

Medulloblastoma (MB) is the most common malignant brain tumor in children. It arises in the cerebellum and has been associated with a variety of genetic alterations, including genes in the Sonic hedgehog (Shh), Notch and Wnt signaling pathways. This study focuses on the Shh pathway, which is activated in a subset of MBs [1] [2] [3]. In this pathway, Shh binds to the receptor, Patched (PTCH), which liberates the Smoothened (SMO) protein, allowing GLI and MYCN transcription factors to turn on target genes, including, in a negative feedback loop, PTCH itself. Mutations in the PTCH gene are observed in 10-20% of sporadic MBs and are associated with a familial predisposition to MB, known as Gorlin’s syndrome [4]. Additional Shh pathway activating mutations have been identified in SMO and Suppressor of Fused (SUFU) genes. Yet these mutations account for less than 25% of tumors that show Shh pathway activation [2]. The purpose of the current study was to begin exploring epigenetic mechanisms by which the Shh pathway could be activated.
Methylation of tumor suppressor genes is increasingly recognized as a causative mechanism in tumorigenesis [5]. In MB, hypermethylation has been consistently identified in several genes, including, but not limited to RASSF1A, a multi-faceted tumor-suppressor [6], Caspase 8 (CASP8), whose disruption alters apoptosis and tissue homeostasis [7] and Hypermethylated in cancer 1 (HIC1) [8], a target of p53 (see [9] for detailed review). Notably, additional studies on CASP8 and HIC1 [10] have also revealed methylation in control samples (either adult or fetal cerebellum), illustrating the importance of using appropriate tissue matched samples for comparison in methylation studies. Moreover, none of these genes display a clear role in the molecular pathogenesis of MB. Hypermethylation has been identified in regulatory components of activating pathways such as Wnt, a pathway strongly associated with tumorigenesis in other cancers [11]. The present study is motivated by the hypothesis that methylation of the PTCH1 promoter is causative in some MB cases.
We focused on PTCH1 for several reasons: 1) PTCH1 is a negative regulator of the Shh pathway. Thus, constrained transcription or translation of this gene would activate Shh activity. 2) Several MB cases display elevated GLI expression and concordant low PTCH1 expression, suggesting a loss of PTCH1 inhibition. 3) In a mouse model of MB in which one allele of PTCH1 was genetically disabled along with the p53 gene, the remaining allele was naturally silenced by methylation [12]. These findings and the overall scarcity of established determinants of MB raise the possibility that methylation of the PTCH1 gene contributes to the generation and/or maintenance of MB.
The current investigation was designed to identify MB samples in which the promoter of the PTCH1 gene was methylated. This was carried out by identifying patient samples in which expression of indicators of Shh pathway activity, GLI1 and MYCN were elevated but PTCH1 expression remained low and subsequently analyzing these candidate samples by bisulfite PCR and DNA sequencing. The samples tested consisted of 21 primary pediatric MBs. MYCN was analyzed because recent studies showed that it is downstream of Shh and necessary for MB formation [13] [14]. GLI1 and PTCH are the gold standard for Shh pathway targets [15] [16]. Expression of these genes was determined in all samples by real-time polymerase chain reaction (RT-PCR). As seen in Table 1, expression analysis revealed that, when compared to unaffected pediatric cerebellum controls, the majority of MB samples showed elevated expression of MYCN (20 of 21, 95%). A subset of these samples exhibited concordant elevated GLI1 expression (6 of 21, 29%). Four MBs with elevated GLI1 exhibited little or no PTCH1 expression. The samples with low PTCH1 and elevated GLI1 and MYCN were analyzed for PTCH1 promoter methylation.
Table 1. Shh pathway target gene expression.
Total RNA was extracted from primary human MBs or unaffected pediatric cerebellum. cDNA derived from the RNA was amplified by RT-PCR with taqman primers and probes (Applied Biosystems, Foster City, CA) specific to human MYCN, GLI1 and PTCH1. 18s ribosomal RNA gene was concurrently amplified for internal normalization. Expression in MB samples was compared to average control cerebellum (CBLM) expression. Four of 21 samples exhibit increased MYCN and GLI1 in addition to low PTCH1 expression.
| Expression level
|
|||
|---|---|---|---|
| Sample | MYCN | GLI1 | PTCH1 |
| MB-8 | HIGH | HIGH | NONE |
| MB-7 | HIGH | HIGH | NONE |
| MB-6 | HIGH | HIGH | NONE |
| MB-11 | MEDIUM | LOW | NONE |
| MB-2 | HIGH | HIGH | MEDIUM |
| MB-4 | HIGH | NONE | LOW |
| MB-3 | HIGH | NONE | LOW |
| MB-1 | HIGH | NONE | NONE |
| MB-9 | HIGH | NONE | NONE |
| MB-17 | HIGH | NONE | NONE |
| MB-14 | HIGH | NONE | NONE |
| MB-13 | HIGH | NONE | NONE |
| MB-18 | HIGH | NONE | NONE |
| MB-10 | MEDIUM | NONE | NONE |
| MB-16 | MEDIUM | NONE | NONE |
| MB-22 | LOW | NONE | NONE |
| MB-21 | LOW | NONE | NONE |
| MB-12 | LOW | NONE | NONE |
| MB-15 | LOW | NONE | NONE |
| MB-20 | NONE | NONE | NONE |
| MB-19 | NONE | NONE | NONE |
| Legend | |||
| Fold Δ vs. CBLM | |||
| HIGH | >10.0 | ||
| MEDIUM | 3.8-10.0 | ||
| LOW | 1.7-3.8 | ||
| NONE | <1.7 | ||
Identification of methylated bases in the PTCH1 promoter was carried out by the bisulfite conversion method and PCR. Bisulfite modification and sequencing was chosen because of its ability to discern between individually methylated and unmethylated cytosine residues and directly compare across samples. To ensure the most likely regions of methylation were captured, the promoter was divided into 7 contiguous regions, starting 983 bases upstream of the PTCH1-1B transcription start site and ending 530 bases into Exon 1B. There are four PTCH1 transcript variants (1, 1A, 1B, and 1C) which differ in their first exon and each have distinct promoters [17] [18]. The promoter for variant 1B was chosen to analyze in detail for this study by virtue of its robust response to Shh and capacity for strong long-term suppression of Shh signaling in contrast to other variants. The analyzed regions surrounding Exon 1B also encompass a CpG island, one of the hallmarks of tumor-associated methylation hotspots [11]. Primers specific for amplifying bisulfite converted genomic DNA were used to amplify each region. In order to avoid bias for methylated or unmethylated DNA, all primers lack CpG cytosines, except in unavoidable cases. In such cases, a mixture of pyrimidines or purines was used in place of cytosine in forward or reverse primers, respectively. Four MB samples (MB-6, MB-7, MB-8, MB-11) were used as template for this method based on their low PTCH1 mRNA expression and evidence of Shh pathway activity. Five unaffected pediatric cerebellum samples were used as negative controls. As a positive control, we used enzymatically methylated DNA purchased from Millipore. Amplified DNA from each region specific sample was TA-cloned into the pCR4-TOPO vector. Five colonies were picked from each clone in order to ensure consistency for each sample. Plasmid DNA was extracted and directly sequenced for all picked colonies.
The results of sequencing analysis suggest there is no methylation present in the promoter immediately upstream of PTCH1-1B in either tumor or control samples. As seen in Figure 1, the tumor samples across all regions exhibited near zero methylated cytosines at any of the CpG sites (<1%) (open boxes). Similarly, cerebellum control samples displayed no methylation in the proximal promoter (0%). Artificially methylated DNA contained methylcytosines at CpG sites in all amplified regions (closed boxes), confirming that bisulfite sequencing was successful. A small proportion of regions were not amplifiable for some samples (unboxed).
Figure 1. Methylation profile of the Patched-1 proximal promoter.
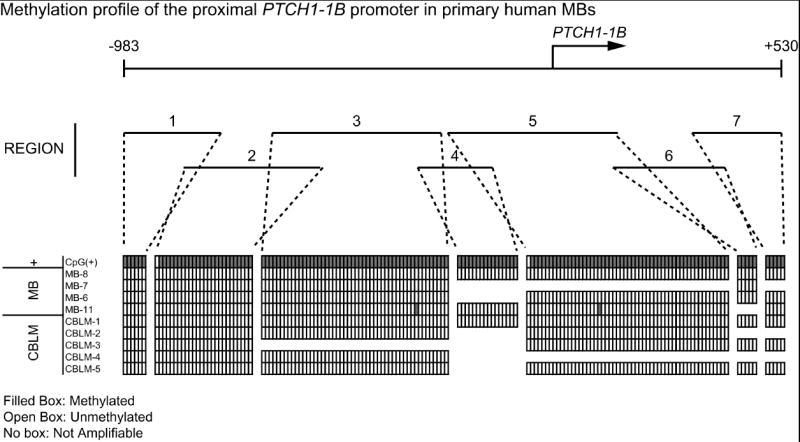
Genomic DNA was extracted from primary MB and pediatric cerebellum samples and treated with sodium bisulfite. Treated samples were PCR amplified with primers specific to contiguous regions of the proximal human PTCH1-1B promoter. Amplified PCR products were cloned into the pCR4-TOPO vector (Invitrogen, Carlsbad, California). Multiple colonies were sequenced for each PCR product. Methylation analysis was carried out using BiQ Analyzer software [25]. The sample CpG+ was bilsulfite converted CpGenome Universal Methylated DNA (Millipore, Billerica, MA). Shown are all CpG dinucleotides present in each amplified region, excluding primers (represented by boxes). Filled boxes represent methylated cytosines and unfilled boxes represent unmethylated cytosines. No boxes represent regions where amplification was unachievable with the current primers. CpG dinucleotides were considered methylated if 3 or greater colonies of 5 at each site were called methylated by BiQ Analyzer. Shown are representative colonies.
The data presented here indicate that despite near absent PTCH1 mRNA expression and robust Shh signaling in a subset of primary human MBs, there is no difference in methylation profiles between MB and unaffected cerebellum samples. Furthermore, there appears to be little if any methylation in either group. Contrary to our hypothesis, this suggests that promoter methylation does not contribute to low PTCH1 mRNA levels in the cases examined. A recent study found that the PTCH1 promoter was methylated in ovarian tumors, but not in basocellular carcinomas, implying differential PTCH1 methylation as a contributing factor in tumorigenesis, depending on tumor type [19]. While we cannot rule out that rare cases of human MB are caused by PTCH1 promoter methylation, the complete absence of methylation in all four samples with elevated GLI1 and low PTCH1 argues against this as a common mechanism. Given that expression analysis of PTCH1 was performed in a region of the gene common to all transcript variants, we also cannot rule out the possibility of alternate or distal promoter methylation, although methylation silencing occurs most often within the immediate flanking region of the start site, particularly for genes with CpG islands in this region [20]. Exon 1B has 70% GC content in the CpG island encompassed in this study [21] and the most robust response to Shh signaling, re-iterating its pertinence as a strong candidate for initial investigation of PTCH1 methylation. Future studies may focus on methylation of Exon 1C, which also contains a CpG island and is sufficient to repress Shh pathway activity [18].
Further investigation into epigenetic control of MB remains important. Other than chromosomal abnormalities, genetic alterations in MB remain ill defined and the causative nature of the majority of cases, elusive. It remains possible that genes other than Patched which negatively regulate the Shh pathway, including NUMB [22], SUFU [23] and GSK3b [24] are methylated in a subset of MB. Importantly, these genes are becoming increasingly relevant in our understanding of multiple types of cancer. Identification of methylated genes which regulate pathways common to many cancers may prove to be key prognostic indicators and therapeutic targets in the near future.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, Russell TL, Ellenbogen RG, Bernstein ID, Beachy PA, Olson JM. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004;64:7794–800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 2.Gilbertson RJ. Medulloblastoma: signalling a change in treatment. The lancet oncology. 2004;5:209–18. doi: 10.1016/S1470-2045(04)01424-X. [DOI] [PubMed] [Google Scholar]
- 3.Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, Allen JC, Zagzag D, Olson JM, Curran T, Wetmore C, Biegel JA, Poggio T, Mukherjee S, Rifkin R, Califano A, Stolovitzky G, Louis DN, Mesirov JP, Lander ES, Golub TR. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–42. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 4.Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, James CD. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997;57:842–5. [PubMed] [Google Scholar]
- 5.Baylin SB. DNA methylation and gene silencing in cancer. Nature clinical practice. 2005;2(Suppl 1):S4–11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- 6.Lusher ME, Lindsey JC, Latif F, Pearson AD, Ellison DW, Clifford SC. Biallelic epigenetic inactivation of the RASSF1A tumor suppressor gene in medulloblastoma development. Cancer research. 2002;62:5906–11. [PubMed] [Google Scholar]
- 7.Ebinger M, Senf L, Wachowski O, Scheurlen W. Promoter methylation pattern of caspase-8, P16INK4A, MGMT, TIMP-3, and E-cadherin in medulloblastoma. Pathol Oncol Res. 2004;10:17–21. doi: 10.1007/BF02893403. [DOI] [PubMed] [Google Scholar]
- 8.Waha A, Waha A, Koch A, Meyer-Puttlitz B, Weggen S, Sorensen N, Tonn JC, Albrecht S, Goodyer CG, Berthold F, Wiestler OD, Pietsch T. Epigenetic silencing of the HIC-1 gene in human medulloblastomas. Journal of neuropathology and experimental neurology. 2003;62:1192–201. doi: 10.1093/jnen/62.11.1192. [DOI] [PubMed] [Google Scholar]
- 9.Lindsey JC, Anderton JA, Lusher ME, Clifford SC. Epigenetic events in medulloblastoma development. Neurosurgical focus. 2005;19:E10. doi: 10.3171/foc.2005.19.5.11. [DOI] [PubMed] [Google Scholar]
- 10.Lindsey JC, Lusher ME, Anderton JA, Bailey S, Gilbertson RJ, Pearson AD, Ellison DW, Clifford SC. Identification of tumour-specific epigenetic events in medulloblastoma development by hypermethylation profiling. Carcinogenesis. 2004;25:661–8. doi: 10.1093/carcin/bgh055. [DOI] [PubMed] [Google Scholar]
- 11.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nature reviews. 2006;6:107–16. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 12.Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, Chen JK, Cooper MK, Taipale J, Olson JM, Beachy PA. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science (New York, NY) 2002;297:1559–61. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 13.Hatton BA, Knoepfler PS, Kenney AM, Rowitch DH, de Alboran IM, Olson JM, Eisenman RN. N-myc Is an Essential Downstream Effector of Shh Signaling during both Normal and Neoplastic Cerebellar Growth. Cancer research. 2006;66:8655–61. doi: 10.1158/0008-5472.CAN-06-1621. [DOI] [PubMed] [Google Scholar]
- 14.Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, Wickramasinghe R, Scott MP, Wechsler-Reya RJ. Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7331–6. doi: 10.1073/pnas.0832317100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee J, Platt KA, Censullo P, Ruiz i Altaba A. Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Development (Cambridge, England) 1997;124:2537–52. doi: 10.1242/dev.124.13.2537. [DOI] [PubMed] [Google Scholar]
- 16.Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, Scott MP. Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes & development. 1996;10:301–12. doi: 10.1101/gad.10.3.301. [DOI] [PubMed] [Google Scholar]
- 17.Kogerman P, Krause D, Rahnama F, Kogerman L, Unden AB, Zaphiropoulos PG, Toftgard R. Alternative first exons of PTCH1 are differentially regulated in vivo and may confer different functions to the PTCH1 protein. Oncogene. 2002;21:6007–16. doi: 10.1038/sj.onc.1205865. [DOI] [PubMed] [Google Scholar]
- 18.Shimokawa T, Rahnama F, Zaphiropoulos PG. A novel first exon of the Patched1 gene is upregulated by Hedgehog signaling resulting in a protein with pathway inhibitory functions. FEBS letters. 2004;578:157–62. doi: 10.1016/j.febslet.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 19.Cretnik M, Musani V, Oreskovic S, Leovic D, Levanat S. The Patched gene is epigenetically regulated in ovarian dermoids and fibromas, but not in basocellular carcinomas. International journal of molecular medicine. 2007;19:875–83. [PubMed] [Google Scholar]
- 20.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. The New England journal of medicine. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 21.Agren M, Kogerman P, Kleman MI, Wessling M, Toftgard R. Expression of the PTCH1 tumor suppressor gene is regulated by alternative promoters and a single functional Gli-binding site. Gene. 2004;330:101–14. doi: 10.1016/j.gene.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 22.Di Marcotullio L, Ferretti E, Greco A, De Smaele E, Po A, Sico MA, Alimandi M, Giannini G, Maroder M, Screpanti I, Gulino A. Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nature cell biology. 2006;8:1415–23. doi: 10.1038/ncb1510. [DOI] [PubMed] [Google Scholar]
- 23.Lee Y, Kawagoe R, Sasai K, Li Y, Russell HR, Curran T, McKinnon PJ. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene. 2007 doi: 10.1038/sj.onc.1210467. [DOI] [PubMed] [Google Scholar]
- 24.Knoepfler PS, Kenney AM. Neural precursor cycling at sonic speed: N-Myc pedals, GSK-3 brakes. Cell cycle (Georgetown, Tex) 2006;5:47–52. doi: 10.4161/cc.5.1.2292. [DOI] [PubMed] [Google Scholar]
- 25.Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics (Oxford, England) 2005;21:4067–8. doi: 10.1093/bioinformatics/bti652. [DOI] [PubMed] [Google Scholar]