

Crystals of a selenomethionine-incorporated YphC–GDP complex have been grown using the hanging-drop vapour-diffusion method and polyethylene glycol as a precipitating agent.
Keywords: GTPase, EngA, YphC
Abstract
The Bacillus subtilis YphC gene encodes an essential GTPase thought to be involved in ribosome binding and whose protein product may represent a target for the development of a novel antibacterial agent. Sequence analysis reveals that YphC belongs to the EngA family of GTPases, which uniquely contain two adjacent GTP-binding domains. Crystals of a selenomethionine-incorporated YphC–GDP complex have been grown using the hanging-drop vapour-diffusion method and polyethylene glycol as a precipitating agent. The crystals belong to space group P212121, with unit-cell parameters a = 62.71, b = 65.05, c = 110.61 Å, and have one molecule in the asymmetric unit. Data sets at three different wavelengths were collected on a single crystal to 2.5 Å resolution at the Daresbury SRS in order to solve the structure by MAD. Ultimately, analysis of YphC in complex with GDP may allow a greater understanding of the EngA family of essential GTPases.
1. Introduction
Members of the GTPase superfamily are critical components of many signalling pathways, in which the cycling between ‘on’ (GTP bound), ‘off’ (GDP bound) and apo states plays an important role in regulatory processes including cell division, cell cycling, signal transduction, mRNA translation and hormone signalling (Bourne et al., 1991 ▶; Vetter & Wittinghofer, 2001 ▶). Despite low sequence identity across the GTPase superfamily, their structures share a common fold comprising of a central β-sheet flanked by α-helices, with the five regions (G1–G5) that show sequence similarity between the different GTPase subfamilies being involved in nucleotide binding (Leipe et al., 2002 ▶). In different GTPases the cycling between the GTP-bound ‘on’ state and the GDP-bound ‘off’ state following GTP hydrolysis has been seen to result in conformational changes in two distinct regions termed the switch I and switch II regions, which include motifs G2 and G3, respectively (Bourne et al., 1991 ▶; Knudsen et al., 2001 ▶).
Genome sequence data has suggested that bacteria possess 11 universally conserved GTPases, many of which have been proposed to interact with the ribosome (Caldon et al., 2001 ▶). Amongst these, the EngA family of GTPases are unique as they contain two GTPase domains joined by a variable-length acidic linker (Caldon et al., 2001 ▶; Hwang & Inouye, 2001 ▶). The EngA family are thought to act as a cellular messenger by forming interactions with the ribosome, with the overexpression of EngA in Escherichia coli restoring the growth of null mutants of an rRNA methyltransferase (RrmJ) which modifies the 23S rRNA in intact 50S ribosomal subunits (Tan et al., 2002 ▶). This family appears to be restricted to bacteria and a number of important parasites such as Plasmodium and Eimeria, but absent in man, yeast and fungi. Studies in Bacillus subtilis and Neisseria have shown that the EngA homologues in these organisms are essential for bacterial survival, with knockouts in the former displaying an increase in cell length, nucleoid condensation and abnormally curved cell shape (Morimoto et al., 2002 ▶; Mehr et al., 2000 ▶). The essentiality of EngA suggests that it might form a promising target for antimicrobial agents.
Structural studies of an EngA homologue in Thermotoga maritima (TmDer) have revealed a domain architecture in which the two GTPase domains flank a C-terminal domain which adopts a fold reminiscent of an RNA-binding KH-domain (Robinson et al., 2002 ▶). In addition, although not added during crystallization, a GDP nucleotide remained bound to the second GTPase domain, whilst in the first GTPase domain two phosphates could be observed whose positions are approximately equivalent to those expected for the β and γ phosphates of GTP. This has led to the suggestion that in the structure of TmDer the first GTPase domain mimics a GTP-bound form of the enzyme (Robinson et al., 2002 ▶). In order to better understand how this important family of GTPases facilitates their function when cycling between GTP- and GDP-bound states, we have cloned, overexpressed, purified, crystallized and collected a MAD data set to 2.5 Å resolution of the B. subtilis EngA homologue YphC in complex with GDP.
2. Materials and methods
2.1. Cloning and overexpression
The coding sequence of yphc was amplified from genomic DNA of the 168 strain of B. subtilis using Pwo DNA polymerase (Roche) and the primers ATGGGTAAACCTGTCGTAGCC (forward) and TTATTTTCTAGCTCTTGCAAATATTTTG (reverse). The resulting YphC gene was ligated into a pETBlue-1 vector using an AccepTor vector kit (Novagen), creating an expression vector pMAT1 which was subsequently extracted and transformed into Escherichia coli Tuner (DE3) (Novagen). In order to produce SeMet-incorporated YphC protein, the transformed E. coli Tuner was grown in LB minimum medium containing 10.5 g l−1 K2HPO4, 1 g l−1 (NH4)2PO4, 4.5 g l−1 KH2PO4, 0.5 g l−1 trisodium citrate·2H2O, 5 g l−1 glycerol, 0.5 g l−1 adenine, guanosine, thymine and uracil, 1 ml l−1 MgSO4·7H2O, 4 mg l−1 thiamine, 40 mg l−1 selenomethionine and 100 mg l−1 of the amino acids Lys, Phe and Thr in addition to 50 mg l−1 Ile, Leu and Val. Growth was carried out at 310 K with vigorous aeration until an OD600 of 0.6 was reached, at which point overexpression of YphC was induced by the addition of 1 mM IPTG; growth then continued at 310 K for 5 h, after which the cells were harvested by centrifugation at 4000g for 20 min at 277 K.
2.2. Purification
Cells containing the overexpressed SeMet-incorporated YphC were disrupted by sonication in buffer A (50 mM Tris–HCl pH 8.0) and the cell debris was removed by centrifugation at 43 000g for 10 min. Analysis of the soluble fraction by SDS–PAGE showed a large overexpression band corresponding to the expected molecular weight of YphC of approximately 48 kDa. The supernatant was collected and loaded onto a DEAE-Sepharose Fast Flow column (Amersham Biosciences) and YphC was eluted with a linear gradient of 0–0.5 M NaCl in buffer A. The fractions containing the highest concentration of YphC [estimated by the method of Bradford (1976 ▶) using the Bio-Rad protein-assay reagent] were combined and 4.0 M (NH4)2SO4 was added to a final concentration of 1.7 M, at which concentration YphC is soluble. The precipitated protein was then removed by centrifugation and the supernatant was loaded onto a Phenyl-ToyoPearl 650S (Tosoh) column and eluted with a reverse gradient of (NH4)2SO4 from 1.2 to 0 M in buffer A. The sample was subsequently subjected to gel filtration using a Hi-Load Superdex 200 column (Amersham Biosciences) equilibrated with 0.5 M NaCl in buffer A and eluted with the same buffer. Peak fractions containing YphC were concentrated to 15 mg ml−1 in a VivaSpin 10 000 Da molecular-weight cutoff concentrator and the buffer exchanged to 10 mM Tris–HCl pH 8.0, which contained no antioxidants. The purity of the SeMet protein was checked by SDS–PAGE and estimated to be over 95%.
2.3. Crystallization and preliminary X-ray analysis
Crystals of SeMet-incorporated YphC were grown using the hanging-drop vapour-diffusion technique, mixing 2.0 µl protein solution (15 mg ml−1 YphC in 10 mM Tris–HCl pH 8.0, 5 mM GDP and 5 mM MgCl2) with 2.0 µl reservoir solution at 290 K. Initial screening of crystallization conditions was conducted using Crystal Screen 1, Crystal Screen 2 and the PEG/Ion Screen (Hampton Research); the most promising hit was produced in PEG/Ion Screen solution No. 1 [0.2 M sodium fluoride and 20%(w/v) PEG 3350]. This condition was subsequently refined to achieve an optimal reservoir solution of 0.4 M sodium fluoride and 14%(w/v) PEG 3350, producing crystals which took approximately one week to grow. For data collection, crystals of the YphC–GDP complex were flash-cooled to 100 K in a cryoprotectant solution consisting of 0.4 M sodium fluoride, 16%(w/v) PEG 3350 and 20%(w/v) glycerol.
A multiple-wavelength anomalous diffraction (MAD) experiment was carried out on a single crystal of selenomethionine-incorporated YphC–GDP complex on station 10.1 at the Daresbury Synchrotron Radiation Source. The three wavelengths for the MAD experiment (peak, inflection and remote) were chosen near the selenium absorption edge based on the fluorescence absorption spectrum obtained from a frozen crystal at 100 K. For each wavelength, a total of 180 images were collected to 2.5 Å using a 1° oscillation width on a MAR CCD 165 detector (Fig. 1 ▶).
Figure 1.

A representative 1° oscillation image of data collected from a YphC–GDP complex crystal using a MAR CCD 165 detector at station 10.1 at the SRS Daresbury Laboratory. The edge of the image corresponds to a resolution of 2.3 Å.
3. Results and discussion
Analysis of the diffraction data using the autoindexing routine in MOSFLM (Leslie, 1992 ▶) showed that the crystals belong to a primitive orthorhombic space group, with unit-cell parameters a = 62.71, b = 65.05, c = 110.61 Å, α = β = γ = 90°, with a monomer in the asymmetric unit giving a V M value of 2.3 Å3 Da−1, which is within the range observed by Matthews for protein crystals (Matthews, 1977 ▶). The data were subsequently processed using the MOSFLM (Leslie, 1992 ▶) and SCALA (Collaborative Computational Project, Number 4, 1994 ▶) packages and analysis of the pattern of systematic absences is consistent with the correct space group being P212121. Data-collection and processing statistics are presented in Table 1 ▶. Given the quality of the derivative data and in order to minimize any potential bias, we have chosen to proceed with the structure determination using the MAD method. Ultimately, it is hoped that a complete solution of the YphC–GDP complex structure will lead to a better understanding of the EngA family and reveal conformational changes between the different nucleotide-bound forms of this important family of GTPases.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Data set | Peak (λ1) | Inflection (λ2) | Remote (λ3) |
|---|---|---|---|
| Wavelength (Å) | 0.9794 | 0.9797 | 1.0094 |
| Resolution (Å) | 30–2.8 (2.95–2.8) | 30–2.5 (2.65–2.5) | 30–2.8 (2.95–2.8) |
| Reflections measured | 70020 | 110152 | 80701 |
| Unique reflections | 11679 | 16253 | 11691 |
| Completeness (%) | 99.9 | 99.9 | 99.9 |
| Redundancy | 6.0 | 6.8 | 6.9 |
| I/σ(I) | 14.6 (5.6) | 17.8 (5.1) | 21.8 (8.2) |
| Rmerge† (%) | 0.13 (0.26) | 0.098 (0.32) | 0.081 (0.21) |
R
merge = 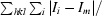
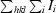 , where I
i and I
m are the observed intensity and mean intensity of related reflections, respectively.
, where I
i and I
m are the observed intensity and mean intensity of related reflections, respectively.
References
- Bourne, H. R., Sanders, D. A. & McCormick, F. (1991). Nature (London), 349, 117–127. [DOI] [PubMed] [Google Scholar]
- Bradford, M. M. (1976). Anal. Biochem.72, 248–254. [DOI] [PubMed] [Google Scholar]
- Caldon, C. E., Yoong, P. & March, P. E. (2001). Mol. Microbiol.41, 289–297. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Hwang, J. & Inouye, M. (2001). J. Biol. Chem.276, 31415–31421. [DOI] [PubMed] [Google Scholar]
- Knudsen, C., Wieden, H. J. & Rodnina, M. V. (2001). J. Biol. Chem.276, 22183–22190. [DOI] [PubMed] [Google Scholar]
- Leipe, D. D., Wolf, Y. I., Koonin, E. V. & Aravind, L. (2002). J. Mol. Biol.317, 41–72. [DOI] [PubMed] [Google Scholar]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Matthews, B. W. (1977). The Proteins, edited by H. Neurath & R. L. Hill, Vol. 3, pp. 468–477. New York: Academic Press.
- Mehr, I. J., Long, C. D., Serkin, C. D & Seifert, H. S. (2000). Genetics, 154, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto, T., Loh, P. C., Hirai, T., Asai, K., Kobayashi, K., Moriya, S. & Ogasawara, N. (2002). Microbiology, 148, 3539–3552. [DOI] [PubMed] [Google Scholar]
- Robinson, V. L., Hwang, J., Fox, E., Inouye, M. & Stock, A. M. (2002). Structure, 10, 1649–1658. [DOI] [PubMed] [Google Scholar]
- Tan, J., Jakob, U. & Bardwell, J. C. (2002). J. Bacteriol.184, 2692–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter, I. R. & Wittinghofer, A. (2001). Science, 294, 1299–1304. [DOI] [PubMed] [Google Scholar]