

Abstract
The Gli family of zinc-finger transcription factors mediates Hedgehog (Hh) signaling in all vertebrates. However, their roles in ventral neural tube patterning, in particular motor neuron induction, appear to have diverged across species. For instance, cranial motor neurons are essentially lost in zebrafish detour (gli1−) mutants, whereas motor neuron development is unaffected in mouse single gli and some double gli knockouts. Interestingly, the expression of some Hh-regulated genes (ptc1, net1a, gli1) is mostly unaffected in the detour mutant hindbrain, suggesting that other Gli transcriptional activators may be involved. To better define the roles of the zebrafish gli genes in motor neuron induction and in Hh-regulated gene expression, we examined these processes in you-too (yot) mutants, which encode dominant repressor forms of Gli2 (Gli2DR), and following morpholino-mediated knockdown of gli1, gli2, and gli3 function. Motor neuron induction at all axial levels was reduced in yot (gli2DR) mutant embryos. In addition, Hh target gene expression at all axial levels except in rhombomere 4 was also reduced, suggesting an interference with the function of other Glis. Indeed, morpholino-mediated knockdown of Gli2DR protein in yot mutants led to a suppression of the defective motor neuron phenotype. However, gli2 knockdown in wild-type embryos generated no discernable motor neuron phenotype, while gli3 knockdown reduced motor neuron induction in the hindbrain and spinal cord. Significantly, gli2 or gli3 knockdown in detour (gli1−) mutants revealed roles for Gli2 and Gli3 activator functions in ptc1 expression and spinal motor neuron induction. Similarly, gli1 or gli3 knockdown in yot (gli2DR) mutants resulted in severe or complete loss of motor neurons, and of ptc1 and net1a expression, in the hindbrain and spinal cord. In addition, gli1 expression was greatly reduced in yot mutants following gli3, but not gli1, knockdown, suggesting that Gli3 activator function is specifically required for gli1 expression. These observations demonstrate that Gli activator function (encoded by gli1, gli2, and gli3) is essential for motor neuron induction and Hh-regulated gene expression in zebrafish.
Keywords: Zebrafish, Hindbrain, Motor neuron, Induction, Rhombomere, Green fluorescent protein, Gli, Transcription factor, Hedgehog, Morpholino
Introduction
The signaling pathway initiated by the hedgehog (Hh) family of secreted proteins plays an essential role in the induction and patterning of numerous cell types during invertebrate and vertebrate development (Ingham and McMahon, 2001). Furthermore, defective Hh signaling has been implicated in tumorigenesis in humans (Wechsler-Reya and Scott, 2001), leading to extensive interest in elucidating the molecular mechanisms underlying Hh-mediated signal transduction. The Hh protein binds to a twelve-pass transmembrane protein Patched, relieving inhibition of a seven-pass transmembrane protein Smoothened and resulting in signaling into the cell. The ultimate transcriptional effectors of Hh signaling within the responding cell are the Gli family of zinc finger transcription factors (reviewed in Lum and Beachy, 2004), although Gli-independent mechanisms have also been described (Krishnan et al., 1997). Glis generally contain an N-terminal repressor domain, a zinc finger DNA-binding domain, and a C-terminal activator domain. The Drosophila gli homolog, cubitus interruptus (ci), is the best-understood member of this family. In the absence of an Hh signal, Ci is proteolytically cleaved between the zinc finger and the activator domains to generate a repressor that blocks expression of Hh target genes. Upon Hh binding, proteolysis of Ci is inhibited, and the full-length protein functions as a transcriptional activator of Hh target genes (Aza-Blanc et al., 1997; reviewed in Lum and Beachy, 2004). The situation in vertebrates is considerably more complex because the function of ci has been expanded and distributed between at least three Gli genes. In the mouse spinal cord, the cellular response to Hh signaling is mediated primarily by the activator function of Gli2 and the repressor function of Gli3, while the activator forms of Gli1 and Gli3 also play smaller but significant roles (Bai et al., 2004; Lei et al., 2004; Motoyama et al., 2003). Glis similarly mediate Hh signaling in the zebrafish neural tube, but Gli1 is the primary activator of Hh signaling, with Gli2 and Gli3 playing minor activator and repressor roles (Karlstrom et al., 1999, 2003; Tyurina et al., 2005).
Despite this general picture of Gli function in neural patterning, the roles of the various vertebrate Gli genes in the formation of particular cell types such as motor neurons have not been fully resolved. In frog, misexpression of Gli1 or Gli2 but not Gli3 can induce ectopic motor neurons, supporting a role for Gli1/2 in motor neuron formation (Ruiz i Altaba, 1998). In the chick spinal cord, inhibition of all Gli activator function blocks motor neuron induction (Persson et al., 2002), and ectopic expression of Gli2 activator can induce ventral neural tube markers (Lei et al., 2004), suggesting that Gli1 and/or Gli2 activator function is necessary and sufficient for motor neuron formation. In contrast, mouse Gli single and double knockouts exhibit no defects in motor neuron induction. Motor neurons are induced normally in the spinal cords of Gli1, Gli2, and Gli3 single mutants, as well as in Gli1;Gli2, and Gli1;Gli3 double mutants (Ding et al., 1998; Matise et al., 1998; Park et al., 2000). Importantly, in Gli2;Gli3 double mutants, which are essentially Gli1;Gli2;Gli3 knockouts since Gli1 expression is lost in these mutants, a small but significant number of spinal motor neurons differentiate although their migration and patterning are affected (Bai et al., 2004; Lei et al., 2004). In Smo;Gli3 double mutants, which completely lack an Hh response, relatively normal numbers of motor neurons are induced at the forelimb level, while no motor neurons differentiate more posteriorly (Wijgerde et al., 2002). These results collectively indicate that while the induction of a majority of spinal motor neurons in mouse requires Gli-dependent Hh signaling, a small number of motor neurons can also be induced by Hh- and Gli-independent processes.
We therefore tested whether Gli function is absolutely necessary for motor neuron induction in zebrafish. We have been investigating the roles of Hh pathway components in motor neuron induction in zebrafish embryos using gain- and loss-of-function approaches (Bingham et al., 2001; Chandrasekhar et al., 1998, 1999). In detour (dtr) mutants (Brand et al., 1996; Karlstrom et al., 1996), induction of midbrain and hindbrain motor neurons (branchiomotor and somatomotor neurons) is completely blocked, while spinal motor neurons are induced normally (Chandrasekhar et al., 1999). Since dtr encodes the zebrafish gli1 homolog (Karlstrom et al., 2003), these results indicate that zebrafish gli1 plays different roles in motor neuron induction in the hindbrain and spinal cord. Therefore, we were particularly interested in examining the roles of zebrafish gli2 and gli3 in motor neuron development. We show here that you-too (yot) mutants (Brand et al., 1996; van Eeden et al., 1996), which encode dominant repressor (DR) forms of Gli2 (Karlstrom et al., 1999; 2003), exhibit severe loss of motor neurons in the hindbrain and spinal cord. However, the motor neuron phenotype appears to be an indirect effect of mutant Gli2DR protein on Gli activator functions in the neural tube, since we show that wild-type gli2 plays only a minor role in motor neuron induction within the spinal cord, and no discernable role in the hindbrain. In contrast, gli3 plays a more prominent role in motor neuron induction in the hindbrain and spinal cord. By combining different gli mutants with gli antisense morpholinos to inhibit the function of multiple gli genes, we demonstrate that, unlike in mouse, Gli activator function (encoded by gli1, gli2, and gli3) is absolutely required for motor neuron induction at all axial levels in the zebrafish neural tube.
Materials and methods
Animals
Maintenance of zebrafish stocks and collection and development of embryos in E3 embryo medium were carried out as described previously (Chandrasekhar et al., 1997, 1999; Bingham et al., 2002; Westerfield, 1995). Throughout the text, the developmental age of the embryos corresponds to the hours elapsed since fertilization (hours post-fertilization, hpf, at 28.5° C).
You-too (yotty17 and yotty119) and slow muscle omitted (smub641) mutants were identified on the basis of the morphology of the somites at 21 hpf (Barresi et al., 2000; van Eeden et al., 1996; Varga et al., 2001). Detour mutants (dtrte370) were identified on the basis of defects in motor neuron development or Hh target gene expression in the hindbrain following immunohistochemistry or in situ hybridization (Chandrasekhar et al., 1999). For analysis of branchiomotor neuron development, the motor neuron-expressed islet1-GFP transgene (Higashijima et al., 2000) was crossed into these mutant backgrounds. Since the islet1-GFP reporter is not suitable for counting neuronal cell bodies, islet antibody labeling was used for quantifying motor neuron populations (see below).
Immunohistochemistry, in situ hybridization, and imaging
Whole-mount immunohistochemistry was performed with the various antibodies as described previously (Bingham et al., 2002; Chandrasekhar et al., 1997). The following antibodies were used: zn5/8 (Trevarrow et al., 1990; 1:10); islet (39.4D5; Korzh et al., 1993; 1:200); 3A10 (Hatta, 1992; 1:500). For fluorescent immunolabeling (zn5 and 3A10), RITC-conjugated secondary antibody (Jackson Immunochemicals) was used.
Synthesis of the digoxygenin- and fluorescein-labeled probes and whole-mount in situ hybridization were carried out as described previously (Bingham et al., 2003; Chandrasekhar et al., 1997; Prince et al., 1998). Two-color in situs were performed essentially as described (Prince et al., 1998) with the following modifications. After deactivating the first in situ reaction with 0.1M glycine, embryos were washed several times in 0.1M glycine before the blocking and antibody incubation steps for the second in situ probe. Fast Red substrate (Sigma) was used for the second reaction, and the substrate was replaced every 45 min until the desired color intensity was reached (usually after 5–6 hours). The following in situ probes were used: fgf3 (Maves et al., 2002); gli1 (Karlstrom et al., 2003); gli2 (Karlstrom et al., 1999); krox20 (Oxtoby and Jowett, 1993); net1a (Lauderdale et al., 1997); nk2.2 (Barth and Wilson, 1995); and ptc1 (Concordet et al., 1996).
Embryos were de-yolked, mounted in glycerol, and examined with an Olympus BX60 microscope. In all comparisons, at least ten wild-type and ten mutant embryos were examined. Confocal imaging was carried out on fixed embryos mounted in 70% glycerol. Images were captured on an Olympus IX70 microscope equipped with a BioRad Radiance 2000 confocal laser system.
Vibratome sectioning
Embryos processed for in situs were removed from 70% glycerol, rehydrated in PBS, and embedded in 7% low melting point agarose. Embryos were oriented to obtain transverse sections in the hindbrain. This process was facilitated in 15 hpf embryos (Fig. 9) by processing them for two-color in situs with fgf3 (and gli2) to label the midbrain–hindbrain boundary. Since fgf3 is also expressed in rhombomere 4 (Maves et al., 2002), the fgf3 staining was developed very weakly to avoid interference with visualization of the gli2 signal in the hindbrain. 50-μm-thick slices were generated using the Vibratome Plus 1000 system. The appearance of the notochord (in caudal r4) and the otic vesicle (spanning r4–r6) was monitored in successive sections and used to assign rhombomere identities to particular sections.
Fig. 9.
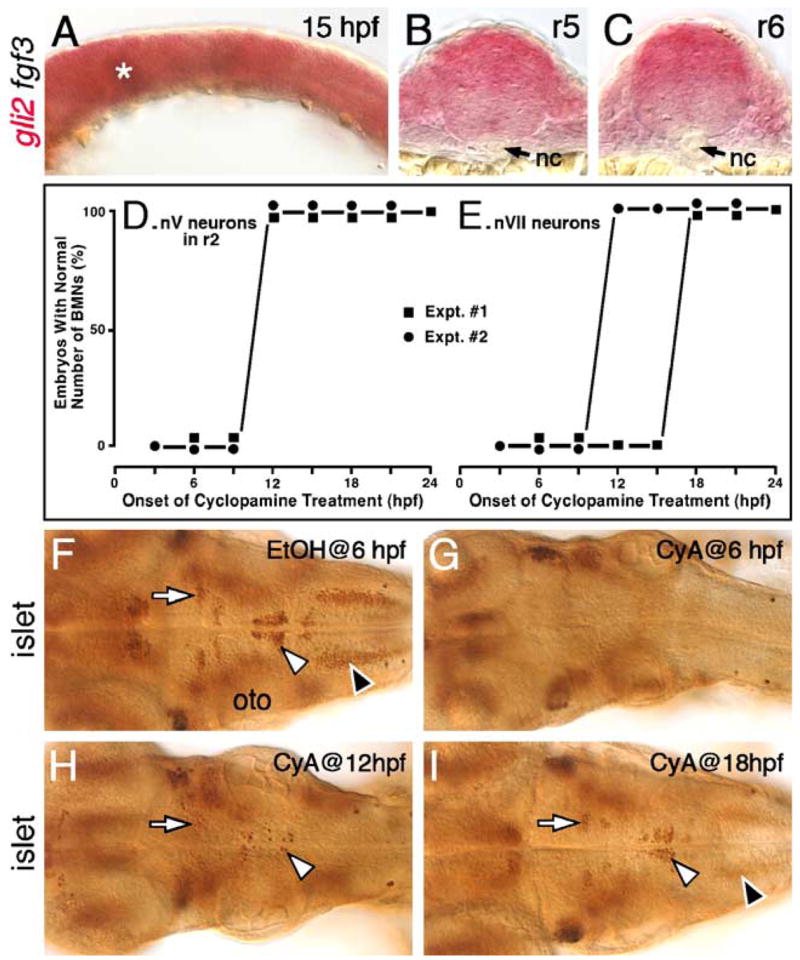
Hh signaling is required before 18 hpf for the induction of branchiomotor neurons. Panel A shows a lateral view, and panels F–I show dorsal views of the hindbrain, with anterior to the left. (A) In a 15-hpf wild-type embryo, gli2 is expressed at all axial levels throughout the dorsoventral extent of the hindbrain. The asterisk marks low level of fgf3 expression at the mid-hindbrain boundary, which was used to orient embryos for sectioning (see Materials and methods for details). (B and C) Cross-sections (dorsal is up) showing that gli2 is expressed in the ventral aspects of the neural tube in rhombomeres 5 and 6, but at lower levels than in the dorsal neural tube. nc, notochord. (D and E) Quantification of embryos with normal numbers of GFP-expressing nV motor neurons in r2 (D) and nVII neurons in r4–r7 (E) following cyclopamine (CyA) treatment beginning at the times indicated (hpf). There is no effect on nV neuron number in r2 when CyA treatment is initiated at 12 hpf or later (2 experiments; 20 embryos per experiment). There is no effect on nVII neuron number when CyA treatment is initiated at 18 hpf or later (2 experiments). (F) In a 48-hpf wild-type embryo treated with ethanol (EtOH) from 6 hpf, islet antibody labeling reveals that the number and organization of branchiomotor neurons, including nV (arrow), nVII (white arrowhead) and nX (black arrowhead), are unaffected. (G) In an embryo treated with CyA from 6 hpf, islet-labeled branchiomotor neurons are absent. (H) In an embryo treated with CyA from 12 hpf, nV neurons in r2 are mostly present (arrow; light staining), nVII neurons are greatly reduced in number (arrowhead), and nX neurons are absent. (I) In an embryo treated with CyA from 18 hpf, the nV (arrow), nVII (white arrowhead), and nX neurons (black arrowhead; out of focus) are mostly unaffected. oto, otocyst.
mRNA and antisense morpholino oligonucleotide injections
Synthesis of capped, full-length dnPKA mRNA (Ungar and Moon, 1996) was carried out as described previously (Chandrasekhar et al., 1998). Synthetic mRNAs were checked for purity and size by gel electrophoresis, estimated by u.v. spectrophotometry, and diluted to 400 ng/μl. mRNA (~1–2 ng/embryo) was injected into 1–8 cell stage embryos as described previously (Chandrasekhar et al., 1998, 1999).
Antisense morpholino oligonucleotides (MOs) against gli1, gli2, and gli3 mRNA sequences were described previously (Karlstrom et al., 2003; Tyurina et al., 2005). Working dilutions of gli1 MO (1.8 ng/nl), gli2 MO (3 ng/nl), and gli3 MO (10 ng/nl) were prepared in Danieu buffer, and morpholinos (5–30 ng/embryo) were injected into 1–4 cell stage embryos. The effective concentration for each morpholino was determined through dose–response experiments (see footnotes of Tables 3 and 4 for more details). Since injection of control (5 base mismatch) morpholinos had no effects on motor neuron development or Hh-regulated gene expression (see Supplementary Fig. S1), uninjected embryos served as controls in all experiments described here.
Table 3.
Gli2 and Gli3 contribute to spinal motor neuron induction@
| Morpholino | Number of islet-labeled cells in the hindbrain
|
Number of islet-labeled cells in ventral spinal cord# |
||
|---|---|---|---|---|
| Wild-type (gli1+)* | dtr mutant (gli1−/−)* | Wild-type (gli1+)* | dtr mutant (gli1−/−)* | |
| none | 294.8 ± 14.6 (6) | 16.7 ± 10 (6) | 22.3 ± 2.3 (6) | 20.4 ± 0.6 (6) |
| gli2 MO§ | 294 ± 25.4 (6) | 5.2 ± 2.8 (6) | 22.7 ± 2.1 (6) | 15.6 ± 1.2 (6) |
| P > 0.05 (NS) | P > 0.05 (NS) | P > 0.05 (NS) | P < 0.001 | |
| none | 287.5 ± 22 (4) | 12 ± 4.5 (4) | 35.2 ± 3.4 (4) | 38.9 ± 4.5 (4) |
| gli3 MO£ | 214.5 ± 5.3 (4) | 14.8 ± 3.3 (4) | 16.2 ± 2.6 (4) | 13.6 ± 2.9 (4) |
| P < 0.001 | P > 0.05 (NS) | P < 0.001 | P < 0.001 | |
While the numbers of control and MO-injected embryos were much higher (>40 for each treatment), we counted motor neuron cell bodies in 4–6 representative embryos for each condition. Mutant embryos were identified by the severe loss of hindbrain motor neurons.
Number of labeled cells per hemisegment.
Number of embryos scored in parenthesis.
Approximately 10 ng of gli2 MO (see Materials and methods) was injected per embryo based upon previous studies (Karlstrom et al., 2003).
Approximately 30 ng of gli3 MO was injected per embryo. In our hands, this high dose was needed to generate the ectopic fkd4 expression phenotype described for gli3 morphants (Tyurina et al., 2005). No non-specific effects were seen, and >90% of injected embryos survived and were healthy when fixed at 36 hpf.
Table 4.
Gli2DR interferes with Gli1 and Gli3 activator functions@
| Morpholino | Number of islet-labeled cells in the hindbrain
|
Number of islet-labeled cells in ventral spinal cord# |
||
|---|---|---|---|---|
| WT heterozygotes (gli2+/DR)* | yot mutant (gli2 DR/DR)* | WT heterozygotes (gli2+/DR)* | yot mutant (gli2 DR/DR)* | |
| none§ | 223 ± 5 (4) | 56.5 ± 4.4 (4) | 34 ± 2.9 (4) | 4.2 ± 1.4 (4) |
| gli1 MO£ | 93.8 ± 8.2 (4) | 2.3 ± 2.1 (4) | 25.9 ± 5.8 (4) | 0.5 ± 0.1 (4) |
| P < 0.001 | P < 0.001 | P < 0.01 | P < 0.05 | |
| none§ | 304.8 ± 23.6 (5) | 84.6 ± 22.9 (5) | 33.6 ± 2.2 (5) | 19.4 ± 0.7 (5) |
| gli3 MO† | 233 ± 33.2 (5) | 118.8 ± 32.1 (5) | 16.4 ± 2.1 (5) | 3.1 ± 1.7 (5) |
| P < 0.01 | P > 0.05 (NS) | P < 0.001 | P < 0.001 | |
While the numbers of control and MO-injected embryos were much higher (>40 for each treatment), we counted motor neuron cell bodies in 4–5 representative embryos for each condition. Embryos were genotyped by PCR.
Number of labeled cells per hemisegment.
Number of embryos scored in parenthesis.
Approximately 5 ng of gli1 morpholino (see Materials and methods) was injected per embryo. In our hands, this amount did not result in an observable loss of nk2.2 expression in wild-type embryos as described previously (Karlstrom et al., 2003), suggesting that it is a suboptimal dose. Injection of larger doses generated deformed embryos, and was not pursued further.
The numbers of hindbrain and spinal motor neurons in control wild-type and mutant embryos in the two experiments (performed about one year apart) are substantially different. We attribute these differences to variations in age of embryos, islet antibody staining intensity, and genetic background. Importantly however, the responses to gli1 and gli3 MO injections are very similar between the two experiments.
Approximately 30 ng of gli3 MO was injected per embryo. See Table 3 for details.
Genotyping of embryos
The tails of fixed embryos were clipped and stored in 70% glycerol. DNA was extracted for genotyping as described previously (Westerfield, 1995) with the following modifications: tail fragments were washed five times in 1 M Tris–HCl, pH 8.2 prior to digestion, and 20 μg of glycogen (Roche) was added to each sample prior to ethanol precipitation for 48 h at −20°C.
A restriction site (NlaIII) disrupted by the yotty119 point mutation (Karlstrom et al., 1999) served as an RFLP marker. PCR primers were designed (Fwd: 5′ ATG ATG CCT CAC GAA GTT CC 3′; Rev: 5′GGC AGA CGT GAT AGG TTC GT 3′) to introduce mutations to silence nearby, endogenous NlaIII sites unaffected by the yotty119 mutation. Undigested PCR products from wild-type and yotty119 mutant alleles were 137 base pairs (bp) in length. NlaIII treatment digested the PCR product from the yotty119 allele into 102 bp and 35 bp fragments, and the larger fragment could be distinguished from the uncut wild-type product on a 3% agarose gel.
Cyclopamine treatment
Embryos were treated with cyclopamine (Taipale et al., 2000) as described previously (Karlstrom et al., 2003) with the following modifications. Embryos were staged and synchronized at 5.25 hpf by discarding any embryos not at 50% epiboly. Cyclopamine stock (10 mM in 95% ethanol) was diluted to a working concentration of 100 μM in E3 medium containing 0.5% DMSO, and treated embryos were incubated in 12-well tissue culture plates.
Quantification of neuronal populations
Islet antibody-labeled nuclei of hindbrain motor neurons and neurons in the ventral spinal cord were counted in strongly labeled preparations. Counts were performed under 40× magnification. Spinal motor neurons were counted on one side in three contiguous segments at the level of the tip of the yolk tube. To determine whether observed differences were statistically significant, we performed one-way, parametric ANOVA analyses (with Bonferroni post-tests) using Graphpad Instat version 3 software (GraphPad Software, Inc.).
Results
Induction of cranial and spinal motor neurons is affected in you-too (gli2) mutants
In zebrafish detour (dtr (gli1−)) mutants (Brand et al., 1996; Karlstrom et al., 1996, 2003), cranial (midbrain and hindbrain) motor neurons are not induced, while spinal motor neurons are induced normally (Chandrasekhar et al., 1999), indicating that gli1 is necessary for cranial motor neuron induction, and suggesting that other gli genes may function redundantly in spinal motor neuron induction. To test this hypothesis, we examined motor neuron development and Hh-regulated gene expression in you-too (yot) mutants (Brand et al., 1996; Karlstrom et al., 1996), which encode dominant repressor forms of Gli2 (Gli2DR) that block Gli1-mediated Hh signaling in reporter assays (Karlstrom et al., 1999; 2003). The characteristic organization in a wild-type embryo of the motor neurons in the midbrain (nIII, nIV) and the branchiomotor neurons in the hindbrain (nV, nVII, nX) has been described previously (Figs. 1A and D; Chandrasekhar et al., 1997; Higashijima et al., 2000). In yotty17 mutants, midbrain motor neurons (nIII, nIV) were mostly missing, and branchiomotor neurons in the hindbrain were significantly reduced (Figs. 1B and E), consistent with the idea that Gli2DR proteins block Gli1-mediated motor neuron induction. In yotty119 mutants, branchiomotor neuron induction was even more severely affected, with the nV neurons in rhombomeres 2 and 3 (r2 and r3), and the nX neurons in the caudal hindbrain almost completely missing (Figs. 1C and F). Similarly, the zn5 antibody-labeled nVI motor neurons in r5 and r6 (Fig. 1G; Chandrasekhar et al., 1997; Trevarrow et al., 1990) were reduced in number in yotty17 mutants (Fig. 1H), and completely lost in yotty119 mutants (Fig. 1I). There were no observable differences in the patterns of neurogenesis (ngn1, ash1a, ash1b, deltaD expression) or cell death (acridine orange labeling) in the hindbrain between wild-type and yot mutant embryos (data not shown), suggesting that motor neuron induction, and not neurogenesis or cell death, is specifically affected in these mutants.
Fig. 1.
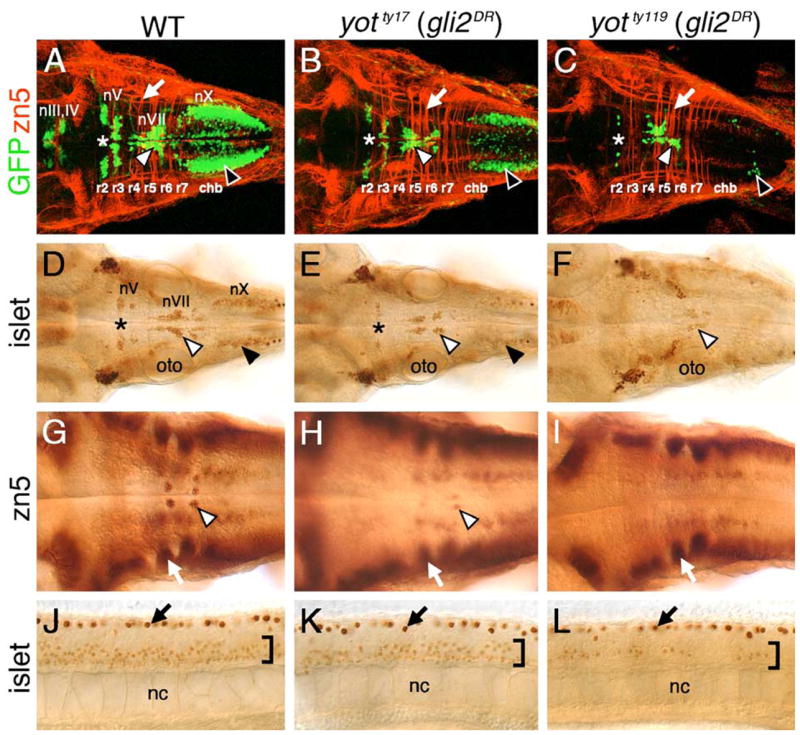
Cranial and spinal motor neuron development is affected in you-too (yot) mutants. Panels A–I show dorsal views of the hindbrain, and panels J–L show lateral views of the trunk, with anterior to the left. Asterisks in A–E indicate the location of nV motor neurons in rhombomere 2. Panels A–C are composite confocal images of embryos and identify GFP-expressing cranial motor neurons in the fluorescein channel, and zn5 antibody-labeled commissural neurons and axons at rhombomere boundaries (arrows) in the rhodamine channel. (A–C) In a 48-h post-fertilization (hpf) wild-type embryo (A), the nIII and nIV somatic motor neurons are located in the midbrain, the trigeminal motor neurons (nV; asterisk) in r2 and r3, the facial motor neurons (nVII; white arrowhead) in r5, r6, and r7, and the vagal motor neurons (nX; black arrowhead) in the caudal hindbrain (chb). The nV neurons are reduced in number in yotty17 mutants (B), and almost absent in yotty119 mutants (C), and, similarly, the nX neurons (black arrowheads) exhibit progressively severe losses in yotty17 and yotty119 mutants. While nVII neurons (white arrowheads) are also reduced in number in both mutant alleles, the reduction is less severe than those for nV and nX neurons. (D–F) At 36 hpf, the islet antibody labels hindbrain motor neurons in characteristic locations (arrowheads, asterisk; see panel A for details) in wild-type embryos (D), and these neurons are found in progressively reduced numbers in yotty17 (E) and yotty119 (F) embryos. (G) In a 48-hpf wild-type embryo, the zn5 antibody labels axons and cell bodies of the nVI abducens motor neurons in rhombomeres 5 and 6 (arrowhead), and the commissural neurons at rhombomere boundaries (arrow). (H and I) In both yot mutants, the commissural neurons (arrows) develop normally, while the nVI neurons are reduced in number in yotty17 mutants (H), and absent in yotty119 mutants (I). (J–L) At 36 hpf, the islet antibody labels motor neurons in the ventral spinal cord (bracket) in wild-type embryos (J), and these neurons are found in progressively reduced numbers in yotty17 (K) and yotty119 (L) embryos. The strongly labeled cells in the dorsal spinal cord (arrows) are Rohon-Beard sensory neurons. oto, otocyst; nc, notochord.
In contrast to dtr (gli1−) mutants, yot (gli2DR) mutants had reduced numbers of islet-expressing primary and secondary motor neurons in the spinal cord (Figs. 1K and L; data not shown), confirming that Gli function plays a role in spinal motor neuron induction. We quantified motor neuron loss in yotty17 and yotty119 mutants (36 hpf) labeled with the islet antibody and showed that these two alleles affect motor neuron induction to different degrees (Table 1). While motor neuron number was reduced ~40–50% in the hindbrain and spinal cord of yotty17 mutants, there was a ~75% loss in yotty119 mutants, suggesting that the yotty119 allele encodes a more potent repressor of motor neuron induction than does the yotty17 allele (Table 1). This is consistent with molecular identification of these alleles showing that both alleles encode truncated Gli2 proteins, with the Gli2ty119 protein missing more of its putative activator domain (Karlstrom et al., 1999). Interestingly, the number of motor neurons in r4–r7 (corresponding to nVI and nVII neurons in the wild-type embryo) was less reduced in yotty119 mutants (only nVII neurons present) than were other branchiomotor neuron populations (nV, nX) (Table 1; Figs. 1A and C). Given that nVII neurons are born in r4 and undergo tangential migration through r5 into r6 and r7 (Bingham et al., 2002), these data suggest that motor neuron induction in r4 is less severely affected than in other rhombomeres in yotty119 mutants.
Table 1.
Motor neuron induction is reduced in the hindbrain and spinal cord of you-too mutants
| Phenotype* | Number of islet antibody-labeled cells
|
||
|---|---|---|---|
| Hindbrain (nV, nVI, nVII neurons)# | Hindbrain (nVI, nVII neurons)§ | Ventral spinal cord (motor neurons)@ | |
| WT (ty17) | 201.2 ± 26.4 | 137.8 ± 18.5 | 21.8 ± 1.3 |
| you-tooty17 | 96.5 ± 14.9 | 72.3 ± 11.4 | 12.7 ± 4.0 |
| Ratio (mut/WT) | 0.48 | 0.53 | 0.58 |
| Wt (ty119) | 190.3 ± 24.9 | 132.3 ± 16.9 | 23.1 ± 1.4 |
| you-tooty119 | 45.3 ± 6.5 | 42.8 ± 7.2 | 4.8 ± 0.8 |
| Ratio (mut/WT) | 0.24 | 0.32 | 0.21 |
Six embryos were scored for each phenotype.
Total number of labeled cells in rhombomeres 2–7.
Total number of labeled cells in r4–r7 (nVI and nVII neurons in WT, nVII neurons in mutants).
Number of labeled cells per hemisegment.
Hh signaling in r4 is largely unaffected in yot mutants
To determine the extent to which Hh signaling occurs normally in r4 of yot mutants, we examined the expression of several Hh target genes in the ventral hindbrain. While nk2.2 (Barth and Wilson, 1995) was expressed without gaps along the anteroposterior (AP) axis in wild-type siblings at 21 hpf (Figs. 2A and C), its expression was almost completely eliminated in yotty119 mutants except in r4 (Figs. 2B and D). There was a slight decrease in the number of nk2.2-expressing cells in r4 of mutants (Fig. 2D), but the boundaries of expression coincided precisely and reproducibly with r4 (n = 30 embryos). Similarly, the expression of patched1 (ptc1; Concordet et al., 1996) was severely reduced throughout the mutant hindbrain except in r4 (Figs. 2E and F). Another Hh target, netrin1a (net1a; Lauderdale et al., 1997), is normally expressed throughout the ventral neural tube, with marked upregulation at rhombomere boundaries (Fig. 2G). In yotty119 mutants, however, net1a expression was reduced in many rhombomeres (especially evident in r5 and r6) with the exception of r4 and adjacent boundaries (Fig. 2H). In cross-sections, ptc1 was expressed ventrally in r4 in mutants, albeit in a smaller number of cells (Figs. 2I and J), while expression was completely missing in adjacent rhombomeres including r6 (Figs. 2K and L), thus highlighting the rhombomere-specific nature of this expression pattern in yot mutants.
Fig. 2.

Hh signaling is significantly normal in rhombomere 4 of yot mutants. Panels A, B, E–H, and M–S show lateral views of hindbrain, with anterior to the left. Panels C and D show dorsal views with anterior to the left. Panels I–L show cross sections of the hindbrain (top is dorsal) at the indicated rhombomere levels. (A–D) In 21 hpf wild-type embryos (A and C), nk2.2 is expressed (arrowhead) in the ventral neural tube at all axial levels. In yotty119 mutants (B and D), nk2.2 expression (arrowhead) is virtually absent at all axial levels, except in rhombomere 4 (r4), delineated by the expression of krox20 in r3 and r5. Within r4 (D), the nk2.2 expression domain is slightly reduced compared to wild-type. (E–H) In 21 hpf wild-type embryos, ptc1 (E) and net1a (G) are expressed (arrowheads) at all axial levels in the ventral neural tube. In yotty119 mutants, ptc1 expression (F) is greatly reduced, with significant expression retained in r4 (arrowhead). Similarly, while net1a expression is substantially reduced in the yotty119 mutant hindbrain (H), it is significantly normal in r4 (arrowhead). (I–L) In rhombomere 4 (r4), ptc1 expression (arrowheads) in the ventral neural tube is comparable between wild-type (I) and yot mutants (J). In contrast, ptc1 expression in r6 (K) (arrowhead) is greatly reduced in yot mutants (L), while its expression in paraxial mesoderm (arrows) appears normal. Asterisk, notochord. (M–O) While nk2.2 is specifically expressed in r4 of mutants (N, arrowhead), nk2.2 expression in all rhombomeres, including r4 (arrowhead), is completely absent in wild-type and mutant embryos treated with cyclopamine (O). (P) In a control 30 hpf wild-type embryo treated with EtOH from 6 hpf, net1a expression is normal, with elevated expression in r4 (arrowhead). (Q) In a wild-type embryo treated with cyclopamine (CyA) from 6 hpf, net1a expression is completely absent at all axial levels. (R) In an embryo treated from 18 hpf, net1a is mostly expressed in r4 (arrowhead). (S) In an embryo treated from 24 hpf, net1a expression is mostly normal, with prominent expression in r4 (arrowhead). Asterisks in P–S mark the anterior end of the notochord in r4.
We next tested whether residual nk2.2 and net1a expression in r4 of yotty119 mutants required Hh signaling using the alkaloid cyclopamine (CyA) to block Hh signaling at the level of the Smoothened receptor (Barresi et al., 2000). In ethanol-treated control embryos, the wild-type (n = 15) and mutant (n = 4) expression patterns were unaffected (Figs. 2M and N). Following CyA treatment beginning at 3 hpf, nk2.2 expression was completely lost along the AP axis, including r4, in wild-type and mutant embryos (Fig. 2O; n = 44), demonstrating that r4-specific expression of ventral neural tube markers in yotty119 mutants requires Hh signaling. Interestingly, blocking Hh signaling at different times had differential effects on the pattern of net1a expression, assayed at 30 hpf. While an early block (CyA treatment beginning at 15 hpf or earlier) blocked net1a expression throughout the hindbrain (Figs. 2P and Q; data not shown; n = 12), CyA treatment from 18 hpf blocked net1a expression in all rhombomeres except in r4 (Fig. 2R; n = 3), while CyA treatment from 24 hpf or later had no significant effect on net1a expression (Fig. 2S; n = 6). These observations indicate that net1a expression in r4 has different temporal requirements for Hh signaling and suggest that retention of Hh target gene expression in r4 of yot mutants may reflect the inability of the mutant Gli2 protein to block early Hh signaling in this rhombomere.
Activation of Hh signaling can alleviate defective motor neuron induction and Hh target gene expression in yot mutants
Since the yot alleles encode dominant repressors of Hh signaling (Karlstrom et al., 2003), we wondered whether ectopic activation of Hh signaling upstream of the Gli transcription factors by overexpression of dominant-negative protein kinase A (dnPKA; Ungar and Moon, 1996) could overcome the repressive effects of these mutations on Hh target gene (nk2.2, net1a) expression and motor neuron induction. When dnPKA RNA was injected into embryos from yotty119+/−;islet1-GFP crosses, there was a dramatic increase in the number of GFP-expressing branchiomotor neurons in all rhombomeres, and in ectopic locations along the dorsoventral axis, in ~80% of wild-type siblings (Figs. 3A and B; Table 2). Consistent with this, nk2.2 expression was upregulated in the ventral neural tube and at ectopic locations throughout the hindbrain in ~88% of dnPKA-injected wild-type embryos (Figs. 3E and F). In marked contrast, no ectopic GFP-expressing cells were seen in dnPKA-injected yotty119 mutants, but there was a small, reproducible increase in the number of nVII neurons, and less frequently nX neurons (Figs. 3C and D; Table 2). Furthermore, while nk2.2 was not expressed in the ventral hindbrain or in ectopic dorsal locations in dnPKA-injected yotty119 mutants, its expression in ventral r4 was slightly but reproducibly upregulated (Figs. 3G and H). Interestingly, when dnPKA RNA was injected into embryos from yotty17+/−; islet1-GFP crosses, net1a was expressed ectopically in the dorsal neural tube at all axial levels in wild-type and yotty17 mutants, but more weakly in mutants (Figs. 3I–L; Table 2). Similarly, ectopic GFP-expressing motor neurons and nk2.2-expressing cells were found in r2 and r4 of dnPKA-injected yotty17 mutants (data not shown). These results suggest that while both yot alleles encode dominant repressors, their inhibitory activities can be overcome to different extents by hyperactivation of Hh signaling, with the ty119 allele being more resistant to this treatment. These observations are consistent with our earlier results that the yot alleles represent a phenotypic series, with the ty119 allele encoding a stronger repressor of motor neuron induction (Table 1), in accordance with reporter assays (Karlstrom et al., 2003).
Fig. 3.
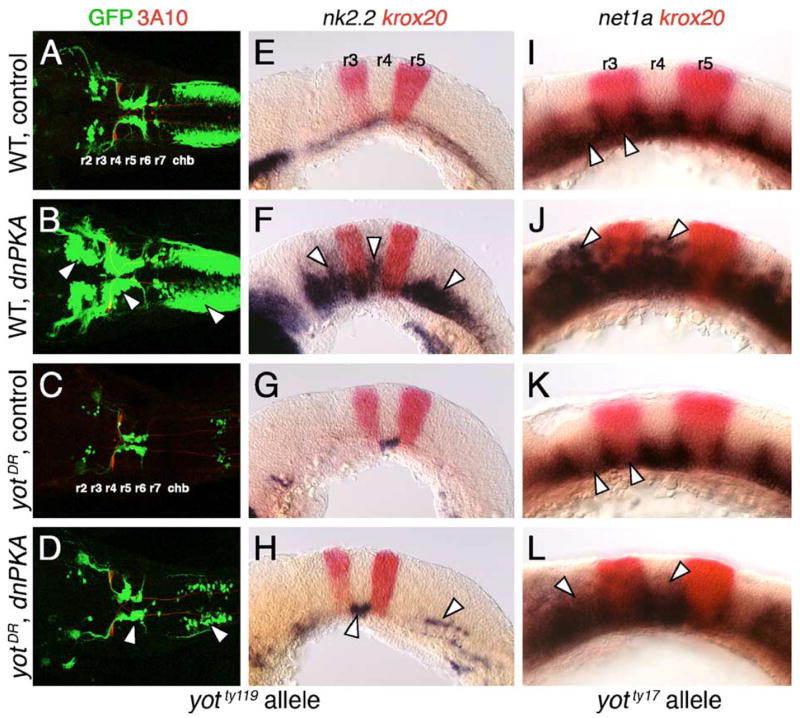
Hh-regulated events in yotty119 mutants are resistant to ectopic Hh pathway activation. Anterior is to left in all panels. Panels A–D show dorsal views, and panels E–L show lateral views. A–D are composite confocal images of embryos, and identify GFP-expressing branchiomotor neurons (green), and 3A10 antibody-labeled Mauthner neurons and axons in rhombomere 4 (red). (A and E) In control wild-type embryos, nk2.2 is expressed ventrally at all axial levels at 21 hpf (E), and branchiomotor neurons are found at their characteristic locations at 36 hpf (A). (B and F) In dnPKA RNA-injected wild-type embryos, nk2.2 is expressed at high levels, and at ectopic locations (arrowheads) in all rhombomeres (F). Branchiomotor neurons are also greatly increased in number (arrowheads), and found at ectopic locations at all axial levels (B). (C and G) In control yotty119 mutants, nk2.2 expression is missing throughout the hindbrain, except within r4 (G), and, concomitantly, there is a severe loss of branchiomotor neurons at all axial levels, except nVII neurons, which originate in r4 (C). (D and H) In dnPKA RNA-injected yotty119 mutants, nk2.2 expression is increased within its normal ventral domain in r4, and slightly within the caudal hindbrain (H, arrowheads). There is a corresponding increase in the number of branchiomotor neurons originating in r4 and the caudal hindbrain (arrowheads), but there is no effect on motor neurons in other rhombomeres (D). (I–L) In a control 21 hpf wild-type embryo (I), net1a is expressed ventrally at all axial levels (arrowheads), with dorsal expansion at rhombomere boundaries. In a dnPKA RNA-injected wild-type embryo (J), net1a expression is expanded dorsally (arrowheads) at all axial levels. In a control yotty17 mutant (K), net1a expression is reduced in all rhombomeres except r4 (arrowheads; compare to I). In a dnPKA RNA-injected yotty17 mutant (L), net1a expression is expanded dorsally in all rhombomeres including r2 and r4 (arrowheads).
Table 2.
Effects of ectopic Hh pathway activation on hindbrain gene expression in yot mutants
| yot allele (marker gene) | Injected RNA | Number of Embryos | Fraction of embryos with ectopic marker expression
|
|
|---|---|---|---|---|
| Wild-type (+/+ and +/−)# | Mutant (−/−)# | |||
| you-tooty17(netrin1a expression at21 hpf) | none | 137 | 0% (0/97) | 0% (0/40) |
| dnPKA* | 128 | 91% (84/92) | 81% (29/36) | |
| you-tooty119(nk2.2 expression at21 hpf) | none | 32 | 0% (20/20) | 0% (12/12) |
| dnPKA* | 60 | 88% (38/43) | 0% (0/17)@ | |
| you-tooty119 (islet1-GFP expression at36 hpf) | none | 27 | 0% (20/20) | 0% (7/7) |
| dnPKA* | 52 | 79% (30/38) | 0% (0/14)@ | |
While dnPKA- injected yot mutants could be readily identified after in situ hybridization, roughly half of the embryos in each experiment was genotyped by PCR. Since there was perfect correlation between PCR genotyping and morphological identification, the data from embryos scored by the two methods were pooled.
Approximately 1 ng of dnPKA RNA (see Materials and methods) was injected per embryo, generating the Hh overexpression phenotype in 80–90% of injected wild-type embryos (Chandrasekhar et al., 1999).
Although no nk2.2-or GFP- expressing cells were found at ectopic locations in the yotty119 mutant hindbrain following dnPKA overexpression, expressing cells were found in the caudal hindbrain of several embryos, where nX neurons are normally found (see Figs. 3D, H). In addition, the number of GFP-expressing nVII neurons was markedly increased, and nk2.2 expression in ventral r4 was consistently upregulated in many of these embryos.
Hh signaling is essential for branchiomotor neuron induction
Given that the yot (gli2DR) mutations affect branchiomotor neuron number, and that Hh signaling may regulate Gli2 activator and repressor functions (Stamataki et al., 2005), we determined the overall requirement for Hh signaling in motor neuron induction, and compared it to the requirement for Gli1, which possesses only activator function (Karlstrom et al., 2003; Tyurina et al., 2005). To do this, we examined branchiomotor neuron induction and Hh-regulated gene expression in slow muscle omitted (smub641) mutants where Hh signaling is blocked due to a loss-of-function mutation in smoothened (smo) (Barresi et al., 2000; Chen et al., 2001; Varga et al., 2001), and in detour (dtrte370) mutants, which carry a loss-of-function mutation in gli1 (Karlstrom et al., 2003).
In smu (smo−) and dtr (gli1−) mutants, the motor neurons in the midbrain (nIII, nIV) and the branchiomotor neurons in the hindbrain (nV, nVII, nX) were almost completely missing (Figs. 4B and C; compare to Fig. 4A), demonstrating that the functions of the Smo receptor and Gli1 transcription factor are essential for their induction (see also Chandrasekhar et al., 1999). Consistent with this, the expression of several Hh-regulated genes in the hindbrain (Figs. 4D, G, J, M) was greatly reduced or completely lost in smu mutants (Figs. 4E, H, K, N; see also Chen et al., 2001; residual net1a and gli1 expression likely due to maternally supplied Smo function). While nk2.2 expression was similarly absent in dtr mutants (Fig. 4F), significant levels of ptc1 and net1a expression were retained (Figs. 4I and L), indicating that some aspects of their expression do not require gli1 function. Furthermore, gli1 expression, which is greatly reduced in smu mutants, was essentially unaffected in dtr mutants (Fig. 4O), indicating that gli1 expression depends on Hh signaling but does not require Gli1 function. These results demonstrate that some Hh-regulated events in the hindbrain (motor neuron induction, and expression of ventral neural tube (nk2.2) and motor neuron (isl1, tag1) markers (data not shown)) require gli1 function, while others (expression of ptc1, net1a, gli1) do not, suggesting that other Glis (encoded by gli2 and/or gli3) may activate some Hh target genes in the hindbrain (Fig. 11; Tyurina et al., 2005). Strikingly, most Hh-regulated events including motor neuron induction occur normally in the dtr mutant spinal cord (Chandrasekhar et al., 1999; Karlstrom et al., 2003; data not shown; see Table 3), suggesting that Glis may function in a redundant fashion in the spinal cord (Fig. 11; Tyurina et al., 2005).
Fig. 4.
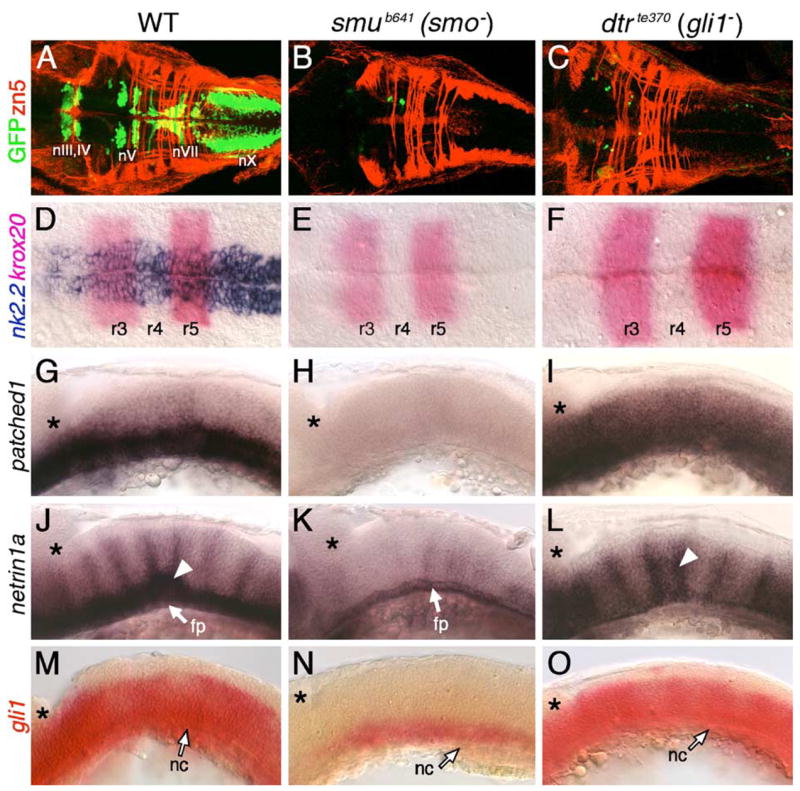
Hh signaling and Gli1 function are essential for branchiomotor neuron development. Panels A–F and G–O show dorsal and lateral views, respectively, of the hindbrain with anterior to the left. Asterisks in G–O mark the cerebellum. (A) In a 48-hpf wild-type embryo, the nIII, nIV, nV, nVII, and nX motor neurons are found in their characteristic positions. (B–C) In slow muscle-omitted (smu; B) and detour (dtr; C) mutants, GFP-expressing cranial motor neurons are almost completely missing, while the zn5-labeled axons at rhombomere boundaries are unaffected. (D) In a 21-hpf wild-type embryo, nk2.2 is expressed throughout the ventral neural tube at all axial levels. (E–F) In smu (E) and dtr (F) mutants, nk2.2 expression is lost, while krox20 expression in r3 and r5 is unaffected. (G–I) In a 24-hpf wild-type embryo (G), ptc1 is expressed in the ventral hindbrain, while it is expressed diffusely at a reduced level in dtr mutants (I), and not expressed at all in smu mutants (H). (J) In a 30-hpf wild-type embryo, net1a is expressed in the ventral hindbrain and at rhombomere boundaries (arrowhead). (K) In smu mutants, net1a expression is mostly lost, with residual expression in the floor plate (fp). (L) In dtr mutants, net1a expression in the ventral hindbrain is greatly reduced, but expression at rhombomere boundaries (arrowhead) is unaffected. (M–O) In a 21-hpf wild-type embryo (M), gli1 is expressed in the ventral two-thirds of the neural tube, while its expression is greatly reduced in smu mutants (N), and is unaffected in dtr mutants (O). The anterior tip of the notochord (nc) is at the level of r4, and is indicated by arrows.
Fig. 11.
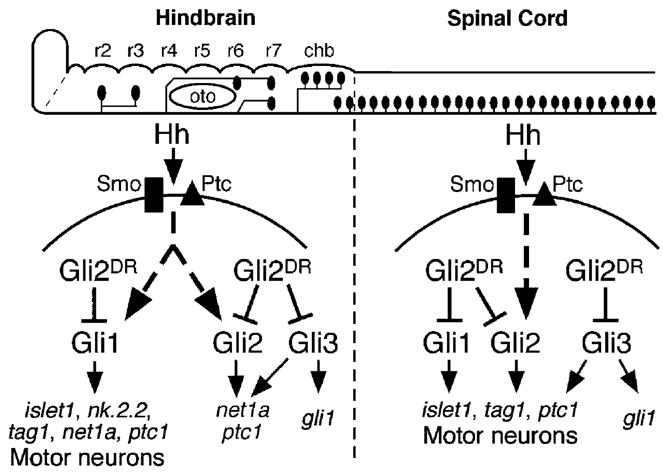
Model to explain motor neuron phenotypes of dtr (gli1−) and yot (gli2DR) mutants, and of gli1, gli2, and gli3 morpholino injection experiments. The hindbrain and spinal cord are shown schematically in lateral view (anterior is to left), with the branchiomotor and spinal motor neurons depicted as black ovals. Rhombomeres 2–7 (r2–r7) and the caudal hindbrain (chb) are indicated. The pathways depict signaling within ventral neural tube cells upon transduction of Hh signal through the Smoothened (Smo, black rectangle)-Patched (Ptc, black triangle) receptor system. In the hindbrain, Hh-mediated induction of motor neurons (and motor neuron markers like nk2.2, islet1, and tag1) is completely dependent on gli1 function. Branchiomotor neuron loss in yot mutants appears to result from the action of mutant Gli2 (Gli2DR) on Gli1 activator function. Complete activation of net1a and ptc1 expression requires activator function of Gli1, Gli2, and Gli3. In the spinal cord, Gli1, Gli2, and Gli3 activator functions contribute to spinal motor neuron induction (and islet1 and tag1 marker gene expression). The significant loss of spinal motor neurons in yot mutants appears to result from the dominant repressor effect of Gli2DR on all Gli activators. Gli1 expression in the hindbrain and spinal cord appears to specifically require Gli3 activator function.
Gli2 plays a minor role in spinal motor neuron induction
Given that Gli2 contributes to the Hh response in the zebrafish midbrain (Karlstrom et al., 2003), we tested whether gli2 plays any role in motor neuron development. To knock down gli2 function, we injected gli2 antisense morpholinos (MOs) into embryos obtained from yotty119+/−;islet1-GFP crosses, which produce on average 25% homozygous mutant yot embryos. There was no difference in the number or distribution of GFP-expressing motor neurons in the midbrain or hindbrain between control (16/16 embryos) and gli2 MO-injected (57/57 embryos) wild-type siblings at 48 hpf (Figs. 5A and B; see Table 3 for quantification using islet antibody). Similarly, there were no effects on nk2.2 expression in the ventral neural tube of 21 hpf control (43/43 embryos) and gli2 MO-injected (25/25 embryos) wild-type siblings (Figs. 5E and F). These results demonstrate that gli2 plays little or no role in branchiomotor neuron development.
Fig. 5.
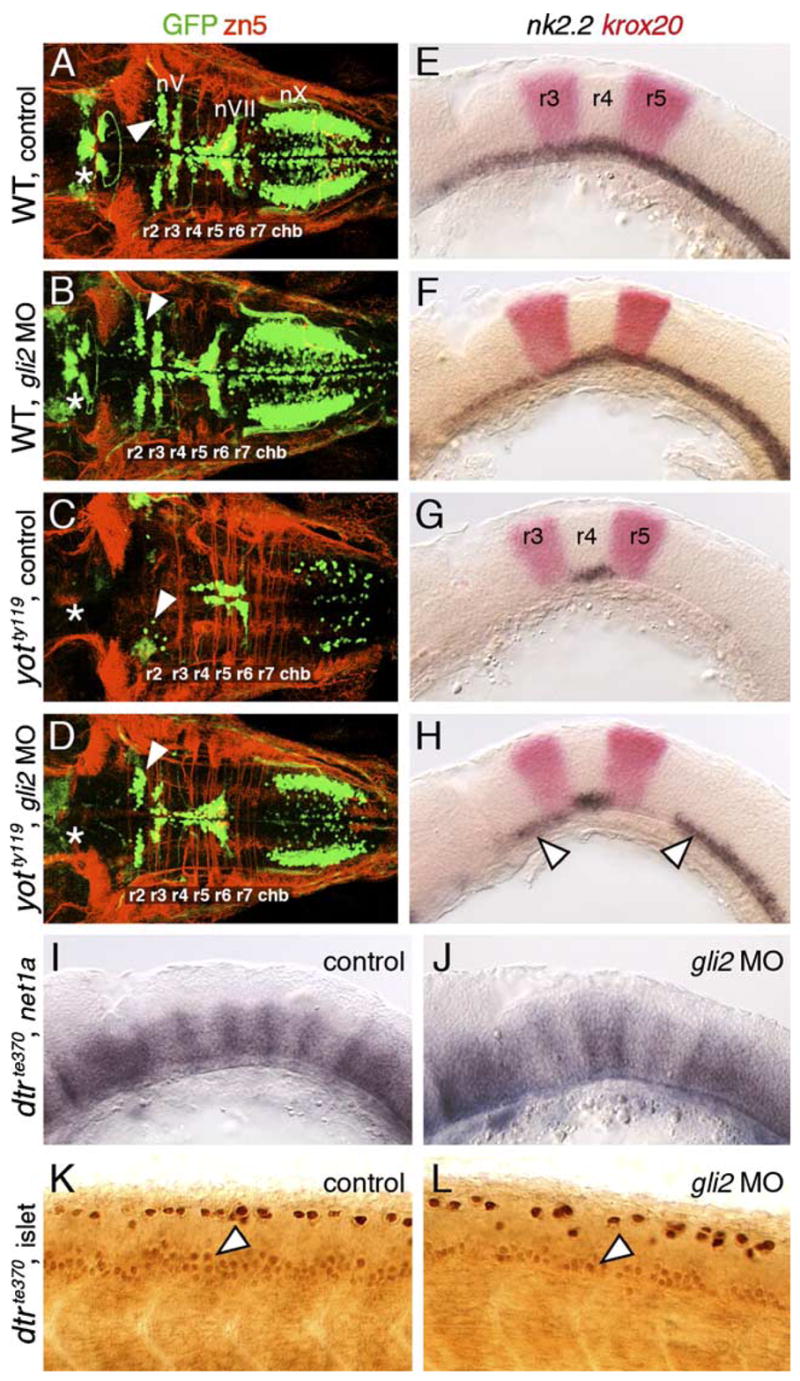
Gli2 contributes to spinal motor neuron induction. Panels A–D show dorsal views, and panels E–L show lateral views of the hindbrain (E–J) and spinal cord (K and L), with anterior to the left. Asterisks in A–D indicate the position of the nIII and nIV motor neurons in the midbrain (A and B), and their absence (C and D). Arrowheads in A–D indicate the nV neurons in r2. (A and E) In control wild-type embryos, nk2.2 expression (E) and organization of cranial motor neurons (A) are identical to those described previously. (B and F) In gli2 MO-injected wild-type embryos, expression of nk2.2 (F) and development of cranial motor neurons (B) are identical to control embryos. (C and G) In control yotty119 mutants, the patterns of loss of nk2.2 expression (G) and cranial motor neurons (C) are similar to those described previously. (D and H) In gli2 MO-injected yotty119 mutants, nk2.2 expression is significantly restored in anterior rhombomeres and the caudal hindbrain (H, arrowheads). Concomitantly, the number of nV neurons (arrowhead) and nX neurons is significantly increased (D), and the pattern looks similar to that in control wild-type embryos. (I and K) In control dtrte370 mutants, net1a is expressed at low levels in the ventral hindbrain, and at higher levels at rhombomere boundaries (I). In the mutant spinal cord (K), islet antibody-labeled motor neurons (arrowhead) are found in similar numbers to wild-type embryos. (J and L) In gli2 MO-injected dtrte370 mutants, net1a expression in the hindbrain (J) shows no significant change (loss). In contrast, the number of motor neurons in the ventral spinal cord (arrowhead) is slightly but reproducibly reduced in MO-injected mutants (L).
Strikingly, loss of GFP-expressing motor neurons in yotty119 mutants was largely rescued upon injection of gli2 MO (Fig. 5D; 19/20 embryos; compare to control: Fig. 5C; 8/8 embryos), and motor neurons were found in all rhombomeres at their characteristic locations. This result confirms that the gli2 MOs effectively block translation of gli2 mRNA. Furthermore, since no full-length Gli2 protein is present in yot mutants, the rescue of the yot mutant phenotype convincingly demonstrates that branchiomotor neurons can be specified in the absence of full length, presumably activator forms of Gli2, as suggested by the normal induction of these neurons in gli2 MO-injected wild-type embryos (Fig. 5B). Although the yot mutant phenotype is rescued, motor neuron numbers, especially evident for nX neurons, were still lower than in wild-type siblings. Furthermore, the nIII and nIV motor neurons in the midbrain never reappeared in gli2 MO-injected mutants. Consistent with these results, nk2.2 expression was restored in large regions of the ventral hindbrain, but never in the midbrain, of gli2 MO-injected yot mutants (Fig. 5H; 8/8 embryos; control, Fig. 5G: 11/11 embryos). Therefore, the incomplete rescue of the mutant phenotype in the midbrain and hindbrain of gli2 MO-injected yot mutants may reflect substantial, but not complete, knockdown of Gli2DR synthesis.
If Hh-regulated events could be mediated redundantly by activator functions of Gli1, Gli2, and Gli3 (see model in Fig. 11), the lack of a phenotype in gli2 MO-injected wild-type embryos may merely reflect the relative contributions of the various Glis in mediating specific events. In this case, we would predict that minor roles for the Gli2 activator would be discernable in the absence of functional Gli1. We therefore examined Hh-regulated events in the hindbrain and spinal cord of detour (gli1−) mutants following gli2 MO injection. The number of islet antibody-labeled motor neurons in the spinal cord was reproducibly and significantly reduced by ~25% in gli2 MO-injected dtr mutants (Fig. 5L; Table 3) compared to those in uninjected and gli2 MO-injected wild-type embryos, and uninjected dtr mutants (Fig. 5K; Table 3). Furthermore, the residual, but substantial, expression of net1a in the dtr mutant hindbrain (Figs. 1L and 5I; n = 15 embryos) was either slightly reduced or unaffected following gli2 MO injection (Fig. 5J; 21/21 embryos), and net1a expression was unaffected in wild-type siblings injected with gli2 MO (n = 40) as seen for nk2.2 (Figs. 5E and F). These observations suggest strongly that although gli2 appears to play no role in motor neuron induction or Hh target gene activation in the hindbrain, Gli2 activator function is needed for inducing some spinal motor neurons.
Dominant repressor Gli2 blocks motor neuron induction by interfering with Gli1 function
If motor neuron loss in yot mutants were a consequence of Gli2DR-mediated interference with Gli1 activator function in hindbrain, and with Gli1 and other Gli activator functions in the spinal cord (see Fig. 11), one would predict that the yot (gli2DR) mutant motor neuron phenotype would be exacerbated upon reduction of Gli1 activator function. We tested this by injecting gli1 MO into embryos obtained from yotty119+/− crosses, and examining motor neuron development with islet antibody labeling (Fig. 6; Table 4). In wild-type (gli2+/+) embryos, injection of a sub-optimal amount of gli1 MO (Karlstrom et al., 2003) did not result in a discernable loss of motor neurons in the hindbrain or spinal cord, and the embryos looked identical to control gli2+/+ embryos (Figs. 6A and I; data not shown; Table 4). In contrast, gli1 MO injection into gli2 heterozygotes (gli2+/DR) led to a substantial loss of hindbrain motor neurons (Fig. 6B), with the pattern of loss greatly resembling that found in control yot (gli2 DR/DR) mutants (Fig. 6C). However, unlike in the hindbrain, spinal motor neuron loss was less severe in gli1 MO-injected gli2 heterozygotes (Fig. 6J) compared to yot mutants (Fig. 6K; Table 4). Finally, as predicted, gli1 MO injection into yot mutants led to the complete loss of motor neurons in the hindbrain and spinal cord (Figs. 6D and L; Table 4), similar to the phenotype of smu mutants and cyclopamine-treated embryos (data not shown). These results suggest strongly that the Gli2DR protein affects motor neuron development by interfering with the activator functions of Gli1, Gli2, and possibly other Glis (Fig. 11).
Fig. 6.

Gli1 contributes to spinal motor neuron induction. Panels A–D show dorsal views, and panels E–L show lateral views, with anterior to the left. (A, E, and I) In an uninjected 36 hpf wild-type (yot+/+ or +/−) embryo, islet antibody labeling (A) reveals the characteristic organization of the nV neurons in r2 (asterisk), the nVII neurons (white arrowhead), the nX neurons (black arrowhead), and the patterned expression of net1a (E) in the ventral hindbrain and at rhombomere boundaries (arrowheads). Spinal motor neurons (arrowhead, I) are found in characteristic numbers. (B, F, and J) In a yotty119+/− heterozygote injected with gli1 MO, nV and nVII neurons are reduced in number (asterisk, arrowhead in B), nX neurons are absent, and net1a expression is significantly reduced in the hindbrain (arrowheads, F). Spinal motor neurons are slightly reduced in number (arrowhead, J). (C, G, and K) In an uninjected yotty119 mutant, nV (asterisk, C), nX, and spinal motor neurons (arrowhead, K) are greatly reduced in number, while the nVII neurons (arrowhead, C) show a moderate reduction. Net1a expression is significantly reduced in the hindbrain (arrowheads, G). (D, H, and L) In a yotty119 mutant injected with gli1 MO, branchiomotor (D) and spinal motor neurons (L) are completely absent. Net1a expression (arrowheads, H) is also severely reduced in the hindbrain. The strongly labeled cells in the dorsal spinal cord (I–L) are Rohon-Beard sensory neurons, which are unaffected by these treatments.
Our earlier observations that significant levels of net1a and ptc1 expression are retained in yot and dtr mutants (Figs. 2 and 4; Fig. 6G) and in gli2 MO-injected dtr mutants (Fig. 5J) suggested that Gli activator-independent mechanisms may be involved in regulating their expression. However, step-wise reduction of Gli activator function by gli1 MO injection into gli2 heterozygotes (gli2+/DR; 15/25 embryos) and yot mutants (gli2 DR/DR; 9/13 embryos) led to additive losses in net1a expression (Figs. 6F and H), suggesting strongly that Gli activator function is necessary for regulating all aspects of net1a (Fig. 6) and ptc1 (see Fig. 10) expression.
Fig. 10.
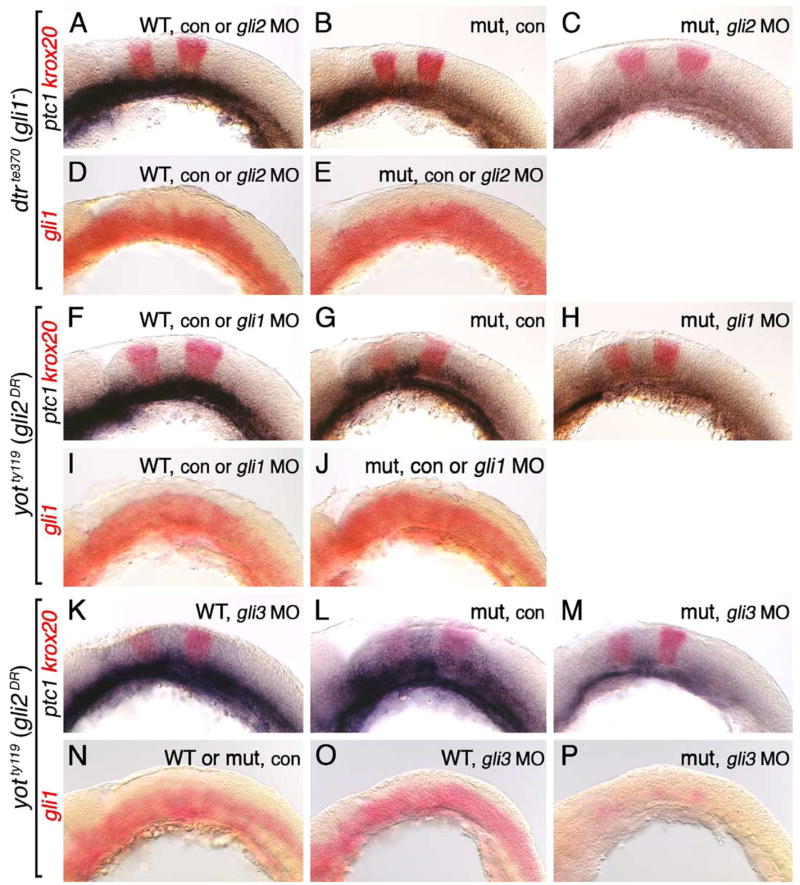
Regulation of gli1 expression requires Gli3, but not Gli1 or Gli2, function. All panels show lateral views of the hindbrain with anterior to the left. Embryos in A–E were obtained from crosses between dtrte370+/− heterozygotes, while those in F–P were obtained from crosses between yotty119+/− heterozygotes. Krox20 expression (red) in ptc1;krox20 double in situ panels identifies rhombomeres 3 and 5. (A) In 21 hpf uninjected or gli2 MO-injected wild-type embryos, ptc1 is expressed at all axial levels in the ventral hindbrain. (B) In an uninjected dtr mutant, ptc1 expression is significantly reduced. (C) In a gli2 MO-injected dtr mutant, ptc1 expression is completely lost in the hindbrain. (D) In a 21-hpf uninjected or gli2 MO-injected wild-type embryo, gli1 is expressed at all axial levels in the ventral half/two-thirds of the hindbrain. (E) Gli1 expression is unaffected in control or gli2 MO-injected dtr mutants. (F–H) Ptc1 expression is normal in uninjected or gli1 MO-injected wild-type embryos (F), is reduced significantly in an uninjected yot mutant (G), and is completely lost from the hindbrain in a gli1 MO-injected yot mutant (H). (I and J) Gli1 is expressed normally in the ventral hindbrain in uninjected or gli1 MO-injected wild-type (I) or yot mutants (J). (K–M) Ptc1 expression is slightly reduced in a gli3 MO-injected 22 hpf wild-type embryo (K), is reduced significantly in an uninjected 24 hpf yot mutant (L), and is almost completely lost in a gli3 MO-injected 22 hpf yot mutant (M). (N–P) Gli1 is expressed normally in the ventral hindbrain in uninjected wild-type or yot mutant embryos (N), and in a gli3 MO-injected wild-type embryo (O). In a gli3 MO-injected yot mutant, gli1 expression is greatly reduced (P).
Gli3 plays a role in branchiomotor and spinal motor neuron induction
Given the redundant functions of gli1 and gli2 in spinal motor neuron induction, we tested whether zebrafish gli3 (Tyurina et al., 2005) also contributed to motor neuron induction. Since the roles of gli1 and gli2 in spinal motor neuron induction were revealed only in Gli-deficient backgrounds (Figs. 5 and 6), we examined the effects of knocking down gli3 function using dtr (gli1−) and yot (gli2DR) mutants. We first verified that gli3 MO injection indeed knocked down gli3 function by showing that ~36% of wild-type embryos injected with 30 ng gli3 MO exhibited ectopic expression of the floor plate marker fkd4 in the dorsal hindbrain, as described previously (Tyurina et al., 2005; data not shown; 24/66 embryos). Interestingly, injection of gli3 MO into wild-type siblings of dtr or yot mutants led to significant reductions in both branchiomotor (Figs. 7B and J; compare to Figs. 7A and I) and spinal motor neurons (Figs. 7F and N; compare to Figs. 7E and M) (Tables 3 and 4), suggesting that gli3 plays a more prominent role than gli2 in motor neuron induction. In dtr (gli1−) mutants injected with gli3 MO, while the severe loss of branchiomotor neurons seen in control embryos was maintained (Figs. 7C and D; Table 3), spinal motor neurons were greatly reduced compared to control mutant embryos (Figs. 7G and H; Table 3). This result suggests an activator role for Gli3 in spinal motor neuron induction. In yot (gli2DR) mutants injected with gli3 MO, there was a small decrease in branchiomotor neuron number compared to control embryos (Figs. 7K and L; Table 4), but a dramatic reduction in spinal motor neuron number over control mutant embryos (Figs. 7O and P; Table 4). This result again suggests an activator function for Gli3 in spinal motor neuron induction, and that this function may be subject to interference from Gli2DR protein. In addition, Gli3 may regulate motor neuron induction indirectly by activating gli1 expression (see Figs. 10 and 11, and Discussion). These results also suggest that Gli3 does not function as a repressor of motor neuron fate in the dorsal neural tube. But we cannot rule out other roles for gli3 in hindbrain development because the fourth ventricle (above the hind-brain) was variably reduced in gli3 MO-injected embryos, and the hindbrain had a mottled appearance suggestive of increased cell death (data not shown). Expression of ptc1 and krox20 (Fig. 10K) however was unaffected in these embryos.
Fig. 7.
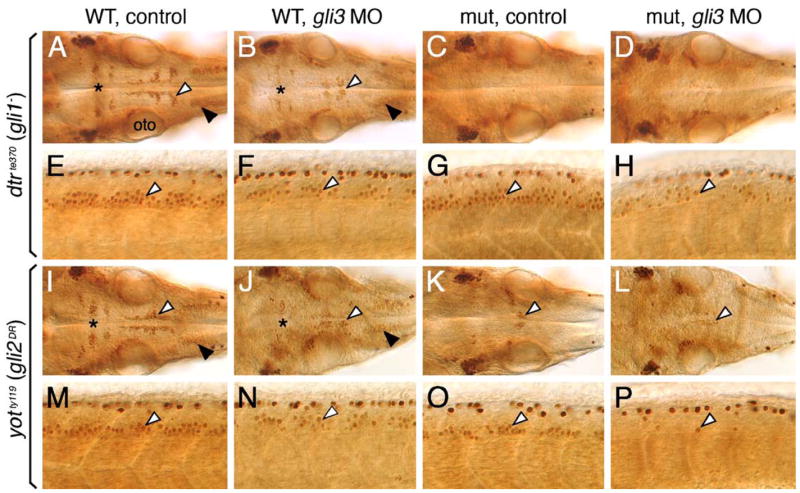
Gli3 plays a role in branchiomotor and spinal motor neuron induction. Panels A–D and I–L show dorsal views of the hindbrain, and panels E–H and M–P show lateral views of the spinal cord, with anterior to the left. (A and E) In an uninjected 36 hpf wild-type (dtr+/+ or +/−) embryo, islet antibody labeling reveals the characteristic organization of the nV neurons in r2 (asterisk), the nVII neurons (white arrowhead), and the nX neurons (black arrowhead) in the hindbrain (A), and the characteristic distribution of motor neurons (arrowhead, E) in the ventral spinal cord. (B and F) In a wild-type sibling injected with gli3 MO, the nV (asterisk), nVII (white arrowhead), and nX (black arrowhead) neurons in the hindbrain (B), and spinal motor neurons (arrowhead, F) are reduced in number. (C, D, G, and H) In an uninjected dtr mutant (C), branchiomotor neurons are essentially absent, and this phenotype is maintained in a gli3 MO-injected dtr mutant (D). In contrast, spinal motor neurons are moderately reduced in number in a gli3 MO-injected dtr mutant (arrowhead, H) compared to an uninjected dtr mutant (arrowhead, G). (I, J, M, and N) In an uninjected wild-type (yot+/+ or +/−) embryo, branchiomotor neurons are found in characteristic numbers (I; see A for details), and their numbers are moderately reduced in a gli3 MO-injected wild-type sibling (J). Similarly, spinal motor neurons are reduced in number in a gli3 MO-injected wild-type embryo (arrowhead, N) compared to an uninjected wild-type sibling (arrowhead, M). (K, L, O, P) In an uninjected yot mutant (K), most branchiomotor neurons, except nVII neurons (arrowhead), are greatly reduced in number or absent, and the number of nVII neurons is slightly decreased in a gli3 MO-injected yot mutant (L). In contrast, spinal motor neurons are greatly reduced in number in a gli3 MO-injected yot mutant (arrowhead, P) compared to an uninjected yot mutant (arrowhead, O). oto, otocyst.
Endogenous Gli2 repressor function does not regulate branchiomotor neuron induction
While wild-type gli2 appears to play no role in branchiomotor neuron induction (Fig. 5; Table 3), the Gli2 dominant repressors encoded by the yot alleles interfere with branchiomotor neuron induction (Figs. 1 and 6; Tables 1 and 4). Therefore, we wondered whether endogenous Gli2 repressor activity normally plays any role in branchiomotor neuron development, analogous to the role of Gli3 repressor function in ventral neural tube patterning in mouse (Litingtung and Chiang, 2000; Wijgerde et al., 2002). In a 21 hpf wild-type hindbrain, the islet1 expression domain (presumptive motor neurons) in rhombomere 4 (r4) and the nk2.2 expression domain (containing presumptive motor neurons) in r5 were located in the ventral-most neural tube, outside the gli2 expression domain (Figs. 8A and E). This dorsal pattern of gli2 expression is evident from 16.5 hpf until 36 hpf (data not shown; Karlstrom et al., 1999), the time period when most of the branchiomotor neurons differentiate (Chandrasekhar et al., 1997; Higashijima et al., 2000). This absence of expression in differentiating motor neurons suggests that gli2 does not directly influence motor neuron induction at these time points. However, since motor neurons are clearly affected in smu, dtr, and yot mutants, this suggests either that (1) gli2 (and therefore Gli2 or Gli2DR proteins) is aberrantly expressed in the ventral hindbrain (in motor neuron progenitors) in these mutants, (2) gli2 indirectly affects motor neuron induction, or (3) Gli-mediated motor neuron differentiation occurs prior to 16.5 hpf at a time when gli2 is expressed in motor neuron progenitors.
Fig. 8.
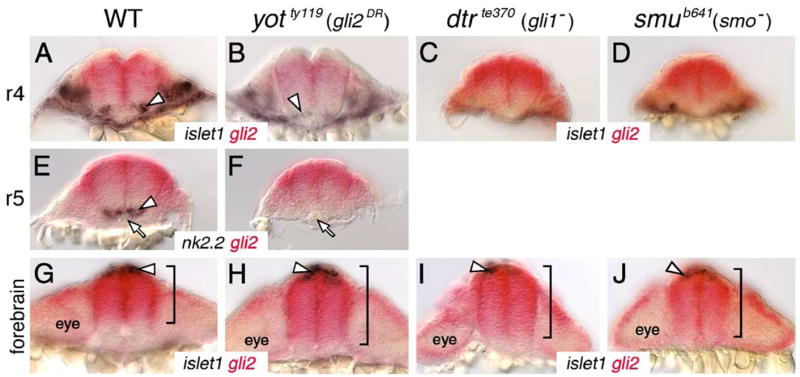
Gli2 expression is affected in the forebrain, but not in the hindbrain, of Hh pathway mutants. All panels show cross sections of the neural tube at the indicated axial levels, with dorsal at the top. Embryos were processed for islet1 (purple); gli2 (red) (A–D, G–J) or nk2.2 (purple); gli2 (red) (E and F) two-color in situs. (A–D) In r4 of a wild-type embryo (A), islet1-expressing motor neurons (arrowhead) are located just ventral to the gli2 expression domain. Similarly, the few islet1-expressing neurons in yot mutants (arrowhead) are located ventral to the gli2 expression domain (B), and gli2 expression does not extend into the ventral-most neural tube in dtr (C) and smu (D) mutants. (E and F) In r5, while nk2.2-expressing cells (arrowhead) are found immediately adjacent to the floor plate and notochord (arrow) in a wild-type embryo (E), nk2.2 expression is absent in yot mutants (F), but the gli2 expression domain does not expand ventrally. (G–J) At the forebrain level, the islet1-expressing epiphysial neurons (arrowhead) are located at the roof of the neural tube. In a 21-hpf wild-type embryo (G), gli2 expression is limited to the dorsal two-thirds of the neural tube (bracket). In contrast, in yotty119 (H), dtrte370 (I), and smub641 (J) mutants, the gli2 expression domain is expanded ventrally to different extents (brackets).
To distinguish between these possibilities, we first tested whether branchiomotor neuron loss in smu, dtr, and yot mutants might result from the ventral expansion of gli2 expression into regions containing motor neuron progenitors. We examined the expression domains of gli2 at various levels along the AP axis in 21 hpf mutant embryos processed for gli2;islet1 double in situ hybridization. There was no significant or reproducible ventral expansion of gli2 expression in the neural tube at hindbrain levels in any rhombomere in any of the three mutant backgrounds (4–6 embryos examined per mutant, 5–6 sections in the hind-brain per embryo). In the various mutants containing few (Fig. 8B) or no islet1-expressing cells (Figs. 8C and D), or no nk2.2-expressing cells (Fig. 8F), gli2 expression was clearly excluded from the ventral neural tube containing the motor neuron progenitor domain. There was no ventral expansion of gli2 expression in the hindbrains of smu (21 and 26 hpf), dtr (18 and 21 hpf), and yot mutants (16.5, 18, 21, and 26 hpf) (data not shown). Together, these data suggest strongly that the loss of branchiomotor neurons in these Hh pathway mutants is not due to the suppression of motor neuron fate by Gli2 repressor function but may simply be due to loss of Gli1 activator function (Figs. 4M–O; see Fig. 11 for model). Interestingly, gli2 expression expands ventrally in the forebrain of Hh pathway mutants. In cross sections of the forebrain at the level of the epiphysis, gli2 was expressed in the dorsal ~65% of the neural tube in wild-type embryos (Fig. 8G; Table 5). In all mutants examined, the gli2 expression domain at the forebrain level was expanded ventrally (and reproducibly) to 75–90% of the extent of the neural tube (Figs. 8H–J; Table 5), demonstrating a significant effect on dorsoventral patterning when Hh signaling is reduced or absent. Therefore, the reduction or loss of Hh target gene expression in the forebrain in Hh pathway mutants may result from Gli2-mediated repression.
Table 5.
Ventral expansion of gli2 expression domain in the forebrain of Hh pathway mutants
| Gene | Ventral margin of gli2 expression domain# |
P | |
|---|---|---|---|
| Wild-type* | Mutant* | ||
| smub641 | 63 ± 8% (5s, 5e) | 92 ± 8% (2s, 2e) | < 0.02 |
| dtrte370 | 63 ± 9% (4s, 4e) | 74 ± 1% (7s, 5e) | < 0.005 |
| yotty119 | 68 ± 5% (6s, 5e) | 83 ± 5% (5s, 5e) | < 0.001 |
Forebrain sections were scored at the level of the epiphysis, which was identified by the presence of islet1-expressing cells (Figs. 8G–J). The dorsoventral height of the neural tube was measured, and the length of the gli2 expression domain (measured from the dorsal surface) was expressed as a fraction (%) of the height of the neural tube.
The first number in the parenthesis indicates the total number of sections scored, while the second number indicates the number of embryos these sections came from.
To test the possibility that Gli2 repressor function in the dorsal neural tube has an indirect effect on Hh-mediated events in the ventral neural tube, we asked whether knockdown of gli2 function could rescue any aspect of Hh signaling, including branchiomotor neuron induction, in smu mutants, in a manner similar to the rescue of motor neurons in Shh;Gli3 and Smo;Gli3 mutant mice (Litingtung and Chiang, 2000; Wijgerde et al., 2002). When gli2 MO was injected into embryos from a smu+/−;islet1-GFP incross, branchiomotor neurons did not reappear in smu mutants, and there was no rescue of nk2.2 or net1a expression in the mutant hindbrain (data not shown), consistent with the idea that Gli2 repressor function in the dorsal hindbrain does not influence Hh-mediated events in the ventral hindbrain.
Finally, we tested the possibility that the loss of motor neurons in yot mutants could result from the ventral expression of gli2 (and Gli2DR) in motor neuron progenitors before 16.5 hpf. Gli2 was expressed extensively in the neural plate and the developing neural tube at 9 and 12 hpf (data not shown), and continued to be expressed in the ventral neural tube at 15 hpf throughout the hindbrain (Figs. 9A–C). These results indicate that aberrant Gli2DR activity in motor neuron progenitors before 15 hpf can account for reduced motor neuron induction in yot mutants and suggest that branchiomotor neurons are specified before 15 hpf, which is several hours earlier than suggested by observations of motor neuron differentiation (Chandrasekhar et al., 1997; Higashijima et al., 2000).
Hh signaling is primarily required before 18 hpf to induce branchiomotor neurons
Given that Gli2DR proteins block motor neuron induction in yot mutants by interfering with Gli1 and Gli3 function, and that gli1 and gli2 are co-expressed in motor neuron progenitors only before 16.5 hpf, we re-examined the idea that the branchiomotor neurons are specified continuously between 15 and 40 hpf (Chandrasekhar, 2004). To determine directly the critical period for motor neuron induction, we asked when different subsets of branchiomotor neurons were specified using the alkaloid cyclopamine (CyA) to block Hh signaling. Wild-type islet1-GFP transgenic embryos were treated with CyA beginning at 3, 6, 9, 12, 15, 18, 21, and 24 hpf. Control (EtOH-treated) and CyA-treated embryos were scored for motor neuron phenotypes at 48 hpf by examining GFP expression (Figs. 9D and E), fixed and processed for islet immunohistochemistry (Figs. 9F–I), and motor neurons in various rhombomeres (corresponding to specific types of branchiomotor neurons) were counted. CyA treatment at 9 hpf or earlier led to the complete loss of all branchiomotor neurons (Figs. 9D–G). However, a few nV neurons (in r2 and r3), and nVII neurons (in r4–r7) were generated between 9 and 12 hpf (Fig. 9H). Indeed, normal numbers of nV neurons were generated in r2 even when Hh signaling was blocked from 18 hpf (Figs. 9D and I). Similarly, a majority of nVII neurons were generated in 18 hpf-treated embryos (Figs. 9E and I), and essentially normal numbers of nV neurons (r2 and r3) and nVII neurons (r4–r7) are generated in 24 hpf-treated embryos (data not shown). These results demonstrate that Hh signaling is required before 24 hpf for generating all branchiomotor neurons, and that a majority of branchiomotor neurons are specified before 18 hpf, consistent with an early inhibitory role for mutant Gli2DR proteins within motor neuron progenitors in yot mutants.
Gli3, but not Gli1 or Gli2, activator function is required for inducing gli1 expression
We showed earlier that while gli1 expression is completely normal in detour (gli1−) mutants, it is severely reduced in smu (smo−) mutants, demonstrating that gli1 expression is regulated by Hh signaling but does not require Gli1 activator function (Fig. 4). Similarly, while Gli1 expression is unaffected in Gli1 knockout mouse embryos (Park et al., 2000), it is completely lost in Gli2;Gli3 double knockout embryos (Bai et al., 2004). To test the potential role of Gli activator function in regulating gli1 expression in zebrafish, we examined ptc1 and gli1 expression in conditions where overall Gli activator function was greatly attenuated (Fig. 10). In the first set of experiments, embryos from dtrte370+/− crosses were injected with gli2 MO that leads to a reduction of motor neurons in the spinal cord (see Fig. 5). In uninjected (21/21 embryos) and gli2 MO-injected (36/36 embryos) wild-type siblings, ptc1 expression in the ventral hindbrain was unaffected (Fig. 10A), indicating that the reduction in Gli activator function was not sufficient to reduce ptc1 expression. In contrast, ptc1 expression was reduced in uninjected dtr mutants (Fig. 10B; 5/5 embryos; see also Fig. 4I), and severely reduced in gli2 MO-injected dtr mutants (Fig. 10C; 14/14 embryos), indicating that the Gli activator functions encoded by gli1 and gli2 are necessary for inducing ptc1 expression. As expected, there was no effect on gli1 expression in gli2 MO-injected wild-type embryos (Fig. 10D). Surprisingly, gli1 expression was completely normal in gli2 MO-injected dtr mutants (Fig. 10E), indicating that a severe reduction in Gli1 and Gli2 activator function could not alter gli1 expression. In a second set of experiments, embryos from yotty119+/− crosses were injected with gli1 MO that leads to a severe loss of motor neurons in the hindbrain and spinal cord (see Fig. 6). While ptc1 expression was normal in control (17/17 embryos) and gli1 MO-injected wild-type (Fig. 10F; 20/20 embryos), the lower level of ptc1 expression seen in uninjected yot mutants (Fig. 10G; 5/5 embryos) was almost eliminated in gli1 MO-injected yot mutants (Fig. 10H; 3/3 embryos). Most importantly, gli1 expression was not affected in gli1 MO-injected yot mutants (compare Figs. 10I and J). These results again indicate that conditions that lead to severe attenuation of overall Gli activator function, such that the expression of Hh target genes net1a (Fig. 6) and ptc1 is severely reduced, are still unable to alter gli1 expression.
Since we have defined an activator function for Gli3 in motor neuron induction (Fig. 7), we next tested whether Gli3 may regulate gli1 expression by injecting gli3 MO into embryos from dtrte370+/− crosses and yotty119+/− crosses. Gli1 was expressed normally in all wild-type and dtr mutant embryos injected with gli3 MO, and the expression patterns were indistinguishable from those in control embryos (69/69 control and 49/49 gli3 MO-treated embryos; data not shown; see Figs. 10D and E for expression patterns). These data indicate that a dose of gli3 MO that knocks down gli3 expression (function) substantially enough to reduce motor neuron induction (Fig. 7) is not able to affect gli1 expression. Similarly, gli1 expression appeared normal in gli3 MO-injected wild-type siblings obtained from yotty119+/− crosses (Figs. 10N and O; 124/124 embryos). Significantly, gli1 expression was greatly reduced throughout the ventral neural tube in yot mutants injected with gli3 MO (Fig. 10P; 37/37 embryos). Since loss of Gli1 and Gli2 activator function alone and in combination had no effect on gli1 expression, these data suggest that the combined effect on Gli3 function produced by Gli2DR-mediated interference and gli3 MO knockdown is responsible for the observed reduction in gli1 expression. While ptc1 expression was slightly reduced in some gli3 MO-injected wild-type siblings (Fig. 10K; 13/36 embryos) compared to control wild-type siblings (data not shown; 28/28 embryos; see Fig. 10F), its expression was almost eliminated in gli3 MO-injected yot mutants (Fig. 10M; 13/15 embryos) compared to control mutant embryos (Fig. 10L; 9/9 embryos). Collectively, these results suggest strongly that Gli activator functions encoded by gli1, gli2, and gli3 are all equally capable of inducing ptc1 expression, whereas Gli3 activator function is specifically required for inducing gli1 expression (Fig. 11).
Discussion
Role of Glis in motor neuron development
In mouse and zebrafish, loss of Smoothened-mediated Hh signaling results in the failure of motor neuron induction (Chen et al., 2001; Varga et al., 2001; Wijgerde et al., 2002; this study). Nevertheless, a small number of motor neurons differentiate but are patterned abnormally in mice lacking all Gli activator function (Bai et al., 2004; Lei et al., 2004). Moreover, while mouse Gli1 function is not required for motor neuron induction (Park et al., 2000), zebrafish gli1 is essential for motor neuron induction in the hindbrain (Chandrasekhar et al., 1999). Given these differing roles for gli1 in Hh-mediated motor neuron induction in zebrafish and mouse, we tested in this study whether other zebrafish glis such as gli2 and gli3 played any role in this process, and particularly whether Gli activator function was essential for motor neuron induction in zebrafish. We have addressed these questions by examining the motor neuron phenotypes of you-too (yot) mutants, which carry mutations in gli2 (Karlstrom et al., 1999, 2003), and of embryos treated with antisense morpholinos to knockdown specific gli function. Our results demonstrate that, unlike mouse, Gli activator function is absolutely required for motor neuron induction at all axial levels in zebrafish.
The you-too (yot) motor neuron phenotype
The yot mutants were originally identified on the basis of defects in somite patterning, midline defects in the spinal cord, and defective retinotectal projections (Brand et al., 1996; Karlstrom et al., 1996; van Eeden et al., 1996). The yot locus encodes Gli2, which contains both C-terminal activator and N-terminal repressor domains (Karlstrom et al., 1999). The yotty119 and yotty17 alleles encode C-terminally truncated proteins that appear to function as dominant repressors (DR) of Hh signaling (Karlstrom et al., 1999, 2003). In contrast, Gli1, encoded by the detour (dtr) locus, appears to lack the N-terminal repressor domain, and functions only as a transcriptional activator (Karlstrom et al., 2003). Consistent with this, extant gli1 mutant alleles encode C-terminally truncated proteins missing the activator domains and exhibit no biological activity in reporter assays (Karlstrom et al., 2003).
In both yot alleles, there is a reduction in motor neuron number in the hindbrain and spinal cord, with the yotty119 allele exhibiting a more severe phenotype (Fig. 1; Table 1), consistent with the stronger dominant suppressor function of this allele in the transcriptional reporter assay (Karlstrom et al., 2003). The yotty119 mutants also exhibit more severe defects than yotty17 mutants in the expression of Hh-regulated genes (Fig. 2; data not shown). Nevertheless, motor neuron number and Hh-regulated gene expression are not noticeably reduced in embryos heterozygous for either allele, except for a slight but consistent defect in nk2.2 expression in the yotty119+/− hindbrain (data not shown). These results suggest that the mutant Gli2DR proteins cannot significantly affect Gli1 and Gli2 activator function when multiple copies of the wild-type genes (gli1 and gli2) are present. Consistent with this idea, hyperactivation of Hh signaling by misexpression of dominant-negative protein kinase A (dnPKA) leads to a substantial recovery of motor neuron number and Hh target gene expression in the hindbrain of yotty17, and to a lesser extent yotty119, mutants (Fig. 3; Table 2), presumably by upregulating the level of Gli1 activator.
Strikingly, expression of many Hh target genes is greatly reduced throughout the yotty119 mutant hindbrain, except in rhombomere 4 (Fig. 2). Residual gene expression in r4 is still Hh-dependent since cyclopamine treatment completely blocks expression in mutant embryos. The cyclopamine inhibitor experiments also reveal that r4-specific expression of Hh target genes (the yotty119 phenotype) can be phenocopied by cyclopamine treatment beginning at 18 hpf, suggesting that the yotty119 mutant phenotype may reflect the failure of Hh signaling at different times in different rhombomeres. Furthermore, ectopic Hh pathway activation through dnPKA overexpression in yotty119 mutants leads to a consistent upregulation of nk2.2 expression within the normal ventral domain, but not at ectopic locations in r4 (Fig. 3). These observations collectively suggest that the Hh signaling environment in r4 is different from that in adjacent rhombomeres, which is not surprising given that r4 develops earlier than adjacent rhombomeres, and functions as an organizer to signal to and regulate the patterning of these compartments (Maves et al., 2002).
Gli1, Gli2, and Gli3 activators contribute to the induction of spinal motor neurons
The normal induction of spinal motor neurons in dtr (gli1−) mutants (Chandrasekhar et al., 1999) suggests that Gli activator function provided by Gli1, Gli2, and Gli3 can function in a redundant fashion in the spinal cord. We tested this idea by knocking down gli1 or gli3 function in yot mutants, which led to the complete loss of motor neurons in the spinal cord (Figs. 6 and 7), indicating that Gli1 and Gli3 activators can contribute to spinal motor neuron induction (Fig. 11).
Since knockdown of gli2 function in wild-type embryos did not reveal any defects in motor neuron development, we sought potential roles for gli2 in sensitized conditions where the total level of Gli activator was reduced. Therefore, we examined the consequences of gli2 knockdown in dtr (gli1−) mutants. While normal numbers of spinal motor neurons develop in dtr mutants (Chandrasekhar et al., 1999; Fig. 5; Table 3), there is a small (~25%) but significant reduction in motor neuron number following gli2 MO injection, demonstrating that Gli2 contributes to spinal motor neuron induction. Since hindbrain motor neurons are essentially absent in dtr mutants (Chandrasekhar et al., 1999), it is unlikely that gli2 plays any role in their formation. In contrast to gli2, knockdown of gli3 led to loss of both branchiomotor (hindbrain) and spinal motor neurons in wild-type embryos, and in the dtr (gli1−) and yot (gli2DR) mutant spinal cords (Fig. 7), indicating that Gli3 activator function plays a more prominent role than Gli2 in motor neuron induction. Moreover, gli2 knockdown in dtr mutants or gli3 knockdown in dtr or yot mutants led to the loss of residual ptc1 expression in the hindbrain (Fig. 10), consistent with the minor activator roles for both Gli2 and Gli3 in Hh signaling shown previously (Karlstrom et al., 2003; Tyurina et al., 2005). Thus the residual expression of net1a (Fig. 5) and ptc1 seen in dtr (gli1−) mutants appears to result from the activator functions of Gli2 and Gli3 (Fig. 11). Importantly, our data now show that Gli3 activator function is also critical for the induction of a distinct ventral cell type, namely motor neurons.
Knockdown of gli2 function alone had no effect on the induction of hindbrain and spinal motor neurons (Fig. 5; Table 3). Since motor neuron number is a measure of floor plate- and notochord-derived Hh activities (Chandrasekhar et al., 1998), this suggests that the development of the floor plate and other ventral cell types does not require Gli2 activity. Consistently, nk2.2-expressing medial and lateral floor plate cells are unaffected in gli2 MO-injected wild-type embryos (Karlstrom et al., 2003). In contrast, mouse gli2 is essential for development of the floor plate and adjacent cells (Ding et al., 1998; Matise et al., 1998), reflective of the functional divergence in Gli2 reported earlier (Karlstrom et al., 2003). Although spinal motor neurons develop normally in mouse Gli2 mutants due to notochord-derived Hh signals (Ding et al., 1998; Matise et al., 1998), it is not known whether mouse Gli2 is required for motor axon outgrowth, in a similar fashion to zebrafish gli2 (Brand et al., 1996; Zeller et al., 2002).
In this report, we have tested whether gli2 and gli3 are required for motor neuron induction. Our data suggest important roles for the activator forms of both Gli2 and Gli3 in this process. In mouse, a Gli1 knock-in (encoding only Gli activator function) can rescue Gli2 mutant phenotypes, indicating that loss of Gli2 activator function is responsible for the phenotypes seen in Gli2 mutants (Bai and Joyner, 2001; Bai et al., 2002). Consistent with this, knockdown of zebrafish gli2 function in smu (smo−) mutants does not abrogate defective Hh-regulated events, including motor neuron induction, suggesting that the repressor function of zebrafish Gli2 does not play a significant role in Hh-regulated patterning in the hindbrain. Nevertheless, we cannot rule out that some of the effects on zebrafish spinal motor neuron development following gli2 knockdown are indirect effects of the repressor function of Gli2. Similarly, while our data suggest strongly that Gli3 activator function contributes to branchiomotor and spinal motor neuron induction (Fig. 7), it is formally possible that Gli3 repressor function may influence motor neuron fate in the hindbrain. These ideas will be tested in the future through gli gain-of-function and gli2;gli3 double knockdown experiments.
Differential requirements for Gli activator function in mouse and zebrafish
While Gli2 repressor function appears to be dispensable for neural tube patterning in both mouse and zebrafish, it appears that Gli activator functions have profoundly different roles during motor neuron development in the two species. We have shown here that zebrafish gli1, gli2, and gli3 can contribute to spinal motor neuron induction. Furthermore, we showed previously that zebrafish gli1 is essential for the development of all motor neurons in the midbrain and hindbrain (Chandrasekhar et al., 1999; A.C., unpublished data), and we show here that gli3 also plays a role in hindbrain motor neuron induction. In sharp contrast, analysis of specific Gli single and double knockouts suggested that Gli activator function is not required for the induction of motor neurons in the mouse spinal cord (Ding et al., 1998; Matise et al., 1998; Park et al., 2000). In mouse Gli2;Gli3 double knockouts (which are essentially Gli1;Gli2;Gli3 triple knockouts, since Gli1 is not expressed in the double mutant), motor neuron progenitors are found in normal numbers, while the number of differentiated motor neurons is much smaller, and they exhibit patterning and migration defects (Bai et al., 2004; Lei et al., 2004). These and other results from Shh;Gli3 (Litingtung and Chiang, 2000) and Smo;Gli3 (Wijgerde et al., 2002) knockout mice suggest that spinal motor neurons can be induced in small numbers by stochastic, Gli/Hh-independent mechanisms, likely through retinoid signaling (Novitch et al., 2003). It should also be noted that since motor neuron development at more rostral levels (in the brainstem) was not examined in any of these compound mutants, Gli activators may yet be found to play a role in motor neuron induction in the mouse head, similar to zebrafish.
Why do Gli knockout mice and gli knockdown or mutant zebrafish exhibit significantly different motor neuron phenotypes? Specifically, mouse Gli1 knockout mice develop motor neurons normally (at all axial levels) and are viable (Park et al., 2000), while zebrafish detour (gli1−) mutants specifically fail to generate cranial motor neurons (Chandrasekhar et al., 1999) and die as embryos (Karlstrom et al., 1996). In Gli1 mutant mice, Gli2 and Gli3 are expressed ventrally, and their activator functions compensate for the loss of Gli1 activity (Bai et al., 2004; Lei et al., 2004). In contrast, while gli2 and gli3 are transiently expressed in the ventral neural tube within the normal gli1 expression domain in dtr (gli1−) mutants (this study; Karlstrom et al., 1999, 2003; Tyurina et al., 2005), the absence of hindbrain motor neurons in this mutant suggests that Gli2 and Gli3 activator functions cannot compensate for the loss of Gli1 activator function. This hypothesis can be tested through gain-of-function experiments (see below). Interestingly, gli3 knockdown results in decreased numbers of hindbrain motor neurons, suggesting a role for Gli3 activator in their induction (Fig. 7). However, given the dtr motor neuron phenotype, the role for zebrafish gli3 in branchiomotor neuron induction is likely to be indirect, since gli3 function is needed for inducing gli1 expression (Fig. 10).
Unlike the dramatic difference in cranial motor neuron phenotypes between mouse and fish Gli1 mutants, there is a significant but subtler difference in spinal motor neuron phenotypes between the mouse Gli “triple” knockout and the zebrafish gli2DR/DR;gli1 MO and gli2DR/DR;gli3 MO embryos. Differentiated, Islet+ motor neurons in the spinal cord are greatly reduced in number, but not completely missing in the Gli2;Gli3 mutant mice (Lei et al., 2004), whereas they are essentially lost in the zebrafish gli2DR/DR;gli1 MO and gli2DR/DR;gli3 MO embryos (Figs. 6 and 7). While the number of Olig2+ spinal motor neuron progenitors in the mutant mice is unaffected, mouse Gli activator function seems to be especially important for the differentiation of these progenitors and patterning of motor neurons (Bai et al., 2004; Lei et al., 2004). Since retinoic acid (RA) signaling can independently generate progenitor domains in the mouse ventral spinal cord (Novitch et al., 2003), it is possible that the small number of motor neurons differentiating in the mutant mice also result from RA-dependent mechanisms. We have not tested whether olig2-expressing spinal motor neuron progenitors (Park et al., 2002, 2004) form normally in zebrafish gli2DR/DR;gli1 MO and gli2DR/DR;gli3 MO embryos, as in the mutant mice. However, it is clear that even if the progenitors are formed, they are unable to differentiate into motor neurons in the absence of Gli1, Gli2, and Gli3 activator function.
While our studies address whether zebrafish gli1, gli2, and gli3 are necessary for motor neuron induction, they provide no insight into whether zebrafish glis have different abilities to induce motor neurons, or whether gli1-, gli2-, and gli3-encoded activator functions are equivalent. Activator forms of mouse Gli2 and Gli3 have distinct abilities to induce ventral cell fates when expressed ectopically in the chick neural tube, with Gli2 generating a broader range of cell types (Lei et al., 2004). In addition, mouse Gli3 expressed from the Gli2 promoter cannot fully rescue the Gli2 knockout phenotype (Bai et al., 2004). In contrast, mouse Gli1 expressed from the Gli2 promoter can fully rescue the Gli2 knockout phenotype (Bai and Joyner, 2001). These results indicate that mouse Glis have varying degrees of overlapping activator functions. It will be of interest to test whether zebrafish Glis similarly share some functionality. Furthermore, given that zebrafish Gli1 is essential for motor neuron induction while mouse Gli1 is not, it would be instructive to test whether mouse Gli1 can participate in motor neuron formation in zebrafish. These experiments will reveal whether the different abilities of the Gli1 orthologs to induce motor neurons reflect differences in activity levels as proposed (Stamataki et al., 2005), or more fundamental differences in their abilities to induce the motor neuron fate.
Early role for Hh signaling in branchiomotor neuron induction
In seeking to explain the loss of branchiomotor neurons in yot mutants, we discovered that these neurons are specified very early during embryogenesis. In zebrafish, the reticulospinal interneurons and a few, large spinal motor neurons are specified by the end of gastrulation and represent the earliest-born (primary) neurons (Mendelson, 1986; Eisen, 1991). By contrast, smaller and more numerous spinal motor neurons are specified in a secondary wave of neurogenesis during somitogenesis and later stages (Kimmel and Westerfield, 1990). The branchiomotor neurons have been generally regarded as later-born secondary neurons since the earliest differentiated neurons appear around 15 hpf, and their numbers increase continuously until 36–40 hpf (Chandrasekhar et al., 1997; Higashijima et al., 2000; Linville et al., 2004). Therefore, we were surprised when the cyclopamine experiments (Fig. 8) revealed that Hh signaling specifies virtually all branchiomotor neurons prior to 15 hpf. A significant number of nV and nVII neurons are specified between 6 and 12 hpf, spanning gastrulation. Blocking Hh signaling after 18 hpf has no effect on the induction of any branchiomotor neuron subtype including the nX neurons, most of which appear between 30 and 40 hpf. Interestingly, BrdU labeling reveals that many nV and nVII neuronal progenitors remain in S-phase at 18 and 21 hpf (A.C., unpublished), several hours after Hh signaling has specified their formation. The temporal lag between the critical period of Hh sensitivity and the appearance of differentiated neurons likely reflects the time needed for motor neuron progenitors to undergo an orderly process of specification, cell-cycle exit, and differentiation involving the expression of numerous regional (e.g., pax6) and cell-type specific (e.g., nk2.2, olig2) transcription factors, in a manner similar to that described for spinal motor neurons in chick and mouse (reviewed in Shirasaki and Pfaff, 2002). In addition to Hh-mediated signaling, retinoic acid (RA) also appears to play a role in inducing branchiomotor neurons, especially the nX motor neurons (Begemann et al., 2004; Linville et al., 2004), mirroring the parallel roles of Hh and RA signaling in the formation of motor neuron progenitor domains in the mouse spinal cord (Novitch et al., 2003). However, our results suggest that RA signaling cannot induce any branchiomotor neurons in the absence of Gli-mediated Hh signaling. It will be of interest to determine the relationship between these signaling pathways during motor neuron induction in zebrafish.
In summary, while a small number of motor neurons can be induced by Hh- and Gli-independent processes in mouse, our data demonstrate an absolute requirement for Gli activator function in the formation of motor neurons at all axial levels in zebrafish. Furthermore, our data have identified separable roles for Gli1, Gli2, and Gli3 activator functions in motor neuron induction. Thus while the overall requirement for Gli function in motor neuron induction is largely conserved between zebrafish and mouse, the roles of particular Gli family members in this process appear to have diverged. Whether this functional divergence reflects changes in gene expression patterns, changes in level of protein activity, and/or changes in protein function remains to be determined.
Acknowledgments
We thank Michael Matise for critically reading the manuscript. We thank Andy McClellan for help with statistical analysis, and Steve Devoto for the smub641 carrier fish. We thank Amy Foerstel, Moe Baccam, Keqing Zhang, Matthew McClure, and Vinoth Sittaramane for excellent fish care, and Stephanie Bingham and Rhituparna Chatterjee for capturing some images. We thank Jim Lauderdale, Kate Lewis, Lisa Maves, and Vicky Prince for providing cDNAs to make in situ probes. The islet, zn5, and 3A10 monoclonal antibodies were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa. This work was supported by an NIH training grant fellowship (NIGMS T32 GM08396 to GV) and NIH grants (NS39994 to ROK and NS40449 to AC).
Appendix A. Supplementary Data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.ydbio.2005.04.010.
References
- Aza-Blanc P, Ramirez-Weber FA, Laget MP, Schwartz C, Kornberg TB. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell. 1997;89:1043–1053. doi: 10.1016/s0092-8674(00)80292-5. [DOI] [PubMed] [Google Scholar]
- Bai CB, Joyner AL. Gli1 can rescue the in vivo function of Gli2. Development. 2001;128:5161–5172. doi: 10.1242/dev.128.24.5161. [DOI] [PubMed] [Google Scholar]
- Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell. 2004;6:103–115. doi: 10.1016/s1534-5807(03)00394-0. [DOI] [PubMed] [Google Scholar]
- Barresi MJ, Stickney HL, Devoto SH. The zebrafish slow-muscle-omitted gene product is required for hedgehog signal transduction and the development of slow muscle identity. Development. 2000;127:2189–2199. doi: 10.1242/dev.127.10.2189. [DOI] [PubMed] [Google Scholar]
- Barth KA, Wilson SW. Expression of zebrafish nk2.2 is influenced by sonic hedgehog/vertebrate hedgehog-1 and demarcates a zone of neuronal differentiation in the embryonic forebrain. Development. 1995;121:1755–1768. doi: 10.1242/dev.121.6.1755. [DOI] [PubMed] [Google Scholar]
- Begemann G, Marx M, Mebus K, Meyer A, Bastmeyer M. Beyond the neckless phenotype: influence of reduced retinoic acid signaling on motor neuron development in the zebrafish hindbrain. Dev Biol. 2004;271:119–129. doi: 10.1016/j.ydbio.2004.03.033. [DOI] [PubMed] [Google Scholar]
- Bingham S, Nasevicius A, Ekker SC, Chandrasekhar A. Sonic hedgehog and tiggy-winkle hedgehog cooperatively induce zebrafish branchiomotor neurons. Genesis. 2001;30:170–174. doi: 10.1002/gene.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham S, Higashijima S, Okamoto H, Chandrasekhar A. The zebrafish trilobite gene is essential for tangential migration of branchiomotor neurons. Dev Biol. 2002;242:149–160. doi: 10.1006/dbio.2001.0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham S, Chaudhari S, Vanderlaan G, Itoh M, Chitnis A, Chandrasekhar A. Neurogenic phenotype of mind bomb mutants leads to severe patterning defects in the zebrafish hindbrain. Dev Dyn. 2003;228:451–463. doi: 10.1002/dvdy.10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M, Heisenberg CP, Warga RM, Pelegri F, Karlstrom RO, Beuchle D, Picker A, Jiang YJ, Furutani-Seiki M, van Eeden FJ, Granato M, Haffter P, Hammerschmidt M, Kane DA, Kelsh RN, Mullins MC, Odenthal J, Nusslein-Volhard C. Mutations affecting development of the midline and general body shape during zebrafish embryogenesis. Development. 1996;123:129–142. doi: 10.1242/dev.123.1.129. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar A. Turning heads: development of vertebrate branchiomotor neurons. Dev Dyn. 2004;229:143–161. doi: 10.1002/dvdy.10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekhar A, Moens CB, Warren JT, Jr, Kimmel CB, Kuwada JY. Development of branchiomotor neurons in zebrafish. Development. 1997;124:2633–2644. doi: 10.1242/dev.124.13.2633. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar A, Warren JT, Jr, Takahashi K, Schauerte HE, van Eeden FJM, Haffter P, Kuwada JY. Role of sonic hedgehog in branchiomotor neuron induction in zebrafish. Mech Dev. 1998;76:101–115. doi: 10.1016/s0925-4773(98)00101-4. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar A, Schauerte HE, Haffter P, Kuwada JY. The zebrafish detour gene is essential for cranial but not spinal motor neuron induction. Development. 1999;126:2727–2737. doi: 10.1242/dev.126.12.2727. [DOI] [PubMed] [Google Scholar]
- Chen W, Burgess S, Hopkins N. Analysis of the zebrafish smoothened mutant reveals conserved and divergent functions of hedgehog activity. Development. 2001;128:2385–2396. doi: 10.1242/dev.128.12.2385. [DOI] [PubMed] [Google Scholar]
- Concordet JP, Lewis KE, Moore JW, Goodrich LV, Johnson RL, Scott MP, Ingham PW. Spatial regulation of a zebrafish patched homologue reflects the roles of sonic hedgehog and protein kinase A in neural tube and somite patterning. Development. 1996;122:2835–2846. doi: 10.1242/dev.122.9.2835. [DOI] [PubMed] [Google Scholar]
- Ding Q, Motoyama J, Gasca S, Mo R, Sasaki H, Rossant J, Hui CC. Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development. 1998;125:2533–2543. doi: 10.1242/dev.125.14.2533. [DOI] [PubMed] [Google Scholar]
- Eisen JS. Motoneuronal development in the embryonic zebrafish. Development. 1991;2:141–147. [PubMed] [Google Scholar]
- Hatta K. Role of the floor plate in axonal patterning in the zebrafish CNS. Neuron. 1992;9:629–642. doi: 10.1016/0896-6273(92)90027-b. [DOI] [PubMed] [Google Scholar]
- Higashijima S, Hotta Y, Okamoto H. Visualization of cranial motor neurons in live transgenic zebrafish expressing green fluorescent protein under the control of the islet-1 promoter/enhancer. J Neurosci. 2000;20:206–218. doi: 10.1523/JNEUROSCI.20-01-00206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- Karlstrom RO, Trowe T, Klostermann S, Baier H, Brand M, Crawford AD, Grunewald B, Haffter P, Hoffmann H, Meyer SU, Muller BK, Richter S, van Eeden FJ, Nusslein-Volhard C, Bonhoeffer F. Zebrafish mutations affecting retinotectal axon pathfinding. Development. 1996;123:427–438. doi: 10.1242/dev.123.1.427. [DOI] [PubMed] [Google Scholar]
- Karlstrom RO, Talbot WS, Schier AF. Comparative synteny cloning of zebrafish you-too: mutations in the Hedgehog target gli2 affect ventral forebrain patterning. Genes Dev. 1999;13:388–393. doi: 10.1101/gad.13.4.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlstrom RO, Tyurina OV, Kawakami A, Nishioka N, Talbot WS, Sasaki H, Schier AF. Genetic analysis of zebrafish gli1 and gli2 reveals divergent requirements for gli genes in vertebrate development. Development. 2003;130:1549–1564. doi: 10.1242/dev.00364. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Westerfield M. Primary neurons of the zebrafish. In: Edelman GM, Cowan WM, editors. Signals and Sense: Local and Global Order in Perceptual Maps. Wiley Interscience; New York: 1990. pp. 561–588. [Google Scholar]
- Korzh V, Edlund T, Thor S. Zebrafish primary neurons initiate expression of the LIM homeodomain protein Isl-1 at the end of gastrulation. Development. 1993;118:417–425. doi: 10.1242/dev.118.2.417. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Pereira FA, Qiu Y, Chen CH, Beachy PA, Tsai SY, Tsai MJ. Mediation of Sonic hedgehog-induced expression of COUP-TFII by a protein phosphatase. Science. 1997;278:1947–1950. doi: 10.1126/science.278.5345.1947. [DOI] [PubMed] [Google Scholar]
- Lauderdale JD, Davis NM, Kuwada JY. Axon tracts correlate with netrin-1a expression in the zebrafish embryo. Mol Cell Neurosci. 1997;9:293–313. doi: 10.1006/mcne.1997.0624. [DOI] [PubMed] [Google Scholar]
- Lei Q, Zelman AK, Kuang E, Li S, Matise MP. Induction of graded hedgehog signaling by a combination of Gli2 and Gli3 activator functions in the developing spinal cord. Development. 2004;131:3593–3604. doi: 10.1242/dev.01230. [DOI] [PubMed] [Google Scholar]
- Linville A, Gumusaneli E, Chandraratna RA, Schilling TF. Independent roles for retinoic acid in segmentation and neuronal differentiation in the zebrafish hindbrain. Dev Biol. 2004;270:186–199. doi: 10.1016/j.ydbio.2004.02.022. [DOI] [PubMed] [Google Scholar]
- Litingtung Y, Chiang C. Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat Neurosci. 2000;3:979–985. doi: 10.1038/79916. [DOI] [PubMed] [Google Scholar]
- Lum L, Beachy PA. The Hedgehog response network: sensors, switches, and routers. Science. 2004;304:1755–1759. doi: 10.1126/science.1098020. [DOI] [PubMed] [Google Scholar]
- Matise MP, Epstein DJ, Park HL, Platt KA, Joyner AL. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development. 1998;125:2759–2770. doi: 10.1242/dev.125.15.2759. [DOI] [PubMed] [Google Scholar]
- Maves L, Jackman W, Kimmel CB. FGF3 and FGF8 mediate a rhombomere 4 signaling activity in the zebrafish hindbrain. Development. 2002;129:3825–3837. doi: 10.1242/dev.129.16.3825. [DOI] [PubMed] [Google Scholar]
- Mendelson B. Development of reticulospinal neurons of the zebrafish: I. Time of origin. J Comp Neurol. 1986;251:160–171. doi: 10.1002/cne.902510203. [DOI] [PubMed] [Google Scholar]
- Motoyama J, Milenkovic L, Iwama M, Shikata Y, Scott MP, Hui CC. Differential requirement for Gli2 and Gli3 in ventral neural cell fate specification. Dev Biol. 2003;259:150–161. doi: 10.1016/s0012-1606(03)00159-3. [DOI] [PubMed] [Google Scholar]
- Novitch BG, Wichterle H, Jessell TM, Sockanathan S. A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron. 2003;40:81–95. doi: 10.1016/j.neuron.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Oxtoby E, Jowett T. Cloning of the zebrafish krox-20 gene (krx-20) and its expression during hindbrain development. Nucleic Acids Res. 1993;21:1087–1095. doi: 10.1093/nar/21.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HL, Bai C, Platt KA, Matise MP, Beeghly A, Hui C, Nakashima M, Joyner AL. Mouse gli1 mutants are viable but have defects in SHH signaling in combination with a gli2 mutation. Development. 2000;127:1593–1605. doi: 10.1242/dev.127.8.1593. [DOI] [PubMed] [Google Scholar]
- Park HC, Mehta A, Richardson JS, Appel B. olig2 is required for zebrafish primary motor neuron and oligodendrocyte development. Dev Biol. 2002;248:356–368. doi: 10.1006/dbio.2002.0738. [DOI] [PubMed] [Google Scholar]
- Park HC, Shin J, Appel B. Spatial and temporal regulation of ventral spinal cord precursor specification by Hedgehog signaling. Development. 2004;131:5959–5969. doi: 10.1242/dev.01456. [DOI] [PubMed] [Google Scholar]
- Persson M, Stamataki D, te Welscher P, Andersson E, Bose J, Ruther U, Ericson J, Briscoe J. Dorsal–ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 2002;16:2865–2878. doi: 10.1101/gad.243402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince VE, Joly L, Ekker M, Ho RK. Zebrafish hox genes: genomic organization and modified colinear expression patterns in the trunk. Development. 1998;125:407–420. doi: 10.1242/dev.125.3.407. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A. Combinatorial Gli gene function in floor plate and neuronal inductions by Sonic hedgehog. Development. 1998;125:2203–2212. doi: 10.1242/dev.125.12.2203. [DOI] [PubMed] [Google Scholar]
- Shirasaki R, Pfaff SL. Transcriptional codes and the control of neuronal identity. Annu Rev Neurosci. 2002;25:251–281. doi: 10.1146/annurev.neuro.25.112701.142916. [DOI] [PubMed] [Google Scholar]
- Stamataki D, Ulloa F, Tsoni SV, Mynett A, Briscoe J. A gradient of Gli activity mediates graded Sonic Hedgehog signaling in the neural tube. Genes Dev. 2005;19:626–641. doi: 10.1101/gad.325905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- Trevarrow B, Marks DL, Kimmel CB. Organization of hindbrain segments in the zebrafish embryo. Neuron. 1990;4:669–679. doi: 10.1016/0896-6273(90)90194-k. [DOI] [PubMed] [Google Scholar]
- Tyurina OV, Guner B, Popova E, Feng J, Schier AF, Kohtz JD, Karlstrom RO. Zebrafish Gli3 functions as both an activator and a repressor in Hedgehog signaling. Dev Biol. 2005;277:537–556. doi: 10.1016/j.ydbio.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Ungar AR, Moon RT. Inhibition of protein kinase A phenocopies ectopic expression of hedgehog in the CNS of wild-type and cyclops mutant embryos. Dev Biol. 1996;178:186–191. doi: 10.1006/dbio.1996.0209. [DOI] [PubMed] [Google Scholar]
- van Eeden FJ, Granato M, Schach U, Brand M, Furutani-Seiki M, Haffter P, Hammerschmidt M, Heisenberg CP, Jiang YJ, Kane DA, Kelsh RN, Mullins MC, Odenthal J, Warga RM, Allende ML, Weinberg ES, Nusslein-Volhard C. Mutations affecting somite formation and patterning in the zebrafish, Danio rerio. Development. 1996;123:153–164. doi: 10.1242/dev.123.1.153. [DOI] [PubMed] [Google Scholar]
- Varga ZM, Amores A, Lewis KE, Yan YL, Postlethwait JH, Eisen JS, Westerfield M. Zebrafish smoothened functions in ventral neural tube specification and axon tract formation. Development. 2001;128:3497–3509. doi: 10.1242/dev.128.18.3497. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya R, Scott MP. The developmental biology of brain tumors. Annu Rev Neurosci. 2001;24:385–428. doi: 10.1146/annurev.neuro.24.1.385. [DOI] [PubMed] [Google Scholar]
- Westerfield M. The Zebrafish Book. University of Oregon; Eugene, OR: 1995. [Google Scholar]
- Wijgerde M, McMahon JA, Rule M, McMahon AP. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev. 2002;16:2849–2864. doi: 10.1101/gad.1025702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller J, Schneider V, Malayaman S, Higashijima S, Okamoto H, Gui J, Lin S, Granato M. Migration of zebrafish spinal motor nerves into the periphery requires multiple myotome-derived cues. Dev Biol. 2002;252:241–256. doi: 10.1006/dbio.2002.0852. [DOI] [PubMed] [Google Scholar]