to be correlated to the severity of heart failure and the abnormalities seen on echocardiogram.8
Characteristic electrocardiogram features include low voltage in the limb leads or loss of anterior forces consistent with anteroseptal infarction.5 There have been case reports of intramural coronary artery stenosis and obstruction in patients with amyloidosis and concurrent heart failure, angina, or arrhythmias,9–12 but many patients with infarct patterns on electrocardiogram do not have major underlying atherosclerotic disease. These infarct patterns may be caused by amyloid-mediated occlusion of smaller intramyocardial arteries. Atrial fibrillation and heart block are also common electrocardiogram findings.13 When present, symptomatic conduction system disease often follows the onset of congestive heart failure and may require pacemaker placement.14
A majority of patients will also have an abnormal echocardiogram at diagnosis. Major features include thickened left and right ventricular walls, abnormal myocardial texture, valvular thickening and regurgitation, atrial enlargement, and pericardial effusion.6 Echocardiogram might also reveal high left-sided filling pressures with a small or diminutive mitral A wave secondary to restricted filling and atrial infiltration.1
The treatment and prognosis of cardiac amyloidosis directly depends on the underlying etiology of amyloid deposition. However, in general calcium channel blockers are contraindicated because of case reports of worsening left ventricular function in patients with cardiac amyloidosis, thought to be related to their negative inotropic effects.15 Beta blockers are often avoided for the same reason.1 Digoxin should be used with caution because it is bound extracellularly by amyloid fibrils, which may cause patients with amyloidosis to be “hypersensitive” to its effects.16 Angiotensin converting enzyme inhibitors or other vasodilating agents are of varying utility depending on the presence or absence of comorbidities such as the nephrotic syndrome or refractory orthostatic hypotension. Pacemakers may be required depending on the degree of conduction system disease.1 Some clinicians also advocate treating patients with warfarin with a goal International Normalized Ratio of 2.0–3.0 because of the higher incidence of intrachamber thrombi seen in cardiac amyloidosis, even in the absence of atrial fibrillation.17
The prognosis for patients with AL amyloidosis is poor. Median survival is 13 months without treatment and can be extended to 17 months with cyclic oral melphalan and prednisone therapy.18,19 However, such treatment rarely results in complete remission of disease or reversal of end organ dysfunction because of amyloid deposits. Only 5% of patients survive for more than 10 years.20 Recently, high-dose intravenous melphalan has been combined with autologous peripheral blood stem cell transplantation resulting in varying degrees of hematologic response, decreased end-organ dysfunction and prolonged survival in some patients.21–23 High-dose thalidomide and dexamethasone have been tried as well, but few patients are able to tolerate the toxicity of this regimen.24 Cardiac transplantation has been used in some patients with AL amyloidosis, but long-term survival is less favorable than in patients transplanted for other conditions.25–28
Transplantation, however, is an integral part of the approach to patients with familial amyloidosis. Because the abnormal protein in familial amyloidosis is in large part produced by the liver, liver transplantation has become the most important (and only definitive) therapeutic intervention in cases of hereditary amyloidosis. Numerous case studies have documented that liver transplantation halts the neurologic progression of the disease29–31 and may even result in improvement.32 However, after liver transplantation there have been reports that cardiac involvement, as measured by progressive thickening of the left ventricle, valvular thickening, and left ventricular ejection fraction, continues to worsen in patients who have measurable cardiac disease before transplantation.3,33,34 This has prompted some centers to perform combined heart and liver transplantation in patients with cardiac involvement from familial amyloidosis.35
The congestive heart failure resulting from senile systemic amyloid may be more responsive to medical management. Olson et al. (1987)36 described a series of 5 patients with pre-mortem diagnoses of senile cardiac amyloidosis. Four of the 5 patients survived a mean of 26 months after diagnosis and improved from New York Heart Association (NYHA) class 3 functional status to NHYA class 2 with standard heart failure regimens.
Although cardiac involvement from secondary amyloidosis is not common, it has been shown to be a marker of poorer prognosis in patients with this disease. Tanaka et al. (2003)37 looked at a series of 42 patients with rheumatic disease who were found to have secondary amyloidosis. Survival at 5 years was only 31.3% in patients with cardiac involvement as opposed to 63.3% in patients without cardiac involvement. Treatment includes standard heart failure regimens as well as treatment aimed at the underlying disease process.
Considering that the ultimate prognosis and treatment directly depends on the underlying cause of the cardiac amyloid, it is imperative to identify the etiology before beginning therapy (see Fig. 4). However, it is oftentimes difficult to distinguish among the various causes of cardiac amyloidosis. Only 85% of patients with AL amyloidosis will have circulating monoclonal proteins in serum or urine. As a result, patients with a clinical picture consistent with cardiac amyloid infiltration but no evidence of a monoclonal gammopathy will require an endomyocardial biopsy to definitively distinguish between AL amyloidosis and familial or senile systemic amyloidosis.38,39 Even if a patient has circulating monoclonal proteins, the diagnosis of primary AL amyloidosis is not certain considering that up to 3% of patients over the age of 70 have a monoclonal gammopathy of undetermined significance (MGUS).40 Lachmann, et al. (2002) showed that almost 10% of patients with presumed AL amyloidosis have evidence of familial amyloidogenic mutations.41 It is even possible, although extremely rare, for a patient to have both senile systemic and AL amyloid infiltration of the heart.42
Figure 4.
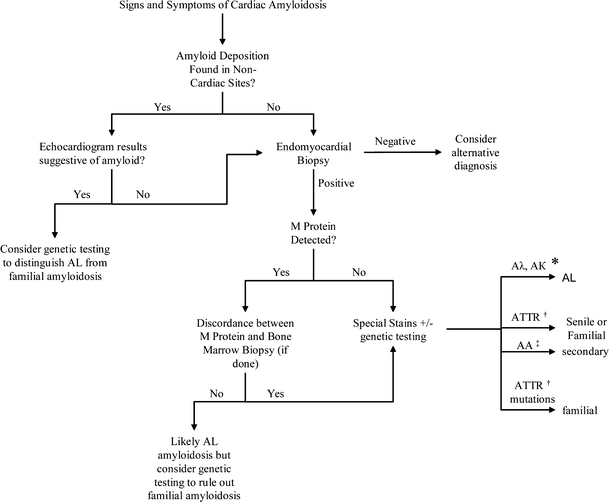
Diagnostic algorithm for determining the cause of cardiac amyloid deposition. Aλ, Aκ* indicate amyloid lambda and amyloid kappa proteins, respectively; ATTR† indicates transthyretin mutation/protein; AA‡ indicates secondary amyloid protein.
Immunologic testing of tissue samples may help to clarify the diagnosis. Strege and colleagues (1998) demonstrated in a case series of 43 patients with systemic amyloid deposition that the underlying etiology could be determined in most cases by staining tissue samples with antibodies directed against the amyloid fibril proteins AA (secondary amyloidosis), Aλ, Aκ (AL amyloidosis), transthyretin (familial amyloidosis or senile amyloidosis), and AB2M (dialysis-associated amyloidosis).42 It is also possible to use electron microscopy to search for amyloid protein in tissue samples and then to identify the specific type of protein using immunoelectron microscopy with staining for κ, λ, apolipoprotein A1, serum amyloid A (SAA), and transthyretin.43 These tests are not widely available, but may be helpful in cases where the diagnosis is in doubt.
Because familial amyloidosis is caused by an autosomal dominant mutation in the transthyretin gene, it is possible to screen for the most common mutations to help distinguish between familial and AL amyloidosis, especially in cases where there is no circulating M protein. Approximately 100 transthyretin mutations have been identified to date.44 Genetic testing has become a common part of the diagnostic algorithm at some institutions and has helped to avoid the administration of chemotherapy to patients who had previously been thought to have AL amyloidosis but actually had a familial form of the disease.41
The clinical picture of syncope, orthostatic hypotension, nephrotic range proteinuria, pseudo-infarct pattern with low voltage on EKG and restrictive pattern on echocardiography strongly suggests the diagnosis of cardiac amyloidosis. As treatment ultimately depends on the cause of amyloid deposition, it is important that clinicians consider the various types of systemic amyloidosis before intiating therapy.
Acknowledgements
The authors would like to thank Dr. Charles Wiener for his help in editing the manuscript.
Conflict of Interest Statement Dr. Garibaldi has no conflicts of interest to report. Dr. Zaas reports receiving honoraria from Pfizer.
References
- 1.Falk RH, Comenzo RL, Skinner M. Medical progress: the systemic amyloidoses. N Engl J Med. 1997;337(13):898–909. [DOI] [PubMed]
- 2.Benson, MD. Amyloidosis. In: Scriver CR, Beaudet AL, Sly WS, et al., eds. The Metabolic and Molecular Bases of Inherited Disease. Vol. 4. New York, NY: McGraw-Hill; 2001:5345–78.
- 3.Dubrey SW, Davidoff R, Skinner M, Bergethon P, Lewis D, Falk RH. Progression of ventricular wall thickening after liver transplantation for familial amyloidosis. Transplantation. 1997;64(1):74–80. [DOI] [PubMed]
- 4.Westermark P, Sletten K, Johansson B, Cornwell GG 3rd. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci U S A. 1990;87(7):2843–5. [DOI] [PMC free article] [PubMed]
- 5.Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmstead County, Minnesota, 1950 through 1989. Blood. 1992;336:466–73. [PubMed]
- 6.Kyle RA. Amyloidosis. Circulation. 1995;91:1269–71. [DOI] [PubMed]
- 7.Kushwaha SS, Fallon JT, Fuster V. Medical progress: restrictive cardiomyopathy. N Eng J Med. 1997;336(4):267–76. [DOI] [PubMed]
- 8.Falk RH, Rubinow A, Cohen AS. Cardiac arrhythmias in systemic amyloidosis: correlation with echocardiographic abnormalities. J Am Coll Cardiol. 1984;3(1):107–13. [DOI] [PubMed]
- 9.Yamano S, Motomiya K, Akai Y, et al. Primary systemic amyloidosis presenting as angina pectoris due to intramyocardial coronary artery involvement: a case report. Heart Vessels. 2002;16(4):157–60. [DOI] [PubMed]
- 10.Mueller PS, Edwards WD, Gertz MA. Symptomatic ischemic heart disease resulting from obstructive intramural coronary amyloidosis. Am J Med. 2000;109(3):181–8. [DOI] [PubMed]
- 11.Smith RL, Hutchins GM. Ischemic heart disease secondary to amyloidosis of intramyocardial arteries. Am J Cardiol. 1979;44(3):413–7. [DOI] [PubMed]
- 12.Hongo M, Yamamoto H, Kohda T, et al. Comparison of electrocardiographic findings in patients with AL (Primary) amyloidosis and in familial amyloid polyneuropathy and anginal pain and their relation to histopathologic findings. Am J Cardiol. 2000;85:849–53. [DOI] [PubMed]
- 13.Kyle RA, Gertz MA. Primary systemic amyloidosis, clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45–59. [PubMed]
- 14.Mathew V, Olson LJ, Gertz MA, et al. Symptomatic conduction system disease in cardiac amyloidosis. Am J Cardiol. 1997;80:1491–2. [DOI] [PubMed]
- 15.Gertz MA, Falk RH, Skinner M, et al. Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents. Am J Cardiol. 1985;55:1645. [DOI] [PubMed]
- 16.Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation. 1981;63(6):1285–8. [DOI] [PubMed]
- 17.Pellikka PA, Holmes DR, Edwards WD, et al. Endomyocardial biopsy in 30 patients with primary amyloidosis and suspected cardiac involvement. Arch Int Med. 1988;148:662–6. [PubMed]
- 18.Skinner M, Anderson JJ, Simms R, et al. Treatment of 100 patients with primary amyloidosis: a randomized trial of melphalan, prednisone, and colchicine versus colchicine only. Am J Med. 1996;100(3):290–8. [DOI] [PubMed]
- 19.Kyle RA, Gertz MA, Greipp PR, et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med. 1997;336(17):1202–7. [DOI] [PubMed]
- 20.Kyle RA, Gertz MA, Greipp PR, et al. Long-term survival (10 years or more) in 30 patients with primary amyloidosis. Blood. 1999;93(3):1062–6. [PubMed]
- 21.Sanchorawala V, Wright DG, Seldin DC, et al. An overview of the use of high-dose melphalan with autologous stem cell transplantation for the treatment of AL amyloidosis. Bone Marrow Transplant. 2001;28(7):637–42. [DOI] [PubMed]
- 22.Skinner M, Sanchorawala V, Seldin DC, et al. High-Dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: An 8-year study. Ann Intern Med. 2004;140:85–93. [DOI] [PubMed]
- 23.Gertz MA, Lacy MQ, Dispenzieri A, et al. Stem cell transplantation for the management of primary systemic amyloidosis. Am J Med. 2002;113(7):549–55. [DOI] [PubMed]
- 24.Palladini G, Perfetti V, Perlini S, et al. The combination of thalidomide and intermediate-dose dexamethasone is an effective but toxic treatment for patients with primary amyloidosis (AL). Blood. 2005;105:2949–51. [DOI] [PubMed]
- 25.Hosenpud JD, Uretsky BF, Griffith BP, et al. Successful intermediate-term outcomes for patients with cardiac amyloidosis undergoing heart transplantation: results of a multicenter survey. J Heart Transplant. 1990;9(4):346–50. [PubMed]
- 26.Pelosi F Jr, Capehart J, Roberts WC. Effectiveness of cardiac transplantation for primary (AL) cardiac amyloidosis. Am J Cardiol. 1997;79(4):532–5. [DOI] [PubMed]
- 27.Hosenpud JD, DeMarco T, Frazier OH, et al. Progression of systemic disease and reduced long-term survival in patients with cardiac amyloidosis undergoing heart transplantation. Follow-up results of a multicenter survey. Circulation. 1991;84(5 Suppl):III338–43. [PubMed]
- 28.Dubrey SW, Burke MM, Khaghani A, et al. Long term results of heart transplantation in patients with amyloid heart disease. Heart. 2001;85:202–7. [DOI] [PMC free article] [PubMed]
- 29.Kobayashi S, Morita H, Asawa T, et al. Peripheral nerve function in patients with familial amyloid polyneuropathy after liver transplantation. Amyloid: Journal of Protein Folding Disorders. 2003;10(1):17–24. [DOI] [PubMed]
- 30.Skinner M, Lewis WD, Jones LA, et al. Liver transplantation as a treatment for familial amyloidotic polyneuropathy. Ann Intern Med. 1994;120(2):133–4. [DOI] [PubMed]
- 31.Zeldenrust S, Gertz M, Uemichi T, et al. Orthotopic liver transplantation for hereditary fibrinogen amyloidosis. Transplantation. 2003;75(4):560–1. [DOI] [PubMed]
- 32.Bergethon PR, Sabin TD, Lewis D, et al. Improvement in the polyneuropathy associated with familial amyloid polyneuropathy after liver transplantation. Neurology. 1996;47(4):944–51. [DOI] [PubMed]
- 33.Pomfret EA, Lewis WD, Jenkins RL, et al. Effect of orthotopic liver transplantation on the progression of familial amyloidotic polyneuropathy. Transplantation. 1998;65(7):918–25. [DOI] [PubMed]
- 34.Stangou AJ, Hawkins PN, Heaton ND. Progressive cardiac amyloidosis following liver transplantation for familial amyloid polyneuropathy: implications for amyloid fibrillogenesis. Transplantation. 1998;66(2):229–33. [DOI] [PubMed]
- 35.Nardo B, Beltempo P, Bertelli R, et al. Combined heart and liver transplantation in four adults with familial amyloidosis: experience of a single center. Transplant Proc. 2004;36(3):645–7. [DOI] [PubMed]
- 36.Olson LJ, Gertz MA, Edwards WD, et al. Senile cardiac amyloidosis with myocardial dysfunction. Diagnosis by endomyocardial biopsy and immunohistochemistry. N Engl J Med. 1987;317(12):738–42. [DOI] [PubMed]
- 37.Tanaka F, Migita K, Honda S, et al. Clinical outcome and survival of secondary (AA) amyloidosis. Clin Exp Rheumatol. 2003;21(3):343–6. [PubMed]
- 38.Olson LJ, Gertz MA, Edwards WD, et al. Senile cardiac amyloidosis with myocardial dysfunction. Diagnosis by endomyocardial biopsy and immunohistochemistry. N Engl J Med. 1987;317(12):738–42. [DOI] [PubMed]
- 39.Kyle RA, Spittell PC, Gertz MA, et al. The premortem recognition of systemic senile amyloidosis with cardiac involvement. Am J Med. 1996;101:395–400. [DOI] [PubMed]
- 40.Gertz MA, Kyle RA, Edwards WD. Recognition of congestive heart failure due to senile cardiac amyloidosis. Biomed Pharmacother. 1989;43(2):101–6. [DOI] [PubMed]
- 41.Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Eng J Med. 2002;346(23):1786–91. [DOI] [PubMed]
- 42.Strege RJ, Saeger W, Linke R. Diagnosis and immunohistochemical classification of systemic amyloidoses. Report of 43 cases in an unselected autopsy series. Virchows Arch. 1998;433:19–27. [DOI] [PubMed]
- 43.Arbustini E, Verga L, Concardi M, et al. Electron and immuno-electron microscopy of abdominal fat identifies and characterizes amyloid fibrils in suspected cardiac amyloidosis. Amyloid: Journal of Protein Folding Disorders. 2002;9(2):108–14. [PubMed]
- 44.Connors LH, Lim A, Prokaeva T, et al. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid: Journal of Protein Folding Disorders. 2003;10(3):160–84. [DOI] [PubMed]